

Abstract
The mechanism of colistin-induced neurotoxicity is still unknown. Our recent study (L. Zhang, Y. H. Zhao, W. J. Ding, G. Z. Jiang, Z. Y. Lu, L. Li, J. L. Wang, J. Li, and J. C. Li, Antimicrob Agents Chemother 59:2189–2197, 2015, http://dx.doi.org/10.1128/AAC.04092-14; H. Jiang, J. C. Li, T. Zhou, C. H. Wang, H. Zhang, and H. Wang, Int J Mol Med 33:1298–1304, 2014, http://dx.doi.org/10.3892/ijmm.2014.1684) indicates that colistin induces autophagy and apoptosis in rat adrenal medulla PC-12 cells, and there is interplay between both cellular events. As an important cellular stress sensor, phosphoprotein p53 can trigger cell cycle arrest and apoptosis and regulate autophagy. The aim of the present study was to investigate the involvement of the p53 pathway in colistin-induced neurotoxicity in PC-12 cells. Specifically, cells were treated with colistin (125 μg/ml) in the absence and presence of a p53 inhibitor, pifithrin-α (PFT-α; 20 nM), for 12 h and 24 h, and the typical hallmarks of autophagy and apoptosis were examined by fluorescence/immunofluorescence microscopy and electron microscopy, real-time PCR, and Western blotting. The results indicate that colistin had a stimulatory effect on the expression levels of the target genes and proteins involved in autophagy and apoptosis, including LC3-II/I, p53, DRAM (damage-regulated autophagy modulator), PUMA (p53 upregulated modulator of apoptosis), Bax, p-AMPK (activated form of AMP-activated protein kinase), and caspase-3. In contrast, colistin appeared to have an inhibitory effect on the expression of p-mTOR (activated form of mammalian target of rapamycin), which is another target protein in autophagy. Importantly, analysis of the levels of p53 in the cells treated with colistin revealed an increase in nuclear p53 at 12 h and cytoplasmic p53 at 24 h. Pretreatment of colistin-treated cells with PFT-α inhibited autophagy and promoted colistin-induced apoptosis. This is the first study to demonstrate that colistin-induced autophagy and apoptosis are associated with the p53-mediated pathway.
INTRODUCTION
Colistin is used as a last-line treatment option against multidrug-resistant (MDR) Gram-negative bacteria, which can cause life-threatening infections (1 – 3). However, its clinical use is limited by potential nephrotoxicity and neurotoxicity (4, 5), the mechanisms of which are still unknown. It has been discovered in a mouse model and neuroblastoma 2a cells that autophagy is involved in colistin-induced nephrotoxicity (6, 7). Apoptosis and autophagy are two common forms of cell death (8, 9). Apoptosis is a prevalent form of programmed cell death (PCD) in multicellular organisms, which is the culmination of coordinately regulated intrinsic and extrinsic pathways involving major protein families, including the Bcl-2 family and caspases (10). Autophagy is a catabolic process of degradation and recycling of dysfunctional cellular components by lysosomal systems (11 – 13). Autophagy participates in organelle turnover and in the bioenergetic management of starvation stresses, pathogen infection, and hypoxia in order to maintain cellular homeostasis (14, 15). A number of stimuli can induce autophagy, apoptosis, or both, and recent studies suggest that autophagy delays or promotes apoptosis under certain conditions (16, 17). For example, treatment of cells with pemetrexed and simvastatin promoted autophagy and inhibited apoptosis (16), while oridonin phosphate induced autophagy and enhanced apoptotic cell death (17). However, the precise mechanisms that determine autophagy, apoptosis, and their interaction remain to be elucidated.
A number of studies in tumor cells (e.g., hepatocellular carcinoma and OVCAR-3 cancer cells) have shown that the p53 tumor suppressor protein, which is an important cellular stress sensor, can trigger cell cycle arrest and apoptosis and also regulate autophagy (18 – 20). Activation of p53 in response to a death stimulus leads to the transcription of genes involved in apoptosis, including PUMA (p53 upregulated modulator of apoptosis), AMPK (AMP-activated protein kinase), and Bax in the nucleus. These in turn activate the intrinsic mitochondrial apoptotic pathway in the cytoplasm via inhibition of the antiapoptotic proteins Bcl-2 and Bcl-XL (19, 20). In addition, p53 appears to play a dual role in the control of autophagy. At basal levels, p53 has an inhibitory effect, and its activation initiates the autophagic process (21, 22). Thus, p53-induced autophagy may either constitute a physiological cellular defense response (23) or contribute to cell death (24). The cellular localization of p53 appears to determine whether a cell will undergo autophagy or apoptosis. Nuclear p53 induces and regulates autophagy, while cytoplasmic p53 inhibits autophagy (22, 25).
