

Abstract
We present clinical practice guidelines for the diagnosis and treatment of homozygous familial hypercholesterolaemia (HoFH) in the Middle East region. While guidelines are broadly applicable in Europe, in the Middle East we experience a range of confounding factors that complicate disease management to a point whereby the European guidance cannot be applied without significant modification. Specifically, for disease prevalence, the Middle East region has an established epidemic of diabetes and metabolic syndrome that can complicate treatment and mask a clinical diagnosis of HoFH. We have also a high incidence of consanguineous marriages, which increase the risk of transmission of recessive and homozygous genetic disorders. This risk is further augmented in autosomal dominant disorders such as familial hypercholesterolaemia (FH), in which a range of defective genes can be transmitted, all of which contribute to the phenotypic expression of the disease. In terms of treatment, we do not have access to lipoprotein apheresis on the same scale as in Europe, and there remains a significant reliance on statins, ezetimibe and the older plasma exchange methods. Additionally, we do not have widespread access to anti-apolipoprotein B therapies and microsomal transfer protein inhibitors. In order to adapt existing global guidance documents on HoFH to the Middle East region, we convened a panel of experts from Oman, Saudi Arabia, UAE, Iran and Bahrain to draft a regional guidance document for HoFH. We also included selected experts from outside the region. This panel statement will form the foundation of a detailed appraisal of the current FH management in the Middle Eastern population and thereby provide a suitable set of guidelines tailored for the region.
Keywords: Homozygous familial hypercholesterolaemia, prevalence, diagnosis, treatment, genetics
Introduction
In the states of The Gulf Co-operation Council (GCC), cardiovascular disease (CVD) is the most common cause of death, accounting for up to 45% of all deaths [1]. The INTERHEART study showed that CVD risk factors like dyslipidaemia and smoking were prevalent in young populations in the region with a mean age of the first presentation of acute myocardial infarction (MI) 10 years younger in the Middle East countries than in other regions [2].
The Gulf Registry of Acute Coronary Events (Gulf RACE)-2 study showed that the Middle East also has a poor record with treatment of elderly-at risk CVD patients, and that adherence to clinical practice guidelines could be much improved [3]. Recently, the Gulf Locals with Acute Coronary Syndrome Events Registry (Gulf COAST), including 3,188 regional patients admitted with a diagnosis of acute coronary syndrome (ACS) found that the Gulf ACS population is characterised as young with a very high-risk profile [4]. The projected future burden of CVD in the Middle East is set to exceed that of other global regions [5]. Therefore, conditions that overtly raise cardiovascular (CV) risk are of particular interest in the region, and clinical practice guidelines are needed.
Homozygous familial hypercholesterolaemia (HoFH) is a rare and serious genetic condition characterised by markedly elevated low-density lipoprotein cholesterol (LDL-C) levels, and often (but not universally) cutaneous and tendon xanthomas and/or corneal arcus caused by deposition of excess cholesterol [6, 7]. If untreated, most HoFH patients will develop overt atherosclerosis before the age of 20 years and are unlikely to survive the past 30 years [6]. In addition to atherosclerotic complications, HoFH patients can also develop aortic and supra aortic valve stenosis, conditions that significantly increase morbidity and mortality [7].
HoFH was originally characterised by its clinical features: plasma cholesterol levels >13 mmol/L (>500 mg/dL), extensive xanthomas, and premature progressive atherosclerotic CVD. Studies in cultured fibroblasts showed that patients with these clinical features had a severe impairment in their ability to bind and internalise LDL particles. Subsequently, this defect was shown to result from mutations in both alleles of the LDL receptor (LDLR) gene [6]. However, findings of recent studies suggest that HoFH can also be a result of mutations in other genes such as the apolipoprotein B (apo B) gene or the proprotein converting subtilisin/kexin type 9 (PCSK-9) gene [8] indicating that the disease is genetically heterogeneous.
Prevalence of HoFH
HoFH is a highly under-recognised condition. In Oman where data are available, HoFH detection rates have been estimated at below 1% of the total population of patients likely to have the disease [9]. However, estimating diagnosis rates and prevalence is difficult. Data on the prevalence of HoFH in the Middle East are lacking, and there are no national registries currently collecting data. Therefore, we must rely mainly on data from outside the region to estimate prevalence and then apply some adjustment to acknowledge the unique family structures in the Middle East. In the past, the overall prevalence of HoFH was estimated to be around 1 in 1 million [6], but recent studies, particularly from The Netherlands, suggest that it may affect as many as 1 in 160,000-300,000 people [9, 10]. For the Middle East, HoFH may have a higher prevalence than in the Western world because consanguineous marriages are more common than in the West [11].
Rough estimates can be made by looking at the numbers of HoFH patients in key hospitals in the region. At the time of writing, the Sultan Qaboos University Hospital, Muscat, Oman, had 5 HoFH patients [12, 13]. If we take the population of Oman as 3.6 million persons, acknowledge 12 Omani hospitals capable of identifying and referring HoFH, and then consider that there may be other hospitals with unreported HoFH cases, then at least the Omani prevalence of HoFH will be higher than the historical estimates of 1 in 1 million. Published data from other countries are needed to complete the picture, and efforts are underway with a research group in the Middle East that includes the Oman Society of Lipid and Atherosclerosis (OSLA) to explore HoFH prevalence.
