

Abstract
Hospitalized patients with severe influenza are at significant risk for morbidity and mortality. MHAA4549A is a human monoclonal immunoglobulin (Ig) G1 antibody that binds to a highly conserved stalk region of the influenza A virus hemagglutinin protein and neutralizes all tested seasonal human influenza A virus strains. Two phase 1 trials examined the safety, tolerability, and pharmacokinetics of MHAA4549A in healthy volunteers. Both single ascending-dose trials were randomized, double blinded, and placebo controlled. Trial 1 randomized 21 healthy adults into four cohorts receiving a single intravenous dose of 1.5, 5, 15, or 45 mg/kg MHAA4549A or placebo. Trial 2 randomized 14 healthy adults into two cohorts receiving a single intravenous fixed dose of 8,400 mg or 10,800 mg of MHAA4549A or placebo. Subjects were followed for 120 days after dosing. No subject was discontinued in either trial, and no serious adverse events were reported. The most common adverse event in both studies was mild headache (trial 1, 4/16 subjects receiving MHAA4549A and 1/5 receiving placebo; trial 2, 4/8 subjects receiving MHAA4549A and 2/6 receiving placebo). MHAA4549A produced no relevant time- or dose-related changes in laboratory values or vital signs compared to those with placebo. No subjects developed an antitherapeutic antibody response following MHAA4549A administration. MHAA4549A showed linear serum pharmacokinetics, with a mean half-life of 22.5 to 23.7 days. MHAA4549A is safe and well tolerated in healthy volunteers up to a single intravenous dose of 10,800 mg and demonstrates linear serum pharmacokinetics consistent with those of a human IgG1 antibody lacking known endogenous targets in humans. (These trials have been registered at ClinicalTrials.gov under registration no. NCT01877785 and NCT02284607).
INTRODUCTION
Influenza viruses propagate within the respiratory tract and can cause annual epidemics during autumn and winter. Classic symptoms include chills, fever, dry cough, muscular aches, and malaise. Most infected individuals recover from influenza virus infections without requiring medical attention; however, influenza poses the highest risk of hospitalization and secondary complications in young children, the elderly, pregnant women, patients with underlying chronic medical conditions, and the immunocompromised (1). Influenza can also cause serious secondary complications requiring hospitalization, such as pneumonia, leading to acute respiratory failure, secondary bacterial respiratory infections, and death. Each year, seasonal influenza results in approximately three to five million cases of severe illness and up to 500,000 deaths worldwide (2). Population-based surveillance data from 2010 to 2013 shows that influenza causes an estimated 4,900 to 27,000 deaths and 114,000 to 624,000 hospitalizations per year in the United States (3, 4). Neuraminidase inhibitors are typically prescribed for patients with influenza virus infection; however, the greatest clinical benefit from these agents occurs within 48 h of onset of symptoms (5). Although there are no approved pharmacological treatments for hospitalized influenza patients, a recent meta-analysis of individual participants from the 2009-2010 influenza A H1N1 virus pandemic suggests that in hospitalized patients, neuraminidase inhibitors significantly reduced mortality in adults by 62% if started within 48 h of symptom onset (6).
Currently, there is a significant unmet medical need in the severely ill hospitalized patient population, as evidenced by the considerable degree of morbidity and mortality in this setting. To address this need, MHAA4549A, a human monoclonal immunoglobulin (Ig) G1 antibody, is being developed as a treatment for hospitalized patients with severe influenza. MHAA4549A was cloned from a single human plasmablast cell isolated from an influenza virus-vaccinated donor (7). This monoclonal antibody can neutralize all tested seasonal human influenza A virus strains by two complementary mechanisms of action. First, MHAA4549A binds hemagglutinin on viral particles, thereby preventing hemagglutinin maturation and blocking hemagglutinin-mediated membrane fusion in the endosome (8). Second, by binding to hemagglutinin on the surface of influenza virus-infected cells, MHAA4549A induces natural killer (NK) cells to lyse influenza virus-infected cells through antibody-dependent cell-mediated cytotoxicity (L. Kamen and E. Kho, unpublished data). In vivo, efficacy of MHAA4549A was demonstrated in mouse models of influenza A virus infection, both as a single agent and in combination with oseltamivir (7). In the absence of influenza virus infection, the MHAA4549A-targeted epitope on the human influenza A virus hemagglutinin glycoprotein is not endogenously expressed in human tissue. As a result, the antibody does not have any known host targets. This paper reports results from two phase 1 studies that assessed the safety, tolerability, and pharmacokinetics (PK) of MHAA4549A in healthy volunteers.
MATERIALS AND METHODS
Participants.
Subjects were required to be healthy adults, 18 years of age or older, with a body mass index (BMI) of ≥18.0 and ≤32.0 kg/m2, weight of 40 to 100 kg, and in good health, as determined by the absence of clinically significant findings from medical history, electrocardiogram, clinical laboratory assessments, and vital sign measurements. Subjects were excluded under the following conditions: if they had a history or clinically significant manifestations of metabolic, hepatic, renal, hematologic, immunological, pulmonary, cardiovascular, gastrointestinal, urologic, neurologic, or psychiatric disorders; if they had a history of anaphylaxis, hypersensitivity, or drug allergies; or if they were current tobacco smokers. Female subjects were excluded if they had a positive pregnancy test result at screening or were breastfeeding during the study. Subjects were also excluded if they had participated in a clinical trial within 4 weeks before initiation of dosing on day 1, had used any experimental small-molecule therapy within the 4 weeks prior to initiation of dosing on day 1 or biologic therapy within the 12 weeks prior to or within five half-lives (t1/2s) of the product, or received any vaccine within 14 days prior to screening.
Study design.
Both trial 1 and trial 2 were phase 1, randomized, double-blind, placebo-controlled, single ascending-dose studies that evaluated the safety, tolerability, and pharmacokinetics of MHAA4549A in healthy volunteers who were enrolled at a single site in a clinic environment in Québec, Canada. Trial 1 was the entry-into-human study, and trial 2 assessed higher doses of MHAA4549A.