Our recent studies have demonstrated that colistin induces autophagy and apoptosis in a tumor cell line, PC-12 cells (26, 27). Specifically, treatment with colistin (125 and 250 μg/ml) for 12 and 24 h stimulated the expression of LC3 (microtubule-associated protein 1 light chain 3), Beclin 1, caspase-3, and Bax (26, 27), and autophagy appeared to be protective against colistin-induced apoptosis. In the present study, the role of p53 in colistin-induced autophagy and apoptosis was examined in PC-12 cells.
MATERIALS AND METHODS
Reagents and compounds.
Fetal bovine serum (FBS) was purchased from Gibco BRL (Gaithersburg, MD). Colistin sulfate (20,195 U/mg; lot 095K1048; Sigma-Aldrich, St. Louis, MO, USA) was dissolved in distilled water. Pifithrin-α (PFT-α) (Gene Operation, USA) was prepared in dimethyl sulfoxide (DMSO). MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphnyl-2H-tetrazolium bromide] and DAPI (4′,6-diamidino-2-phenylindole) were obtained from Sigma-Aldrich. The Hoechst 33258 staining kit, lysis buffer, and an immunofluorescence staining kit with Alexa Fluor 488-labeled goat anti-rabbit IgG were purchased from Beyotime (Shanghai, China). A BCATM protein assay kit was obtained from Wuhan Boster Bio-engineering Limited Co. (Wuhan, Hubei, China). Primary antibodies against LC3-II/I and caspase-3 were purchased from Cell Signaling Technology (Beverly, MA). Primary antibodies against Bax, phospho-mTOR, and phospho-AMPKα1/2 were purchased from Immunoway Biotechnology Company (Newark, NJ, USA). Rabbit anti-rat DRAM (damage-regulated autophagy modulator), PUMA, and p53 were obtained from Santa Cruz Biotechnology (TX, USA). Anti-β-actin and horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG were purchased from Beijing Zhongshan Golden Bridge Biotechnology Co. Ltd. (Beijing, China).
Cell culture.
PC-12 cells were purchased from the Cell Bank of the Type Culture Collection, Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS at 37°C in a humidified atmosphere containing 5% CO2 (26).
MTT assay for cell viability determination.
PC-12 cells were seeded in 96-well plates at a density of 0.5 × 105 cells/well. Twenty-four hours after cell seeding, cells were either treated with increasing concentrations of PFT-α (0 to 80 nM) for 24 h or pretreated with PFT-α (20 nM) for 1 h, followed by colistin (125 μg/ml) treatment for 24 h (28). Cell viability was assessed by the MTT assay as described previously (26).
LC3 immunofluorescence microscopic examination.
Cells were seeded in 24-chamber culture slides and treated with 20 nM PFT-α for 1 h, followed by colistin treatment (125 μg/ml) for 12 h. Cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 10 min, and then blocked with 5% bovine serum albumin (BSA) for 1 h. Cells were subsequently incubated with the LC3 antibody (1:400 dilution) overnight, followed by the Alexa Fluor 488-labeled goat anti-rabbit secondary antibody and 2.5 μg/ml of DAPI solution for 20 min. The stained neurons were magnified 400× and examined using a Nikon Eclipse TE 2000U fluorescence microscope (28). Ten images were collected, and the percentage of cells positive for LC3 immunofluorescence was calculated by counting 100 cells for each experimental group (n = 3).
Hoechst 33258 staining for nuclear morphology.
Cells were seeded in 24-chamber culture slides and then either incubated with colistin (125 μg/ml) or PFT-α (20 nM) alone or preincubated with 20 nM PFT-α for 1 h, followed by treatment with colistin (125 μg/ml) for 12 h or 24 h. Subsequently, PC-12 cells were washed twice with PBS and then stained with Hoechst 33258, and fluorescence was visualized using a Nikon Eclipse TE 2000U (26). Ten images at ×400 magnification were collected, and the percentage of apoptotic cells was calculated by counting 100 cells for each experimental group (n = 3).
Electron microscopy for autophagy and apoptosis.
Cells were seeded in cell culture bottles (25 cm2) at a density of 1 × 107 cells/bottle and treated for 12 h or 24 h as described above. Cells were collected and fixed with 2.5% glutaraldehyde. Subsequently, the samples were postfixed in 1% osmium tetroxide at 4°C for 30 min, dehydrated by increasing concentrations of acetone, and embedded in epoxy resins. Ultrathin sections were contrasted with uranyl acetate and lead citrate (29). Samples were examined for autophagy and apoptosis with a GEM-1200ES transmission electron microscope (JEOL Ltd., Tokyo, Japan). Identification of autophagic vacuoles with characteristic double or multiple membranes intracellularly using conventional electron microscopy remains the gold standard for assessing autophagy in cultured cells (29). Chromatin concentration, edge accumulation, and cytoplasmic vacuolization are used to identify apoptotic cells.
Quantitative real-time PCR assay.