The Importance of HoFH Awareness
The true prevalence of HoFH is important. Cardiologists, dermatologists, paediatricians, endocrinologists, plastic surgeons and general practitioners need to understand that they may well see a case during their clinical career. They need to be educated and equipped in terms of clinical criteria to recognise and respond to a case. In the Middle East in particular, there is an epidemic of type 2 diabetes mellitus (T2DM) [14]. This presents a particular problem for the diagnosis of HoFH in that patients who present with premature CVD typical of HoFH may be considered as diabetic or metabolic syndrome cases in the first instance, and hence not undergo appropriate lipid profiling and onward specialist referral. For example, along with high LDL-C levels, HoFH patients may present with high triglyceride levels, which may direct the clinician towards a diagnosis of metabolic syndrome. In contrast to all other hyperlipidaemias in which obesity, hypertension and T2DM are highly prevalent, these conditions are generally uncommon in FH [15]. If a delayed or wrong diagnosis is made, then a non-response to lipid-lowering therapies should trigger a re-evaluation for HoFH, including a detailed assessment of the family history.
The origins of the Middle East panel
A recently published position paper from a consensus panel of the European Atherosclerosis Society (EAS) has highlighted the need for early identification of patients with HoFH, and the importance of prompt referral to specialist centres to enable appropriate treatment to be initiated as early as possible [8]. These recommendations are a useful guide to the diagnosis and treatment of HoFH. However, for the Middle East, there are clinical, genetic and practical factors specific to the region that need to be taken into account before we can apply the principles of the EAS guidelines. The Middle Eastern HoFH guidelines panel of regional HoFH experts was convened to discuss existing guidance on HoFH and to evaluate the considerations that need to be made to identify and treat the disease in Middle-Eastern hospitals and clinics.
Diagnosis of HOFH
Recent European guidelines suggest that diagnosis can be made on the basis of LDL-C levels, physical signs, family history and, where available, genetic confirmation [8].
At present, it is understood that the diagnosis rate of HoFH in the Middle East is very low [16], which is a concern given the particular circumstances that put the population at elevated risk of inheriting the disease.
LDL-C
Historically, HoFH has mostly been diagnosed on the basis of clinical and metabolic characteristics. Patients with HoFH have markedly elevated LDL-C levels [6, 7]. Importantly, the range of LDL-C levels observed in patients with HoFH is quite broad and can overlap with ranges found in other types of hypercholesterolaemia (Fig. 1) [8].
Fig. (1).
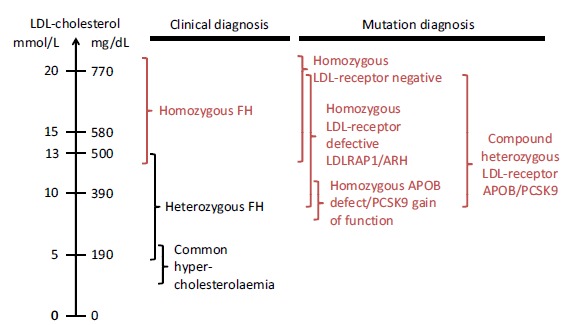
Low-density lipoprotein cholesterol (LDL-C) ranges in patients with familial hypercholesterolaemia (FH). Reproduced from Cuchel et al. Eur Heart J 2014; 35: 2146-57 [8], under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/).
Typically HoFH can be suspected if a patient presents with an untreated LDL-C >13 mmol/L (500 mg/dL) or treated LDL-C >8 mmol/L (300 mg/dL) [8]. Recent data from clinical trials indicate that in some instances LDL-C levels may be as low as 4 mmol/L (155 mg/dL), so even in patients without classically high LDL-C, a diagnosis of HoFH can still be made [10].
Al-Rasadi et al. have published diagnosis criteria for the Omani FH population, which suggest using the Simon Broome criteria [17]. These criteria suggest diagnosis of FH at total cholesterol (TC) above 7.5 (290 mg/dL) or LDL-C levels >4.9 mmol/L (190 mg/dL) in adults and TC >6.7 mmol/L or LDL-C >4.0 mmol/L in children under 16 years, combined in both cases with a confirmed family history compatible with the diagnosis of FH [18]. However, these criteria are not specific to HoFH and should not be used in the HoFH setting. Similarly, guidance from the Dutch Lipid Clinic Network [9] and the Make Early Diagnosis to Prevent Early Deaths (MEDPED) criteria (USA) [19] use clinical parameters beyond LDL-C, but are also not specific to HoFH.
Xanthomas
The high levels of cholesterol to which HoFH patients are exposed from birth, lead to accumulation of cholesterol in tendons, cutaneous tissues and cardiovascular tissues usually earlier than 10 years of age [7]. Thus, in addition to highly elevated LDL-C levels, other clinical characteristics of HoFH include early onset cutaneous and tendon xanthomas (Fig. 2), corneal arcus, widespread and severe atherosclerosis, aortic and supra aortic valve disease as well as early onset coronary heart disease [7].