Trial 1 consisted of four cohorts (A to D) with at least 16 subjects receiving MHAA4549A (Genentech, Inc., South San Francisco, CA) (Fig. 1). Trial 2 consisted of two cohorts (A and B) with at least 8 subjects receiving MHAA4549A. Patients were allocated to MHAA4549A and placebo in ratios of 4:2 in cohort A and 4:1 in cohorts B to D in trial 1 and in ratios of 4:4 in cohort A and 4:2 in cohort B in trial 2, according to randomization schedules generated by the SAS PLAN procedure (SAS Institute, Inc., Cary, NC). Both the site investigator and the subject were blinded to the treatment assignment until the study was completed. Sponsor-selected staff were also blinded to study drug assignment throughout the trial to facilitate ongoing monitoring of safety and tolerability. Designated personnel at the site prepared and generated the study medications and were aware of the randomization codes.
FIG 1.
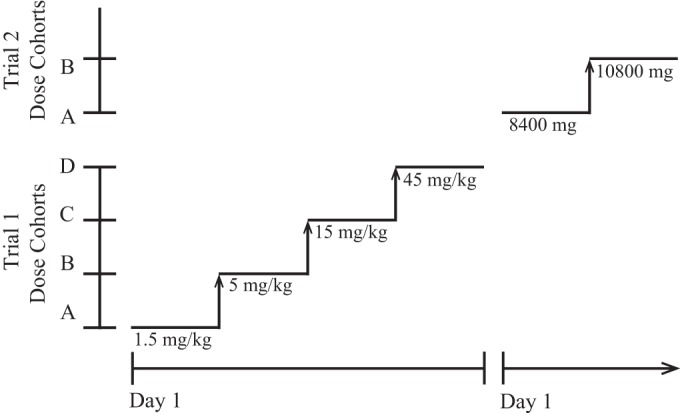
Study design.
Prior to entering the trial, subjects had a screening visit to establish eligibility within 35 days before study drug administration. Subjects were confined to the research facility from day −1 prior to drug administration on day 1 until at least 24 h postdose (day 2). Return visits to assess safety and pharmacokinetic outcomes were scheduled for days 4 (±1 day), 8 (±1 day), 15 (±2 days), 29 (±3 days), 57 (±7 days), 85 (±7 days), and 120 (±7 days).
As previously described (7), MHAA4549A was isolated from a single influenza virus-vaccinated donor and screened by an enzyme-linked immunosorbent assay (ELISA) for its ability to bind hemagglutinin subtypes and neutralize all human influenza A virus isolates tested. MHAA4549A is a recombinant antibody that was expressed in suspension cultures of Chinese hamster ovary (CHO) cells genetically engineered to synthesize the antibody. After the protein was manufactured in bioreactors, the supernatant containing the antibody was separated from the CHO cells by centrifugation, and filtration and was subsequently purified using affinity and ion exchange chromatography. The antibody was then formulated using ultrafiltration and diafiltration with steps to inactivate and remove potential adventitious viruses.
MHAA4549A (50 mg/ml in placebo solution) and placebo (10 mM sodium succinate, 240 mM sucrose, 0.02% [wt/vol] polysorbate 20, pH 5.5) were diluted in 0.9% normal saline and administered as single intravenous (i.v.) infusions over approximately 1 h in trial 1. In trial 2, the infusion time was approximately 2 h. Subjects in trial 1 received a single weight-based i.v. dose of either MHAA4549A or placebo for each cohort as follows: A, 1.5 mg/kg; B, 5 mg/kg; C, 15 mg/kg; and D, 45 mg/kg. Subjects in trial 2 received a single fixed i.v. dose of either MHAA4549A or placebo for each cohort as follows: A, 8,400 mg (∼105 mg/kg); B, 10,800 mg (∼135 mg/kg).
The clinical study protocols, any relevant associated documents, and informed consent forms were reviewed and approved by an institutional review board prior to subject screening and enrollment. All patients provided written, informed consent. All clinical work was conducted in compliance with Good Clinical Practices as referenced in the International Conference on Harmonization guidelines (ICH E6), Good Laboratory Practices as referenced in the ICH guidelines, local regulatory requirements, and the recommendations laid down in the most recent version of the Declaration of Helsinki. These trials were registered at ClinicalTrials.gov under registration numbers NCT01877785 and NCT02284607.
Safety assessments.
Safety assessments, including repeated measures of laboratory testing, vital signs, electrocardiograms (ECGs), physical examinations, and adverse events (AEs), were performed before, during, and following study drug infusion up to day 120. Subjects were confined at the clinical research facility from day −1 through day 2 (at least 24 h after dosing). AEs were assessed every 30 min (±15 min) until infusion was completed. After infusion, AEs were assessed at 30, 60, and 120 min (±15 min) and at regular intervals throughout the confinement period and at each subsequent study visit.
For both trials, an internal safety monitoring committee reviewed all available safety data in order to make dose escalation decisions, in consultation with the principal investigator. Safety data reviewed for dose escalations included AEs, ECGs, vital signs, and clinical laboratory results from all previously dosed subjects through day 8. AEs were graded according to the following categories: mild, moderate, or severe. Treatment-emergent adverse events (TEAEs) were defined as AEs that occurred on or after the date and time of the start of drug infusion. TEAEs were captured until study completion at approximately 120 days following study drug administration. Any serious adverse events (SAEs) that occurred were also collected and reported within 24 h.
Pharmacokinetic assessments.
For both trials, serum samples for PK analyses were collected predose (within 60 min prior to drug administration), at 30 min, and at 4 h after the end of infusion on day 1 and on days 2, 4 (±1), 8 (±1), 15 (±2), 29 (±3), 57 (±7), 85 (±7), and 120 (±7) or at the early-termination visit.