Total cellular RNA was isolated from the control and treated cells, and 1 mg was reverse transcribed into single-stranded cDNA using a Golden 1st-strand cDNA synthesis kit (HaiGene, Harbin). The levels of p53, DRAM, PUMA, and Bax mRNA were determined using real-time PCR with a SYBR premix Ex Taq kit (Perfect Real Time; TaKaRa) on an Applied Biosystems 7500 real-time PCR system thermocycler. The sequences of the oligonucleotide primers are shown in Table 1. The data obtained from real-time PCR were analyzed by the 2−ΔΔCT method (30).
TABLE 1.
Sequences of the oligonucleotide primers used for gene transcription analysis by real-time PCR
| Gene | Primer sequence | Product length (bp) |
|---|---|---|
| p53 | Forward, 5′-GCTGAGTATCTGGACGACAGG-3′ | 164 |
| Reverse, 5′-AGCGTGATGATGGTAAGGATG-3′ | ||
| DRAM | Forward, 5′-CAGCCTTCATCATCTCCTACG-3′ | 191 |
| Reverse, 5′-GGTCTCGTTCTGCTTCTCCA-3′ | ||
| PUMA | Forward, 5′-ACCTCAACGCGCAGTACGA-3′ | 143 |
| Reverse, 5′-CTAGTTGGGCTCCATTTCTGG-3′ | ||
| Bax | Forward, 5′-TGCCCGAGTTCTACTACAGACC-3′ | 125 |
| Reverse, 5′-AGAGTTTGCCTGAGACCCAAT-3′ | ||
| β-Actin | Forward, 5′-ACCGCAAATGCTTCTAAACC-3′ | 192 |
| Reverse, 5′-CCAATCTCGTCTTGTTTTATGC -3′ |
Western blotting.
Western blotting was carried out as described previously (26). Briefly, blots were blocked and then immunolabeled with primary antibodies for LC3 (1:1,000 dilution), caspase-3 (1:1,000 dilution), DRAM, PUMA, Bax, phospho-AMPKα1/2, phospho-mTOR, p53, and β-actin (1:1,000 dilution) at room temperature for 1.5 h. The membrane was subsequently incubated with HRP-conjugated secondary antibody (1:2,000 dilution) at room temperature for 2 h. An enhanced chemiluminescence (ECL) detection system was used to detect immunoblots, and the protein bands were analyzed by densitometry using Image J (version 1.42; National Institutes of Health, USA).
Statistical analysis.
Data were obtained from three or five independent experiments and are presented as means ± standard deviations (SD). Statistical analysis was conducted by one-way analysis of variance (ANOVA) using SPSS (version 13.0; SPSS, Chicago, IL, USA) to determine differences among experimental groups, with a P value of <0.05 being statistically significant.
RESULTS
Determination of cell viability.
To confirm the role of p53 in colistin-induced autophagy and apoptosis, a highly selective and potent synthetic p53 inhibitor, PFT-α, was used in this study. In order to select an appropriate dose for this compound, PC-12 cells were treated for 24 h with a range of PFT-α concentrations (0 to 80 nM), and cell viability was assessed via an MTT assay. As shown in Fig. 1A, cell viability decreased in a concentration-dependent manner, with significant cell death observed at concentrations greater than 20 nM PFT-α. Therefore, 20 nM was considered the optimal concentration of PFT-α for treating cells in subsequent experiments.
FIG 1.

Assessment of cell viability of PC-12 cells following drug treatment by MTT assay. (A) Effects of increasing concentrations of PFT-α (0 nM to 80 nM) after 12 h of exposure. (B) Effects of colistin (125 μg/ml), PFT-α (20 nM), and colistin (125 μg/ml) plus PFT-α (20 nM) at the indicated times. Error bars represent standard deviations (SD) of the means (n = 5). **, P < 0.01 for treated groups versus the untreated control group; ##, P < 0.01 for the intergroup comparison among the treated groups.
To determine whether PFT-α had an inhibitory effect on colistin-induced cell death in PC-12 cells, the cells were treated with either PFT-α alone, colistin alone (125 μg/ml), or colistin plus PFT-α for up to 24 h. As shown in Fig. 1B, significant differences in cell viability relative to the 0-h control group were observed after 6 h for cells treated with colistin only and after 12 h for cells treated with PFT-α only (P < 0.01). Pretreatment of cells with PFT-α did not have any significant effects on the loss of cell viability in colistin-treated cells. Thus, 20 nM PFT-α was not able to inhibit cell death induced by 125 μg/ml colistin in PC-12 cells.
Morphological analysis of colistin-induced autophagy and apoptosis in the presence of PFT-α.