Fig. (2).
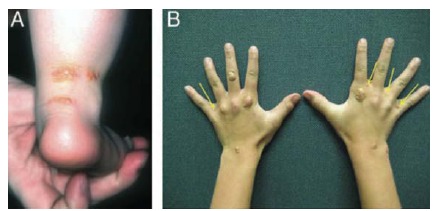
Tendon xanthomas characteristic of homozygous familial hypercholesterolaemia (HoFH). Cutaneous and tuberous xanthomas in homozygous familial hypercholesterolaemia. Original photographs by Prof. Eric Bruckert and Prof. Frederick Raal. Reproduced from Cuchel et al. Eur Heart J 2014; 35: 2146-57 [8], under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/).
Although xanthoma are a classic clinical sign of HoFH and are included in international guidelines for HoFH management, their appearance is not universal; therefore their absence does not preclude an HoFH diagnosis. Importantly, these xanthomas lead many HoFH patients to present in the first instance to dermatologists or plastic surgeons, thereby providing opportunities for prompt diagnosis.
Family History
Family history can provide additional clues to aid the diagnosis of HoFH. Factors that can be taken into account include myocardial infarction at age ≤60 years in a first-degree relative (or ≤50 years in a second-degree relative), xanthoma, persistent and treatment-resistant elevated LDL-C levels, and genetically confirmed heterozygous FH (HeFH) or HoFH. In the Middle East, tracing family histories may be easier than in some other regions because family trees are more clearly defined, and many individuals undergo medical examination as part of the requirements for marriage. Where there is a confirmed family history of FH, pre-natal counselling should be provided to ensure that prospective parents understand the likelihood of producing offspring with HoFH, and the risks and burdens that the disease can bring. As such, diagnostic teams should also establish if the parents of a patient are related. People marrying within a small town or village (i.e. a small genetic pool) create what is termed a ‘founder effect’ – a term derived from founding settlers of new communities (e.g. white South Africans), who multiplied within their own gene pool [20, 21].
Genetic Testing
In cases where the LDL-C level is extremely high, a clinical diagnosis of HoFH can be quite clear cut. However, at the lower ranges, the situation is less certain, and genetic testing is important for an unequivocal FH diagnosis. The application of genetic testing for HoFH across the world is variable. Genetic testing is widespread in Europe, and relatively uncommon in the United States. In the Middle East, genetic testing is available in a limited number of specialist centres or referral units. Some units out-source genetic testing to Europe.
Once a case is identified, the testing centre will usually direct the genetic testing of siblings and parents. In some countries (e.g. The Netherlands and the UK), a positive genetic test triggers ‘cascade screening’ where all living relatives (parents, siblings, cousins, uncles, aunts etc.) are tested for FH-related mutations [22]. In the Middle East, cascade screening is not routinely performed, and is usually reserved for very severe HoFH cases. Application of cascade screening in the region is impeded by a complicated hospital system in which there are marked differences in treatment patterns and availability between military and non-military hospitals [23].
Although genetic testing may provide a definitive diagnosis of HoFH, in some patients extensive testing may fail to yield genetic confirmation [6, 7]. This is because up to 20% of clinically defined FH patients will have a mutation that has not yet been identified [24]. HoFH is most commonly caused by mutations in both alleles of the LDLR gene, in individuals with parents who each have heterozygous FH. However, recently, mutations in genes coding for apo B, PCSK9 and LDL receptor adapter protein 1 (LDLRAP1), have been identified as causal in some patients with a severe phenotype resembling HoFH [8, 13, 25].
HoFH patients may be ‘true homozygotes’, with the same mutation in both alleles of the same gene, or ‘compound heterozygotes’ with different mutations in the two alleles of the same gene. Patients may also carry mutations in two different genes affecting LDL receptor function (Fig. 3). The severity of the mutation is a major factor governing the severity of the phenotype. Patients with mutations resulting in defective LDL-R usually have less elevated LDL-C than patients with mutations resulting in a complete absence of LDL-R activity [8]. Generally, the pattern of mean LDL-C levels by genotype increase as follows: HeFH < double heterozygote (e.g. LDL+PCSK9 gain-of-function or apo B mutation) < homozygous apo B or PCSK9 gain-of-function mutation < homozygous LDLRAP1 or LDLR-defective mutations < compound heterozygote LDLR-defective+LDLR-negative mutations < homozygous LDLR-negative mutations [8].
Fig. (3).
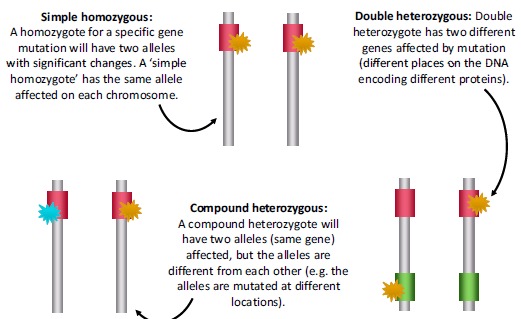
Genetic diversity of homozygous familial hypercholesterolaemia (HoFH).