A validated ELISA with a lower limit of quantification of 0.2 μg/ml was used to determine serum MHAA4549A concentrations. The assay used a murine monoclonal anti-idiotypic antibody specific for MHAA4549A to coat ELISA plates, which allowed the specific capture and quantification of MHAA4549A in serum samples. The following PK parameters were assessed for serum samples: observed maximum concentration (Cmax), area under the concentration-time curve extrapolated to infinity (AUC0–∞), apparent terminal half-life (t1/2), apparent clearance (CL), and volume of distribution at steady-state (Vss).
Immunogenicity assessments.
Serum samples for antitherapeutic antibody (ATA) analysis for both trials were collected prior the start of the infusion and on days 29 (±3) and 85 (±7) posttreatment, as well as on day 120 (±7) or at an early-termination visit. The presence of ATAs in those samples was initially detected with a validated MHAA4549A-specific bridging ELISA that used the formation of a complex between ATAs and two MHAA4549A conjugates: MHAA4549A-biotin, which enabled the capture of ATAs on streptavidin-coated ELISA plates, and MHAA4549A-digoxigenin (DIG), which allowed detection of captured complexes by subsequent addition of an anti-DIG antibody conjugated with horseradish peroxidase (HRP). Color development after addition of HRP substrate indicated the potential presence of ATAs in that sample. Samples with signals above an established assay threshold were considered positive in the screening assay. The threshold for the screening assay was established during assay validation using a panel of 100 serum samples from healthy donors and was set to yield an untreated-positive rate (UTPR) or false-positive rate of approximately 5% (9). Positive samples from the screening assay were further tested in a validated confirmatory assay. In the confirmatory assay, each sample was preincubated with an excess concentration of unlabeled MHAA4549A prior to performance of the same steps as described in the screening assay procedure. An ATA sample was defined as confirmed positive if competitive binding with MHAA4549A significantly reduced its signal in the assay. The percent reduction in assay signal required for a sample to be confirmed positive was established using the same panel of 100 serum samples from healthy donors used to set the screening assay threshold, as described above. The confirmatory assay threshold was established to yield a UTPR of approximately 1% (10). Subsequently, titer values were determined for all samples that were confirmed as positive by retesting those samples serially diluted in the same screening assay described above.
Statistical methods.
The sample size for this trial was based on the dose escalation rules described in the study design section and not based on any statistical criteria. A sufficient number of subjects were screened to ensure that approximately six subjects were enrolled in cohort A and five subjects were enrolled in cohorts B to D for trial 1 and that eight subjects were enrolled in cohort A and six subjects were enrolled in cohort B for trial 2. It was determined that four subjects dosed with active drug in all cohorts should be sufficient to characterize the single-dose safety and tolerability of MHAA4549A.
The analysis of safety and tolerability was based on the safety population, defined as all subjects who received any amount of MHAA4549A or placebo. Descriptive statistics were calculated for demographic continuous variables, and frequency counts and percentages were tabulated for categorical variables. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 16.0 (trial 1) or version 17.1 (trial 2). TEAEs were summarized by number and percentage of subjects experiencing TEAEs and by number of events and were tabulated by system organ class, preferred term, severity, causality, and treatment.
Descriptive statistics and change-from-baseline results were tabulated for continuous clinical laboratory results (serum chemistry, hematology, and urinalysis), vital sign results, and ECGs and were presented by treatment and time point for each test. Baseline was defined as the last results obtained prior to dosing. For categorical variables (urinalysis test), the number of subjects (frequency and percentage) was tabulated by results (e.g., negative, positive, or trace).
The pharmacokinetic population was the group of subjects who received MHAA4549A and had an adequate serum PK profile for MHAA4549A. PK data were available from day 1 (predose) to day 120 from 16 PK-evaluable patients across the four dose groups of MHAA4549A (trial 1) and 8 PK-evaluable patients across the two dose groups of MHAA4549A (trial 2). The PK parameters derived from the serum concentration-time profiles following administration of MHAA4549A were calculated using noncompartmental methods. The linear-up, log-down calculation method and plasma model for i.v. infusion were used to derive the PK parameters. Descriptive statistics were calculated for PK parameters including the means and standard deviations (SD).
Data analysis was performed using SAS, version 9.2 (SAS Institute, Inc., Cary, NC), and PK analyses were performed using Phoenix WinNonlin, versions 6.2 (trial 1) and 6.4 (trial 2) (Pharsight Corporation, Mountain View, CA).
RESULTS
Subject characteristics.
In trial 1, 21 of 53 volunteers screened were deemed eligible for the study and randomized to receive MHAA4549A or placebo. The first subject was dosed in July 2013, and the last subject completed the study in December 2013. The safety population consisted of 7 male and 14 female subjects, of whom 16 received one dose of active treatment and 5 received placebo. In trial 2, 27 of 58 volunteers screened were deemed eligible for the study, and 14 subjects were randomized to receive MHAA4549A or placebo. The first subject was dosed in November 2014, and the last subject completed the study in March 2015. The safety population consisted of seven male and seven female subjects, of whom eight received one dose of active treatment and six received placebo. In both trials, all subjects who were dosed completed the study. All subjects who received a single dose of MHAA4549A were included in the PK analyses (trial 1, n = 16; trial 2, n = 8). In both trials, no notable differences were observed among treatment groups or between subjects who received MHAA4549A and those who received placebo with respect to demographic parameters (Table 1).
TABLE 1.