We have previously observed that treatment of PC-12 cells with colistin resulted in apoptotic cell death, and this appears to be attenuated by autophagy (26). In the present study, the effects of PFT-α on apoptosis and autophagy in colistin-treated cells were investigated via fluorescence microscopy using an LC3 antibody to detect LC3-II/I in autophagosomes and Hoechst 33258 to assess nuclear morphology. Visualization of LC3 immunofluorescence revealed no detectable fluorescence in the untreated control cells and those treated with PFT-α alone for 12 h (Fig. 2A, control and PFT-α groups), indicating the absence of autophagic vacuoles from these cells. Quantitative analysis (Fig. 2B) revealed no significant difference in the frequency of autophagy between the two groups. In contrast, different sizes and stages of autophagic vacuoles were visible by immunofluorescence staining with the LC3 antibody after treatment with colistin for 12 h (Fig. 2A, colistin group), and the proportion of cells positive for LC3 was significantly higher than that in the control group (P < 0.01). In cells treated with colistin plus PFT-α, immunofluorescence was not readily observed (Fig. 2A, colistin plus PFT-α group), and the number of LC3 immunopositive cells was significantly less relative to the colistin-treated group (Fig. 2B). These results show that PFT-α was able to reduce colistin-induced autophagy.
FIG 2.

Morphological analysis of PFT-α on colistin-induced autophagy and apoptosis in PC-12 cells. (A) Fluorescence images of cells treated with colistin alone (125 μg/ml), PFT-α alone (20 nM), or PFT-α (20 nM) plus colistin (125 μg/ml) showing LC3 immunofluorescence (green) and DAPI staining (blue). The appearance of punctuated staining (marked by the white arrows) indicates autophagosome-associated LC3. (B) Proportion of cells (percentage of total) treated with colistin, PFT-α, or PFT-α plus colistin showing LC3 immunofluorescence. The bar graph represents the mean percentages ± SD of LC3 immunopositive cells (n = 3). **, P < 0.01 for treated groups versus the untreated control group; ##, P < 0.01 for intergroup comparisons among the treated groups. (C) Effect of drugs on the nuclear morphology of PC-12 cells for 24 h. Changes in nuclear morphology were observed by Hoechst 33258 staining. Arrows indicate apoptotic cells. (D) Proportion of cells (percentage of total) treated with either colistin, PFT-α, or PFT-α plus colistin showing apoptotic nuclei. The bar graph represents the mean percentage ± SD of apoptotic cells (n = 3). **, P < 0.01 for treated groups versus the untreated control group (12 h); ##, P < 0.01 for intergroup comparison among the treated groups (12 h); aa, P < 0.01 for treated groups versus the untreated control group (24 h); bb, P < 0.01 for intergroup comparison among the treated groups (24 h). (E) Electron microscopy images of the untreated control cells and those treated with colistin alone, PFT-α alone, or PFT-α plus colistin. Autophagosomes, chromatin condensation, and cytoplasmic vacuolization in the treated groups are marked with black, white, and yellow arrows, respectively. Changes in the organelles following treatment with either colistin or PFT-α plus colistin include dilation of chondriosome swelling and cytoplasmic vacuolization.
Analysis of nuclear morphology following drug treatment at 12 h and 24 h by Hoechst 33258 staining showed hypercondensed nuclei and chromatin condensation in colistin-treated cells (Fig. 2C), indicating that these cells underwent apoptotic cell death (31). Determination of the proportion of cells undergoing apoptosis (Fig. 2D) revealed at 12 h of treatment a significant difference between the colistin-treated and control groups (P < 0.01) but not between the PFT-α-treated and control groups. Treatment with colistin plus PFT-α at 12 h and 24 h appeared to increase the proportion of apoptotic cells relative to the corresponding colistin-treated group, suggesting that PFT-α augmented colistin-induced apoptosis.
In addition to fluorescence microscopy, electron microscopy was employed to further characterize the morphological changes induced by colistin in the absence and presence of PFT-α in PC-12 cells (29, 32). As shown in Fig. 2E, different sizes and stages of autophagic vacuoles were evident in cells treated with colistin for 12 h, and these vacuoles predominantly contained cytoplasmic organelles and autophagosomes. A further 12-h treatment with colistin resulted in nuclear chromatin condensation, edge accumulation, and cytoplasmic vacuolization. Similar observations were obtained for the group treated with colistin plus PFT-α for 24 h, indicating that PFT-α had no significant effects on the cellular changes induced by colistin at this time point. In cells treated with PFT-α alone for 12 h and 24 h, and also in cells treated with colistin plus PFT-α for 12 h, autophagosomes and nuclear chromatin condensation were not apparent, but dilation of chondriosome swelling was evident.
Real-time PCR assay and Western blotting of key biomarkers in the p53 pathway and autophagy.