However, there is considerable overlap in the observed untreated LDL-C levels according to genotype [10], so an individual compound heterozygote for LDLR, for example, may have very severe hypercholesterolaemia, and equally, a simple LDLR homozygote may have lower untreated LDL-C levels. In many instances, a patient with a negative genetic test for HoFH may still have homozygous mutations, but these mutations have not been identified within the current panel of known HoFH-associated mutations. Therefore, if the spectrum of mutations causing FH in a certain population is not known/identified, genetic testing, while valuable, cannot yet be considered a 100% reliable means of identifying HoFH patients in such patients. Next-generation sequencing techniques may alleviate or eradicate this limitation.
Genetic testing, where available still needs to be accompanied by comprehensive clinical and family history profiles [24]. A positive genetic test is definitive for HoFH. It is possible that cascade testing in the immediate family of an index patient may be made easier if the index mutation is known, and if the most common mutations in the Middle East region could be profiled.
Another disorder of lipid metabolism, sitosterolaemia (or phytosterolaemia), may have a similar clinical presentation to HoFH. A definitive diagnosis of sitosterolaemia can be confirmed by genetic analysis. In common with HoFH, any genetically determined metabolic disorder is likely to be more common in regions with lower genetic admixture than those with very few consanguineous marriages [26].
Summary and Recommendations
Our recommendations for diagnosis of HoFH are similar to those set out in the European guidelines (Table 1) [8].
Table 1. Summary recommendations for the diagnosis of homozygous familial hypercholesterolaemia (HoFH).
| Genetic confirmation of two mutant alleles at the LDLR, APOB, PCSK9, or LDLRAP1 gene |
| Or An untreated LDL-C >13 mmol/L (500 mg/dL) or treated LDL-C ≥8 mmol/L (300 mg/dL)* Cutaneous or tendon xanthomas before age 10 years Or Untreated LDL-C consistent with heterozygous FH in both parents |
| *These LDL-C levels are only indicative, and lower levels, especially in children or in treated patients, do not exclude HoFH |
HoFH can be suspected if LDL-C levels are >13 mmol/L (500 mg/dL) in untreated patients, or >8 mmol/L (300 mg/dL) in treated patients.
Confirmation of diagnosis can be made if the elevated LDL-C is accompanied by either cutaneous or tendon xanthomas before the age of 10 years, or untreated elevated LDL-C levels consistent with HeFH in both parents.
Genetic testing can independently confirm the diagnosis of HoFH, but a negative result does not rule out the possibility of FH.
Detection rates for HoFH in the Middle East are low, and efforts must be made by the preventative cardiology and lipidology communities to educate dermatologists, general practitioners, plastic surgeons and cardiologists on the true prevalence and recognition of the disease.
Genetic counselling should be given to couples where there is a risk of HoFH in their offspring, and patients should be advised to limit the number of pregnancies for reasons of both genetic transmission and maternal cardiovascular risk. Hormonal contraception should be avoided [8].
As standard of care, patients should be screened for other underlying problems that might cause hyperlipidaemia, such as diabetes, thyroid function disorders, impaired kidney function, drug treatments and sitosterolaemia (this is also probably under-diagnosed).
Reproduced from Cuchel et al. Eur Heart J 2014; 35: 2146-57 [8], under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/).
Natural history and cardiovascular complications of HOFH and the importance of screening for subclinical disease
In patients with HoFH, plasma levels of LDL-C are markedly elevated from birth and this high lipid burden underlies the development of atherosclerotic cardiovascular disease complications in these patients. HoFH is characterised by accelerated atherosclerosis, typically (but not exclusively) affecting the aortic root [8]. HoFH patients often experience their first major cardiovascular event during adolescence, and possibly even earlier if they are LDLR-negative and/or untreated [8]. In young children, early signs and symptoms are often those associated with aortic stenosis and regurgitation, as a result of cholesterol accumulation at valvular leaflets [8].
Even when cholesterol levels are reduced, valvular and supra-valvular aortic diseases may still progress due to haemodynamic stress and fibrosis [27], therefore regular screening for subclinical carotid, aortic and coronary heart disease is indicated. A number of screening methods may be considered including Doppler echocardiography, computed tomography coronary angiography, magnetic resonance imaging, trans-oesophageal echocardiography, stress testing, invasive coronary angiography [8] and genetic testing. At present, these methods are not routinely used in the Middle East to screen for aortic involvement in undiagnosed patients with a confirmed family history. A number of studies in children with FH has shown some evidence of endothelial dysfunction detected by flow-mediated dilation (FMD) [28, 29]. In addition, significant improvement of endothelial dysfunction was demonstrated by statins treatment in hypercholesterolaemic adults and in children with FH [30-32]. However, most of the guidelines underscore the value of non-invasive imaging of atherosclerosis in assessing and managing asymptomatic FH subjects [33, 34].
