

Abstract
The mechanisms underlying the drug resistance of Leishmania spp. are manifold and not completely identified. Apart from the highly conserved multidrug resistance gene family known from higher eukaryotes, Leishmania spp. also possess genus-specific resistance marker genes. One of them, ARM58, was first identified in Leishmania braziliensis using a functional cloning approach, and its domain structure was characterized in L. infantum. Here we report that L. infantum ARM58 is part of a gene cluster at the telomeric end of chromosome 34 also comprising the neighboring genes ARM56 and HSP23. We show that overexpression of all three genes can confer antimony resistance to intracellular amastigotes. Upon overexpression in L. donovani, ARM58 and ARM56 are secreted via exosomes, suggesting a scavenger/secretion mechanism of action. Using a combination of functional cloning and next-generation sequencing, we found that the gene cluster was selected only under antimonyl tartrate challenge and weakly under Cu2+ challenge but not under sodium arsenite, Cd2+, or miltefosine challenge. The selective advantage is less pronounced in intracellular amastigotes treated with the sodium stibogluconate, possibly due to the known macrophage-stimulatory activity of this drug, against which these resistance markers may not be active. Our data point to the specificity of these three genes for antimony resistance.
INTRODUCTION
Parasitic protozoa of the genus Leishmania, order Trypanosomatida, are responsible for the various clinical manifestations of leishmaniasis. The disease is transmitted by the bite of sandflies, with 2 million new infections occurring per year, and occurs in 98 countries on five continents (1). Leishmania spp. exist in two morphologically distinct life cycle stages. In the transmitting sandflies, the flagellated and elongated promastigotes proliferate attached to the epithelium of the digestive tract until they reach a high density. A switch in the surface molecule composition then causes detachment, and the so-called metacyclic promastigotes are free to invade a mammalian host when the sandfly takes a blood meal. Inside the mammalian skin, the parasites are quickly taken up by antigen-presenting cells, such as dendritic cells, neutrophilic granulocytes, and macrophages, and establish themselves in these cells as small, ovoid, aflagellated amastigotes. The resulting destruction of infected macrophages causes inflammatory immune responses and the concomitant immune pathologies.
Depending on the infecting species and on the host's immune status, Leishmania-related pathologies range from localized, ulcerating skin lesions (cutaneous leishmaniasis [CL]) and diffuse cutaneous lesion formation (diffuse cutaneous leishmaniasis [DCL]) to mucocutaneous leishmaniasis (MCL) and, lastly, a generalized infection known as kala azar (visceral leishmaniasis [VL]). VL is invariably lethal in untreated cases, while MCL can also have lethal outcomes due to secondary infections of the nasopharyngeal area.
As there is no vaccine against Leishmania and the transmitting sandflies are not deterred by conventional bed nets, efforts to control the disease center on the chemotherapeutic treatment of infected people. Treatment options include pentavalent antimonial (stibogluconate [SbV]) agents, miltefosine, paromomycin, pentamidine, and amphotericin B. Even after 8 decades of use, SbV is still the first-line drug for the treatment of leishmaniasis in many countries where it is endemic.
SbV acts as a prodrug which needs to be reduced to the active antimonyl tartrate (SbIII) (2). Host cells and amastigotes but not promastigotes are able to reduce the prodrug to SbIII. SbV can also activate macrophages into producing microbicidal molecules (3), adding to its antiparasitic activity. Since the 1980s, there have been reports about the spread of resistance to antimonials in northeastern India, rendering SbV-based drugs useless in the ongoing control campaigns there (4 – 6).
SbV resistance mechanisms include (i) the downregulation of uptake transporters, such as AQP1 (7 – 9); (ii) the upregulation of ABC transporters, such as the P-glycoprotein MRPA in Leishmania (10, 11) or in macrophages (12); and (iii) the production of increased levels of trypanothione for the efflux or sequestration of SbIII (13, 14). An analysis of resistance marker expression in clinical isolates of Leishmania infantum from the Mediterranean Basin showed diverse patterns of expression and coexpression of the above-mentioned resistance genes and several others (15); among these are the SbIII/miltefosine resistance gene P299, which was identified by functional cloning from a L. infantum genomic DNA (gDNA) cosmid library under miltefosine selection (16).
Similarly, another antimony resistance gene, ARM58, was identified in L. braziliensis using a functional cloning approach (17). ARM58 is exclusively found in the genus Leishmania. However, a neighboring gene with a domain structure highly similar to that of ARM58, initially dubbed ARM58 related (ARM58rel), has orthologs in both Trypanosoma cruzi and T. brucei. Studies performed in L. infantum showed that both proteins are composed of four related domains of unknown function collectively referred to as DUF1935. In ARM58, the first and the second DUF1935 domains are important for full function of the protein, while the third domain, containing a putative transmembrane domain, is essential for conferring SbIII resistance. Replacement of the third DUF1935 domain with the corresponding third DUF1935 domain of ARM58rel caused a loss of function. Conversely, the third domain of ARM58 conferred antimony resistance activity to ARM58rel (18). In that study, an mCherry-ARM58 fusion protein was localized near the flagellar pocket. ARM58, ARM58rel, and the small 23-kDa heat shock protein HSP23 are neighboring genes near the telomeric end of chromosome 34 (L. infantum) or chromosome 20 (L. braziliensis). This is significant because HSP23-null mutants are hypersensitive to SbIII in vitro, indicating a role for HSP23 in SbIII tolerance (19).
In this paper, we show that all three genes in the telomeric cluster, ARM58, ARM58rel, and HSP23, are able to confer antimony resistance to intracellular L. donovani amastigotes when they are overexpressed. ARM58 and ARM58rel, which is named ARM56 in the following, are secreted via exosomes when they are overexpressed, hinting at a sequestration mechanism at the heart of ARM58/ARM56-mediated antimony resistance.
MATERIALS AND METHODS
Parasite strains and isolates.
Leishmania infantum clone 35.11 was derived from isolate MHOM/FR/91/LEM and was provided by A. Sulahian (20). L. donovani 1SR is a laboratory strain and was a gift from D. Zilberstein (21). Trypanosoma cruzi strain Y and the HG39 cell line were gifts from T. Jacobs.
Parasite cultivation.
Promastigotes were grown at 25°C in supplemented M199 medium as described previously (16). Recombinant promastigotes were cultured in the presence of G418 (50 μg ml−1; Carl Roth) or nourseothricin (ClonNAT; 150 μg ml−1; Werner Bioreagents). Axenic amastigotes were grown at 37°C with 5% CO2 in supplemented M199 medium (pH 5.5) as described previously (22).
Trypanosoma cruzi strain Y parasites were grown intracellularly in HG39 host cells at 37°C with 5% CO2 in modified RPMI medium (Sigma-Aldrich), with 5% heat-inactivated fetal calf serum (FCS), 2 mM glutamine, 10,000 U penicillin, and 10 mg ml−1 streptomycin. Routine passage was performed every 3 to 4 days by using sanguineous trypomastigotes released into the supernatant of infected HG39 cells to infect new HG39 cells. Parasite numbers were determined by harvesting sanguineous trypomastigotes from the host cell supernatant, inactivation with ProClin 300 preservative (Sigma-Aldrich), and counting using a CASY cell counter (Roche).
In vitro infections.
Bone marrow-derived macrophages (BMMs) were isolated from the femurs of C57BL/6 mice and differentiated in Iscove's modified Dulbecco's medium (IMDM) supplemented with 10% heat-inactivated FCS, 5% horse serum, and 30% L929 cell-conditioned medium (23). After differentiation, the BMMs were harvested, washed, and seeded into 6-well plates (Sarstedt) at a density of 6 × 105 cells per well. The macrophages were incubated for 48 h at 37°C in 5% CO2 to allow adhesion. Adherent BMMs were infected with stationary-phase promastigotes (24) at a multiplicity of infection (MOI) of 5:1. After 4 h of incubation at 37°C in IMDM supplemented as described above, free parasites were washed off with phosphate-buffered saline (PBS), and incubation was continued for another 72 h in IMDM at 37°C in 5% CO2. At 24 h postinfection, 160 μg ml−1 of sodium stibogluconate or 30 μM miltefosine (Sigma-Aldrich) was added. Control cells were left untreated. After the medium was removed, the gDNA of the infected cells was isolated using a mag kit (LGC Group, Berlin, Germany). The quantification of parasites was performed by a quantitative real-time PCR (qPCR) targeting the parasite and host cell actin-coding genes with a dually labeled probe and total parasite/host gDNA as the template (64). The parasite load (in percent) was defined as the ratio of the amount of parasite actin DNA to the amount of mouse actin DNA, with the value for the vector control at 24 h being set at 100%. For drug treatment experiments, the relative parasite load (in percent) was defined as the parasite load after treatment as a percentage of the parasite load in untreated cells.
Electrotransfection.
Electrotransfection of Leishmania promastigotes was carried out as previously described (22). Briefly, late-log-phase promastigotes were harvested and washed twice in ice-cold PBS and once in electroporation buffer (25). Cells were then suspended at a density of 1 × 108 ml−1 in electroporation buffer. Plasmid or cosmid DNA (20 to 50 μg) was mixed with 0.4 ml of the cell suspension, and the mixture was immediately subjected to electroporation, using a Bio-Rad Gene Pulser apparatus, with three pulses at 3.750 V/cm and 25 μF in a 4-mm electroporation cuvette. Mock transfection (without plasmid) was performed identically to obtain negative-control strains for antibiotic selection. Following electroporation, cells were kept on ice for 10 min before they were diluted in 10 ml of supplemented medium 199. After 24 h, G418 (50 μg ml−1) or nourseothricin (150 μg ml−1) was added to select for recombinant cell populations.
Drug selection and recovery of cosmid DNA.
Dose inhibition experiments for cadmium acetate (Cd2+), copper acetate (Cu2+), SbIII, miltefosine, and sodium arsenite (AsIII) were performed by seeding promastigotes at 5 × 105 ml−1 in supplemented medium M199 containing the toxic compounds at various concentrations. The cell density was monitored after 72 h and plotted against the toxic compound concentration. The 50% inhibiting concentrations (IC50s) were established graphically by plotting the log10 concentration against the cell density and determining the point of intersection with a line where y was equal to 50%.
L. infantum promastigotes transfected with the L. infantum genomic DNA cosmid library (16) were seeded at a density of 1× 106 cells ml−1 in medium with the respective compounds at the IC50. Untreated populations were propagated as controls. Parasites were passaged into fresh medium with the chemicals at 2- to 3-day intervals for 34 days. Selected parasites were harvested for cosmid isolation.
Late-stationary-phase promastigotes transfected with the L. infantum genomic DNA cosmid library were added to 5 × 107 BMMs at a multiplicity of infection of 8:1 in a vented T25 flask (Sarstedt). After 4 h, free parasites were removed. At 24 h postinfection, 160 μg ml−1 sodium stibogluconate (SbV) or 35 μM miltefosine (IC50) was added. At 72 h postinfection, the infected cells were washed twice with prewarmed PBS. Then, 3 ml of 0.01% SDS in PBS, prewarmed to 37°C, was added to each flask. After 10 min at 37°C in 5% CO2, 2 volumes of supplemented M199 medium was added to each flask. The cell lysates were spun down at 4°C and 1,250 × g for 10 min. The supernatant was discarded, while the pellet was resuspended in 2 ml of medium M199 and passed three times through a 0.4-mm hypodermic needle. The released amastigotes were then seeded into 10 ml supplemented medium M199 containing 50 μg ml−1 G418 for the maintenance of cosmids and kept at 25°C for conversion back into promastigotes. This selection regimen was repeated three times.
