

Abstract
Aspergillus fumigatus intrinsic fluconazole resistance has been demonstrated to be linked to the CYP51A gene, although the precise molecular mechanism has not been elucidated yet. Comparisons between A. fumigatus Cyp51Ap and Candida albicans Erg11p sequences showed differences in amino acid residues already associated with fluconazole resistance in C. albicans. The aim of this study was to analyze the role of the natural polymorphism I301 in Aspergillus fumigatus Cyp51Ap in the intrinsic fluconazole resistance phenotype of this pathogen. The I301 residue in A. fumigatus Cyp51Ap was replaced with a threonine (analogue to T315 at Candida albicans fluconazole-susceptible Erg11p) by changing one single nucleotide in the CYP51A gene. Also, a CYP51A knockout strain was obtained using the same parental strain. Both mutants' antifungal susceptibilities were tested. The I301T mutant exhibited a lower level of resistance to fluconazole (MIC, 20 μg/ml) than the parental strain (MIC, 640 μg/ml), while no changes in MIC were observed for other azole- and non-azole-based drugs. These data strongly implicate the A. fumigatus Cyp51Ap I301 residue in the intrinsic resistance to fluconazole.
INTRODUCTION
Aspergillus fumigatus is the most common hyphomycete to cause disease in humans (1 – 3). It is intrinsically resistant to ketoconazole and fluconazole but normally susceptible to the other available azole antifungal agents (itraconazole, posaconazole, voriconazole, and isavuconazole) (4 – 8). The molecular mechanism for fluconazole intrinsic resistance has not been described yet. However, a hypothetical molecular mechanism has been proposed by Edlind et al., who linked A. fumigatus fluconazole intrinsic resistance with a naturally occurring amino acid substitution in Cyp51Ap (14-α sterol demethylase A) (9). These authors carried out an in silico comparison of the Candida albicans Erg11p and A. fumigatus Cyp51Ap sequences and found that among the residues most commonly implicated in fluconazole resistance in C. albicans (Y132, T315, S405, G464, and R467) (10, 11), only the T315 residue is not conserved in A. fumigatus Cyp51Ap and is naturally replaced by a nonpolar isoleucine (I301). In C. albicans, the replacement of the polar T315 residue by the nonpolar alanine (T315A) is enough to confer fluconazole resistance on the yeast (10).
The aim of this study was to molecularly confirm that the natural polymorphism I301 in the Cyp51Ap is necessary and sufficient to explain the intrinsic reduced fluconazole susceptibility of A. fumigatus. An A. fumigatus mutant harboring the I301T substitution was generated, and susceptibilities to fluconazole and other antifungals were tested. Also, a CYP51A-defective mutant was obtained using the same parental strain in order to compare their antifungal susceptibility patterns.
MATERIALS AND METHODS
Strains.
Aspergillus fumigatus akuBKU80 (12) was considered the wild-type strain, and its DNA was used as the template for all PCRs. It was the recipient strain for electroporation assays. Escherichia coli TOP10 (Promega) was used to propagate all plasmids.
Genetic constructs.
A transformation plasmid named LMDM-P87 was generated. It contains a mutated T973C CYP51A gene (that leads to the amino acid substitution I301T in Cyp51Ap) with its intact 5′ flanking region. The CYP51A 3′ untranslated region (UTR) is interrupted by a hygromycin B resistance cassette (hph) between nucleotides 126 and 127 upstream of the CYP51A stop codon. Plasmid LMDM-P87 was obtained in two sets of three PCRs each (Fig. 1). The first set was aimed to introduce the mutation T973C into CYP51A. In the first reaction of this set, primers A7 and A19 were used to amplify a 1,452-bp fragment which contained a 462 bp of the CYP51A promoter region plus 990 bp of the first portion of its coding sequence (5′ UTR and 5′ of the open reading frame [ORF]). The second PCR employed oligonucleotides A18 and A17, which amplify a 790-bp fragment including 664 bp of the 3′ portion of the CYP51A ORF and 126 bp upstream of the CYP51A stop codon (3′ UTR). Primer A18 carries the mutation T973C in the middle of its sequence. Primer A19 is reverse complementary with A18, and both generate an overlapping region of 35 bp which was used in the final fusion PCR, performed as follows. The two previously generated fragments (1,452 bp and 790 bp) were used as templates together with the primers A7 and A17. The resulting 2.2-kb product was cloned into the pGEM-T Easy vector (Promega) to obtain plasmid LMDM-P75. Both strands of the complete 2.2-kb construct were sequenced to confirm the presence of the mutation, as described previously (13). In parallel, a second set of PCRs was performed. The hph cassette was fused to a 0.9-kb fragment of the 3′ UTR region of CYP51A starting 127 bp upstream of the CYP51A stop codon. The resistance cassette was used as a selection marker for recombinants, while the 0.9-kb fragment was used later as a flanking region for homologous integration together with the 5′ UTR-CYP51A. In the first PCR amplification of this set, the 1.4-kb hph cassette was obtained from plasmid pUM102 (A. fumigatus CYP51AΔhph) (a kind gift of Emilia Mellado) (14) using primers H1 and HF2. The second PCR was performed using primers HF1 and H2 in order to obtain the 0.9-kb fragment of the 3′ UTR of the CYP51A gene described before. HF1 and HF2 are reverse complementary primers designed to allow the fusion of the described fragments in the next reaction. The last PCR of this set was done using the 1.4-kb (hph) and the 0.9-kb fragments as templates and primers H1 and H2. The last oligonucleotides include a SacI site on both ends. The resulting construct (2.3 kb) was cloned into a pGEM-T Easy vector to obtain the plasmid LMDM-P82.