Should atherosclerotic CVD be present, then surgical procedures may be indicated in aortic valve replacement in cases of severe left ventricular outflow obstruction and reconstruction of the aortic root might also be necessary [35]. Coronary angiography should be reserved for those with symptoms or signs of ischaemia, because of the risk associated with catheterisation of patients with coronary ostial stenosis [35].
Summary and Recommendations
Screening for coronary calcium score should be conducted every 3 years [considering that the negative predicted value is low in young patients who have the usual risk factors for CVD (e.g. diabetes mellitus, hypertension and smoking)].
Screening for plaque formation should be conducted every 5 years using low radiation computerised tomographic angiography (provided that radiation dose does not exceed 3-5 milliSievert). Use of carotid Doppler to image carotid plaque and velocity every 6 months is a reasonable surrogate in between computerised tomographic scans. If the initial computerised tomographic angiography at time of diagnosis is already abnormal with existing plaque, the time interval between scans can be reduced.
Carotid intima media thickness should ideally be assessed every 6 months, but there is need for consistent technician/radiologist training to achieve this.
Stress testing is not recommended for assessment of atherosclerotic plaques.
If progression of subclinical disease is seen, intensification of treatment is warranted.
Treatment
Current Treatment Options for HoFH
Principles of Treatment
Reducing elevated LDL-C levels is the fundamental principle of the treatment of HoFH. Current guideline LDL-C targets in HoFH are <2.5 mmol/L (<100 mg/dL) [N.B. the target levels in children are somewhat higher, <3.5 mmol/L (<135 mg/dL)], or <1.8 (<70 mg/dL) in adults with atherosclerotic CVD [8, 9]. Importantly, the rarity of HoFH means that there is no prospect of robust therapeutic outcomes data [9]. In most studies the only data collected on cardiac events are reported as adverse events. Some trials are now looking at further surrogates for CVD effects other than LDL-C such as changes in atherosclerotic plaque size and morphology. In the meantime, reduction in LDL-C levels, which are firmly correlated with CV outcomes in other conditions [9, 36], need to be accepted as a marker for therapeutic activity.
Until the 1980s, the only available treatment options for patients with HoFH were adherence to a strict low-fat diet and use of minimally effective lipid-modifying drugs. These approaches have not demonstrated sufficient efficacy to control LDL-C to desired targets. Since the 1980s, and particularly with the advent of statins, a variety of different therapeutic strategies have been explored, including pharmacotherapy, apheresis and surgical approaches.
Statins
Statins have been the mainstay of treatment of HoFH, and have been shown to reduce mortality [37]. In some instances, where permission is granted by the institute and insurer, statins may be used in children; the cut-off age may vary between countries (8, 10 or 12 years). Licensed treatment options for younger children are limited to dietary control, resins and apheresis. Recent European guidance on statin use in children with FH underscores the need to start therapy at 8 years of age [38], but in some instances this may be too late.
Statins work by in part increasing the expression of LDL-Rs via a feedback loop that detects reduced cholesterol levels caused by the inhibition of HMG-CoA reductase in the liver [39]. In HoFH, the LDL-R is either non-functional or severely impaired [6]; therefore, any drug with a LDL-R-dependent mode of action would be expected to have limited efficacy. Indeed, most HoFH patients achieve only modest reductions in plasma levels of LDL-C (10-25%) when receiving statins [8]. Further reductions in LDL-C levels can be achieved by the addition of other cholesterol-lowering medications, including ezetimibe, bile acid sequestrants, and niacin, but their use may be limited by tolerability and availability [8]. Additionally, a relatively large proportion of patients prescribed statins (estimates 10.5-23%) can exhibit intolerance to the drugs, manifested as muscle symptoms [40]. Statin intolerance has been recently defined as the inability to tolerate at least two low-dose statins with symptomatic or biomarker associations with dose titration or therapy withdrawal [41]. In these patients, alternative therapies should be sought. Statins have also been associated with the emergence of new onset T2DM [42], which may be a consideration, particularly in patients already suffering from, or at risk of, T2DM.
Statin Combination with Ezetimibe
Several studies have confirmed the efficacy of ezetimibe in combination with statin in patients with HoFH [43-49]. A study demonstrated at least 14.0% to 20.5% reduction in LDL-C when ezetimibe was co-administered with a moderate (40 mg) or maximal (80 mg) dose of statin therapy compared with maximal therapy with statins alone [44].
In patients with HoFH, the effect of statins can be significantly limited by the inability of these patients to effectively upregulate the LDL receptor. This seems not to be the case in ezetimibe, where the LDL-C lowering is mainly achieved through the inhibition of intestinal cholesterol absorption. The IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) was the first trial to demonstrate incremental cardiovascular events reduction when adding ezetimibe to statin therapy. Over a 7-year period, the addition of ezetimibe to simvastatin 40 mg reduced the primary end point – a composite of cardiovascular death, MI, unstable angina requiring rehospitalisation, coronary revascularisation, or stroke – by 6.4% when compared with patients who received simvastatin alone (p=0.016). The absolute reduction in risk over 7 years was 2.0%, with 32.7% in the ezetimibe/simvastatin arm experiencing an event in the primary end point set compared with 34.7% in the simvastatin arm [50].