Cosmid DNA was isolated from Leishmania promastigotes by alkaline lysis following the protocol for plasmid DNA minipreparation (26). After phenol-chloroform-isoamyl alcohol (25:24:1) extraction, ammonium acetate was added to 0.45 M, the cosmid DNA was precipitated by addition of 2.5 volumes of ethanol, and the pellet was dissolved in Tris-EDTA (TE; pH 8.0) buffer for 24 h. The DNA was precipitated again by addition of 2.5 volumes of ethanol and finally dissolved in TE buffer.
Electroporation of Escherichia coli.
Approximately 100 ng of cosmid DNA recovered from Leishmania was mixed on ice with Escherichia coli XL1-Blue electroporation-competent cells (Life Technologies). Electroporation was performed following the manufacturer's protocol, and transformed bacteria were plated on LB agar under ampicillin selection.
Selected cosmids were subjected to next-generation sequencing in a MiSeq sequencer (Illumina). Library preparation was performed with a Nextera XT library kit (Illumina) following the manufacturer's protocol.
Sequence reads in the fastq format were aligned to the L. infantum genome (DNA) sequence (TriTrypDB-26_LinfantumJPCM5_Genome.fasta) using the Assembler module of the MacVector software package and the Bowtie (version 2.0) algorithm. Settings were fast alignment, end to end, 6 bases no gap, 4 threads, paired-end alignment, and an insert size of 200 to 1,500 bp.
Dose inhibition experiments.
Dose inhibition curves for antimonyl tartrate (catalog no. 383376; Sigma-Aldrich) were carried out as previously described (18). Briefly, promastigotes were seeded at 5 × 105 ml−1 in supplemented medium M199 with various concentrations of the drug. After 72 h, the cell density was determined using a CASY cell counter (Roche). Cell densities were normalized against the density of untreated cells.
Plasmids.
L. infantum ARM58 (L. infantum J34.0220), L. infantum ARM58rel (=ARM56, J34.0210), and the domain-swapping (DS) variants ARM58-DS and ARM56-DS (18) were expressed in the pCLN plasmid. HSP23 was expressed in the pCLS plasmid (19). Point mutations in ARM58 were created using a previously described strategy (27). A list of the primers used for targeted mutagenesis can be found in Table S1 in the supplemental material.
Recombinant protein expression in E. coli and protein purification.
The expression of ARM58 and ARM56 was done as described previously (28). The (His)10-tagged proteins in the bacterial lysates were purified by metal chelate affinity chromatography as described previously (29).
Immunization and antibody preparation.
The purified proteins were used to immunize laying hens. The immunization and the purification of IgY antibodies have been described previously (30). The purified antibodies were tested against recombinant ARM58 and ARM56 and against lysates of L. donovani and L. infantum. Immunization of laying hens was performed in accordance with §10a of the German Animal Protection Law and registered with the Amt für Gesundheitlichen Verbraucherschutz, Behörde für Umwelt und Gesundheit, Freie und Hansestadt Hamburg.
Western blot analysis.
Discontinuous SDS-PAGE and transfer by Western blotting were performed following standard protocols. Membranes were treated with blocking buffer (5% milk powder and 0.1% Tween 20 in Tris-buffered saline), with IgY raised against ARM58 (1:200) and ARM56 (1:400) in blocking buffer, and with anti-chicken IgY-alkaline phosphatase (1:2,000; Dianova) in blocking buffer as the secondary antibody. The blots were developed with Nitro Blue Tetrazolium chloride (Roth) and 5-bromo-4-chloro-3-indolyl phosphate (Roth).
Nondenaturing PAGE.
Nondenaturing protein extraction, nondenaturing gradient gel electrophoresis, and Western blotting of the gels were performed as described previously (31, 32). Briefly, 1 × 107 L. donovani promastigotes were harvested by centrifugation, washed twice with cold PBS, and resuspended in 40 μl extraction buffer (15% glycerol, 0.5 mM 1,10-phenanthroline, 10 mM Tris-HCl, pH 8.0, 70 mM KCl). After three freeze-thaw cycles, a small amount of 2-mm ceramic beads (<10 beads) was added to the tubes. After vigorous shaking for 20 s, the cell lysates were subjected to centrifugation (16,000 × g, 10 min, 4°C) and the supernatant, which contained the soluble protein fraction, was mixed (5:1, vol/vol) with loading buffer (50% glycerol, 0.1% bromophenol blue). The samples were run on a 4 to 18% polyacrylamide (2.5 to 6% glycerol) gradient gel in 0.5× Tris-borate-EDTA buffer (26) (24 h, 20 V/cm, 4°C). Following this, the gel was incubated at 60°C in transfer buffer (48 mM Tris, 39 mM glycine, 0.5% SDS, 20% methanol, 10 mM dithiothreitol) for 30 min, followed by transfer by Western blotting and immunological detection.
Cell fractionation.
Leishmania promastigotes were fractionated according to a published protocol (28, 33). Briefly, 108 stationary-growth-phase L. infantum promastigotes were washed three times in 15 ml MES (morpholineethanesulfonic acid) buffer (20 mM MOPS [morpholinepropanesulfonic acid], pH 7.0, 0.25 M saccharose, 3 mM EDTA). After the last centrifugation, the cells were suspended at 109 cells/ml in MES buffer and mixed with an equal volume of MES with 2 mg ml−1 digitonin, 1 μM phenylmethylsulfonyl fluoride (PMSF), and 0.5 mM 1,10-phenanthroline, followed by incubation at room temperature for 5 min. The samples were centrifuged at 10,000 × g for 5 min at 4°C. The supernatant was collected as the cytoplasmic fraction. The pellet was resuspended in 0.1 ml of MES with 20 mM sodium phosphate, pH 7.0, 3 mM EDTA, and 2 mg ml−1 digitonin and subjected to sonication (3 s, 50 W). The samples were centrifuged for 5 min at 10,000 × g, and the supernatant was collected as the intermediate fraction. The pellet was resuspended in 20 mM sodium phosphate, pH 7.0, and collected as the mitochondrial fraction. Samples were precipitated with 4 volumes of cold acetone for 2 h at −20°C. The samples were centrifuged at 16,000 × g for 10 min at 4°C. The supernatant was discarded, and the pellet was left to dry. The samples were then solubilized in SDS sample buffer and subjected to SDS-PAGE followed by Western blotting.
Secretome analysis.
Secreted proteins were isolated following the method described previously (34). Briefly, 2 × 108 late-log-phase L. donovani promastigotes (pCLN and pCLNARM58) were washed thrice in ice-cold PBS, seeded into 4 ml of PBS solution with 5% saccharose, and kept at 26°C or 37°C for 2 h. The cells were then sedimented by centrifugation (500 × g, 4°C) and resuspended in 200 μl PBS with the protease inhibitors 1,10 ortho-phenanthroline (10 μM) and PMSF (5 μM). The supernatant was filtered through a 0.20-μm-pore-size membrane and concentrated using an Amicon filter system (molecular mass cutoff, 100,000 Da; Millipore). The concentrated secretomes and the cell pellets were stored at −80°C.
Exosome isolation.
Exosome isolation was performed by a modification of a previously published protocol (35). Late-log-phase L. donovani promastigotes (1.2 × 109) were washed thrice in ice-cold PBS, seeded into 15 ml of 5% sucrose in PBS, and kept at 37°C for 2 h. The cells were then sedimented by centrifugation at 500 × g (4°C). The pellet was resuspended in PBS and analyzed microscopically to exclude cell destruction. The medium was filtered (pore size, 0.20 μm), and exosomes were concentrated (molecular mass cutoff, 100,000 Da; Amicon Ultra-15 centrifugal filter devices; Millipore). The concentrated exosomes were loaded on a sucrose cushion (2 ml of ice-cold PBS over 750 μl of ice-cold 1 M sucrose, 20 mM Tris-HCl, pH 7.4) in a 5.2-ml Beckman Coulter Ultra-Clear tube. The tubes were subjected to ultracentrifugation for 2 h at 100,000 × g (4°C) and washed with ice-cold PBS, followed by centrifugation in 1.5-ml tubes (Microfuge tube; Beckman Coulter) for 1 h at 125,000 × g (4°C). The exosomes were resuspended in 20 mM Tris-HCl (7.4) and stored at 4°C.
Trypsin digestion.
Verification that the exosomes were intact was performed by a modification of a previously published protocol (36, 37). After centrifugation, the intact exosomes were suspended either in 20 mM Tris-HCl (pH 7.4) with 40 μg ml−1 trypsin (Promega) or in Tris-HCl (pH 7.4) with 0.1% Triton X-100 and 40 μg ml−1 trypsin. The samples were then incubated at 37°C for 2 h. Trypsin was inactivated by adding PMSF at a final concentration of 22 mM. The sample was mixed vigorously for 10 s, followed by precipitation with 4 volumes of cold acetone for 2 h at −20°C. The samples were centrifuged at 16,000 × g for 10 min at 4°C. The supernatant was discarded, and the samples were air dried. The samples were then subjected to SDS-PAGE and Western blotting.
Transmission electron microscopy of promastigotes.
L. donovani promastigotes (2.5 × 108) were washed three times with PBS, centrifuged (600 × g) for 8 min at 4°C, and afterwards fixed for 30 min with 2.5% glutaraldehyde (Electron Microscopy Sciences) and 4% paraformaldehyde (Electron Microscopy Sciences) in PBS. The cells were washed with PBS three times for 5 min each time and postfixed with 2% OsO4 in PBS for 40 min on ice in the dark. Afterwards the cells were washed two times with water and treated with 0.5% uranyl acetate in water for 30 min. After rinsing thoroughly with water, the cells were dehydrated at room temperature in 50%, 60%, 70%, 80%, and 95% ethanol for 5 min each and then three times in 100% ethanol for 10 min each time. The cells were immersed overnight at room temperature in an Epoxy resin-ethanol mix (50%, vol/vol; catalog number 8619.2; Roth) and finally embedded in fresh 100% Epoxy resin. After polymerization at 60°C for 48 h, the embedded cell pellet was sectioned into 60-nm sections using a Leica EM UC7 ultramicrotome, and sections were collected on 300-mesh copper grids (Plano GmbH). The sections were examined with a Tecnai Spirit transmission electron microscope at 80 kV (FEI).
Transmission electron microscopy of exosomes.
After the second centrifugation of the exosomes (see above), the samples were fixed in PBS with 4% paraformaldehyde. The sample was incubated for 5 min on Formvar-coated carbon grids (Plano) and washed once with distilled water. The samples were given contrast with 0.5% uranyl acetate for 5 min. Transmission electron microscopy of Leishmania promastigotes was performed as described previously (38).
Flow cytometry with SYTOX.