FIG 1.

Construction of the LMDM-P87 plasmid employed to generate the A. fumigatus CYP51A T973C mutant strain. Striped and gray boxes represent the CYP51A coding and UTR sequences, respectively. Unfilled boxes indicate the hygromycin resistance cassette (hph) obtained from the pUM102 plasmid. Black arrows symbolize oligonucleotide primers. The cross symbol represents the introduced T973C mutation. SacI restriction sites are indicated with scissors (primers H1 and H2).
To generate the complete transformation vector, the 2.3-kb fragment with an hph cassette was released from the LMDM-P82 by SacI digestion by following the manufacturer's instructions (Promega). Simultaneously, LMDM-P75 was linearized with SacI and dephosphorylated using calf intestinal alkaline phosphatase (CIAP; Promega) according to the manufacturer's protocol. Afterwards, the 2.3-kb fragment was ligated with T4 DNA ligase (Promega) to the linearized LMDM-P75 to create LMDM-P87. The complete vector sketch can be seen in Fig. 1. Primer sequences are described in Table 1.
TABLE 1.
Oligonucleotide primers used in this work
| Primer | Sequence (5′–3′)a | Orientation | Use |
|---|---|---|---|
| A7b | TCATATGTTGCTCAGCGG | Sense | LMDM-P87 construction and evaluation of the transforming vector integration |
| A19 | GCATAATCCAGGCGCTGGTGGACGAAGACGAATGC | Antisense | LMDM-P87 construction |
| A18 | GCATTCGTCTTCGTCCACCAGCGCCTGGATTATGC | Sense | LMDM-P87 construction |
| A17 | GGCCAGTAAGGTCTGAATAAG | Antisense | LMDM-P87 construction |
| H1 | TTTGAGCTCGTTAACTGATATTGAAGGAGCATTTTTTGGGC | Sense | LMDM-P87 construction |
| H2 | ACGGAGCTCCATCGAACCTCTCGTGTGACTATG | Antisense | LMDM-P87 construction |
| HF1 | AGAGTAGATGCCGACCGGGAACCAGTTAACCCTGAAGTGTTGTTGCCTATACTGAG | Sense | LMDM-P87 construction |
| HF2 | CTCAGTATAGGCAACAACACTTCAGGGTTAACTGGTTCCCGGTCGGCATCTACTCT | Antisense | LMDM-P87 construction |
| P450-1c | ATGGTGCCGATGCTATGG | Sense | CYP51A knockout cassette amplification |
| P450-2c | CTGTCTCACTTGGATGTG | Antisense | CYP51A knockout cassette amplification |
| A10 | ATTGCCGCAGAGATGTCC | Antisense | Evaluation of transforming vector integration and CYP51A expression |
| HS3 | ACATGGCGTGATTTCATATGCGCG | Sense | Evaluation of transforming vector integration |
| HS4 | TGGTCAAGACCAATGCGGAGCATA | Antisense | Evaluation of transforming vector integration |
| A14 | CCAGAGAGACTTTGACACAG | Sense | Evaluation of transforming vector integration |
| A1b | CTTCTTTGCGTGCAGAGA | Sense | Evaluation of CYP51A expression |
Transformations.
Two linear PCR fragments were used for A. fumigatus akuBKU80 transformation: (i) the cassette containing the mutated CYP51A gene together with the hph selection marker (LMDM-P87) and (ii) the CYP51A knockout cassette (14). Fragments were PCR amplified with the primer pairs A7/H2 and P450.1/P450.2, respectively. Transformation experiments by electroporation were carried out as described before (13) using 0.3 μg of the PCR fragments. Transformants were selected with 350 μg/ml of hygromycin B (HygB; InvivoGen) in minimal medium (MM) (15) and subcultured for further analysis. The incorporation of the hph cassette was phenotypically confirmed by plating the strains in duplicate in MM with 350 μg/ml of HygB.