Previous studies have shown that p53 mediates PC-12 cell apoptosis by promoting the expression of caspase-3 (27) and autophagy-mediated apoptosis through activation of the mTOR/AMP kinase pathway and DRAM expression (19, 33 – 35). In order to investigate the role of p53 in colistin-induced apoptosis, the expression levels of p53 and its target genes and proteins were examined by real-time PCR and Western blotting, respectively. Analysis of mRNA levels of p53, DRAM, PUMA, and Bax showed elevated expression of them after 12 h of treatment of cells with colistin relative to the untreated control group, with the highest expression levels detected at 24 h (Fig. 3A, a to d). Pretreatment of colistin-treated cells with PFT-α significantly decreased the mRNA expression levels of these genes at 12 h. PFT-α alone had no detectable effects on mRNA levels at both 12 h and 24 h relative to the control group.
FIG 3.

Real-time PCR assay and Western blotting of key biomarkers in the p53 pathway and autophagy. (A) Detection of p53, DRAM, PUMA, and Bax mRNA levels by real-time PCR. The bar graph represents the mean percentages ± SD of apoptotic cells (n = 3). **, P < 0.01 for treated groups versus the untreated control group; ##, P < 0.01 for intergroup comparison among the treated groups. (B) Western blotting of LC3, caspase-3, cleaved caspase-3, DRAM, PUMA, Bax, p-AMPK, and p-mTOR. Samples containing 30 μg protein were loaded onto 15% (for LC3), 12% (for caspase-3 and cleaved caspase-3), or 10% (for DRAM, PUMA, Bax, p-mTOR, and p-AMPK) SDS-PAGE gels. β-Actin was used as a loading control. (a) Blots showing protein levels. (b to h) Quantitative data of protein levels. The bar graph represents the mean percentages ± SD of protein expression levels (n = 3). **, P < 0.01 for treated groups versus the untreated control group; ##, P < 0.01 for colistin only versus PFT-α plus colistin. (C) Blots showing levels of nuclear p53 and cytoplasmic p53. Detection was carried out using 10% SDS-PAGE gels, and β-actin was used as a loading control. (b and c) Quantitative data of p53 levels in each cellular fraction. The bar graph represents the mean percentages ± SD of protein expression levels (n = 3). **, P < 0.01 for treated groups versus the untreated control group; ##, P < 0.01 for colistin only versus PFT-α plus colistin.
Western blotting revealed that colistin alone induced a significant increase in the expression levels of DRAM, PUMA, Bax, and phospho-AMPK (p-AMPK), but not p-phospho-mTOR (mTOR), which was suppressed, relative to the untreated control group after 12 h of treatment, while PFT-α pretreatment of colistin-treated cells had the opposite effect on protein expression at the same time point (Fig. 3B). In the PFT-α-treated group, the levels of all target proteins were comparable to those in the control group, indicating that PFT-α alone had no major effects on the expression of these proteins.
The levels of nuclear and cytoplasmic p53 were also analyzed by Western blotting following different treatments. As shown in Fig. 3C, colistin induced the highest expression of nuclear p53 after 12 h of treatment. On the contrary, colistin induced the highest expression of cytoplasmic p53 at 24 h (Fig. 3C, b). At both 12 and 24 h, levels of nuclear and cytoplasmic p53 in the PFT-α group were similar to those in the control group. In the colistin plus PFT-α group, PFT-α pretreatment significantly decreased the expression of p53 proteins relative to the colistin group in both nuclei and cytoplasm (Fig. 3C).
DISCUSSION
As a tumor suppressor gene, p53 is a major checkpoint that plays a critical role in mammalian cells (36). Under normal circumstances, p53 is maintained at minimal levels in cells (37). Intracellular genotoxic stresses (e.g., oncogene activation, DNA damage, and hypoxia) activate p53, which in turn initiates cellular tumor suppression programs, including cell cycle arrest or apoptosis (37). In addition, p53 has been shown to play a critical role in autophagy modulation (38). Upon activation, autophagy may promote or inhibit apoptosis depending on the level of the insult, and the interplay between these two cellular processes has been the subject of intensive relationship (17, 39). Associated with colistin therapy, nephrotoxicity and neurotoxicity remain poorly characterized adverse effects. In terms of the relationship of colistin-induced neurotoxicity and p53, Dai et al. reported that the gene expression of p53, Bax, and caspase-8 significantly increased in the model of colistin-induced neurotoxicity using the mouse neuroblastoma 2a (N2a) cell line (all P values of < 0.01) (7). A recent study has shown that p53 plays a pathological role in the progression of colistin-induced apoptosis in PC-12 cells (27). We have further shown that colistin treatment also induces autophagy under these conditions (26). However, whether p53 mediates colistin-induced autophagy and apoptosis in these cells remains to be determined. In order to address this and elucidate the possible mechanism(s) involved in initiating these events, we used a highly selective and potent synthetic p53 inhibitor, pifithrin-α (PFT-α) (40), in the present study. In order to elucidate the possible role of p53 in the colistin-induced autophagy and apoptosis in PC-12 cells, LC3 immunofluorescence and downstream apoptotic events (caspase-3 activation and nuclear condensation) were examined. As depicted in Fig. 2C and D and 3B, the data indicate that PFT-α was able to inhibit the cellular events of p53-mediated autophagy induced by colistin, which appears to increase the propensity of cells to undergo apoptosis.
