

Abstract
We characterized clinically occurring and novel mutations in the β subunit of RNA polymerase in Clostridium difficile (CdRpoB), conferring rifamycin (including rifaximin) resistance. The Arg505Lys substitution did not impose an in vitro fitness cost, which may be one reason for its dominance among rifamycin-resistant clinical isolates. These observations were supported through the structural modeling of CdRpoB. In general, most mutations lacked in vitro fitness costs, suggesting that rifamycin resistance may in some cases persist in the clinic.
TEXT
The nonabsorbed rifamycin antibiotic rifaximin has been considered an adjunctive therapy to reduce the recurrence of Clostridium difficile infection following vancomycin treatment (1, 2). Rifaximin, which is approved for the treatment of traveler's diarrhea, inhibits DNA transcription by selectively binding to the β subunit of RNA polymerase (RpoB). Substitutions in the rifamycin resistance-determining region (RRDR) of RpoB confer resistance to rifamycins, including rifaximin, in clinical isolates of C. difficile (3, 4). An arginine to lysine substitution at position 505 (i.e., Arg505Lys) in C. difficile (CdRpoB) is the most common mutation among rifamycin-resistant clinical isolates (3, 5, 6). Other mutations in clinical isolates also occur at His502, Ser488, and Ser550. However, it is unknown whether fitness costs influence the spectra of rifamycin resistance alleles among C. difficile isolates.
Fitness cost is a leading factor that affects the clinical prevalence of specific resistance alleles (7, 8). In the present study, we characterized clinically occurring and novel rifamycin resistance mutations in terms of their impacts on the growth and competitive fitness of C. difficile and by in silico structural modeling of the CdRpoB (Fig. 1).
FIG 1.

Model of CdRpoB with bound rifaximin. Mutational sites conferring rifaximin resistance are shown in red. Rifaximin is shown with yellow carbon atoms.
The rifamycin-susceptible C. difficile strains were CD43 and CD1679 (both epidemic ribotype 027) and were kindly provided by Scott Curry at the University of Pittsburgh. They were cultivated in brain heart infusion tryptone yeast (BHITY) broth or agar at 37°C in a Whitley A35 anaerobic workstation (Don Whitley Scientific). The MIC of rifaximin was defined as the lowest concentration of drug that prevented growth on BHITY agar (9). Spontaneous mutants were recovered by plating aliquots of overnight cultures onto selective agars containing rifaximin at 4× the MIC. Mutations were identified in an ∼200-bp PCR amplicon containing the RRDR (3). The competitive fitness (W) of rifaximin-resistant mutants was determined by pairwise competition between the wild-type parents and their respective derivative mutants (7, 8). Briefly, aliquots from overnight cultures of wild-type and mutant bacteria were coinoculated in BHITY broth at a 10:1 ratio (ca. 104:103 CFU/ml) and grown for 24 h. The numbers of mutant and wild-type bacteria at the start and at the end of the experiments were determined by plating onto selective agar containing 4× the rifaximin MIC and on nonselective BHITY agar (7, 8). W was calculated from ln[NR(24)/NR(0)]/ln[NS(24)/NS(0)], where NR(t) and NS(t) indicate the numbers of resistant and sensitive bacteria, respectively, at time t (0 or 24 h) (8). Doubling times in BHITY broth were calculated in GraphPad Prism 5 from automated optical density readings at 600 nm (OD600) over 48 h at 37°C in a BioTek 2 microplate reader (10). Effects on virulence were assessed in the hamster model of C. difficile infection as described previously (11) using a spore inoculum of ∼200 spores. Animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Texas at Arlington and adhered to the USDA Animal Welfare Act (9 CFR, parts 1 to 3). Using the RpoB sequence of C. difficile CD630, a homology model of CdRpoB was generated from the X-ray crystal structure of Escherichia coli RNA polymerase in complex with rifampin (PDB no. 4KMU) in the Schrödinger molecular modeling suite (12, 13). Changes in the relative binding affinities for rifaximin in the mutated CdRpoB model were calculated using the Prime MM-GBSA software in the Schrödinger molecular modeling suite (14). To assess the impact on RpoB DNA interaction, the DNA and C-chain RpoB from the Thermus thermophilus X-ray crystal structure (PDB no. 4GZY) were aligned with the CdRpoB homology model in the Schrödinger/Maestro alignment software (15). Next, the DNA subunit was transferred into the CdRpoB model and refined by restrained minimization to a convergence of heavy atom root mean square deviation (RMSD) of 0.6 Å. Further method details may be found in the supplemental material.