Integration confirmation.
Genomic DNA from HygB-resistant transformants and the parental strain were obtained (14). Two multiplex PCRs were performed to confirm the homologous recombination of the mutated CYP51A/hph cassette and the knockout cassette (Fig. 2A). The first multiplex reaction was performed with two primer pairs, with the aim of verifying the integration of the hph cassette in the A. fumigatus genome. Primers A7 and A10 were used as a PCR control, as they hybridize the 5′ UTR and the ORF of the CYP51A, respectively. Primers HS3 and HS4 were used to amplify a fragment of the hph cassette. Thus, two PCR fragments were expected when the hph cassette was integrated (1,066 bp and 386 bp). In contrast, only the 1,066-bp fragment would be amplified in nontransformant strains. The second multiplex PCR was meant to confirm the homologous recombination of the mutated CYP51A, using the primers A14, HS3, and HS4. Primer A14 was designed to hybridize the CYP51A 5′ flanking region 603 bp downstream the CYP51A start codon, which is outside the construction cloned into LMDM-P87. Homologous or ectopic recombination would be confirmed by the amplification of two fragments (3.5 kb and 386 bp) or one (386 bp) band, respectively. No bands were expected for nontransformant strains.
FIG 2.

Recombination confirmation. (A) Schematic representation of the gene constructions and primer relative positions. Striped and gray boxes represent the CYP51A coding and UTR sequences, respectively. Unfilled boxes indicate the hygromycin resistance cassette (hph). Black boxes show the CYP51A UTRs not included in the constructions. Lines (in LMDM-P87 and pUM-102) represent the pGEM-T Easy vector. Arrows symbolize oligonucleotide primers. The cross symbol represents the introduced T973C mutation. Dotted lines represent the sizes of the PCR fragments. (B) Multiplex PCRs aimed to confirm homologous recombination events in the studied mutants. Lanes 1 to 5 show the results of the multiplex PCR designed to verify the hph cassette integration (primers A7/A10 and HS3/HS4). Lanes 6 to 10 show the amplification products of the multiplex PCR meant to confirm the homologous recombination of the mutated CYP51A (primers A14, HS3, and HS4). M, 100-bp ladder. Lanes 1 and 6, wild-type akuBKU80; lanes 2 and 7, LMDM-P87; lanes 3 and 8, LMDM-1030; lanes 4 and 9, LMDM-32; lanes 5 and 10, pUM-102. (C) Sequencing chromatograms showing the mutation T973C in the CYP51A gene of the LMDM-1030 strain.
PCRs.
PCR amplifications were performed in a 25-μl volume by following the Pegasus DNA polymerase (PBL, Buenos Aires, Argentina) manufacturer's instructions in an Applied Biosystems thermocycler (Tecnolab-AB, Buenos Aires, Argentina). The thermocycler was programed for one initial step of 2 min at 94°C followed by 30 cycles of 30 s at 95°C, 30 s at the primer pair's melting temperature (Tm), and 1 min per kilobase of the expected PCR product at 72°C and then a final cycle of 10 min at 72°C.
Confirmation of the expression of the mutated CYP51A.
Total RNA was extracted with RNAzol (RNAzolRT; MRC Inc.) from mutant strains, and reverse transcription (RT) was performed with avian myeloblastosis virus (AMV) reverse transcriptase enzyme (Promega, Argentina) according to the manufacturer's protocol. The obtained cDNA was used as the template for a PCR performed with primers A1 and A10 (flanking the 70-bp intron of CYP51A).
Antifungal susceptibility testing.
Susceptibility testing was performed by following the broth microdilution reference method published in document M38-A2 of the Clinical and Laboratory Standards Institute (CLSI) (16). Itraconazole, posaconazole, voriconazole, fluconazole, amphotericin B, and caspofungin (all purchased from Sigma-Aldrich, Argentina) were tested. Concentration ranges of fluconazole were modified from what is standardized to 640 to 1.25 μg/ml to establish differences in fluconazole susceptibilities between the wild-type and mutant strains. Moreover, fluconazole and voriconazole susceptibility were also evaluated by disk diffusion following CLSI document M51-A (17) using commercial disks (Oxoid, Argentina) and by agar diffusion using fluconazole MIC test strips (fluconazole 256-μg MIC test strips; Liofilchem SRL). Susceptibility tests were performed in triplicate on three different days.