Subject demographics and baseline characteristics
| Parameter | Value in trial 1 with:a |
Value in trial 2 with:a |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MHAA4549A |
Placebo | MHAA4549A |
Placebo | |||||||
| 1.5 mg/kg | 5 mg/kg | 15 mg/kg | 45 mg/kg | Overall | 8,400 mg | 10,800 mg | Overall | |||
| No. of subjects randomized | 4 | 4 | 4 | 4 | 16 | 5 | 4 | 4 | 8 | 6 |
| Age (yr) | 43 ± 7a | 41 ± 12 | 43 ± 14 | 40 ± 24 | 41 ± 14 | 51 ± 15 | 41 ± 15 | 34 ± 3 | 37 ± 11 | 49 ± 12 |
| No. of female subjects (%) | 3 (75) | 2 (50) | 3 (75) | 2 (50) | 10 (63) | 4 (80) | 2 (50) | 2 (50) | 4 (50) | 3 (50) |
| No. of White subjects (%) | 4 (100) | 4 (100) | 4 (100) | 4 (100) | 16 (100) | 5 (100) | 4 (100) | 4 (100) | 8 (100) | 6 (100) |
| Height (cm) | 166 ± 14 | 165 ± 5 | 166 ± 15 | 167 ± 3 | 166 ± 10 | 164 ± 5 | 163 ± 9 | 168 ± 6 | 166 ± 7 | 168 ± 5 |
| Weight (kg) | 63 ± 17 | 75 ± 9 | 66 ± 7 | 65 ± 7 | 67 ± 11 | 65 ± 3 | 69 ± 9 | 69 ± 9 | 69 ± 8 | 75 ± 11 |
| BMI (kg/m2) | 23 ± 2 | 28 ± 3 | 24 ± 4 | 23 ± 3 | 25 ± 3 | 24 ± 1 | 26 ± 3 | 24 ± 3 | 25 ± 3 | 27 ± 4 |
Values are means ± SD unless otherwise noted. Trial 1, entry-into-human study; trial 2, high-dose study.
Safety.
In trial 1, 23 TEAEs were reported by 13 of 21 (61.9%) subjects who received one dose of the study medication or placebo (safety population) (Table 2; see also Table S1 in the supplemental material). Nineteen of these TEAEs were reported by 10 of 16 (62.5%) subjects who had received MHAA4549A, and four TEAEs were reported by 3 of 5 (60.0%) subjects who received placebo. The total numbers of TEAEs reported were similar in all cohorts (four TEAEs reported per cohort), with the exception of cohort B, where seven TEAEs were observed. The TEAEs were reported by 2 to 3 of the 4 subjects who received active treatment in each of the cohorts.
TABLE 2.
Summary of adverse events in trial 1
| Parameter | Value for the parameter with: |
|||||
|---|---|---|---|---|---|---|
| MHAA4549A |
Placebo | |||||
| 1.5 mg/kg | 5 mg/kg | 15 mg/kg | 45 mg/kg | Overall | ||
| No. of subjects dosed | 4 | 4 | 4 | 4 | 16 | 5 |
| No. of subjects with at least 1 TEAE (%) | 2 (50) | 3 (75) | 2 (50) | 3 (75) | 10 (63) | 3 (60) |
| No. of TEAEs | 4 | 7 | 4 | 4 | 19 | 4 |
| Treatment-related | 0 | 0 | 4 | 2 | 6 | 0 |
| Mild | 3 | 6 | 4 | 4 | 17 | 4 |
| Moderate | 1 | 1 | 0 | 0 | 2 | 0 |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 |
| Serious | 0 | 0 | 0 | 0 | 0 | 0 |
| No. of subjects who discontinued due to TEAE | 0 | 0 | 0 | 0 | 0 | 0 |
| No. of deaths | 0 | 0 | 0 | 0 | 0 | 0 |
| Notable adverse eventsa (no. of subjects [%]) | ||||||
| Headache | 0 | 2 (50) | 1 (25) | 1 (25) | 4 (25) | 1 (20) |
| Oropharyngeal pain | 1 (25) | 0 | 0 | 1 (25) | 2 (13) | 0 |
| Alanine aminotransferase increased | 0 | 1 (25) | 0 | 0 | 1 (6) | 1 (20) |
| Blood creatine phosphokinase increased | 0 | 0 | 0 | 0 | 0 | 1 (20) |
Table S1 in the supplemental material lists all adverse events for trial 1.
In trial 2, 34 TEAEs were reported by 12 of 14 (85.7%) subjects who received one dose of the study medication or placebo (safety population) (Table 3; see also Table S2 in the supplemental material). Twenty-four of these TEAEs were reported by 7 of 8 (87.5%) subjects who had received MHAA4549A, and 10 TEAEs were reported by 5 of 6 (83.3%) subjects who received placebo. The total numbers of subjects reporting TEAEs were similar in all cohorts. The TEAEs were reported by 3 of the 4 subjects receiving MHAA4549A in cohort A (8,400 mg) and by 4 of the 4 subjects receiving MHAA4549A in cohort B (10,800 mg). No notable trends were observed among dose levels or between subjects who received MHAA4549A and those who received placebo.
TABLE 3.
Summary of adverse events in trial 2
| Parameter | Value for the parameter with: |
|||
|---|---|---|---|---|
| MHAA4549A |
Placebo | |||
| 8,400 mg | 10,800 mg | Overall | ||
| No. of subjects dosed | 4 | 4 | 8 | 6 |
| No. of subjects with at least 1 TEAE (%) | 3 (75) | 4 (100) | 7 (88) | 5 (83) |
| No. of TEAEs | 15 | 9 | 24 | 10 |
| Treatment-related | 6 | 3 | 9 | 1 |
| Mild | 14 | 7 | 21 | 10 |
| Moderate | 1 | 2 | 3 | 0 |
| Severe | 0 | 0 | 0 | 0 |
| Serious | 0 | 0 | 0 | 0 |
| No. of subjects who discontinued due to TEAE | 0 | 0 | 0 | 0 |
| No. of deaths | 0 | 0 | 0 | 0 |
| Notable adverse eventsa (no. of subjects [%]) | ||||
| Headache | 3 (75) | 1 (25) | 4 (50) | 2 (33) |
| Nasopharyngitis | 1 (25) | 2 (50) | 3 (38) | 2 (33) |
| Blood creatine phosphokinase increased | 0 (0) | 1 (25) | 1 (13) | 0 (0) |
Table S2 in the supplemental material lists all adverse events for trial 2.