In both strains, rifaximin resistance arose at a frequency of 10−8, which is consistent with prior reports (16); rifaximin MICs against all mutants were >1,024 μg/ml, indicating that high-level rifaximin resistance is achievable in a single mutational step (Table 1). Most studies adopt a breakpoint of ≥32 μg/ml to signify rifamycin resistance (5, 17). Several mutants possessed clinically occurring mutations, including His502Asn, His502Tyr, Arg505Lys, Ser488Tyr, Asp492Tyr, Ser550Phe, and Ser550Tyr (3, 5, 6). We also identified previously unreported changes, including Ser507Leu, Gln489Leu, Gly510Arg, and Leu584Phe.
TABLE 1.
Impact of rifaximin resistance alleles on the fitness and growth of C. difficile
| Strain | MIC (μg/ml) | Substitution | Fitness (W)a | Doubling time (min) |
|---|---|---|---|---|
| CD43 (parent) | 0.125 | None | 1.00 | 97.3 ± 4.8 |
| CD43-D1 | >1,024 | Gln489Leu | 0.84 ± 0.05 | 89.4 ± 4.1 |
| CD43-D2 | >1,024 | Asp492Tyr | 0.80 ± 0.01 | 100.3 ± 9.2 |
| CD43-D3 | >1,024 | Asp492Tyr | 0.80 ± 0.08 | 118.2 ± 5.1 |
| CD43-D4 | >1,024 | Asp492Gly | 1.2 ± 0.12 | 92.3 ± 6.0 |
| CD43-A1 | >1,024 | His502Tyr | 1.20 ± 0.13 | NDb |
| CD43-A2 | >1,024 | His502Asn | 1.00 ± 0.13 | ND |
| CD43-D5 | >1,024 | Arg505Lys | 0.99 ± 0.008 | 95.3 ± 10.1 |
| CD43-D6 | >1,024 | Arg505Lys | ND | 110.0 ± 5.1 |
| CD43-D7 | >1,024 | Gly510Arg | 0.85 ± 0.12 | 90.3 ± 5.5 |
| CD43-D8 | >1,024 | Ser488Tyr | 0.85 ± 0.02 | ND |
| CD43-D9 | >1,024 | Ser550Tyr | 0.67 ± 0.05 | 104.2 ± 18.4 |
| CD43-D10 | >1,024 | Ser550Phe | 1.26 ± 0.13 | ND |
| CD1769 (parent) | 0.0625 | None | 1.00 | 97 ± 9.5 |
| CD1769-D1 | >1,024 | Asp492Tyr | ND | 98.9 ± 9.2 |
| CD1769-D2 | >1,024 | His502Arg | 0.91 ± 0.3 | 102.1 ± 10.7 |
| CD1769-D3 | >1,024 | Arg505Lys | 1.02 ± 0.15 | 102.0 ± 4.4 |
| CD1769-D4 | >1,024 | Ser507Leu | 0.57 ± 0.07 | 267.5 ± 37.1 |
| CD1769-D5 | >1,024 | Leu584Phe | 1.24 ± 0.07 | ND |
By convention, the fitness of the wild type is designated as 1. MICs were determined using two independent cultures in duplicates. A minimum of three independent replicates were performed to calculate W and doubling times.
ND, not determined.