RESULTS
To establish the role of the I301 residue of Cyp51Ap in the intrinsic fluconazole resistance of A. fumigatus, two mutant strains were generated. One harbors the mutation T793C in CYP51A, which leads to the I301T amino acid substitution, while the other is a CYP51A-defective strain. These mutants were named LMDM-1030 and LMDM-32, respectively. The homologous recombination was confirmed by multiplex PCR using the primers described in Materials and Methods. The mutant strains LMDM-1030 and LMDM-32 showed the genomic integration of the hph cassette (Fig. 2B, lanes 4 and 5, respectively). For both mutants, two PCR bands were observed (386 bp and 1,066 bp) corresponding to the amplification of the DNA region between primers HS3/HS4 and A7/A10, respectively. Similar results were obtained with LMDM-P87 and pUM-102 DNAs, which were used as reaction controls. On the other hand, parental strain akuBKU80 showed only one 1,066-bp band. The homologous recombination of both constructions in LMDM-1030 and LMDM-32 was confirmed by a second multiplex PCR. Two PCR bands were obtained when DNAs from both mutants were used (Fig. 2B, lanes 8 and 9). LMDM-1030 showed 3.5-kb and 386-bp bands which correspond to the amplification using primers A14 and HS4 and the pair HS4 and HS3, respectively. The smaller band shows the presence of hph. The 3.5-kb band demonstrates that the construction was integrated, replacing the wild-type CYP51A gene, since the A14 primer hybridizes the CYP51A 5′ UTR but in a region not included in the construction (A14 hybridizes 603 bp upstream of the start codon). Moreover, the size of the amplicon demonstrates that the hph cassette was integrated in the 3′ UTR, 126 bp upstream of the CYP51A stop codon. For LMDM-32, the multiplex PCR also showed two bands but with different sizes (2.4 kb and 386 bp). The smaller band demonstrates the hph cassette integration as described before, while the 2.4-kb band shows that the hph cassette was integrated inside the CYP51A ORF region. As expected, when A. fumigatus akuBKU80 DNA was used, no amplification was obtained (Fig. 2B, lane 6). The incorporation of the T973C mutation in the CYP51A gene of the LMDM-1030 mutant strain was confirmed by sequencing (Fig. 2C).
The naturally occurring polymorphism T301I at A. fumigatus Cyp51Ap is responsible for the fluconazole resistance phenotype.
The CYP51A-defective strain LMDM-32 and the T973C mutant LMDM-1030 were morphologically indistinguishable from the parental strain, A. fumigatus akuBKU80. However, azole MICs were substantially different. It was clear that the deletion of CYP51A in LMDM-32 decreased the azole MICs 32- to 4-fold for fluconazole and the other azole drugs tested, respectively. In contrast, LMDM-1030 MICs were significantly lower only for fluconazole (32-fold) (Table 2). As expected, there were no susceptibility differences to nonazole antifungals between mutants and parental strains (Table 2). The fluconazole and voriconazole susceptibility differences between the wild-type and mutant strains were confirmed by disk diffusion susceptibility testing. The parental strain showed no inhibition zone when fluconazole disks were used, while the inhibition zone diameter for both mutant strains was 19 mm. Turning to voriconazole, the parental and LMDM-1030 strains showed the same inhibition diameter (32 mm), whereas the knockout mutant showed an inhibition zone of 48 mm (Table 2 and Fig. 3). Moreover, fluconazole MIC differences between akuBKU80 and both mutants were also verified by agar diffusion using Liofilchem MIC test strips. Using this methodology, the akuBKU80 strain showed a MIC of >256 μg/ml, while both mutants exhibited a MIC of 8 μg/ml (64-fold lower) (Table 2 and Fig. 4).
TABLE 2.
Susceptibility testing results of the Aspergillus fumigatus strains used in this study
| Strain | MIC (μg/ml) of antifungal agenta |
|||||
|---|---|---|---|---|---|---|
| FLC | ITC | PCZ | VRC | AMB | CSF | |
| A. fumigatus akuBKU80 | 640.00 (>256.00/0) | 0.12 | 0.25 | 0.12 (ND/32) | 0.50 | 0.06 |
| LMDM-1030 | 20.00 (8.00/19) | 0.06 | 0.25 | 0.12 (ND/32) | 0.50 | 0.06 |
| LMDM-32 | 20.00 (8.00/19) | 0.03 | 0.06 | 0.03 (ND/48) | 0.50 | 0.06 |
Geometric means of at least 3 repetitions performed on different days. In parentheses are the MICs and diameters obtained by agar diffusion for fluconazole and voriconazole (Liofilchem MIC test strips/inhibition diameter, in millimeters). FLC, fluconazole; ITC, itraconazole; PCZ, posaconazole; VRC, voriconazole; AMB, amphotericin B; CSF, caspofungin; ND, not done.
FIG 3.

Agarose gel electrophoresis showing the detection of CYP51A transcripts. Lanes 1 and 2, PCR products using genomic DNAs; lanes 3 and 4, PCR amplification using cDNAs as the template. Lanes 1 and 3, A. fumigatus akuBKU80; lanes 2 and 4, LMDM-1030.
FIG 4.