As a transcription factor, p53 primarily induces the expression of its target genes, including DRAM, PUMA, and Bax. DRAM is an inducer of autophagy-mediated apoptosis, and localization of DRAM in the mitochondria leads to autophagy (41). As an effector of p53-mediated apoptosis, DRAM is a p53 target gene inducing macroautophagy (34). As a Bcl-2 homology 3 (BH3)-only family member, PUMA has been implicated as a p53-upregulated modulator of apoptosis (42). It activates Bax, which in turn initiates caspase-3 activation via the intrinsic mitochondrial apoptotic pathway, and this has been shown to be involved in neuronal apoptosis (43, 44). Hence, the p53-PUMA-Bax system has been postulated to be a classic signal for the induction of cell death via apoptosis (45). The relationship between the p53 signaling pathway and autophagy was established by the discovery of DRAM and PUMA (34, 46). In the present study, colistin appeared to induce the p53 signaling pathway based on the upregulation of DRAM, PUMA, and Bax expression. These changes were in line with the LC3 immunofluorescence data (Fig. 2A and B) and activation of caspase-3 at 12 h and 24 h (Fig. 3B), suggesting that DRAM, PUMA, and Bax play their respective roles in colistin-induced autophagy and apoptosis.
It is known that autophagy induced by p53 is associated with activation of AMPK (AMP-activated protein kinase) and inhibition of mTOR (mammalian target of rapamycin), an autophagy suppressor (21, 22, 47 – 49). To determine whether the p53-AMPK-mTOR pathway is involved in colistin-induced autophagy, the levels of the phosphorylated forms of AMPK and mTOR (activated forms) were examined by Western blotting. As depicted in Fig. 3B, the levels of phospho-AMPK (p-AMPK) and phospho-mTOR (p-mTOR) indicate that the p53-AMPK-mTOR pathway is involved in colistin-induced autophagy. It can be speculated that the increase in p-AMPK levels leads to the activation of p53, which can then inactivate p-mTOR through binding of the TSC1/TSC2 complex to the protein and initiate autophagy.
Recent studies have shown that p53 has a dual role in modulating autophagy based on its localization in the cell. Nuclear p53 facilitates autophagy by transactivating its target genes, while cytoplasmic p53 inhibits autophagy via protein-protein interactions in the mitochondria. In Fig. 2C and 3, the majority of p53 had translocated to the nucleus at 12 h to transactivate its target genes and cytoplasmic p53 also increased after 12 h in cells treated with colistin, although a further increase was observed after 24 h, when the majority of cells underwent apoptosis. Since this is the time when PUMA, Bax, and caspase-3 were maximally expressed (Fig. 3A and B), it is highly probable that cytoplasmic p53, which is normally held inactive through its binding to BCL-XL, is released by the inhibition of BCL-XL by PUMA, and its accumulation induces Bax oligomerization and mitochondrial translocation to activate the mitochondrial apoptotic pathway. In this case, p53 may interact with Bax at the mitochondria, which in turn leads to the activation of caspase-3. A summary scheme of the present study is displayed in Fig. 4.
FIG 4.
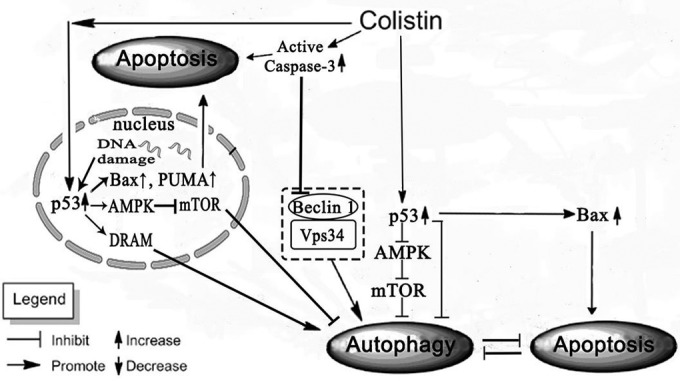
Schematic diagram of the proposed mechanism of p53 mediating colistin-induced autophagy and apoptosis in PC-12 cells.
In general, our data indicate that p53 plays a key role in colistin-induced autophagy and apoptosis in PC-12 cells. Specifically, inhibition of p53 by PFT-α was able to attenuate autophagy after 12 h of treatment with colistin, and this seems to promote apoptosis. Autophagy is usually apparent well before cells undergo complete demise as a stress response to cellular insult, and depending on the level of the insult, it can have an inhibitory effect on apoptosis, especially when the cellular stress has not reached a critical level that signals the point of no return (50). It is highly likely that inhibition of autophagy via the upstream p53 pathway increases the tendency of cells to activate the apoptotic pathway. The intracellular localization of p53 seems to play an important role in mediating this event, since an increase in the level of cytoplasmic p53 can facilitate the downstream apoptotic events via the mitochondria, which is a major checkpoint that determines whether or not cells undergo apoptosis. Further studies are warranted to elucidate the specific mechanisms of cellular localization of p53 in colistin-induced cytotoxicity in neuronal cells.