L. donovani promastigotes were washed twice in ice-cold PBS and fixed in 70% ethanol for 30 min at 4°C. The cells were sedimented by centrifugation at 500 × g (4°C) and washed with 1 ml 50 mM EDTA. The cells were centrifuged again and resuspended in 1 ml PBS with 20 μg ml−1 RNase A. The samples were incubated for 20 min at 37°C. After incubation, the samples were centrifuged at 13,000 × g for 2 min and the pellet was resuspended in 500 μl citrate buffer (45 mM MgCl2, 30 mM sodium citrate, 20 mM MOPS, pH 7, 0.1% Triton X-100) with 0.3 μl SYTOX (5 mM; Life Technologies). The samples were incubated for 20 min at 26°C in the dark and washed twice with PBS with 5% fetal calf serum. The samples were centrifuged at 13,000 × g for 4 min (4°C), and the pellet was resuspended in 1 ml of PBS and 4% paraformaldehyde (PFA). The stained samples were subjected to fluorescence-activated cell sorting (FACS) analysis (excitation λ = 488 nm, emission λ = 515 nm [fluorescein isothiocyanate {FITC}]) using a BD Accuri flow cytometer. Ten thousand events were analyzed using FlowJo (version 10) software.
Flow cytometry with PI-annexin V.
L. donovani promastigotes (2 × 107) were washed twice in ice-cold PBS and resuspended in 1 ml binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 5 mM CaCl2) containing 10 μg ml−1 propidium iodide (PI; Sigma-Aldrich) and annexin V (Life Technologies) following the manufacturer's recommendations. The samples were incubated at room temperature for 10 min in the dark, after which the cells were centrifuged and resuspended in 1 ml PBS with 4% paraformaldehyde. The stained cells were subjected to FACS analysis as described above (excitation λ = 488 nm, emission λ = 515 nm with FITC and emission λ = 617 nm with PI).
Indirect immunofluorescence.
Log-phase promastigotes (1 × 107 cells) were sedimented by centrifugation at 750 × g, washed twice with PBS, and suspended in 1 ml PBS. Aliquots (3 × 106 cells) were applied on glass microscope slides. After drying, the cells were fixed for 10 min in ice-cold methanol and air dried. The slides were washed (0.1% Triton X-100 in PBS), followed by incubation in permeabilization solution (0.1% Triton X-100, 50 mM NH4Cl in PBS) for 10 min. The slides were then washed three times with washing buffer followed by incubation with blocking solution (2% bovine serum albumin, 0.1% Triton X-100 in PBS). The slides were then incubated for 1 h with anti-ARM58 or anti-ARM56 IgY and antitubulin antibody (mouse) diluted 1:200, 1:100, or 1:4,000 in blocking solution. The slides were washed thrice and then incubated for 1 h with anti-chicken immunoglobulin coupled to FITC (1:250; Dianova), anti-mouse IgG coupled to Alexa Fluor 594 (1:250; Invitrogen), and 4′,6-diamidino-2-phenylindole (DAPI; 1:100). The slides were washed thrice and then covered with Mowiol mounting medium and coverslips. The slides were left to harden for 24 h at 4°C. Fluorescence microscopy was performed on a Life Technologies EVOS FL Auto cell imaging system.
RESULTS
Detection of ARM58.
Anti-ARM58 IgY showed a specific recognition of the (His)10-ARM58 protein expressed in E. coli (Fig. 1A, lane 1) as a band with an apparent molecular mass of 80 kDa, exceeding the hypothetical mass by 22 kDa. In the promastigote lysate, a band of the same apparent molecular mass was also observed (Fig. 1A, lane 3). The band increased in intensity under ARM58 overexpression and was mostly soluble (Fig. 1A, lanes 7 to 10). Exposure to SbIII did not alter the abundance of ARM58.
FIG 1.
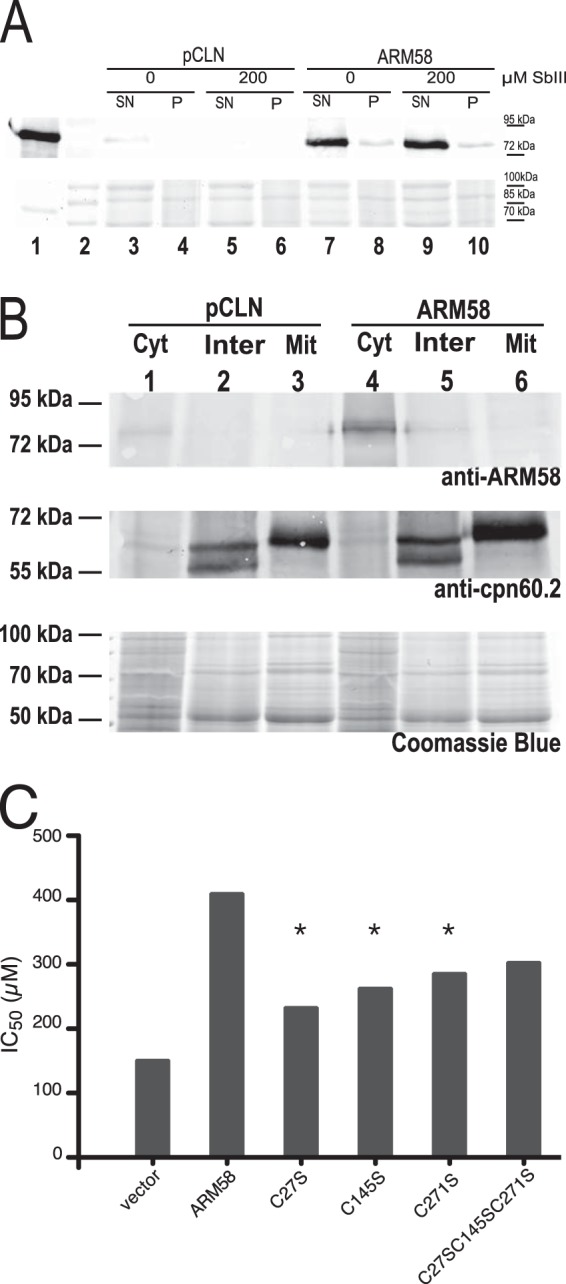
(A) Detection of ARM58 as a recombinant protein and in lysates of L. infantum transfected with pCLN or AMR58. Promastigotes were challenged with 200 μM trivalent antimony. After 72 h the cells were lysed and separated into the soluble supernatant (SN) and the insoluble pellet (P). After SDS-PAGE and Western blotting, ARM58 was detected with anti-ARM58 IgY and rabbit anti-chicken immunoglobulin-alkaline phosphatase. Coomassie blue staining (bottom) was done in parallel as a loading control. Lane 2, marker. (B) Detection of ARM58 in the cytoplasmic (Cyt), intermediate (Inter), or mitochondrial (Mit) fractions; anti-CPN60.2 was used as a control for mitochondrial localization. Coomassie staining was performed in parallel as a loading control. (C) Determination of the IC50 of SbIII for L. infantum promastigotes overexpressing ARM58 with the C27S, C145S, and/or C271S mutation. Promastigotes were challenged with different concentrations of SbIII for 72 h, and the IC50 was determined. *, P < 0.05 (n = 4).
ARM58 contains a putative transmembrane domain in the third DUF1935. To test whether ARM58 associates with the membrane fraction, we next performed cell fractionation following a protocol involving a two-step digitonin lysis of promastigotes, yielding cytoplasmic, intermediate, and mitochondrial fractions, with the mitochondrial fractions including membrane-associated proteins (33). Anti-CPN60.2 was used as a marker for the mitochondrial fraction (28). While CPN60.2 was mostly found in the mitochondrial fraction, with moderate amounts being found in the intermediate fraction, ARM58 was detected exclusively in the cytosolic fraction both in strains carrying pCLN and in strains carrying ARM58 (Fig. 1B). This finding excludes the possibility of a membrane association for ARM58.
Cysteine-to-serine exchanges in ARM58.
It is known that AsIII and SbIII ions can bind to cysteine side chains. For instance, it was shown that the ArsA protein of Escherichia coli forms a triple coordinated complex between AsIII or SbIII and three cysteine residues (39). Comparing the sequences of ARM58 and ARM56, we detected conserved cysteine residues in all four DUF1935 domains. To test whether they are involved in SbIII resistance, we performed targeted Cys-to-Ser mutagenesis at three sites in ARM58. The mutations were C27S, C145S, and C271S, which are located in the first three domains of DUF1935, respectively (DUF1935.1, DUF1935.2, and DUF1935.3, respectively). We expressed the mutant genes as episomes in L. infantum promastigotes and challenged them with various concentrations of SbIII. After 72 h, the cell densities were recorded and normalized against the density of untreated cells to determine the 50% inhibitory concentration (IC50).
Overexpression of ARM58 caused tolerance to SbIII higher than that in the control strain, confirming earlier results (18). The ARM58 transgenes with single cysteine-to-serine exchanges (C27S, C145S, C271S) showed intermediate effects, with the strongest effect being caused by the C27S mutation, indicating a partial contribution of the cysteine residues to the ARM58 function. However, the substitution of all three cysteine residues did not further reduce the ARM58 function (Fig. 1C), arguing that binding of SbIII to Cys27, Cys145, and Cys271 is not at the core of the ARM58 function in drug resistance.
Infection and treatment in macrophages.
ARM58, ARM56, and HSP23 are neighboring genes in all Leishmania species (L. infantum 34.0220, 34.0210, and 34.0230, respectively). These three genes were isolated as part of the same cosmid after SbIII selection of L. braziliensis promastigotes, with ARM58 being identified to be the gene responsible for SbIII resistance and ARM56 (ARM58rel) lacking this function (17). It was recently shown that HSP23 also contributes to the SbIII resistance of promastigotes (19). We therefore decided to test all three genes for their ability to confer resistance against SbV and miltefosine to intracellular amastigotes. Specifically, we compared the overexpression of ARM56, ARM58, and HSP23 and the coexpression of both ARM58 and ARM56 for their ability to confer resistance to SbV compared with that in control parasites. BMMs were infected with the overexpressing parasites or controls. Treatment was started 24 h after infection. Relative parasite loads were evaluated at 72 h postinfection. The results are shown in Fig. 2A. Compared with the vector controls, parasites overexpressing ARM58 (P < 0.01), ARM56 (P < 0.001), and HSP23 (P < 0.05) showed higher parasite loads after treatment, indicating a protective effect due to the overexpression. While the effect of ARM58 was described earlier, ARM56-mediated SbV resistance was not expected from the earlier analysis using promastigotes and SbIII (18). For this reason, we changed the old designation, ARM58rel (where rel indicates related), to ARM56. HSP23 also conferred resistance upon overexpression, showing that the telomeric end of chromosome 34 in Old World leishmania carries a cluster of antimony resistance genes. In spite of this, the combined overexpression of both ARM58 and ARM56 has no synergistic effect, indicating that both gene products have similar functionalities in infected macrophages.
FIG 2.