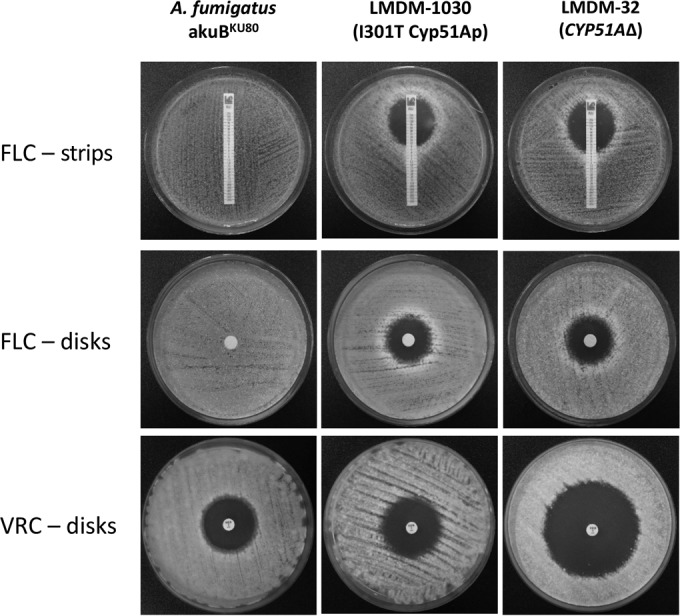
Diffusion susceptibility testing using fluconazole (FLC) disks and Liofilchem MIC test strips and voriconazole (VRC) disks for A. fumigatus LMDM-1030 (I301T Cyp51Ap mutant), A. fumigatus akuBKU80 (parental strain), and A. fumigatus LMDM-32 (CYP51AΔ).
CYP51A gene expression in LMDM-1030.
The construction transformed into LMDM-1030 carried a mutated CYP51A ORF with a 3′ UTR modification (hph insertion). The genomic integration of the construction could alter the CYP51A gene transcription into mRNA, producing a decreased azole MIC phenotype as observed in the CYP51A knockout strains (14). LMDM-1030 and LMDM-32 strains showed low fluconazole MICs. Thus, we decided to evaluate the CYP51A expression during hyphal growth in order to determine whether a loss of CYP51A expression might cause or contribute to the fluconazole MIC change. Total RNAs from the akuBKU80 and LMDM-1030 strains were extracted, and reverse transcription reactions were carried out. Afterwards, genomic DNA and cDNA from both strains were used as templates for PCRs performed with primers A1 and A10. These oligonucleotides hybridize areas surrounding the intron of the CYP51A gene. Hence, fragments of 350 bp and 425 bp were observed when cDNA and DNA from both strains were used as templates, confirming the presence of CYP51A mRNA in both strains (Fig. 3).
DISCUSSION
It is well known that A. fumigatus is intrinsically resistant to fluconazole and ketoconazole and normally susceptible to the other available azole drugs (5, 8). However, clinical secondary azole resistance was described and mostly associated with several amino acid substitutions in Cyp51Ap (4, 13, 14, 18 – 26). In 2005, Mellado et al. reported a CYP51AΔ A. fumigatus mutant which was azole hypersusceptible, confirming the linkage between CYP51A and azole resistance (14). It is also clear that each particular amino acid substitution in Cyp51Ap (or the combination of them) leads to a particular azole MIC pattern and that most of the reported CYP51A mutant strains showed itraconazole resistance, itraconazole-posaconazole cross-resistance, isavuconazole-voriconazole cross-resistance, or pan-triazole cross-resistance (4, 13, 14, 18 – 26). Despite the advances in elucidating the secondary azole resistance mechanisms in A. fumigatus, the molecular basis of intrinsic fluconazole resistance was never studied. The first hypothesis regarding this subject was proposed by Edlind et al., who studied the A. fumigatus Cyp51Ap sequence (9). They suggested that the Cyp51Ap I301 residue could be implicated in fluconazole resistance since the T315A substitution in C. albicans Erg11p produced a similar phenotype. Later, Diaz-Guerra et al. gave the first laboratory clue linking fluconazole resistance with Cyp51Ap. They described that amino acid substitutions at the G54 residue of Cyp51Ap led to itraconazole resistance but 4- to 5-fold fluconazole MIC reductions, possibly due to a better interaction between fluconazole and Cyp51Ap (13).
In this work, we obtained two A. fumigatus mutants, one harboring a I301T substitution in Cyp51Ap and the other a CYP51A deletion mutant. Both mutants showed a 32-fold decrease in fluconazole MIC. To establish that fluconazole susceptibility in the engineered A. fumigatus mutant is due to the I301T change and not due to the loss of CYP51A, the expression of this gene during hyphal growth was confirmed by reverse transcription. The MICs obtained for the other tested azole drugs also confirm that the I301T substitution is necessary and sufficient to explain fluconazole MIC reduction. The LMDM-1030 strain alone showed voriconazole, posaconazole, and itraconazole MICs similar to those obtained for its parental strain, mimicking C. albicans susceptibility patterns (pan-azole susceptibility) (27).