With the exception of Ser507Leu, Asp492Tyr, and Ser550Tyr, most mutations did not impose fitness costs on C. difficile (Table 1). In vivo studies indicated that the clinically occurring Arg505Lys was as virulent as the wild type in terms of the time to mortality in the hamster model of C. difficile infection. Interestingly, the clinically occurring mutations Asp492Tyr and Ser550Tyr that imposed moderate (20%) and significant (33%) in vitro fitness costs did not appear to affect in vivo virulence (Fig. 2; see also Fig. S1 in the supplemental material). This may also imply that the hamster model of C. difficile infection may be inadequate to assess subtle differences in fitness costs due to its remarkable susceptibility to C. difficile (18).
FIG 2.
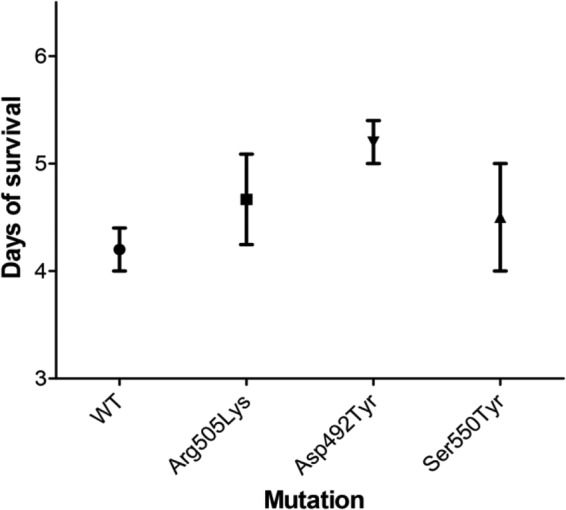
Comparison of virulence of rifaximin-resistant mutants in hamsters. WT, parent strain CD43; Arg505Lys, mutant strain CD43-D5; Asp492Tyr, mutant strain CD43-D3; Ser550Tyr, mutant strain CD43-D9. No significant differences exist between the means as determined by one-way analysis of variance (ANOVA) (P = 0.28). The number of animals in each group was n = 5 for WT, n = 6 for Arg505Lys, n = 5 for Asp492Tyr, and n = 4 for Ser550Tyr. CD43 mutants bearing His502Asn and His502Tyr strains were unavailable at the time of the experiments.
Modeling of the CdRpoB with bound rifaximin suggests that arginine-505 engages in an energetically favorable, Pi-stacking interaction with the polyene moiety (16Z, 18E) in the central scaffold of rifaximin (Fig. 3A). Therefore, a change to lysine-505 results in the loss of the Pi-stacking interaction, leading to rifamycin resistance. According to our computational predictions, a ca. 40 kcal/mol relative energetic cost to rifaximin binding occurs with lysine-505. From the DNA-bound model, arginine-505 interacts with the phosphate backbone via a charge-charge interaction (Fig. 3B). Due to the cationic nature of lysine-505, the charge-charge interaction with bound DNA is maintained. We suggest that the low fitness costs of Arg505Lys may correspond to minimal effects on DNA transcription. Similarly, histidine-502 mutations, including His502Asn and His502Tyr, are predicted to disrupt an active site hydrogen bond network involving glutamine-489 and a phenolic group on rifaximin (see Fig. S2A in the supplemental material). This leads to a conformational change in the rifaximin binding site and an energetic cost between 20 and 30 kcal/mol (see Fig. S2B and C). Based on our DNA-bound model, the histidine-502 residue does not directly engage DNA when bound, which may explain the low fitness cost in C. difficile (see Fig. S2D). The effects of other mutations on rifaximin binding are shown in Table S1 in the supplemental material.
FIG 3.

(A) CdRpoB model with rifaximin bound, highlighting the interaction with arginine-505. (B) CdRpoB model with DNA bound, highlighting the interaction with arginine-505.