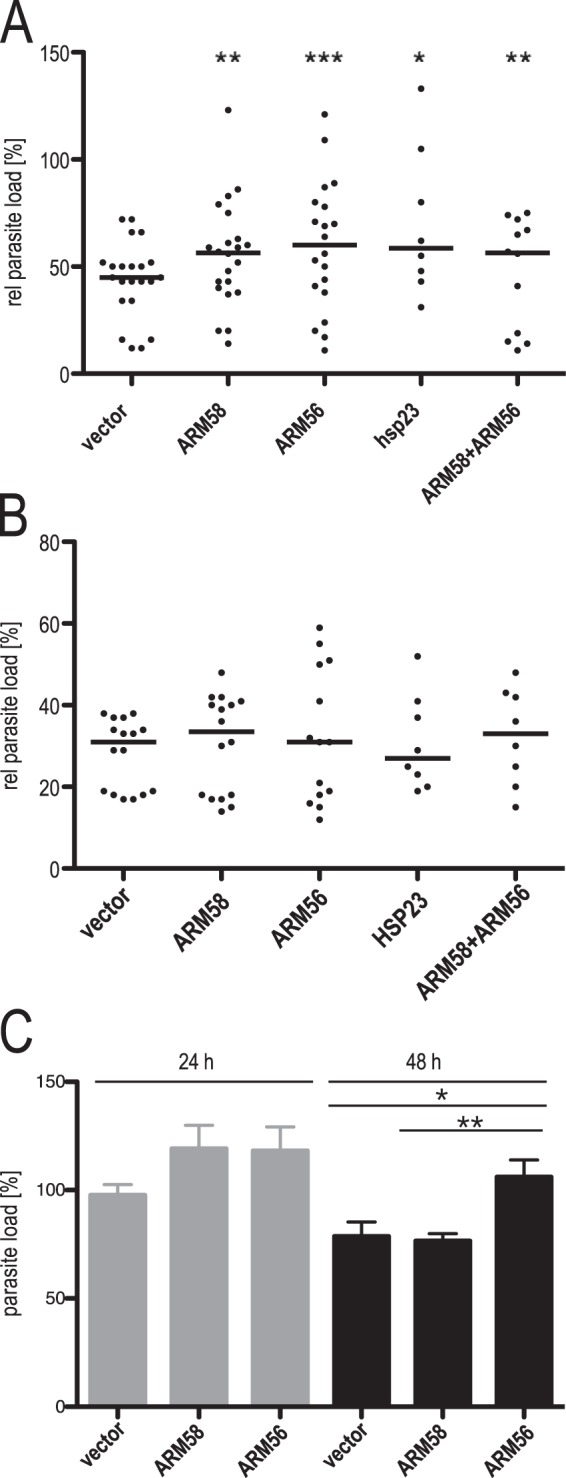
(A) In vitro infection of bone marrow-derived macrophages and treatment with sodium stibogluconate. L. donovani parasites transfected with the vector, ARM58, ARM56, HSP23, or the ARM58-ARM56 transgene were used to infect BMMs at an MOI of 5:1. After 24 h, 160 μg ml−1 sodium stibogluconate was added and incubation was continued for 48 h. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (n = 6 to 24). Parasite load quantification was done by qPCR as described in Materials and Methods; the relative (rel) parasite load was defined as the parasite load after treatment as a percentage of the parasite load in untreated cells. (B) In vitro infection of bone marrow-derived macrophages and treatment with miltefosine. L. donovani parasites transfected with the vector, ARM58, ARM56, HSP23, or the ARM58-ARM56 transgene were used to infect BMMs at a multiplicity of infection of 5:1. At 24 h after infection, miltefosine was added to the cultures at 30 μM, and incubation was continued for 48 h. Parasite load quantification was done by qPCR as described in Materials and Methods; the relative parasite load was defined as the parasite load after treatment as a percentage of the parasite load in untreated cells (n ≥ 6). (C) Parasite load quantification after in vitro infection of BMMs with L. donovani with pCLN and the ARM58 and ARM56 transgenes. Parasite load quantification was done by qPCR as described in Materials and Methods, with the value for control parasites at 24 h being set equal to 100%. *, P < 0.05; **, P < 0.01 (n = 8).
In contrast to the SbV resistance observed, none of the tested genes conferred elevated tolerance to the drug miltefosine (Fig. 2B), underscoring the antimony specificity of the gene cluster.
In L. donovani, resistance against antimony is linked to general fitness and virulence (40). We therefore tested whether overexpression of ARM58 or ARM56 has an impact on in vitro infectivity. Bone marrow-derived macrophages (BMMs) were infected with L. donovani carrying an empty vector, ARM58, or ARM56. After 4 h, the cells were washed and new medium was added. We quantified the relative parasite load at 24 and 48 h after infection. Parasites overexpressing ARM56 or ARM58 showed a moderate increase in the parasite load after the first 24 h (Fig. 2C). Even at 48 h postinfection, ARM56-overexpressing leishmania produced higher parasite loads than the empty vector controls. From this we conclude that overexpression of ARM56 indeed gives a slight advantage to L. donovani during the intracellular stage.
Antibody detection of ARM58 and ARM56.
Using the specific anti-ARM58 and anti-ARM56 IgY antibodies, we were able to detect both proteins in Leishmania donovani promastigote lysates. ARM58 and ARM56 were detected by their cognate antibodies as bands with apparent molecular masses of approximately 80 kDa (Fig. 3A, lanes 1 to 3), both of which exceed the hypothetical molecular masses of 58 kDa and 56 kDa, respectively. We conclude that both proteins showed retarded migration in SDS-polyacrylamide gels. This is not due to a posttranslational modification(s), as the bacterially expressed (His)10-ARM58 migrated at the same rate (Fig. 1A).
FIG 3.
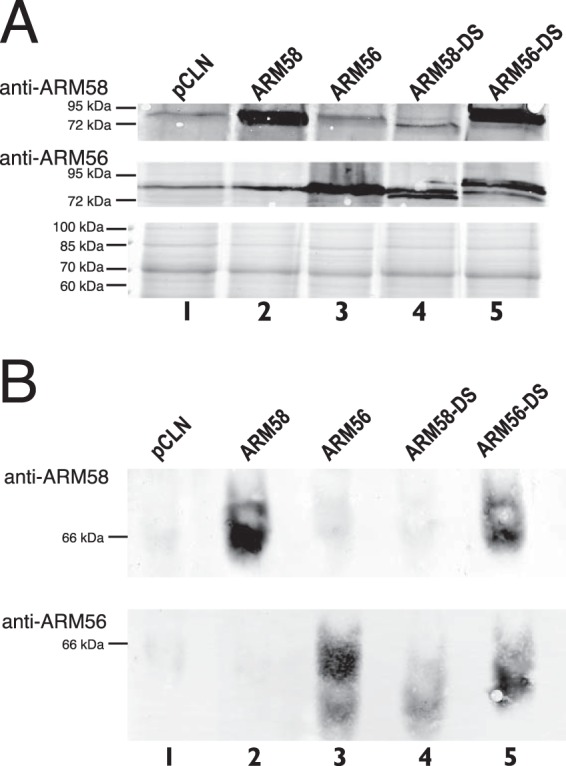
(A) Detection of ARM58 and ARM56 in protein lysates of L. donovani overexpressing ARM58, ARM56, ARM58-DS, and ARM56-DS. The proteins were analyzed by SDS-PAGE and Western blotting with anti-ARM58 (top) or anti-ARM56 (middle). Coomassie blue staining was done in parallel as a loading control (bottom). (B) As described in the legend to panel A, but proteins were isolated under nondenaturing conditions, separated on a nondenaturing polyacrylamide gradient gel, denatured, and transferred to a PVDF membrane. The membranes were then probed with anti-ARM58 (top) or anti-ARM56 (bottom).
ARM58-DS overexpression was not detected by anti-ARM58 IgY (Fig. 3A, top, lane 4), indicating that the lack of the third DUF1935 domain in this domain-swapping mutant interferes with antigenic recognition. In contrast, ARM56-DS overexpression was detected by anti-ARM58 as a band of approximately 90 kDa (Fig. 3A, top, lane 5), further supporting the view that ARM58 antigenicity is centered upon the third DUF1935 domain.
The anti-ARM56 antibodies also have a strong affinity for the ARM56 third DUF1935 domain, as they recognize the fusion protein ARM58-DS. However, the recognition is not solely dependent on DUF1935.3, as the ARM56-DS fusion protein was also recognized (Fig. 3A, middle, lanes 4 and 5).
Having specific antibodies against both proteins, we were now able to address the question of whether one or both proteins may be part of multiprotein complexes. We performed protein extraction under nondenaturing conditions and performed nondenaturing polyacrylamide gel electrophoresis followed by Western blotting (31, 32). ARM58 and ARM56 migrated as proteins of between 55 and 60 kDa, corresponding to their predicted molecular masses (Fig. 3B, lanes 1 to 3). Again, recognition by anti-ARM58 antibodies depended on the presence of the ARM58 DUF1935.3 (Fig. 3B, lane 4), while anti-ARM56 antibodies recognized more than one DUF1935 (Fig. 3B, lane 5). We did not detect slower-migrating bands indicating complex formation.
Secretion analysis of ARM58 and ARM56.
The ARM58 fusion protein was detected near the flagellar pocket, suggesting an involvement in cross-membrane traffic (18). We first wanted to determine whether ARM58 is secreted by the parasite. For that, supernatants of L. donovani promastigotes cultivated at 25°C or 37°C were collected, subjected to ultrafiltration/concentration, and analyzed by SDS-PAGE and Western blotting along with lysates of the cell pellets. The cell pellets contained detectable amounts of ARM58, with a strong increase being detected for the ARM58-overexpressing parasites independently of the temperature (Fig. 4A, lanes 5 to 8). Only the supernatants of ARM58-overexpressing cells showed detectable levels of the protein in Western blots (Fig. 4A, lanes 1 to 4), indicating that ARM58 becomes secreted upon overexpression. Control blots were probed with anti-HSP90 as an exosome marker protein and anti-HSL-U1 as a nonexosomal protein (36) as positive and negative controls, respectively (Fig. 4A, middle and bottom).
FIG 4.

(A) Detection of ARM58 in L. donovani promastigotes and in the secreted protein fraction. L. donovani parasites transfected with pCLN or ARM58 transgenes were incubated for 2 h at 25° or 37°C in serum-free medium. After centrifugation, medium supernatants and sedimented cells were concentrated by acetone precipitation and dissolved in SDS sample buffer, followed by SDS-PAGE and Western blotting. Immunological staining was performed with anti-ARM58, anti-HSP90 (secreted fraction marker), and anti-HSL-U1 (nonsecreted) antibodies. (B) Testing of secretome proteins for sensitivity to trypsin in the presence and absence of Triton X-100 detergent. Antibody detection was as described in the legend to panel A. (C) Antitubulin and DAPI staining of the promastigotes after harvesting of exosomes from cell supernatant. Cells were fixed and stained, followed by fluorescence microscopy. k, kinetoplast; n, nucleus. (D) Ultrastructure of the flagellar pocket of L. donovani promastigotes at 37°C, showing vesicles (V) in the flagellar pocket (FLP) and the flagellum (FL). (E) Isolated exosomes visualized by negative staining and transmission electron microscopy. (F) Detection of ARM58 and ARM56 in exosomes. L. donovani with pCLN, ARM58, ARM56, ARM58-DS, and ARM56-DS transgenes were incubated for 2 h in serum-free medium at 37°C. Culture supernatants were collected and subjected to exosome enrichment. Exosome fractions were then treated with trypsin alone or a combination of trypsin and Triton X-100. Before and after trypsin digestion, samples were precipitated with acetone, redissolved in SDS sample buffer, and analyzed by SDS-PAGE and Western blotting. Proteins were detected with anti-ARM58, anti-ARM56, anti-HSP90 (exosomal marker), and anti-HSL-U1 (nonexosomal protein) antibodies. −/−, nondigested exosome preparations; +/+, exosome preparations subjected to trypsin proteolysis in the presence of Triton X-100 detergent; +/−, exosome preparations subjected to trypsin proteolysis in the absence of Triton X-100 detergent.