Apheresis
Where available, lipoprotein apheresis (LA) is an important adjunctive treatment for HoFH. There is evidence that in the long term it can contribute to plaque regression or stabilisation and improve prognosis [51]; however, it is time-consuming, costly [8], and can be an unpleasant experience for patients. Nevertheless, apheresis is an effective technique, and can be used in patients where licensed drugs are not available (e.g. children), where pharmacotherapy is contraindicated (e.g. pregnancy and breastfeeding) [52] or to limit statin-induced myopathy in those patients who show signs of statin intolerance.
If apheresis is being used in conjunction with other lipid-lowering therapies, and LDL-C levels are approaching the established targets, then physicians may consider adjusting the apheresis schedule or stopping the procedure in the interest of patient convenience. However, in doing so, the patient may lose some of the “pleiotropic” effects of apheresis, which include beneficial effects on lipoprotein a (Lp(a)), clotting factors, and possibly on circulating levels of pro-inflammatory cytokines [51].
LA that selectively removes apolipoprotein B100-containing lipoproteins from plasma, including Lp(a), is considered the current gold standard among apheresis therapies particularly when used alongside pharmacotherapy [51, 53]; however, in the Middle East, LA is largely inaccessible apart from in a few centres with access to advanced technology, and most centres that offer apheresis employ the older plasma exchange method (PEX), which is not selective for apoB-containing lipoproteins. Even for PEX, access may be limited, and patients may need to be prioritised for therapy. Despite its limitations, PEX, like LA, has been shown to improve the survival of patients with HoFH [54], and should ideally be initiated before the age of 5 years where available [8].
For both PEX and LA, LDL-C levels undergo rebound to baseline levels over 2-4 days [55], therefore apheresis should be applied weekly or biweekly [56].
Guidelines from the US National Lipid Association (NLA) that initiation of therapy in children is vital, and that although high dose statins may be somewhat effective, most patients will require LA; liver transplantation may also be considered and gene therapy is a potential new therapy in development [57].
Liver Transplantation and Surgical Procedures
Liver transplantation can achieve a substantial improvement in LDL-C levels but it is associated with many drawbacks, including a high risk of post-operative complications and mortality and a need for lifelong immunosuppressive therapy [58]. Partial ileal bypass or portocaval shunting is not recommended but may be considered if there is limited access to more effective treatments or for patients with very severe disease [8].
New Therapeutic Approaches for HoFH
Despite treatment with various types of lipid-lowering treatments, including lipoprotein apheresis, the majority of HoFH patients do not achieve their LDL-C target treatment goals [9]. While statins have been the mainstay of therapy, the 10-25% reductions in LDL-C associated with using these drugs are not sufficient to achieve target LDL-C levels.
Efforts are underway to understand the cardiovascular benefits of ‘lower is better’ [59, 60]. Therefore, even in the face of very high baseline LDL-C, physicians should be seeking to achieve guideline targets of <2.5 mmol/L adult values; <1.8 mmol/dL in adults with atherosclerotic cardiovascular disease. This may appear to be ambitious for patients with HoFH. However, research into lipid-lowering therapies has not halted with the apparent success of statins and apheresis.
The introduction of lipid-lowering agents, with novel mechanisms of action, may result in improvements in the management of HoFH. Lomitapide, an oral inhibitor of the microsomal triglyceride transfer protein, and mipomersen, an injectable apolipoprotein B antisense oligonucleotide, are approved by the United States Food and Drug Administration as adjunctive therapy for HoFH [61, 62]. Lomitapide is also approved by the European Medicines Agency. After trying diet/lifestyle and statins, these novel agents, if licensed in the patient’s country, could be added in after apheresis or instead of apheresis for adult patients, and in accordance with the product label [61, 62]. Mipomersen is not licenced for use with apheresis in the US [62]. Theoretically, mipomersen will be available in the Middle East but its use is not currently supported through a compassionate use programme or clinical trials.
Certain factors may affect patients willingness to comply with treatments, e.g. for lomitapide, the need to adhere to a low-fat diet, to avoid steatorrhoea [61, 62], and for mipomersen the need to inject the drug and the onset of injection site reactions and flu-like symptoms [63]. Hepatic steatosis evident with lomitapide and mipomersen may be monitored via magnetic resonance imaging in patients considered to be at risk, on the understanding that in the Middle East in particular, non-alcoholic fatty livers are relatively common [64].
Other new approaches currently being studied for the treatment of HoFH include the use of PCSK9 inhibitors, cholesteryl ester transfer protein (CETP) inhibitors and gene therapy. There is virtually no experience of these agents in Middle Eastern patients at this time.