The precise manner in which the I301T substitution impacts fluconazole susceptibility could be explained taking into account that in A. fumigatus there are two homologous CYP51 genes (28). In both Cyp51p genes, four of the five residues most frequently linked with fluconazole resistance in C. albicans are conserved (Y132, T315, S405, G464, and R467) (10, 11). Consequently, these amino acids would not account for fluconazole resistance in A. fumigatus. The fifth residue (T315) is conserved only in Cyp51Bp, while in Cyp51Ap, it is naturally replaced (I301). The LMDM-1030 mutant harbors in both Cyp51p proteins a threonine, as in C. albicans; thus, both enzymes would be inhibited by fluconazole. The T315 residue in the C. albicans 14-α sterol demethylase is crucial for the correct enzyme-substrate and enzyme-drug interactions near the heme group (11, 29). Analogously, the A. fumigatus Cyp51Ap I301 residue is placed in the center the α-I loop (D280-Q312), which was proposed as essential for drug-enzyme interaction (30 – 35). Recently, Hargrove et al. reported A. fumigatus Cyp51Bp crystal structure complexes obtained with and without voriconazole. They confirmed that residue T315 (equivalent to I301 in Cyp51Ap) is part of the N-terminal portion of one of the substrate recognition sequence (SRS4) which showed fungus-specific features not observed in Cyp51p from other kingdoms. These data demonstrated the importance of SRS4 in the specific inhibition of sterol biosynthesis in fungi by azole drugs (31). Moreover, at the beginning of 2016, Liu et al. reported a three-dimensional (3D) structural model of A. fumigatus Cyp51Ap based on a crystal structure of the homologous Saccharomyces cerevisiae enzyme (Erg11p) (32). Itraconazole, voriconazole, and posaconazole were docked to wild-type and mutant Cyp51Ap, and their models demonstrate that the S297 residue (part of the αI helix) is adjacent to a heme group and would interact with ligands and azoles (32). When the Cyp51Ap αI helix is represented as a helical-wheel diagram, the I301 residue is the closest amino acid to S297 and would also interact with azoles (data not shown). The experimental data that we present in this work support the results obtained by Liu et al. and Edlind et al. (9, 32) and strongly implicate the Cyp51Ap I301 residue in the intrinsic resistance of A. fumigatus to fluconazole. Moreover, this knowledge may help to understand how the drugs interact with Cyp51Ap and in the development of new antifungals.
ACKNOWLEDGMENTS
This study was supported in part by the Science, Technology and Productive Innovation Ministry (MinCyT; Argentina) grant PICT2013/1571 to G.G.-E. C.D. and F.L. have a fellowship from CONICET (Argentina). D.M. has a fellowship from MinCyT (Argentina). M.S.C. has a postdoctoral fellowship from CONICET.
REFERENCES
- 1.Doligalski CT, Benedict K, Cleveland AA, Park B, Derado G, Pappas PG, Baddley JW, Zaas DW, Harris MT, Alexander BD. 2014. Epidemiology of invasive mold infections in lung transplant recipients. Am J Transplant 14:1328–1333. doi: 10.1111/ajt.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pappas PG, Alexander BD, Andes DR, Hadley S, Kauffman CA, Freifeld A, Anaissie EJ, Brumble LM, Herwaldt L, Ito J, Kontoyiannis DP, Lyon GM, Marr KA, Morrison VA, Park BJ, Patterson TF, Perl TM, Oster RA, Schuster MG, Walker R, Walsh TJ, Wannemuehler KA, Chiller TM. 2010. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET). Clin Infect Dis 50:1101–1111. doi: 10.1086/651262. [DOI] [PubMed] [Google Scholar]
- 3.Steinbach WJ, Marr KA, Anaissie EJ, Azie N, Quan SP, Meier-Kriesche HU, Apewokin S, Horn DL. 2012. Clinical epidemiology of 960 patients with invasive aspergillosis from the PATH Alliance registry. J Infect 65:453–464. doi: 10.1016/j.jinf.