In trial 1, the most commonly reported TEAEs in subjects who received MHAA4549A were headache and oropharyngeal pain, reported by 4 and 2 subjects, respectively (Table 2; see also Table S1 in the supplemental material). In trial 2, the most commonly reported TEAEs were headache, reported by 4 subjects who received MHAA4549A (3 in cohort A and 1 in cohort B) and nasopharyngitis, reported by 3 subjects who received MHAA4549A (1 in cohort A and 2 in cohort B) (Table 3; see also Table S2). The latter TEAEs were deemed unrelated to MHAA4549A. For both trials, all other TEAEs were each reported by no more than 1 subject who received MHAA4549A.
The severity of TEAEs in trial 1 was primarily mild, with only two moderate TEAEs that were deemed unrelated to MHAA4549A (Table 2). In trial 2, the severity of TEAEs was also primarily mild, with only three moderate TEAEs reported that were deemed unrelated to MHAA4549A (Table 3). There were no deaths, and no subjects in either trial reported pregnancies, severe AEs, or SAEs during the studies.
Clinical laboratory tests, vital signs, ECG measurements.
A number of shifts from baseline were observed for clinical laboratory tests over the course of both studies, but there were no observed trends with respect to either dose level or time. None of these shifts in either study was considered to be clinically significant, except for four clinical laboratory results that were considered to be TEAEs. In trial 1, an alanine aminotransferase (ALT) increase was recorded for one subject who received 5 mg/kg MHAA4549A (Table 2). The ALT increase occurred on day 85 and measured 72 U/liter (normal range, 0 to 41 U/liter). The test was repeated 3 and 10 days later, measuring 54 U/liter (considered not clinically significant by the principal investigator) and 18 U/liter, respectively. In addition, a creatine phosphokinase (CK) increase was recorded for one subject who received placebo in the MHAA4549A 45 mg/kg-cohort. The CK increase occurred on day 120 and measured 417 U/liter (normal range, 24 to 170 U/liter). A repeat test was performed 1 day later, measuring 482 U/liter. A second repeat test was performed 10 days after the initial out-of-range result and yielded a normal result of 163 U/liter.
In trial 2, a CK increase was recorded twice for one subject who received 10,800 mg of MHAA4549A (Table 3). Troponin I results were normal, and both increases were associated with intense physical activity. The principal investigator deemed both CK increases as unrelated to the study drug. The first CK increase occurred on day 15, was measured at 3,197 U/liter, and was judged to be clinically significant by the principal investigator. The test performed on day 29 yielded a result of 196 U/liter, and the result was judged by the principal investigator as not clinically significant. On day 57, the CK test result was 996 U/liter (judged as clinically significant by the principal investigator), and the test was repeated 6 days later with a result of 305 U/liter (considered not clinically significant). By the end of the study, these results overall were judged to be not clinically significant.
No safety issues were observed with respect to vital sign results or ECG measurements. Overall, there were no relevant differences in mean values and shifts from baseline over time with respect to dose level of MHAA4549A compared to results with placebo.
Pharmacokinetics.
In both trials, serum MHAA4549A concentrations exhibited a biphasic disposition, with an initial rapid distribution phase followed by a slow elimination phase (Fig. 2). MHAA4549A appeared to show linear pharmacokinetics, with a mean half-life of approximately 23 days (range, 21.9 to 24.6 days) in trial 1 and 21.5 days (range, 21.4 to 21.6 days) in trial 2.
FIG 2.
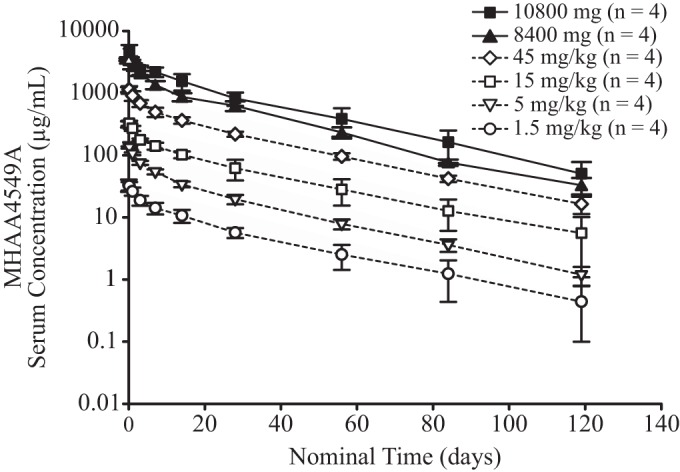
Observed group mean (±SD) MHAA4549A serum concentration-versus-time profile following single intravenous administration. Observed MHAA4549A serum concentration-time profiles were plotted on the basis of group mean values. Error bars represent the standard deviations of MHAA4549A concentrations at each nominal time point.
PK parameters following single i.v. dose administrations of MHAA4549A for trial 1 and trial 2 are summarized in Table 4. For trial 1 and trial 2, the CL values were similar across the dose groups, with a mean of approximately 182 ml/day and 159 ml/day, respectively, indicating that MHAA4549A has dose-proportional PK in the tested dose ranges (1.5 mg/kg to 10,800 mg [∼135 mg/kg]).
TABLE 4.
Summary statistics of serum pharmacokinetic parameters of MHAA4549A in all subjects following single intravenous administration in trial 1 and trial 2
| Dose (n = 4)a | Value for the parameter (mean ± SD)b |
||||
|---|---|---|---|---|---|
| Cmax (μg/ml) | CL (ml/day) | t1/2 (day) | AUC0–∞ (day · μg/ml) | Vss (ml) | |
| 1.5 mg/kg | 33.5 ± 7.20 | 186 ± 73.9 | 22.6 ± 5.04 | 539 ± 149 | 5,170 ± 825 |
| 5 mg/kg | 144 ± 8.52 | 204 ± 42.3 | 21.9 ± 1.49 | 1,850 ± 241 | 5,400 ± 805 |
| 15 mg/kg | 334 ± 30.9 | 186 ± 51.7 | 24.6 ± 4.27 | 5,620 ± 1,490 | 5,600 ± 948 |
| 45 mg/kg | 1,180 ± 139 | 152 ± 20.9 | 23.6 ± 2.04 | 19,500 ± 2,100 | 4,670 ± 424 |
| 8,400 mg | 3,570 ± 179 | 167 ± 16.5 | 21.6 ± 2.01 | 50,700 ± 4,830 | 4,590 ± 661 |
| 10,800 mg | 4,780 ± 1,160 | 151 ± 45.0 | 21.4 ± 2.92 | 76,400 ± 22,200 | 4,170 ± 573 |
n, number of subjects at each dose.