We next tested if ARM58 is secreted as a free protein or is surrounded by a membrane as part of a vesicle payload. We used trypsin digestion in the presence or absence of the detergent Triton X-100 to see whether secreted ARM58 is protected against proteolysis by a membrane. Figure 4B shows that overexpressed, secreted ARM58 was protected against trypsin digestion but became susceptible to proteolysis under Triton X-100-mediated membrane disruption. Together, these results show that overexpressed ARM58 is part of the secretome and is surrounded by a membrane, indicating vesicular export.
Leishmania exosomes are able to modulate the host immune response by exporting parasite cytoplasmic proteins into the host cell cytoplasm (35). We therefore tested whether ARM58 is part of the L. donovani exosomes. For this, we isolated exosomes from the culture supernatants of L. donovani following an establish protocol (36).
In addition to ARM58, we also overexpressed the product of the neighboring ARM56 gene and the two domain-swapping variants, ARM58-DS and ARM56-DS.
We carefully examined the promastigotes by immunofluorescence microscopy for morphological integrity after collection of the supernatant, to exclude the possibility of contamination of the supernatant with cellular debris (Fig. 4C). We also performed transmission electron microscopy on the heat-stressed promastigotes to control the ultrastructure near the flagellar pocket. The flagellar pocket was filled with exosome-like vesicles (Fig. 4D), reflecting the increased release of exosomes at 37°C (36).
We further analyzed the exosome preparations by transmission electron microscopy. As seen in Fig. 4E, the exosomal fraction consists of spherical, membranous particles of approximately 100 nm in diameter, typical for Leishmania exosomes (36).
Exosome preparations were subjected to trypsin proteolysis in the presence and absence of Triton X-100 detergent and then analyzed by SDS-PAGE and Western blotting. The blots were then probed with anti-ARM58, anti-ARM56, HSP90, and anti-HSL-U1 antibodies (Fig. 4F).
Mutants overexpressing ARM58 or ARM56 and domain swap mutants thereof were detectable in the nondigested controls. HSP90 was detected in the controls but was also detected after trypsin digestion without Triton X-100 treatment, confirming its status as an exosomal protein. HSL-U1 was not detectable at all in any sample, showing that cytosolic proteins are largely absent from the exosome preparations. Overexpressed ARM58 was present in the exosome preparation and protected against trypsin alone but not trypsin plus Triton X-100, confirming its localization within membranous vesicles. Similarly, ARM56 was partly protected against trypsin lysis in the absence of Triton X-100, indicating at least partial vesicular localization. The ARM58-DS mutants carrying the ARM56 third DUF1935 domain in place of its own third domain showed a protection pattern similar to that shown by ARM56, indicating partial vesicular localization. The ARM56 with the third domain of ARM58, in contrast, showed the same protection as ARM58 proper, confirming the important role of the third domain for the protein's function (18).
The results show that (i) both ARM58 and ARM56 are secreted from L. donovani via vesicles similar to exosomes, (ii) ARM58 is protected against trypsin more efficiently than ARM56, and (iii) the third DUF1935 domain of ARM58 directs the protein to the exosomal compartment.
Subcellular localization of ARM58 and ARM56.
Previous studies showed that an mCherry-ARM58 fusion protein expressed in L. infantum localized to the flagellar pocket (18). Because we knew that overexpressed ARM58 and ARM56 are released via exosomes and because we had specific antibodies, we decided to localize both proteins in wild-type L. donovani promastigotes and axenic amastigotes. L. donovani promastigotes or axenic amastigotes were fixed onto microscope slides with cold methanol. Fixed cells were decorated with antitubulin (Alexa Fluor) combined with anti-ARM58 or anti-ARM56 (FITC). DAPI was used to stain the nucleus and kinetoplast (Fig. 5). Anti-ARM58 (FITC) showed an almost exclusive localization in the flagellum and in the flagellar pocket in the promastigotes. The same staining pattern was observed in axenic amastigotes, where ARM58 was detected in the rudimentary flagellum and in the flagellar pocket. The flagellar localization may hint at additional functions of the protein in cell motility or cytoskeletal structures.
FIG 5.

Subcellular localization of ARM58 and ARM56. L. donovani promastigotes, L. donovani axenic amastigotes, and Trypanosoma cruzi trypomastigotes were fixed and stained with anti-ARM58 or anti-ARM56 (green), DAPI (blue), and anti-alpha-tubulin (red). Images were taken with an epifluorescence microscope. n, nuclei; k, kinetoplast.
In contrast, ARM56 was a cytoplasmic protein with no specific localization in the cell but a higher abundance in the anterior half of both promastigotes and axenic amastigotes.
ARM58 is specific to the genus Leishmania, but ARM56 has an ortholog in T. cruzi with a predicted molecular mass of 55 kDa (TcCLB.506407.50). We therefore performed indirect immunofluorescence microscopy on T. cruzi Y strain sanguineous trypomastigotes (Fig. 5, bottom). As expected, anti-ARM58 did not recognize structures in T. cruzi, confirming the lack of an ARM58 ortholog in trypanosomes (17). Also as expected, anti-ARM56 staining showed a localization in the cytoplasm, similar to the findings for Leishmania promastigotes. This confirms the existence of an ARM56 ortholog in T. cruzi and raises questions regarding its possible involvement there in therapy resistance.
In summary, we can confirm that (i) ARM58 is a specific protein in Leishmania spp., (ii) the protein is localized in the flagellum and in the flagellar pocket, (iii) ARM56 is localized in the cytoplasm of L. donovani promastigotes and axenic amastigotes, and (iv) an ortholog of ARM56 resides in the cytoplasm of T. cruzi sanguineous trypomastigotes.
Hypodiploidity analysis in DNA.
It is known that both miltefosine and antimony can cause DNA fragmentation in Leishmania spp. as a prelude to cell death (41, 42). We wanted to know whether ARM58 overexpression can protect L. donovani promastigotes against the miltefosine- and/or SbIII-induced loss of DNA content. For this, we cultivated L. donovani promastigotes carrying pCLN or ARM58 transgenes with miltefosine or trivalent antimony. After 72 h, cells were fixed, stained with SYTOX, and analyzed for DNA content by fluorescence-activated cell sorting. Treatment of the control strain bearing the empty vector pCLN with miltefosine (Fig. 6A) or SbIII (Fig. 6B) caused a marked increase of cells with a DNA content of <2n, also known as hypodiploidity, to 59.5% and 44.4%, respectively. Overexpression of ARM58 did not protect L. donovani against miltefosine-induced hypodiploidity (52.4%; Fig. 6C). However, SbIII-induced cell death was effectively prevented (8.05%) by ARM58 overexpression (Fig. 6D), underscoring the specificity of ARM58 as an antimony resistance gene.
FIG 6.
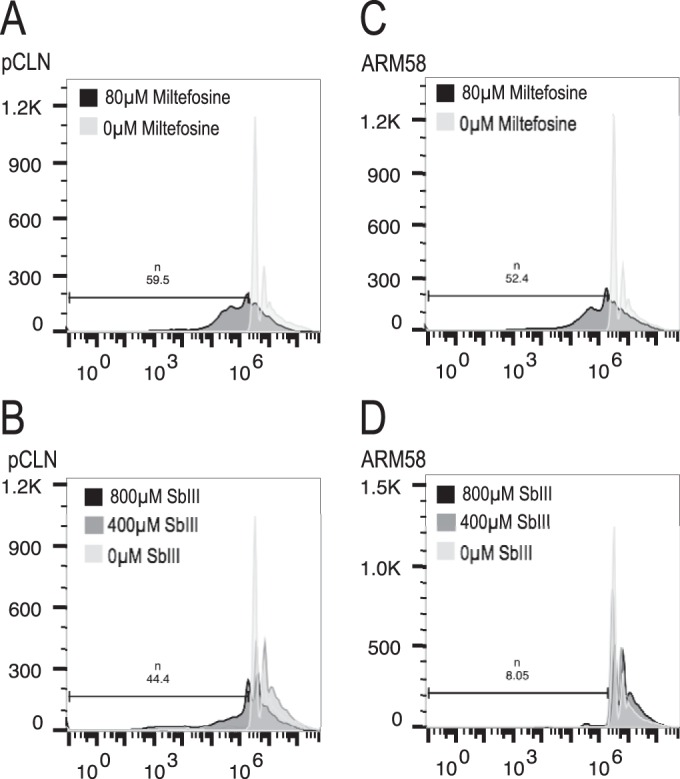
Hypodiploidity detection in L. donovani with pCLN or ARM58 transgenes after a 72-h challenge with 80 μM miltefosine (A,C) or 400 or 800 μM trivalent antimony (B, D). Promastigotes were then stained with SYTOX and subjected to FACS analysis to determine the DNA content. The bars indicate the gates used, with the fraction size (%) being shown on the top.
Quantification of cell death markers.
It is known that both miltefosine and SbIII can induce programmed cell death (PCD) or necrosis (43 – 46). We used propidium iodide (PI) and annexin V to stain promastigotes after miltefosine or SbIII exposure. The former is a non-membrane-permeant nucleic acid stain specific for ruptured cells, and the latter decorates phosphatidylserine lipids in the outer membrane leaf that indicate PCD. We tested whether ARM58 overexpression can protect L. donovani promastigotes from miltefosine- and/or SbIII-induced PCD and/or rupture. For this, we cultivated L. donovani promastigotes (control and ARM58+/+/+ promastigotes) with miltefosine (80 μM) or SbIII (400 or 800 μM) for 72 h. The cells were then stained with annexin V or PI and analyzed by fluorescence-activated cell sorting. Treatment of the control strain (pCLN) with miltefosine caused early or late PCD in 45% of the cells (Table 1). Similar but weaker dose-dependent effects were observed when the control promastigotes were challenged with 400 μM and 800 μM SbIII. Overexpression of ARM58 did not protect L. donovani against miltefosine-induced cell death. In contrast, we observed a higher tolerance at 400 μM and 800 μM SbIII. With 400 μM SbIII, ARM58-overexpressing cells were fully protected against PCD or rupture. However, with 800 μM SbIII, there was an increase of cells in early PCD (6.34%), but no advanced PCD or cell destruction was observed. This finding confirms our previous results where overexpression of ARM58 protects promastigotes against DNA fragmentation under SbIII exposure but not under miltefosine challenge.
TABLE 1.
Quantification of cell death markersa
| Sample no. | Construct | Miltefosine concn (μM) | SbIII concn (μM) | % cellsb: |
|||
|---|---|---|---|---|---|---|---|
| Annexin V−, PI− | Annexin V+, PI− | Annexin V+, PI+ | Annexin V−, PI+ | ||||
| 1 | pCLN | 0 | 99.0 | 0 | 1 | 0 | |
| 2 | ARM58 | 0 | 99 | 0 | 0 | 1 | |
| 3 | pCLN | 80 | 50.4 | 27.1 | 17.8 | 4.76 | |
| 4 | ARM58 | 80 | 36.80 | 33.30 | 26.70 | 3.16 | |
| 5 | pCLN | 0 | 99 | 0 | 0.60 | 0 | |
| 6 | ARM58 | 0 | 99 | 0 | 0.60 | 0 | |
| 7 | pCLN | 400 | 89.5 | 4.6 | 2.4 | 3.4 | |
| 8 | ARM58 | 400 | 97.3 | 0.6 | 1.2 | 1.0 | |
| 9 | pCLN | 800 | 75.9 | 5.7 | 8.2 | 10.2 | |
| 10 | ARM58 | 800 | 90.6 | 6.3 | 1.6 | 1.5 | |
L. donovani promastigotes with pCLN or ARM58 transgenes were challenged with 80 μM miltefosine or 400 to 800 μM SbIII for 72 h. Cells were stained with the cell death markers annexin V and propidium iodide (PI), and 10,000 events were analyzed by fluorescence-assisted cell sorting. Raw data can be found in Fig. S1 in the supplemental material.