Conclusions.
This is the first study to demonstrate that colistin-induced autophagy and apoptosis are associated with the p53 regulatory network. The tendencies of colistin-induced autophagy and apoptosis may be mediated by the intracellular localization of p53. Our study provides important mechanistic insights into understanding the effect of colistin on mammalian cells.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (grants 31272613 and 31472240) and the Natural Science Fund of Heilongjiang Province (grant C201424).
We declare that we do not have any conflict of interest related to this work.
REFERENCES
- 1.Talbot GH, Bradley J, Edwards JE, Gilbert D, Scheld M, Bartlett JG. 2006. Bad bugs need drugs: an update on the development pipeline from the antimicrobial availability task force of the Infectious Diseases Society of America. Clin Infect Dis 42:657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 2.Cho YS, Yim H, Yang HT, Hur J, Chun W, Kim JH, Lee BC, Seo DK, Park JM, Kim D. 2012. Use of parenteral colistin for the treatment of multiresistant Gram-negative organisms in major burn patients in South Korea. Infection 40:27–33. doi: 10.1007/s15010-011-0192-7. [DOI] [PubMed] [Google Scholar]
- 3.Loho T, Dharmayanti A. 2015. Colistin: an antibiotic and its role in multiresistant Gram-negative infections. Acta Med Indones 47:157–168. [PubMed] [Google Scholar]
- 4.Sourli L, Luque S, Grau S, Berenguer N, Segura C, Montero MM, Alvarez-Lerma F, Knobel H, Benito N, Horcajada JP. 2013. Trough colistin plasma level is an independent risk factor for nephrotoxicity; a prospective observational cohort study. BMC Infect Dis 13:380. doi: 10.1186/1471-2334-13-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falagas EM, Kasiakou SK. 2005. Colistin: the revival of polymyxins for the management of multidrug resistant Gram-negative bacterial infections. Clin Infect Dis 40:1333–1341. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]
- 6.Dai CS, Li JC, Tang SS, Li J, Xiao XL. 2014. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob Agents Chemother 58:4075–4085. doi: 10.1128/AAC.00070-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai C, Tang S, Velkov T, Xiao X. 28 August 2015. Colistin-induced apoptosis of neuroblastoma-2a cells involves the generation of reactive oxygen species, mitochondrial dysfunction, and autophagy. Mol Neurobiol; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leist M, Jäättelä M. 2001. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 9.Bursch W. 2001. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ 8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 10.Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, Los M. 2009. Apoptosis and cancer: mutations within caspase genes. J Med Genet 46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]
- 11.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. 2010. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klionsky DJ, Emr SD. 2000. Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gozuacik D, Kimchi A. 2004. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 14.Notte A, Leclere L, Michiels C. 2011. Autophagy as a mediator of chemotherapy induced cell death in cancer. Biochem Pharmacol 82:427–434. doi: 10.1016/j.bcp.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. 2008. Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang KE, Kim YS, Jung JW, Kwon SJ, Park DS, Cha BK, Oh SH, Yoon KH, Jeong ET, Kim HR. 2015. Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. Oncotarget 6:29482–29496. doi: 10.18632/oncotarget.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Wang Y, Wang S, Gao Y, Zhang X, Lu C. 2015. Oridonin phosphate-induced autophagy effectively enhances cell apoptosis of human breast cancer cells. Med Oncol 32:365. doi: 10.1007/s12032-014-0365-1. [DOI] [PubMed] [Google Scholar]
- 18.Bensaad K, Vousden KH. 2007. p53: new roles in metabolism. Trends Cell Biol 17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. 2008. A dual role of p53 in the control of autophagy. Autophagy 4:810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 20.Ghavami S, Mutawe MM, Hauff K, Stelmack GL, Schaafsma D, Sharma P, McNeill KD, Hynes TS, Kung SK, Unruh H, Klonisch T, Hatch GM, Los M, Halayko AJ. 2010. Statin-triggered cell death in primary human lung mesenchymal cells involves p53-PUMA and release of Smac and Omi but not cytochrome c. Biochim Biophys Acta 1803:452–467. doi: 10.1016/j.bbamcr.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Beth L, John A. 2008. p53: the Janus of autophagy? Nat Cell Biol 10:637–639. doi: 10.1038/ncb0608-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tasdemir E, Maiuri CM, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu CL, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. 2008. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. 2007. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Investig 117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crighton D, Wilkinson S, Ryan KM. 2007. DRAM links autophagy to p53 and programmed cell death. Autophagy 3:72–74. doi: 10.4161/auto.3438. [DOI] [PubMed] [Google Scholar]