PK parameters were summarized using noncompartmental analysis. Cmax, observed maximum concentration; CL, clearance; t1/2, apparent terminal half-life; AUC0–∞, area under the time-concentration curve extrapolated to infinity; Vss, volume of distribution at steady-state.
Immunogenicity.
No subject in either trial developed ATAs after the administration of MHAA4549A. In trial 2, one patient who received 10,800 mg of MHAA4549A tested positive for ATAs at baseline and negative at subsequent postbaseline time points.
DISCUSSION
The safety, tolerability, and pharmacokinetics of MHAA4549A were examined in two phase 1 studies. Both trials demonstrated that MHAA4549A was safe and well tolerated in healthy subjects following a single i.v. administration of 1.5, 5, 15, or 45 mg/kg or fixed doses of 8,400 mg and 10,800 mg. None of the TEAEs were severe or serious, and the majority were mild in intensity. No deaths or pregnancies were reported over the course of the study. There were no notable differences with respect to the number of subjects experiencing TEAEs between the different dose groups, and there were no safety concerns regarding clinical laboratory tests, ECGs, and vital signs. No treatment-induced anti-MHAA4549A antibodies were detected for any subject dosed in these studies. The relatively low immunogenicity profile of MHAA4549A is likely because it is a fully human antibody cloned from a single human plasmablast cell (7). The single-dose regimen used in these studies may also have contributed to the observed low incidence of ATAs in these studies since such a regimen is less likely to stimulate the immune system sufficiently to elicit an antibody response (11).
The serum pharmacokinetics of MHAA4549A was linear (dose independent) in healthy subjects over the dose range of 1.5 mg/kg to ∼135 mg/kg (10,800-mg fixed dose) with a terminal half-life of approximately 20 to 30 days. This PK profile in healthy subjects is consistent with that of a human IgG1 antibody that lacks known endogenous targets in humans (12). Given the high doses of the drug and its extended half-life, a single dose of MHAA4549A should provide sufficiently high levels of drug throughout the typical course of an influenza virus infection. This is supported by evidence that MHAA4549A neutralized a panel of influenza A virus human isolates with in vitro median inhibitory concentration (IC50) values ranging from 1.3 nM to 45.1 nM (7). At 2 weeks after the administration of a single i.v. dose of 45 mg/kg (the anticipated efficacious dose), the observed MHAA4549A serum concentration was ∼366 μg/ml, which was ∼55- to 1,879-fold of the IC50. The long half-life may also contribute to the drug's potential for seasonal protection as a prophylactic agent (13).
For these trials, two different dosing approaches were chosen: trial 1 doses were weight based, and trial 2 doses were fixed. Fixed dosing is recommended for monoclonal antibodies due to their low pharmacokinetic variability relative to safety and efficacy variability (12, 14). Similar PK observations between trial 1 and trial 2 studies support the fixed-dosing strategy. Given the many practical advantages and potentially larger therapeutic window of MHAA4549A, fixed dosing will be used in future trials of MHAA4549A.
In trial 1, the selection of the first-in-human doses was based on multiple factors, including the in vivo, nonclinical, pharmacologically active doses and exposure in mouse infection models, expected human PK, and nonclinical safety factors. A broad dose range (1.5 to 45 mg/kg as single i.v. doses) was selected to ensure a safe starting dose to assess the safety of MHAA4549A across a range of drug exposures and to provide PK data for further clinical development. The decision to assess high doses in trial 2 was based on the hypothesis that severely ill patients hospitalized with influenza virus infection have higher viral loads and longer durations of viral shedding (15). These patients may require higher doses of MHAA4549A to achieve sufficient occupancy of virus binding sites in the upper and lower respiratory compartments, and a high concentration of IgG would be necessary in the circulating plasma to enable passive transudation into the site of action for influenza virus, the respiratory mucosa (16, 17). The initial dose selection for trial 2 was based on an exploratory exposure-response analysis of a phase 2a influenza virus challenge study in healthy volunteers (clinical trial NCT01980966), which indicated that higher MHAA4549A nasal exposure was associated with better efficacy. Simulations from a semiquantitative pharmacokinetic model developed from the challenge study suggested that 8,400 mg was the minimum dose that would show a separation of nasal exposure from a dose of 3,600 mg (K. Patel, C. Kirkpatrick, M. Stroh, P. Smith, and R. Deng, presented at the 55th Interscience Conference on Antimicrobial Agents and Chemotherapy [ICAAC], San Diego, CA, 17 to 21 September 2015), which led to a significant decrease in viral shedding in the upper respiratory tract (J. Tavel, J. McBride, R. Deng, M. Derby, T. Burgess, N. Chai, S. Park, M. Xu, and L. Swem, presented at the European Scientific Working Group on Influenza, Riga, Latvia, 14 to 17 September 2014). The 10,800-mg dose was chosen based on serum exposures predicted by a preliminary population PK analysis of results from trial 1, which indicated that the MHAA4549A exposures in a typical 70- to 80-kg subject receiving a 10,800-mg dose would be comparable to those of a 40-kg subject receiving the 8,400-mg dose. The use of high doses of antibodies for treatment of infectious diseases is not unprecedented. For example, high-dose intravenous immunoglobulin (hIVIG), a blood product prepared from the serum of donors, is dosed up to 2 g/kg/month as an immunomodulatory agent (18).