Annexin V-negative (annexin V−) and PI-negative (PI−) cells are viable, annexin V-positive (annexin V+) and PI-negative cells are in early PCD, annexin V-positive and PI-positive (PI+) cells are in PCD, and annexin V-negative and PI-positive cells are ruptured.
Cos-Seq analysis.
ARM58 was identified by functional cloning from a cosmid DNA library (17). Its neighboring gene, ARM56, was implicated in SbV resistance in this study. Neighboring these two genes is the small heat shock protein gene HSP23, which was also found to have an impact on SbIII sensitivity (19). This apparent clustering of drug resistance genes at the telomeric end of chromosome 34 may indicate a need for a synchronized gene copy number regulation, e.g., by indirect repeat-directed episome formation (47). Since this clustering is found in all Leishmania and Viannia species, it cannot result from the relatively recent introduction of antimonial drugs. Therefore, we performed a series of functional cloning selections under challenge with a variety of cytotoxic compounds.
L. infantum promastigotes were transfected with the DNA from an L. infantum gDNA cosmid library (16). The recombinant population was split and subjected to selection in duplicate with SbIII, AsIII, Cu2+, Cd2+, and miltefosine at the respective growth-inhibiting IC50. Control populations were maintained without any selection or with G418 antibiotic selection. After 10 in vitro passages, the selected parasites were harvested for cosmid DNA reisolation. The cosmid preparations from each sample were used for electroporation into E. coli to exclude Leishmania gDNA contaminations and recovered from the recombinant E. coli populations. The cosmid DNA samples were then subjected to fragmentation and next-generation sequencing by a technique recently described as Cos-Seq (48). The resulting sequence reads were aligned to the L. infantum chromosome sequences to detect overrepresentation of the chromosome 34 5′ telomeric end, where ARM58, ARM56, and HSP23 are encoded. Figure 7 shows the distribution of aligned next-generation sequencing reads between base pairs 30,000 and 90,000 of chromosome 34. In the unselected sample (Fig. 7A), the region was represented with an average read density of 2 times. After SbIII selection, coverage of the region between base pairs 38,000 and 85,000 increased to 100 times (Fig. 7B). This region included all three genes of interest (GOIs). Selection under arsenite (AsIII), in contrast, did not result in any representation of this gene locus in the selected cosmids (Fig. 7C). Selection under divalent copper ions (Cu2+), known inducers of the cellular stress response in higher eukaryotes (49, 50), resulted in a marginally elevated representation of the GOIs in the selected cosmids (Fig. 7D). No coverage representing a full cosmid was observed after cadmium (Cd2+) (Fig. 7E) or miltefosine (Fig. 7F) selection, confirming that ARM58 does not mediate miltefosine resistance (18). Taken together, the results indicate that the three GOIs implicated in stress and drug resistance are not markers for general resistance to toxic substances but, rather, are specific for antimony-induced and, possibly, for copper-induced cell stress.
FIG 7.
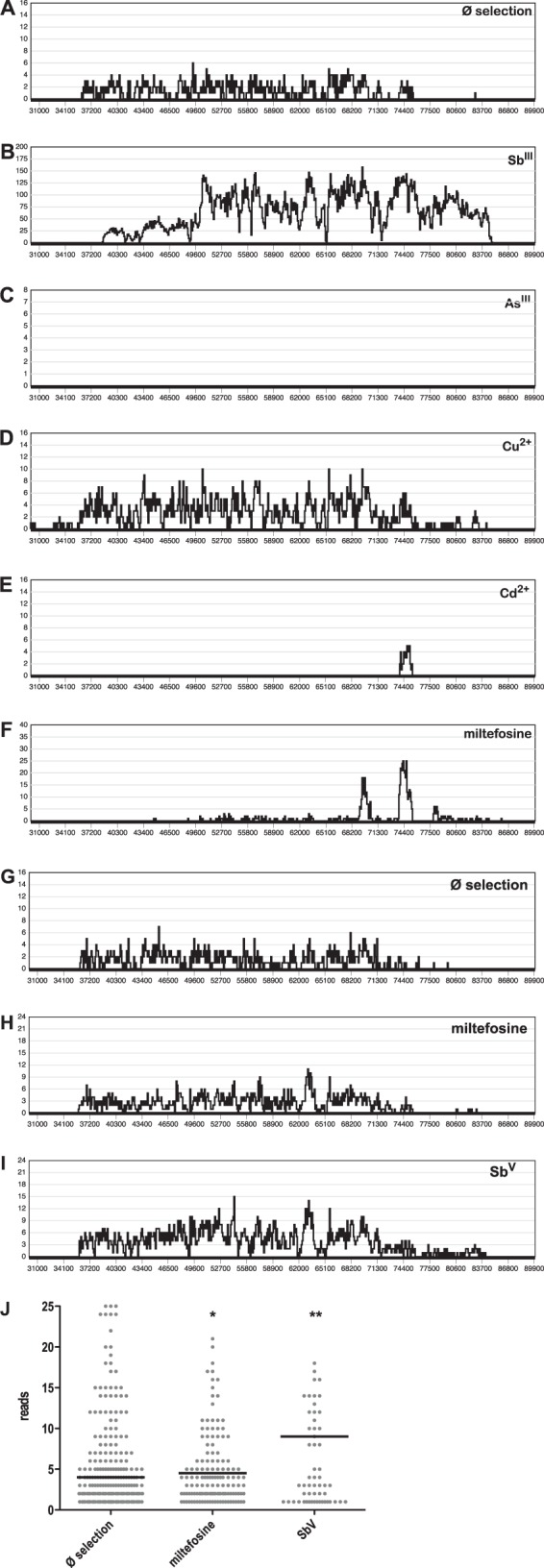
Cos-Seq analysis of L. infantum carrying an L. infantum gDNA cosmid library selected under standard growth conditions (Ø selection) (A) and with antimonyl tartrate (SbIII) (B), sodium arsenite (AsIII) (C), copper acetate (Cu2+) (D), cadmium acetate (Cd2+) (E), and miltefosine (F) at their IC50s. The same transfected population was also used to infect bone marrow-derived macrophages, which were selected by in vitro infection only (G), under miltefosine (H), or under sodium stibogluconate (SbV) (I). In vitro macrophage passages under conditions of drug selection were repeated three times. Cosmid DNA was recovered from all selected populations and subjected to next-generation sequencing. Sequence reads were then aligned with the sequence of L. infantum chromosome 34. In panels A to I, the alignment frequency (y axis) is plotted against chromosomal positions (x axis). (J) Scatter graph analysis of alignment frequencies at positions 36000 to 75000 on chromosome 34 for next-generation sequencing reads from selected intracellular amastigotes (G to I). The black bars represent the median alignment frequencies. *, P < 0.05; **, P < 0.01.
We also tested whether the GOIs are selected in intracellular amastigotes under miltefosine or SbV selection. For this, bone marrow-derived macrophages were infected with the cosmid-bearing population of L. infantum for 24 h before a 48-h treatment at the IC50 of miltefosine or SbV. Parasites were recovered from the macrophages and grown as promastigotes under G418 selection until stationary growth phase. The process was repeated twice for a total of three selection rounds, after which cosmid DNA was isolated from the triple-selected populations. Cosmids were passaged through E. coli to exclude the possibility of Leishmania gDNA contamination and then subjected to next-generation sequencing. Figure 7 shows the read frequency for cosmid DNA inserts corresponding to positions 30,000 to 90,000 of chromosome 34 for cosmids selected by in vitro infection only (Ø selection; Fig. 7G), under miltefosine (Fig. 7H), or under SbV (Fig. 7I). The graphs show a slight increase in the read frequency for the cosmids selected under SbV. This was confirmed by statistical comparison of the read frequencies for the region between positions 36,000 and 76,000, which includes the GOIs. As seen in Fig. 7J, the read frequency after miltefosine selection was barely elevated with weak significance (P = 0.0319, two-tailed U test). However, the median read frequency for SbV-selected cosmids (median read frequency, 9.0) was 2-fold increased compared with that for the control population (median read frequency, 4.0) (P = 0.0063, two-tailed U test). The results indicate that amplification of the GOIs has a stronger effect on antimony resistance in axenic promastigotes than in intracellular amastigotes, indicating that resistance is directed against the active principle, SbIII. The protective effect for intracellular amastigotes may be masked by the known ability of SbV to stimulate macrophage activity.
DISCUSSION
Contrary to predictions (18), ARM58 is not membrane associated regardless of the expression levels. Obviously, the predicted transmembrane domain in the third DUF1935 is not functional, leaving ARM58 as a soluble protein with a specific intracellular localization near the flagellar pocket and in the flagellum itself. The latter finding complements the findings of earlier work using an overexpressed mCherry-ARM58 fusion protein (18). The related ARM56, in contrast, shows a general cytoplasmic distribution.
Flagellar localization is also not a prerequisite for its function as a detoxifying protein. As shown previously (18), the mCherry-ARM58 fusion protein is not found in the flagellum, yet it protects against SbIII.
Both ARM58 and ARM56 display electrophoretic mobility, indicating a considerably larger protein: 80 kDa rather than the 58 kDa or 56 kDa calculated from their amino acid sequence. This effect is not seen during nondenaturing pore gradient electrophoresis, where ionization and net charge cannot influence the results. Both proteins have an isoelectric point in the acidic range (pI 4.96 and pI 4.88, respectively). Similar apparent molecular mass aberrations in SDS-PAGE were reported in the past for other proteins; e.g., the heat shock transcription factors of Saccharomyces cerevisiae (51) and Drosophila melanogaster (52) have calculated molecular masses of 93 kDa and 77 kDa, respectively, and electrophoretic mobilities corresponding to 150 kDa (53) and 115 kDa (54), respectively, and their isoelectric points are also in the acidic range at 4.63. This appears to be a general phenomenon (55).
Upon overexpression, we also found ARM58 in the L. donovani secretome, more specifically, inside vesicular exosomes, along with ARM56. Neither protein was detected in the exosome fraction in earlier studies (35, 36, 56), confirming our observation that only overexpressed ARM58 and ARM56 enter the exosomes and thereby link overexpression, exosomal transport, and drug resistance.
It is not uncommon for proteins associated with antimony resistance to be found in the exosomal protein fraction. At least four ABC transporter family proteins in L. major, F06.0080, F15.0890, F27.0980, and F29.0620, are secreted in this way (36). Another putative stibogluconate resistance protein in L. major encoded by the genes F31.09320, F31.09330, F31.09350, and F31.09360 is also exported via exosomes. Enzymes such as trypanothione synthetase (L. major F27.1870) and trypanothione reductase (L. major F05.0350) are also part of the exosome load and involved in antimony metabolism. In addition to this, two heat shock proteins that have been associated with antimony resistance, HSP90 (57) and HSP70 (58), are also prominent in exosomes. Whether the exosomal transport of these proteins is functionally related to their role as antimony resistance markers is a question that remains to be solved.
Our in vitro infectivity results also showed that overexpression of ARM56 can increase in vitro infectivity. This finding supports earlier observations with field isolates of drug-resistant parasites, where drug resistance and general fitness correlated (40).