The apparent lack of fitness costs for clinically occurring resistance alleles suggests that rifamycin-resistant mutants may in some cases persist in patients and clinical settings. Indeed, Curry et al. (3) reported the isolation of rifamycin-resistant C. difficile in patients who previously received a rifamycin antibiotic in the preceding 6 months prior to the onset of C. difficile infection. The isolates recovered contained the change Arg505Lys and the double substitution Ser488Thr/Arg505Lys or Arg505Lys/Ile548Met (see Fig. 1 for amino acid sites relative to rifaximin). Interestingly, from the initial study period from 2001 to 2002 to the second study period in 2005, Curry et al. (3) observed a 10% decrease in the proportion of rifamycin-resistant C. difficile isolates, which they suggested was due to a decrease in rifamycin exposure and increased infection control measures. Carman et al. (17) also reported the rise of rifaximin resistance during therapy, which resulted from two strains carrying either His502Tyr or His502Tyr/Pro496Ser substitutions. About 45 days after therapy, the two rifaximin-resistant isolates were still present at the time of recurrence. From our studies, we predict that the mutations in the Curry et al. (3) and Carman et al. (17) studies either lacked or were associated with low fitness costs. However, it is unclear why some rifamycin-resistant clinical isolates contain double resistance mutations in CdRpoB (3, 5, 19) and if any of these resistance changes may also be compensatory. Nonetheless, our study suggests that in some cases high-level rifamycin resistance in C. difficile may be maintained in clinical settings even without selection pressure.
Supplementary Material
ACKNOWLEDGMENTS
Molecular graphics figures were generated with the University of California, San Francisco Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
J.G.H. acknowledges the receipt of funding from the National Center for Complementary and Integrative Health at the National Institutes of Health under grant 5R01AT006732. U.T.D. acknowledges the receipt of funding from the University of Texas System Louis Stokes Alliance for Minority Participation (LSAMP) and the American Society for Microbiology Undergraduate Research Fellowship (ASM-URF). K.E.H. acknowledges the ITHS Rising Stars Career Development Program at the University of Washington, which is supported by grants UL1TR000423, KL2TR000421, and TL1TR000422 from the NIH National Center for Advancing Translational Sciences through the Clinical and Translational Science Awards Program (CTSA).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01137-16.
REFERENCES
- 1.Johnson S, Schriever C, Galang M, Kelly CP, Gerding DN. 2007. Interruption of recurrent Clostridium difficile-associated diarrhea episodes by serial therapy with vancomycin and rifaximin. Clin Infect Dis 44:846–848. doi: 10.1086/511870. [DOI] [PubMed] [Google Scholar]
- 2.Garey KW, Jiang ZD, Bellard A, Dupont HL. 2009. Rifaximin in treatment of recurrent Clostridium difficile-associated diarrhea: an uncontrolled pilot study. J Clin Gastroenterol 43:91–93. doi: 10.1097/MCG.0b013e31814a4e97. [DOI] [PubMed] [Google Scholar]
- 3.Curry SR, Marsh JW, Shutt KA, Muto CA, O'Leary MM, Saul MI, Pasculle AW, Harrison LH. 2009. High frequency of rifampin resistance identified in an epidemic Clostridium difficile clone from a large teaching hospital. Clin Infect Dis 48:425–429. doi: 10.1086/596315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang JS, Jiang ZD, Garey KW, Lasco T, Dupont HL. 2013. Use of rifamycin drugs and development of infection by rifamycin-resistant strains of Clostridium difficile. Antimicrob Agents Chemother 57:2690–2693. doi: 10.1128/AAC.00548-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Connor JR, Galang MA, Sambol SP, Hecht DW, Vedantam G, Gerding DN, Johnson S. 2008. Rifampin and rifaximin resistance in clinical isolates of Clostridium difficile. Antimicrob Agents Chemother 52:2813–2817. doi: 10.1128/AAC.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pecavar V, Blaschitz M, Hufnagl P, Zeinzinger J, Fiedler A, Allerberger F, Maass M, Indra A. 2012. High-resolution melting analysis of the single nucleotide polymorphism hot-spot region in the rpoB gene as an indicator of reduced susceptibility to rifaximin in Clostridium difficile. J Med Microbiol 61:780–785. doi: 10.1099/jmm.0.041087-0. [DOI] [PubMed] [Google Scholar]