PCSK9 inhibitors work by binding to the LDL-R, and shortening the lifespan of this receptor, thereby decreasing LDL-R function [65]. Therefore, like statins, they rely on a functioning LDL-R, and may have limited efficacy in HoFH. Indeed, among HoFH patients in a trial of evolocumab (a PCSK9 monoclonal antibody), the drug resulted in a mean reduction in LDL-C levels from baseline of 23% after 12 weeks of treatment [66], which is considerably less than the 40-50% observed for lomitapide [67]. Current evidence on CETP inhibitors suggests that effectiveness of these agents are variable in HoFH, and that current trials used patient populations with lower baseline LDL-C values than commonly encountered in HoFH, making clinical efficacy difficult to evaluate [68].
Despite 23 years of research [69], gene therapy for HoFH involving a single transfected vector is still some way off. Some initial successes have been observed with partial hepatectomy and re-infusion of engineered autologous hepatocytes; and particularly with LDLR-adenovirus-mediated transfer [69]. Cost may turn out to be a major limiting factor to adoption of these drugs.
Summary and Recommendations
Suggested LDL-C targets in HoFH are <2.5 mmol/L (<100 mg/dL) [N.B. the target levels in children are higher, <3.5 mmol/L (<135 mg/dL)], or <1.8 mmol/L (<70 mg/dL) in adults with atherosclerotic CVD [8, 9].
First-line treatment: diet/lifestyle + maximum tolerated dose of high efficacy statins ± ezetimibe ± resin. Smoking should be avoided and exercise should be undertaken. If a genetic confirmation of HoFH is available, first-line treatment should be LA (preferred) or PEX (Fig. 4).
If target is not achieved after 3 months (US National Lipid Association [NLA] guidelines recommend 6 months) apheresis is recommended; in the Middle East PEX is most widely available; however, LA is preferred, where available. PEX cannot be used in young children, below 25 kg in weight. Apheresis should be started by age of 5 years and no later than 8 years [8]. The Middle East Guidelines Panel recognises the difficulty in accessing apheresis therapy in the region, but apheresis is preferable to statins in the young, in whom they are not licensed. A separate algorithm is provided for children (Fig. 4B).
If target is not achieved, new lipid-lowering therapies, such as lomitapide could be added [with statins ± ezetimibe ± resin; also lomitapide (but not mipomersen) can be used with apheresis] (Fig. 4). Lomitapide and mipomersen are not yet approved in the Middle East but this can be expected in the future. A paediatric trial for lomitapide is planned, but the drug remains unlicensed in children in both the US and Europe.
-
Pregnancy and breastfeeding
Pharmacotherapy is contra-indicated for pregnant and breastfeeding mothers.
Patients with HoFH should undergo thorough cardiovascular assessment before becoming pregnant.
It is advisable for HoFH patients to have apheresis during pregnancy to benefit both mother and baby. If a patient is not already on apheresis, the decision to start apheresis would depend on major cardiovascular risk factors and LDL-C level. If a patient has existing CVD they should be advised to undergo apheresis. If access to apheresis is an issue then the medical team should explore compassionate avenues.
Medical teams should be aware that exposure to high LDL-C commences in utero, and hence the cumulative LDL-C exposure over the lifetime of the offspring is increased both for the FH mother and FH child.
Fig. (4).
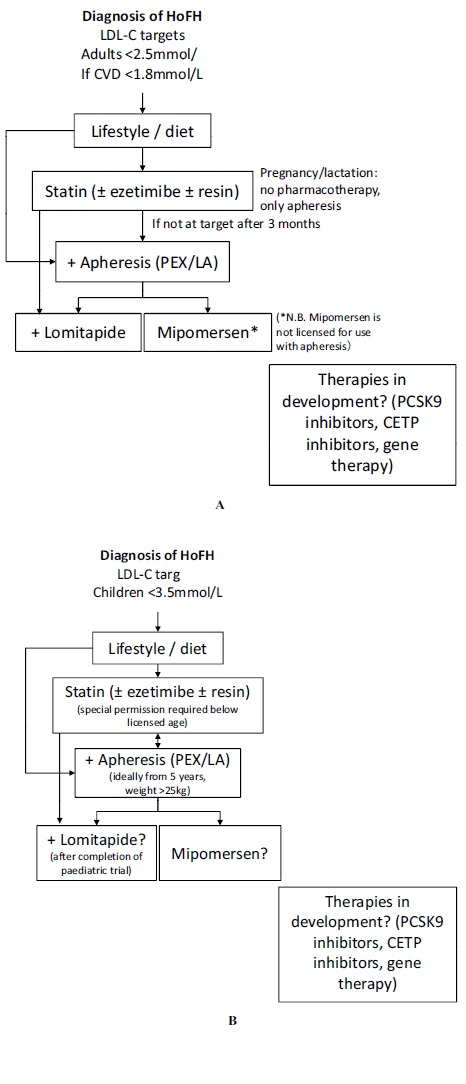
Algorithm for the treatment of homozygous familial hypercholesterolaemia (HoFH). Caption: PEX, plasma exchange; LA, lipoprotein apheresis; PCSK9, proprotein convertase subtilisin/kexin type 9; CETP, cholesteryl ester transfer protein. Adapted from Cuchel et al. Eur Heart J 2014; 35: 2146-57 [8], under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/). Mipomersen licensed in US from age 12 years.