Another interesting finding is that three genes involved in antimony tolerance, ARM58, ARM56, and HSP23, form a cluster at the telomeric end of chromosome 34. The importance of ARM56 and HSP23 was overlooked in our earlier work that focused on the SbIII tolerance of promastigotes and where we confirmed the role of ARM58 only in an in vitro infection model (17, 18). ARM56 and HSP23 were also implicated in SbV resistance through the use of in vitro infection of BMMs in this study. While a function in antimony tolerance of promastigotes was also described for HSP23 (19), ARM56 appears to exert its function only in intracellular amastigotes, confirming the differences between SbIII tolerance in promastigotes and the SbV resistance of intracellular amastigotes (59).
Given that spontaneous gene amplification in Leishmania involves recombination between repeat sequences in the genome (47), the grouping of three genes involved in antimony resistance might be seen as a way to ensure simultaneous amplification for protection against various stresses. However, our Cos-Seq analysis (Fig. 7) of the gene locus argues against this. Only selection under SbIII and, to a much lesser extent, Cu2+ favored the maintenance of cosmids coding for the three genes. No such selection was observed under challenge with another trivalent half metal, AsIII, nor was it observed under selection with Cd2+ or with the antileishmanial drug miltefosine. This indicates a rather specific protective effect and does not support the idea of a chemoresistance gene cluster.
Cos-Seq analysis (48) of the locus after selection of intracellular amastigotes under SbV and miltefosine showed that selection was far less stringent. However, the 3 in vitro infection/selection cycles of 48 h each and not more than 6 generations in total cannot be expected to yield the same stringent selection as the in vitro selection of promastigotes over 30 days and ∼60 generations. A part may also be explained by the fact that SbV acts not only as a prodrug with SbIII as an effector but also by stimulating macrophages into microbicidal activity (3). The latter effect of SbV would not be affected by an SbIII-specific resistance marker, explaining the weaker effect in the in vitro infection system observed here and in previous work (17). Moreover, the analysis confirms the results in Fig. 2. In both test systems, neither ARM58 nor its neighboring genes protected against miltefosine treatment. A very recent paper (48) describing a study using Cos-Seq to identify resistance marker genes in Leishmania also showed strong selection of the triple-gene locus under SbIII but not under miltefosine, confirming our findings presented in this and in earlier (17) papers.
We observed an unusual electrophoretic mobility of ARM58 and ARM56 in denaturing SDS-polyacrylamide electrophoresis. This was not due to posttranslational modifications, since (His)10-ARM58 expressed in E. coli showed the same mobility. Moreover, the same proteins migrate with Stoke's radii corresponding to <65 kDa in nondenaturing electrophoresis, i.e., corresponding to their predicted molecular mass. This analysis also shows that neither ARM58 nor ARM56 is part of stable multisubunit protein complexes. It is also interesting that the third DUF1935 domain of ARM58 largely determines the specificity of anti-ARM58 antibodies, hinting at a highly immunogenic structure. Cysteine side chains have been reported to be involved in chelation of half metals (39). Three cysteine residues conserved between ARM58 and ARM56 were replaced with serines, singly or in combination. While the single exchanges caused a partial loss of activity, the triple amino acid exchange showed no further reduction, indicating that sequestration via cysteine side chains is not essential for ARM58 function in antimony resistance.
We could also show that ARM58 is not a general antagonist of antimony-induced PCD. Rather, the protection against cell death was specific for SbIII and did not extend to PCD induced by miltefosine. This ties in with the known effect of ARM58 overexpression in lowering the intracellular antimony concentration (18).
We have repeatedly attempted to replace the two ARM58 alleles from the L. infantum and L. donovani genomes, using both classical, two-step gene replacement (38, 60) and simultaneous, double-allele gene replacement (61) approaches. No viable double-allele gene replacement mutants were obtained either in the presence or in the absence of a functional ARM58 transgene (D. Zander, unpublished data; P. Tejera Nevado, unpublished data). Therefore, the question whether ARM58 is an essential gene will require novel, inducible mutagenesis approaches, such as an application of a clustered regularly interspaced short palindromic repeat/caspase 9 gene deletion (62) or dimerizable Cre recombinase-mediated recombination (63).
Another important question still open is the impact of ARM58, ARM56, and HSP23 on therapeutic failure in the field. Here, a collaborative effort to quantify resistance gene expression in field samples of L. infantum has been initiated.
Supplementary Material
ACKNOWLEDGMENTS
For the Cos-Seq approach to functional cloning, we thank Marc Ouellette for freely sharing his ideas and concepts. We thank Kathrin Schuldt, Jürgen Sievertsen, and Stefan Lorenz for help and advice with next-generation sequencing and associated data analysis. We are also grateful for the indirect contributions by Janika Bartsch and to Katharina Bartsch for a critical reading of the manuscript.
The work received partial funding under European Regional Development Fund grant no. BWF/H/52228/2012/13.10.10-1/3.4,6 (2012 to 2013) and under the European Union's Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. 603240 (NMTrypI—New Medicines for Trypanosomatidic Infections; http://www.nmtrypi.eu/) (2014 to 2016).
Funding Statement
The work was partly funded by grant 603240 from the European Union 7th Framework Programme.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00544-16.
REFERENCES
- 1.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. 2012. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7:e35671. doi: 10.1371/journal.pone.0035671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frezard F, Demicheli C, Ferreira CS, Costa MA. 2001. Glutathione-induced conversion of pentavalent antimony to trivalent antimony in meglumine antimoniate. Antimicrob Agents Chemother 45:913–916. doi: 10.1128/AAC.45.3.913-916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mookerjee Basu J, Mookerjee A, Sen P, Bhaumik S, Sen P, Banerjee S, Naskar K, Choudhuri SK, Saha B, Raha S, Roy S. 2006. Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob Agents Chemother 50:1788–1797. doi: 10.1128/AAC.50.5.1788-1797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerin PJ, Olliaro P, Sundar S, Boelaert M, Croft SL, Desjeux P, Wasunna MK, Bryceson AD. 2002. Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis 2:494–501. doi: 10.1016/S1473-3099(02)00347-X. [DOI] [PubMed] [Google Scholar]
- 5.Croft SL, Sundar S, Fairlamb AH. 2006. Drug resistance in leishmaniasis. Clin Microbiol Rev 19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mittal MK, Rai S, Ashutosh, Ravinder, Gupta S, Sundar S, Goyal N. 2007. Characterization of natural antimony resistance in Leishmania donovani isolates. Am J Trop Med Hyg 76:681–688. [PubMed] [Google Scholar]
- 7.Gourbal B, Sonuc N, Bhattacharjee H, Legare D, Sundar S, Ouellette M, Rosen BP, Mukhopadhyay R. 2004. Drug uptake and modulation of drug resistance in Leishmania by an aquaglyceroporin. J Biol Chem 279:31010–31017. doi: 10.1074/jbc.M403959200. [DOI] [PubMed] [Google Scholar]
- 8.Decuypere S, Rijal S, Yardley V, De Doncker S, Laurent T, Khanal B, Chappuis F, Dujardin JC. 2005. Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob Agents Chemother 49:4616–4621. doi: 10.1128/AAC.49.11.4616-4621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandal S, Maharjan M, Singh S, Chatterjee M, Madhubala R. 2010. Assessing aquaglyceroporin gene status and expression profile in antimony-susceptible and -resistant clinical isolates of Leishmania donovani from India. J Antimicrob Chemother 65:496–507. doi: 10.1093/jac/dkp468. [DOI] [PubMed] [Google Scholar]
- 10.El Fadili K, Messier N, Leprohon P, Roy G, Guimond C, Trudel N, Saravia NG, Papadopoulou B, Legare D, Ouellette M. 2005. Role of the ABC transporter MRPA (PGPA) in antimony resistance in Leishmania infantum axenic and intracellular amastigotes. Antimicrob Agents Chemother 49:1988–1993. doi: 10.1128/AAC.49.5.1988-1993.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreira DS, Monte-Neto RL, Andrade JM, Santi AM, Reis PG, Frezard F, Murta SM. 2013. Molecular characterization of the MRPA transporter and antimony uptake in four New World Leishmania spp. susceptible and resistant to antimony. Int J Parasitol Drugs Drug Resist 3:143–153. doi: 10.1016/j.ijpddr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukherjee B, Mukhopadhyay R, Bannerjee B, Chowdhury S, Mukherjee S, Naskar K, Allam US, Chakravortty D, Sundar S, Dujardin JC, Roy S. 2013. Antimony-resistant but not antimony-sensitive Leishmania donovani up-regulates host IL-10 to overexpress multidrug-resistant protein 1. Proc Natl Acad Sci U S A 110:E575–E582. doi: 10.1073/pnas.1213839110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Legare D, Papadopoulou B, Roy G, Mukhopadhyay R, Haimeur A, Dey S, Grondin K, Brochu C, Rosen BP, Ouellette M. 1997. Efflux systems and increased trypanothione levels in arsenite-resistant Leishmania. Exp Parasitol 87:275–282. doi: 10.1006/expr.1997.4222. [DOI] [PubMed] [Google Scholar]
- 14.Wyllie S, Mandal G, Singh N, Sundar S, Fairlamb AH, Chatterjee M. 2010. Elevated levels of tryparedoxin peroxidase in antimony unresponsive Leishmania donovani field isolates. Mol Biochem Parasitol 173:162–164. doi: 10.1016/j.molbiopara.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeddi F, Mary C, Aoun K, Harrat Z, Bouratbine A, Faraut F, Benikhlef R, Pomares C, Pratlong F, Marty P, Piarroux R. 2014. Heterogeneity of molecular resistance patterns in antimony-resistant field isolates of Leishmania species from the western Mediterranean area. Antimicrob Agents Chemother 58:4866–4874. doi: 10.1128/AAC.02521-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choudhury K, Zander D, Kube M, Reinhardt R, Clos J. 2008. Identification of a Leishmania infantum gene mediating resistance to miltefosine and SbIII. Int J Parasitol 38:1411–1423. doi: 10.1016/j.ijpara.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Nühs A, Schäfer C, Zander D, Trübe L, Tejera Nevado P, Schmidt S, Arevalo J, Adaui V, Maes L, Dujardin J-C, Clos J. 2014. A novel marker, ARM58, confers antimony resistance to Leishmania spp. Int J Parasitol Drugs Drug Resist 4:37–47. doi: 10.1016/j.ijpddr.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schäfer C, Tejera Nevado P, Zander D, Clos J. 2014. ARM58 overexpression reduces intracellular antimony concentration in Leishmania infantum. Antimicrob Agents Chemother 58:1565–1574. doi: 10.1128/AAC.01881-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hombach A, Ommen G, MacDonald A, Clos J. 2014. A small heat shock protein is essential for thermotolerance and intracellular survival of Leishmania donovani. J Cell Sci 127:4762–4773. doi: 10.1242/jcs.157297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garin YJ, Sulahian A, Pratlong F, Meneceur P, Gangneux JP, Prina E, Dedet JP, Derouin F. 2001. Virulence of Leishmania infantum is expressed as a clonal and dominant phenotype in experimental infections. Infect Immun 69:7365–7373. doi: 10.1128/IAI.69.12.7365-7373.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenzweig D, Smith D, Opperdoes F, Stern S, Olafson RW, Zilberstein D. 2008. Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J 22:590–602. [DOI] [PubMed] [Google Scholar]