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Effron G, Dilger A, Alcazar-Fuoli L, Park S, Mellado E, Perlin DS. 2008. Rapid detection of triazole antifungal resistance in Aspergillus fumigatus. J Clin Microbiol 46:1200–1206. doi: 10.1128/JCM.02330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregson L, Goodwin J, Johnson A, McEntee L, Moore CB, Richardson M, Hope WW, Howard SJ. 2013. In vitro susceptibility of Aspergillus fumigatus to isavuconazole: correlation with itraconazole, voriconazole, and posaconazole. Antimicrob Agents Chemother 57:5778–5780. doi: 10.1128/AAC.01141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfaller MA, Diekema DJ, Ghannoum MA, Rex JH, Alexander BD, Andes D, Brown SD, Chaturvedi V, Espinel-Ingroff A, Fowler CL, Johnson EM, Knapp CC, Motyl MR, Ostrosky-Zeichner L, Sheehan DJ, Walsh TJ. 2009. Wild-type MIC distribution and epidemiological cutoff values for Aspergillus fumigatus and three triazoles as determined by the Clinical and Laboratory Standards Institute broth microdilution methods. J Clin Microbiol 47:3142–3146. doi: 10.1128/JCM.00940-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Tudela JL, Alcazar-Fuoli L, Mellado E, Alastruey-Izquierdo A, Monzon A, Cuenca-Estrella M. 2008. Epidemiological cutoffs and cross-resistance to azole drugs in Aspergillus fumigatus. Antimicrob Agents Chemother 52:2468–2472. doi: 10.1128/AAC.00156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh TJ, Anaissie EJ, Denning DW, Herbrecht R, Kontoyiannis DP, Marr KA, Morrison VA, Segal BH, Steinbach WJ, Stevens DA, van Burik JA, Wingard JR, Patterson TF. 2008. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis 46:327–360. doi: 10.1086/525258. [DOI] [PubMed] [Google Scholar]
- 9.Edlind TD, Henry KW, Metera KA, Katiyar SK. 2001. Aspergillus fumigatus CYP51 sequence: potential basis for fluconazole resistance. Med Mycol 39:299–302. doi: 10.1080/mmy.39.3.299.302. [DOI] [PubMed] [Google Scholar]
- 10.Lamb DC, Kelly DE, Schunck WH, Shyadehi AZ, Akhtar M, Lowe DJ, Baldwin BC, Kelly SL. 1997. The mutation T315A in Candida albicans sterol 14alpha-demethylase causes reduced enzyme activity and fluconazole resistance through reduced affinity. J Biol Chem 272:5682–5688. [DOI] [PubMed] [Google Scholar]
- 11.Marichal P, Koymans L, Willemsens S, Bellens D, Verhasselt P, Luyten W, Borgers M, Ramaekers FC, Odds FC, Bossche HV. 1999. Contribution of mutations in the cytochrome P450 14alpha-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology 145(Part 10):2701–2713. [DOI] [PubMed] [Google Scholar]
- 12.da Silva Ferreira ME, Kress MR, Savoldi M, Goldman MH, Hartl A, Heinekamp T, Brakhage AA, Goldman GH. 2006. The akuB(KU80) mutant deficient for nonhomologous end joining is a powerful tool for analyzing pathogenicity in Aspergillus fumigatus. Eukaryot Cell 5:207–211. doi: 10.1128/EC.5.1.207-211.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz-Guerra TM, Mellado E, Cuenca-Estrella M, Rodriguez-Tudela JL. 2003. A point mutation in the 14alpha-sterol demethylase gene cyp51A contributes to itraconazole resistance in Aspergillus fumigatus. Antimicrob Agents Chemother 47:1120–1124. doi: 10.1128/AAC.47.3.1120-1124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mellado E, Garcia-Effron G, Buitrago MJ, Alcazar-Fuoli L, Cuenca-Estrella M, Rodriguez-Tudela JL. 2005. Targeted gene disruption of the 14-alpha sterol demethylase (cyp51A) in Aspergillus fumigatus and its role in azole drug susceptibility. Antimicrob Agents Chemother 49:2536–2538. doi: 10.1128/AAC.49.6.2536-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pontecorvo G, Roper JA, Hemmons LM, MacDonald KD, Bufton AW. 1953. The genetics of Aspergillus nidulans. Adv Genet 5:141–238. [DOI] [PubMed] [Google Scholar]
- 16.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of filamentous fungi, approved standard M38-A2, 2nd ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 17.Clinical and Laboratory Standards Institute. 2010. Method for antifungal disk diffusion susceptibility testing of non-dermatophyte filamentous fungi. Approved guidelines. CLSI document M51-A. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 18.Bellete B, Raberin H, Morel J, Flori P, Hafid J, Manhsung RT. 2010. Acquired resistance to voriconazole and itraconazole in a patient with pulmonary aspergilloma. Med Mycol 48:197–200. doi: 10.3109/13693780902717018. [DOI] [PubMed] [Google Scholar]