- 25.Feng Z, Zhang H, Levine AJ, Jin S. 2005. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Zhao YH, Ding WJ, Jiang GZ, Lu ZY, Li L, Wang JL, Li J, Li JC. 2015. Autophagy regulates colistin-induced apoptosis in PC-12 cells. Antimicrob Agents Chemother 59:2189–2197. doi: 10.1128/AAC.04092-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang H, Li JC, Zhou T, Wang CH, Zhang H, Wang H. 2014. Colistin-induced apoptosis in PC12 cells: involvement of the mitochondrial apoptotic and death receptor pathways. Int J Mol Med 33:1298–1304. doi: 10.3892/ijmm.2014.1684. [DOI] [PubMed] [Google Scholar]
- 28.Syu-ichi K, Kaori, Ayako T, Shin Y, Masaaki I. 2015. Pifithrin-alpha has a p53-independent cytoprotective effect on docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma HepG2 cells. Toxicol Lett 232:393–402. doi: 10.1016/j.toxlet.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 29.Swanlund JM, Kregel KC, Oberley TD. 2010. Investigating autophagy: quantitative morphometric analysis using electron microscopy. Autophagy 6:270–277. doi: 10.4161/auto.6.2.10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Zhu BS, Xing CG, Lin F, Fan XQ, Zhao K, Qin ZH. 2011. Blocking NF-κB nuclear translocation leads to p53-related autophagy activation and cell apoptosis. World J Gastroenterol 17:478–487. doi: 10.3748/wjg.v17.i4.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Pizzol Júnior JP, Sasso-Cerri E, Cerri PS. 2015. Apoptosis and reduced microvascular density of the lamina propria during tooth eruption in rats. J Anat 227:487–496. doi: 10.1111/joa.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, Zhang J, Zhu X, Wang P, Wang X, Li D. 2015. Progesterone reduces inflammation and apoptosis in neonatal rats with hypoxic ischemic brain damage through the PI3K/Akt pathway. Int J Clin Exp Med 8:8197–8203. [PMC free article] [PubMed] [Google Scholar]
- 34.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. 2006. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 35.Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. 2009. Stimulation of autophagy by the p53 target gene sestrin2. Cell Cycle 8:1571–1576. doi: 10.4161/cc.8.10.8498. [DOI] [PubMed] [Google Scholar]
- 36.Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 37.Jin S, Levine AJ. 2001. The p53 functional circuit. J Cell Sci 114(Part 23):4139–4140. [DOI] [PubMed] [Google Scholar]
- 38.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. 2014. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang W, Zhang X, Hao J, Shen J, Fang J, Dong W, Wang D, Zhang X, Shui W, Luo Y, Lin L, Qiu Q, Liu B, Hu Z. 2014. SIRT1 protects against apoptosis by promoting autophagy in degenerative human disc nucleus pulposus cells. Sci Rep 4:7456. doi: 10.1038/srep07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gu ZT, Li L, Wu F, Zhao P, Yang H, Liu YS, Geng Y, Zhao M, Su L. 2015. Heat stress induced apoptosis is triggered by transcription independent p53, Ca2+ dyshomeostasis and the subsequent Bax mitochondrial translocation. Sci Rep 5:11497. doi: 10.1038/srep11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu K, Lou J, Wen T, Yin J, Xu B, Ding W, Wang A, Liu D, Zhang C, Chen D, Li N. 2013. Depending on the stage of hepatosteatosis, p53 causes apoptosis primarily through either DRAM-induced autophagy or BAX. Liver Int 33:1566–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valente LJ, Grabow S, Vandenberg CJ, Strasser A, Janic A. 7 December 2015. Combined loss of PUMA and p21 accelerates c-MYC-driven lymphoma development considerably less than loss of one allele of p53. Oncogene doi: 10.1038/onc.2015.457. [DOI] [PubMed] [Google Scholar]
- 43.Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. 2010. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci 30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh AP, Walls KC, Klocke BJ, Toms R, Strasser A, Roth KA. 2009. The proapoptotic BH3-only, Bcl-2 family member, Puma is critical for acute ethanol-induced neuronal apoptosis. J Neuropathol Exp Neurol 68:747–756. doi: 10.1097/NEN.0b013e3181a9d524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skírnisdóttir I, Seidal T. 2012. The apoptosis regulators p53, bax and PUMA: relationship and impact on outcome in early stage (FIGO I-II) ovarian carcinoma after post-surgical taxane-based treatment. Oncol Rep 27:741–747. doi: 10.3892/or.2011. [DOI] [PubMed] [Google Scholar]
- 46.Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. 2009. PUMA and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 16:1135–1145. doi: 10.1038/cdd.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. 2010. Autophagy regulation by p53. Curr Opin Cell Biol 22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Kim J, Kundu M, Viollet B, Guan KL. 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mihaylova MM, Shaw RJ. 2011. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. 2007. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]