- 22.Krobitsch S, Brandau S, Hoyer C, Schmetz C, Hübel A, Clos J. 1998. Leishmania donovani heat shock protein 100: characterization and function in amastigote stage differentiation. J Biol Chem 273:6488–6494. doi: 10.1074/jbc.273.11.6488. [DOI] [PubMed] [Google Scholar]
- 23.Racoosin EL, Swanson JA. 1989. Macrophage colony-stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J Exp Med 170:1635–1648. doi: 10.1084/jem.170.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Racoosin EL, Beverley SM. 1997. Leishmania major: promastigotes induce expression of a subset of chemokine genes in murine macrophages. Exp Parasitol 85:283–295. doi: 10.1006/expr.1996.4139. [DOI] [PubMed] [Google Scholar]
- 25.Laban A, Wirth DF. 1989. Transfection of Leishmania enriettii and expression of chloramphenicol acetyltransferase gene. Proc Natl Acad Sci U S A 86:9119–9123. doi: 10.1073/pnas.86.23.9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 27.Hombach A, Ommen G, Chrobak M, Clos J. 2013. The Hsp90-Sti1 interaction is critical for Leishmania donovani proliferation in both life cycle stages. Cell Microbiol 15:585–600. doi: 10.1111/cmi.12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlüter A, Wiesgigl M, Hoyer C, Fleischer S, Klaholz L, Schmetz C, Clos J. 2000. Expression and subcellular localization of Cpn60 protein family members in Leishmania donovani. Biochim Biophys Acta 1491:65–74. doi: 10.1016/S0167-4781(00)00028-2. [DOI] [PubMed] [Google Scholar]
- 29.Clos J, Brandau S. 1994. pJC20 and pJC40—two high-copy-number vectors for T7 RNA polymerase-dependent expression of recombinant genes in Escherichia coli. Protein Expr Purif 5:133–137. doi: 10.1006/prep.1994.1020. [DOI] [PubMed] [Google Scholar]
- 30.Hubel A, Brandau S, Dresel A, Clos J. 1995. A member of the ClpB family of stress proteins is expressed during heat shock in Leishmania spp. Mol Biochem Parasitol 70:107–118. doi: 10.1016/0166-6851(95)00012-P. [DOI] [PubMed] [Google Scholar]
- 31.Westwood JT, Clos J, Wu C. 1991. Stress-induced oligomerization and chromosomal relocalization of heat-shock factor. Nature 353:822–827. doi: 10.1038/353822a0. [DOI] [PubMed] [Google Scholar]
- 32.Ommen G, Chrobak M, Clos J. 2010. The co-chaperone SGT of Leishmania donovani is essential for the parasite's viability. Cell Stress Chaperones 15:443–455. doi: 10.1007/s12192-009-0160-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rey-Ladino JA, Joshi PB, Singh B, Gupta R, Reiner NE. 1997. Leishmania major: molecular cloning, sequencing, and expression of the heat shock protein 60 gene reveals unique carboxy terminal peptide sequences. Exp Parasitol 85:249–263. doi: 10.1006/expr.1996.4137. [DOI] [PubMed] [Google Scholar]
- 34.Twu O, de Miguel N, Lustig G, Stevens GC, Vashisht AA, Wohlschlegel JA, Johnson PJ. 2013. Trichomonas vaginalis exosomes deliver cargo to host cells and mediate host:parasite interactions. PLoS Pathog 9:e1003482. doi: 10.1371/journal.ppat.1003482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silverman JM, Clos J, Horakova E, Wang AY, Wiesgigl M, Kelly I, Lynn MA, McMaster WR, Foster LJ, Levings MK, Reiner NE. 2010. Leishmania exosomes modulate innate and adaptive immune responses through effects on monocytes and dendritic cells. J Immunol 185:5011–5022. doi: 10.4049/jimmunol.1000541. [DOI] [PubMed] [Google Scholar]
- 36.Silverman JM, Clos J, de'Oliveira CC, Shirvani O, Fang Y, Wang C, Foster LJ, Reiner NE. 2010. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J Cell Sci 123:842–852. doi: 10.1242/jcs.056465. [DOI] [PubMed] [Google Scholar]
- 37.Bifeld E, Chrobak M, Zander D, Schleicher U, Schonian G, Clos J. 2015. Geographical sequence variation in the Leishmania major virulence factor P46. Infect Genet Evol 30:195–205. doi: 10.1016/j.meegid.2014.12.029. [DOI] [PubMed] [Google Scholar]
- 38.Krobitsch S, Clos J. 1999. A novel role for 100 kD heat shock proteins in the parasite Leishmania donovani. Cell Stress Chaperones 4:191–198. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhattacharjee H, Li J, Ksenzenko MY, Rosen BP. 1995. Role of cysteinyl residues in metalloactivation of the oxyanion-translocating ArsA ATPase. J Biol Chem 270:11245–11250. doi: 10.1074/jbc.270.19.11245. [DOI] [PubMed] [Google Scholar]
- 40.Vanaerschot M, De Doncker S, Rijal S, Maes L, Dujardin JC, Decuypere S. 2011. Antimonial resistance in Leishmania donovani is associated with increased in vivo parasite burden. PLoS One 6:e23120. doi: 10.1371/journal.pone.0023120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sereno D, Roy G, Lemesre JL, Papadopoulou B, Ouellette M. 2001. DNA transformation of Leishmania infantum axenic amastigotes and their use in drug screening. Antimicrob Agents Chemother 45:1168–1173. doi: 10.1128/AAC.45.4.1168-1173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paris C, Loiseau PM, Bories C, Breard J. 2004. Miltefosine induces apoptosis-like death in Leishmania donovani promastigotes. Antimicrob Agents Chemother 48:852–859. doi: 10.1128/AAC.48.3.852-859.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee N, Bertholet S, Debrabant A, Muller J, Duncan R, Nakhasi HL. 2002. Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ 9:53–64. doi: 10.1038/sj.cdd.4400952. [DOI] [PubMed] [Google Scholar]
- 44.Sudhandiran G, Shaha C. 2003. Antimonial-induced increase in intracellular Ca2+ through non-selective cation channels in the host and the parasite is responsible for apoptosis of intracellular Leishmania donovani amastigotes. J Biol Chem 278:25120–25132. doi: 10.1074/jbc.M301975200. [DOI] [PubMed] [Google Scholar]
- 45.Khademvatan S, Gharavi MJ, Rahim F, Saki J. 2011. Miltefosine-induced apoptotic cell death on Leishmania major and L. tropica strains. Korean J Parasitol 49:17–23. doi: 10.3347/kjp.2011.49.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marinho FDA, Goncalves KC, Oliveira SS, Oliveira AC, Bellio M, d'Avila-Levy CM, Santos AL, Branquinha MH. 2011. Miltefosine induces programmed cell death in Leishmania amazonensis promastigotes. Mem Inst Oswaldo Cruz 106:507–509. doi: 10.1590/S0074-02762011000400021. [DOI] [PubMed] [Google Scholar]
- 47.Ubeda JM, Raymond F, Mukherjee A, Plourde M, Gingras H, Roy G, Lapointe A, Leprohon P, Papadopoulou B, Corbeil J, Ouellette M. 2014. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol 12:e1001868. doi: 10.1371/journal.pbio.1001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gazanion E, Fernandez-Prada C, Papadopoulou B, Leprohon P, Ouellette M. 2016. Cos-Seq for high-throughput identification of drug target and resistance mechanisms in the protozoan parasite Leishmania. Proc Natl Acad Sci U S A 113:E3012–E3021. doi: 10.1073/pnas.1520693113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindquist S. 1992. Heat-shock proteins and stress tolerance in microorganisms. Curr Opin Genet Dev 2:748–755. doi: 10.1016/S0959-437X(05)80135-2. [DOI] [PubMed] [Google Scholar]
- 50.Lindquist S. 1986. The heat-shock response. Annu Rev Biochem 55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- 51.Sorger P, Pelham HRB. 1988. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54:855–864. doi: 10.1016/S0092-8674(88)91219-6. [DOI] [PubMed] [Google Scholar]
- 52.Clos J, Westwood JT, Becker PB, Wilson S, Lambert K, Wu C. 1990. Molecular cloning and expression of a hexameric Drosophila heat shock factor subject to negative regulation. Cell 63:1085–1097. doi: 10.1016/0092-8674(90)90511-C. [DOI] [PubMed] [Google Scholar]
- 53.Sorger PK, Pelham HR. 1987. Purification and characterization of a heat-shock element binding protein from yeast. EMBO J 6:3035–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu C, Wilson S, Walker B, Dawid I, Paisley T, Zimarino V, Ueda H. 1987. Purification and properties of Drosophila heat shock activator protein. Science 238:1247–1253. doi: 10.1126/science.3685975. [DOI] [PubMed] [Google Scholar]
- 55.de Jong WW, Zweers A, Cohen LH. 1978. Influence of single amino acid substitutions on electrophoretic mobility of sodium dodecyl sulfate-protein complexes. Biochem Biophys Res Commun 82:532–539. doi: 10.1016/0006-291X(78)90907-5. [DOI] [PubMed] [Google Scholar]
- 56.Silverman JM, Chan SK, Robinson DP, Dwyer DM, Nandan D, Foster LJ, Reiner NE. 2008. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol 9:R35. doi: 10.1186/gb-2008-9-2-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vergnes B, Gourbal B, Girard I, Sundar S, Drummelsmith J, Ouellette M. 2007. A proteomics screen implicates HSP83 and a small kinetoplastid calpain-related protein in drug resistance in Leishmania donovani clinical field isolates by modulating drug-induced programmed cell death. Mol Cell Proteomics 6:88–101. [DOI] [PubMed] [Google Scholar]
- 58.Brochu C, Haimeur A, Ouellette M. 2004. The heat shock protein HSP70 and heat shock cognate protein HSC70 contribute to antimony tolerance in the protozoan parasite leishmania. Cell Stress Chaperones 9:294–303. doi: 10.1379/CSC-15R1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brochu C, Wang J, Roy G, Messier N, Wang XY, Saravia NG, Ouellette M. 2003. Antimony uptake systems in the protozoan parasite Leishmania and accumulation differences in antimony-resistant parasites. Antimicrob Agents Chemother 47:3073–3079. doi: 10.1128/AAC.47.10.3073-3079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cruz A, Coburn CM, Beverley SM. 1991. Double targeted gene replacement for creating null mutants. Proc Natl Acad Sci U S A 88:7170–7174. doi: 10.1073/pnas.88.16.7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ommen G, Lorenz S, Clos J. 2009. One-step generation of double-allele gene replacement mutants in Leishmania donovani. Int J Parasitol 39:541–546. doi: 10.1016/j.ijpara.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 62.Zhang WW, Matlashewski G. 2015. CRISPR-Cas9-mediated genome editing in Leishmania donovani. mBio 6:e00861. doi: 10.1128/mBio.00861-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Collins CR, Das S, Wong EH, Andenmatten N, Stallmach R, Hackett F, Herman JP, Muller S, Meissner M, Blackman MJ. 2013. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol Microbiol 88:687–701. doi: 10.1111/mmi.12206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bifeld E, Tejera Nevado P, Eick J, Bartsch J, Clos J. 9 June 2016. A versatile qPCR assay to quantify trypanosomatidic infections of host cells and tissues. Med Microbiol Immunol. doi: 10.1007/s00430-016-0460-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.