- 19.Camps SM, van der Linden JW, Li Y, Kuijper EJ, van Dissel JT, Verweij PE, Melchers WJ. 2012. Rapid induction of multiple resistance mechanisms in Aspergillus fumigatus during azole therapy: a case study and review of the literature. Antimicrob Agents Chemother 56:10–16. doi: 10.1128/AAC.05088-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denning DW, Park S, Lass-Florl C, Fraczek MG, Kirwan M, Gore R, Smith J, Bueid A, Moore CB, Bowyer P, Perlin DS. 2011. High-frequency triazole resistance found in nonculturable Aspergillus fumigatus from lungs of patients with chronic fungal disease. Clin Infect Dis 52:1123–1129. doi: 10.1093/cid/cir179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howard SJ, Webster I, Moore CB, Gardiner RE, Park S, Perlin DS, Denning DW. 2006. Multi-azole resistance in Aspergillus fumigatus. Int J Antimicrob Agents 28:450–453. doi: 10.1016/j.ijantimicag.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 22.Howard SJ, Cerar D, Anderson MJ, Albarrag A, Fisher MC, Pasqualotto AC, Laverdiere M, Arendrup MC, Perlin DS, Denning DW. 2009. Frequency and evolution of azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg Infect Dis 15:1068–1076. doi: 10.3201/eid1507.090043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mann PA, Parmegiani RM, Wei SQ, Mendrick CA, Li X, Loebenberg D, DiDomenico B, Hare RS, Walker SS, McNicholas PM. 2003. Mutations in Aspergillus fumigatus resulting in reduced susceptibility to posaconazole appear to be restricted to a single amino acid in the cytochrome P450 14alpha-demethylase. Antimicrob Agents Chemother 47:577–581. doi: 10.1128/AAC.47.2.577-581.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Cuenca-Estrella M, Rodriguez-Tudela JL. 2004. Substitutions at methionine 220 in the 14alpha-sterol demethylase (Cyp51A) of Aspergillus fumigatus are responsible for resistance in vitro to azole antifungal drugs. Antimicrob Agents Chemother 48:2747–2750. doi: 10.1128/AAC.48.7.2747-2750.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellado E, Garcia-Effron G, Alcazar-Fuoli L, Melchers WJ, Verweij PE, Cuenca-Estrella M, Rodriguez-Tudela JL. 2007. A new Aspergillus fumigatus resistance mechanism conferring in vitro cross-resistance to azole antifungals involves a combination of cyp51A alterations. Antimicrob Agents Chemother 51:1897–1904. doi: 10.1128/AAC.01092-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verweij PE, Mellado E, Melchers WJ. 2007. Multiple-triazole-resistant aspergillosis. N Engl J Med 356:1481–1483. doi: 10.1056/NEJMc061720. [DOI] [PubMed] [Google Scholar]
- 27.Pfaller MA, Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mellado E, Diaz-Guerra TM, Cuenca-Estrella M, Rodriguez-Tudela JL. 2001. Identification of two different 14-alpha sterol demethylase-related genes (cyp51A and cyp51B) in Aspergillus fumigatus and other Aspergillus species. J Clin Microbiol 39:2431–2438. doi: 10.1128/JCM.39.7.2431-2438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother 42:241–253. doi: 10.1093/jac/42.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Debnath S, Addya S. 2014. Structural basis for heterogeneous phenotype of ERG11 dependent azole resistance in C. albicans clinical isolates. Springerplus 3:660. doi: 10.1186/2193-1801-3-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hargrove TY, Wawrzak Z, Lamb DC, Guengerich FP, Lepesheva GI. 2015. Structure-functional characterization of cytochrome P450 sterol 14alpha-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J Biol Chem 290:23916–23934. doi: 10.1074/jbc.M115.677310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu M, Zheng N, Li D, Zheng H, Zhang L, Ge H, Liu W. 2016. cyp51A-based mechanism of azole resistance in Aspergillus fumigatus: illustration by a new 3D structural model of Aspergillus fumigatus CYP51A protein. Med Mycol 54:400–408. doi: 10.1093/mmy/myv102. [DOI] [PubMed] [Google Scholar]
- 33.Oliveira C, Okay VTS, Melhem MS, Walderez SM, del Negro GM. 2013. The new mutation L321F in Candida albicans ERG11 gene may be associated with fluconazole resistance. Rev Iberoam Micol 30:209–212. doi: 10.1016/j.riam.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 34.van Nistelrooy JG, van den Brink JM, van Kan JA, van Gorcom RF, de Waard MA. 1996. Isolation and molecular characterisation of the gene encoding eburicol 14 alpha-demethylase (CYP51) from Penicillium italicum. Mol Gen Genet 250:725–733. [DOI] [PubMed] [Google Scholar]
- 35.Warrilow AG, Parker JE, Kelly DE, Kelly SL. 2013. Azole affinity of sterol 14alpha-demethylase (CYP51) enzymes from Candida albicans and Homo sapiens. Antimicrob Agents Chemother 57:1352–1360. doi: 10.1128/AAC.02067-12. [DOI] [PMC free article] [PubMed] [Google Scholar]