The discovery of antibodies to protect against bacteria and toxins dates back to the early 1890s and eventually led to “serum therapy” (13, 19 – 21), a polyclonal antibody admixture from immunized animals or immune human donors. However, serum therapy caused a series of adverse reactions, including hypersensitivity and serum sickness, an antigen-antibody complex disease. Once antibiotics and antivirals were introduced, serum therapy lost popularity due to product batch heterogeneity, pathogen transmission, and immunogenicity risks (19, 20, 22 – 24). In contrast, MHAA4549A is a monoclonal antibody, and in the absence of influenza virus infection, its epitope on the human influenza A virus hemagglutinin glycoprotein is not endogenously expressed in humans; therefore it is expected to be safe (7, 19).
Currently, there is no approved therapeutic for the treatment of hospitalized patients with severe influenza virus infection. MHAA4549A has several advantages as a therapeutic for the treatment of influenza. Monoclonal antibodies are known to have relatively low toxicity profiles and high specificity and are not expected to alter the host flora or select for resistance among nontarget microorganisms (19, 25). In addition, the metabolism of therapeutic monoclonal antibodies, unlike that of small molecules, does not involve CYP450 enzymes, thereby reducing the risk of drug-drug interactions. The kidney does not have a major role in eliminating intact IgGs because of their large molecular size (26). Potential differences in hepatic or renal functions between the hospitalized influenza population and healthy volunteers should have a negligible effect on the metabolism and clearance of monoclonal antibodies (27).
In a nonclinical study where mice were infected with a high, lethal dose of the influenza virus A/Puerto Rico/8/34 (PR8) variant and treated with subefficacious doses of MHAA4549A, the neuraminidase inhibitor oseltamivir, or a combination of the two therapies at 72 h postinfection, the combination of MHAA4549A with oseltamivir significantly improved survival and body weight over results with either treatment alone (7). Since MHAA4549A is unlikely to interact with other influenza virus antivirals, such as neuraminidase inhibitors, combination therapy allows for a dual mechanism of action and may have added benefit in hospitalized patients with severe influenza (7, 13, 19, 20, 25, 28).
In conclusion, our results from the two phase 1 studies showed that the anti-influenza A virus monoclonal antibody MHAA4549A was safe and well tolerated in healthy adult volunteers up to a single i.v. dose of 10,800 mg. MHAA4549A did not induce an ATA response in treated subjects and demonstrated linear serum pharmacokinetics. Further studies in patients with influenza virus infection are needed to establish efficacy and additional safety outcomes.
Supplementary Material
ACKNOWLEDGMENTS
We thank all of the subjects and the investigators who participated in this study.
J.J.L., R.D., M.A.D., P.H., M.A., M.M., T.B., P.K., E.N., and J.A.T. are or were employed at Genentech, Inc., a member of the Roche Group, and own Roche stock. M.A.D. is retired. R.L., S.C., and I.P. are employees of inVentiv Health Clinical.
Editing and writing support was provided by Deborah Solymar (Genentech, Inc., South San Francisco, CA, USA) and was funded by Genentech, Inc.
Funding Statement
This study was supported by Genentech, Inc. Genentech, Inc., was involved in the study design, data interpretation, and the decision to submit for publication in conjunction with the authors.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00607-16.
REFERENCES
- 1.Ison MG, de Jong MD, Gilligan KJ, Higgs ES, Pierson J, Hayden FG. 2010. Endpoints for testing influenza antiviral treatments for patients at high risk of severe and life-threatening disease. J Infect Dis 201:1654–1662. doi: 10.1086/652498. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2014. Influenza (seasonal). Fact sheet no. 211. World Health Organization, Geneva, Switzerland. http://www.who.int/mediacentre/factsheets/fs211/en/.
- 3.Reed C, Chaves SS, Daily Kirley P, Emerson R, Aragon D, Hancock EB, Butler L, Baumbach J, Hollick G, Bennett NM, Laidler MR, Thomas A, Meltzer MI, Finelli L. 2015. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS One 10:e0118369. doi: 10.1371/journal.pone.0118369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hadler JL, Yousey-Hindes K, Pérez A, Anderson EJ, Bargsten M, Bohm SR, Hill M, Hogan B, Laidler M, Lindegren ML, Lung KL, Mermel E, Miller L, Morin C, Parker E, Zansky SM, Chaves SS. 2016. Influenza-related hospitalizations and poverty levels—United States, 2010–2012. MMWR Morb Mortal Wkly Rep 65:101–105. doi: 10.15585/mmwr.mm6505a1. [DOI] [PubMed] [Google Scholar]
- 5.Aoki FY, Macleod MD, Paggiaro P, Carewicz O, El Sawy A, Wat C, Griffiths M, Waalberg E, Ward P, Study Group IMPACT. 2003. Early administration of oral oseltamivir increases the benefits of influenza treatment. J Antimicrob Chemother 51:123–129. doi: 10.1093/jac/dkg007. [DOI] [PubMed] [Google Scholar]