- 7.Hurdle JG, O'Neill AJ, Ingham E, Fishwick C, Chopra I. 2004. Analysis of mupirocin resistance and fitness in Staphylococcus aureus by molecular genetic and structural modeling techniques. Antimicrob Agents Chemother 48:4366–4376. doi: 10.1128/AAC.48.11.4366-4376.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Neill AJ, Huovinen T, Fishwick CW, Chopra I. 2006. Molecular genetic and structural modeling studies of Staphylococcus aureus RNA polymerase and the fitness of rifampin resistance genotypes in relation to clinical prevalence. Antimicrob Agents Chemother 50:298–309. doi: 10.1128/AAC.50.1.298-309.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu X, Alam MZ, Feng L, Tsutsumi LS, Sun D, Hurdle JG. 2014. Prospects for flavonoid and related phytochemicals as nature-inspired treatments for Clostridium difficile infection. J Appl Microbiol 116:23–31. doi: 10.1111/jam.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, Hurdle JG. 2014. The Clostridium difficile proline racemase is not essential for early logarithmic growth and infection. Can J Microbiol 60:251–254. doi: 10.1139/cjm-2013-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherian PT, Wu X, Yang L, Scarborough JS, Singh AP, Alam ZA, Lee RE, Hurdle JG. 2015. Gastrointestinal localization of metronidazole by a lactobacilli-inspired tetramic acid motif improves treatment outcomes in the hamster model of Clostridium difficile infection. J Antimicrob Chemother 70:3061–3069. doi: 10.1093/jac/dkv231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molodtsov V, Nawarathne IN, Scharf NT, Kirchhoff PD, Showalter HD, Garcia GA, Murakami KS. 2013. X-ray crystal structures of the Escherichia coli RNA polymerase in complex with benzoxazinorifamycins. J Med Chem 56:4758–4763. doi: 10.1021/jm4004889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schrödinger. 2015. Biologics Suite 2015-3: Bioluminate, version 2.0. Schrödinger, LLC, New York, NY. [Google Scholar]
- 14.Schrödinger. 2015. Schrödinger Release 2015-3: Prime, version 4.1. Schrödinger, LLC, New York, NY. [Google Scholar]
- 15.Weixlbaumer A, Leon K, Landick R, Darst SA. 2013. Structural basis of transcriptional pausing in bacteria. Cell 152:431–441. doi: 10.1016/j.cell.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchese A, Salerno A, Pesce A, Debbia EA, Schito GC. 2000. In vitro activity of rifaximin, metronidazole and vancomycin against Clostridium difficile and the rate of selection of spontaneously resistant mutants against representative anaerobic and aerobic bacteria, including ammonia-producing species. Chemotherapy 46:253–266. doi: 10.1159/000007297. [DOI] [PubMed] [Google Scholar]
- 17.Carman RJ, Boone JH, Grover H, Wickham KN, Chen L. 2012. In vivo selection of rifamycin-resistant Clostridium difficile during rifaximin therapy. Antimicrob Agents Chemother 56:6019–6020. doi: 10.1128/AAC.00974-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hutton ML, Mackin KE, Chakravorty A, Lyras D. 2014. Small animal models for the study of Clostridium difficile disease pathogenesis. FEMS Microbiol Lett 352:140–149. doi: 10.1111/1574-6968.12367. [DOI] [PubMed] [Google Scholar]
- 19.Polivkova S, Krutova M, Petrlova K, Benes J, Nyc O. 2016. Clostridium difficile ribotype 176—a predictor for high mortality and risk of nosocomial spread? Anaerobe 40:35–40. doi: 10.1016/j.anaerobe.2016.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.