- 6.Muthuri SG, Venkatesan S, Myles PR, Leonardi-Bee J, Al Khuwaitir TS, Al Mamun A, Anovadiya AP, Azziz-Baumgartner E, Báez C, Bassetti M, Beovic B, Bertisch B, Bonmarin I, Booy R, Borja-Aburto VH, Burgmann H, Cao B, Carratala J, Denholm JT, Dominguez SR, Duarte PA, Dubnov-Raz G, Echavarria M, Fanella S, Gao Z, Gérardin P, Giannella M, Gubbels S, Herberg J, Iglesias AL, Hoger PH, Hu X, Islam QT, Jiménez MF, Kandeel A, Keijzers G, Khalili H, Knight M, Kudo K, Kusznierz G, Kuzman I, Kwan AM, Amine IL, Langenegger E, Lankarani KB, Leo YS, Linko R, Liu P, Madanat F, Mayo-Montero E, McGeer A, et al. 2014. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: a meta-analysis of individual participant data. Lancet Respir Med 2:395–404. doi: 10.1016/S2213-2600(14)70041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura G, Chai N, Park S, Chiang N, Lin Z, Chiu H, Fong R, Yan D, Kim J, Zhang J, Lee WP, Estevez A, Coons M, Xu M, Lupardus P, Balazs M, Swem LR. 2013. An in vivo human-plasmablast enrichment technique allows rapid identification of therapeutic influenza A antibodies. Cell Host Microbe 14:93–103. doi: 10.1016/j.chom.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Chai N, Swem LR, Reichelt M, Chen-Harris H, Luis E, Fouts A, Lupardus P, Wu TD, Li O, McBride J, Laurence M, Xu M, Tan M-W. 2016. Two escape mechanisms of influenza A virus to a broadly neutralizing stalk-binding antibody. PLoS Pathog 12(6):e1005702. doi: 10.1371/journal.ppat.1005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, Parish T, Scott G, Shankar G, Shores E, Swanson SJ, Taniguchi G, Wierda D, Zuckerman LA. 2004. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods 289:1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Wakshull E, Coleman D. 2011. Confirmatory immunogenicity assays, p 103–117. In Tovey M. (ed), Detection and quantification of antibodies to biopharmaceuticals: practical and applied considerations. John Wiley & Sons, Hoboken, NJ. [Google Scholar]
- 11.Koren E, Smith HW, Shores E, Shankar G, Finco-Kent D, Rup B, Barrett YC, Devanarayan V, Gorovits B, Gupta S, Parish T, Quarmby V, Moxness M, Swanson SJ, Taniguchi G, Zuckerman LA, Stebbins CC, Mire-Sluis A. 2008. Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J Immunol Methods 333:1–9. doi: 10.1016/j.jim.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Bai S, Jorga K, Xin Y, Jin D, Zheng Y, Damico-Beyer LA, Gupta M, Tang M, Allison DE, Lu D, Zhang Y, Joshi A, Dresser MJ. 2012. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet 51:119–135. doi: 10.2165/11596370-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13.Shriver Z, Trevejo JM, Sasisekharan R. 2015. Antibody-based strategies to prevent and treat influenza. Front Immunol 6:315. doi: 10.3389/fimmu.2015.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang DD, Zhang S, Zhao H, Men AY, Parivar K. 2009. Fixed dosing versus body size-based dosing of monoclonal antibodies in adult clinical trials. J Clin Pharmacol 49:1012–1024. doi: 10.1177/0091270009337512. [DOI] [PubMed] [Google Scholar]
- 15.Lee N, Chan PK, Hui DS, Rainer TH, Wong E, Choi KW, Lui GC, Wong BC, Wong RY, Lam WY, Chu IM, Lai RW, Cockram CS, Sung JJ. 2009. Viral loads and duration of viral shedding in adult patients hospitalized with influenza. J Infect Dis 200:492–500. doi: 10.1086/600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leyva-Grado VH, Tan GS, Leon PE, Yondola M, Palese P. 2015. Direct administration in the respiratory tract improves efficacy of broadly neutralizing anti-influenza virus monoclonal antibodies. Antimicrob Agents Chemother 59:4162–4172. doi: 10.1128/AAC.00290-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner DK, Clements ML, Reimer CB, Snyder M, Nelson DL, Murphy BR. 1987. Analysis of immunoglobulin G antibody responses after administration of live and inactivated influenza A vaccine indicates that nasal wash immunoglobulin G is a transudate from serum. J Clin Microbiol 25:559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jolles S, Sewell WA, Misbah SA. 2005. Clinical uses of intravenous immunoglobulin. Clin Exp Immunol 142:1–11. doi: 10.1111/j.1365-2249.2005.02834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casadevall A, Dadachova E, Pirofski LA. 2004. Passive antibody therapy for infectious diseases. Nat Rev Microbiol 2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- 20.Pai JC, Sutherland JN, Maynard JA. 2009. Progress towards recombinant anti-infective antibodies. Recent Pat Antiinfect Drug Discov 4:1–17. doi: 10.2174/157489109787236319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Both L, Banyard AC, van Dolleweerd C, Wright E, Ma JK, Fooks AR. 2013. Monoclonal antibodies for prophylactic and therapeutic use against viral infections. Vaccine 31:1553–1559. doi: 10.1016/j.vaccine.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Both L, Banyard AC, van Dolleweerd C, Horton DL, Ma JK, Fooks AR. 2012. Passive immunity in the prevention of rabies. Lancet Infect Dis 12:397–407. doi: 10.1016/S1473-3099(11)70340-1. [DOI] [PubMed] [Google Scholar]
- 23.Goudsmit J, Marissen WE, Weldon WC, Niezgoda M, Hanlon CA, Rice AB, Kruif JD, Dietzschold B, Bakker AB, Rupprecht CE. 2006. Comparison of an anti-rabies human monoclonal antibody combination with human polyclonal anti-rabies immune globulin. J Infect Dis 193:796–801. doi: 10.1086/500470. [DOI] [PubMed] [Google Scholar]
- 24.Marasco WA, Sui J. 2007. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol 25:1421–1434. doi: 10.1038/nbt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saylor C, Dadachova E, Casadevall A. 2009. Monoclonal antibody-based therapies for microbial diseases. Vaccine 27(Suppl 6):G38–G46. doi: 10.1016/j.vaccine.2009.09.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chames P, Regenmortel MV, Weiss E, Baty D. 2009. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng R, Jin F, Prabhu S, Iyer S. 2012. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin Drug Metab Toxicol 8:141–160. doi: 10.1517/17425255.2012.643868. [DOI] [PubMed] [Google Scholar]
- 28.Ramos EL, Mitcham JL, Koller TD, Bonavia A, Usner DW, Balaratnam G, Fredlund P, Swiderek KM. 2015. Efficacy and safety of treatment with an anti-M2e monoclonal antibody in experimental human influenza. J Infect Dis 211:1038–1044. doi: 10.1093/infdis/jiu539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.