Research recommendations
Having evaluated the evidence to produce these guidelines, the Middle East HoFH guidelines panel can suggest a range of topics that are worthy of further examination to improve understanding, awareness and management of HoFH. These research suggestions are as follows:
Genetic counselling and neonatal screening. This will be very important given the high prevalence consanguinity in the Middle East region. Pre- and neo-natal counselling are important in families with FH diagnoses.
Real world observational studies are required to compare the clinical performance and pharmacoeconomics of all therapies used to treat HoFH, including apheresis. These studies will need to be designed to acknowledge the established difficulties in obtaining CV and survival outcomes data in rare diseases.
A Middle Eastern HoFH registry is urgently needed to understand the true prevalence of HoFH across the region. A new initiative has recently been announced by the EAS, to try to set up a global registry of FH and HoFH; they are in discussion with other groups around the world, including the International Atherosclerosis Society (IAS), the FH Foundation in the US, and Prof. Gerald Watts in Australia. The initiative is being led by Prof. Kausik Ray, at Imperial College London, UK.
Screening programmes are required to improve diagnosis and improve the quality of the data in the proposed registry.
Drug metabolism studies are needed including covariate analysis, by age, ethnicity, etc. It may also be useful to understand the effects that lomitapide has on risks of non-alcoholic fatty liver disease, which is prevalent in the Middle East.
ACKNOWLEDGEMENTS
We acknowledge the assistance of Eastmond Medicomm Ltd in the preparation of this manuscript. This editorial and organisational support was funded by Aegerion Pharmaceuticals Inc.
CONFLICT OF INTEREST
• Abdullah Al-Ashwal
o Speaking and consultancy: None
o Research grants: None
• Fahad Alnouri
o Speaking and consultancy: Aegerion
o Research grants: None
• Hani Sabbour
o Speaking and consultancy: Aegerion, Amgen, Astra- Zeneca, Pfizer
o Research grants: None
• Abdulraof Al-Mahfouz
o Speaking and consultancy: MSD
o Research grants: None
• Nasreen Al-Sayed
o Speaking and consultancy: MSD
o Research grants: None
• Maryam Razzaghy-Azar
o Speaking and consultancy: None
o Research grants: None
• Faisal Al-Allaf
o Speaking and consultancy: None
o Research grants: National Science, Technology and Innovation Plan (NSTIP); grant number (08-BIO34-10)
• Khalid Al-Waili
o Speaking and consultancy: AstraZeneca, Pfizer
o Research grants: None
• Yajnavalka Banerjee
o Speaking and consultancy: None
o Research grants: None
• Jacques Genest
o Speaking and consultancy: Aegerion, Amgen, Sanofi
o Research grants: Aegerion, Amgen, Sanofi, Pfizer, Valeant
• Raul D Santos
o Speaking and consultancy: Amgen, Biolab, Boehringer-Ingleheim, Eli-Lilly, Genzyme, Jansen, Merck, Praxis, Pfizer, Sanofi/Regeneron, Torrent
o Research grants: Amgen, Genzyme, Pfizer
• Khalid Al-Rasadi (corresponding author)
o Speaking and consultancy: AstraZeneca, Pfizer, Sanofi, Aegerion
o Research grants: None.
REFERENCES
- 1.Husseini A., Abu-Rmeileh N.M., Mikki N., et al. Cardiovascular diseases, diabetes mellitus, and cancer in the occupied Palestinian territory. Lancet. 2009;373(9668):1041–1049. doi: 10.1016/S0140-6736(09)60109-4. [DOI] [PubMed] [Google Scholar]
- 2.Gehani A.A., Al-Hinai A.T., Zubaid M., et al. Association of risk factors with acute myocardial infarction in Middle Eastern countries: the INTERHEART Middle East study. Eur. J. Prev. Cardiol. 2014;21(4):400–410. doi: 10.1177/2047487312465525. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed E., Alhabib K.F., El-Menyar A., et al. Age and clinical outcomes in patients presenting with acute coronary syndromes. J. Cardiovasc. Dis. Res. 2013;4(2):134–139. doi: 10.1016/j.jcdr.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zubaid M, Thani KB, Rashed W, et al. Design and Rationale of Gulf locals with Acute Coronary Syndrome Events (Gulf Coast) Registry. Open Cardiovasc Med 014. 8:88–93. doi: 10.2174/1874192401408010088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almahmeed W, Arnaout MS, Chettaoui R, et al. Coronary artery disease in Africa and the Middle East.Ther Clin Risk Mana 012; 8:65–72. doi: 10.2147/TCRM.S26414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein J.K., Hobbs H.H., Brown M.S. Familial Hypercholesterolemia. In: Scriver CR, Beaudet. In: Sly WS, Valle D, editors. The Metabolic Basis of Inherited Diseas. New York: McGraw- Hill; 2001. pp. 2863–913. [Google Scholar]
- 7.Raal F.J., Santos R.D. Homozygous familial hypercholesterolaemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223(2):262–268. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Cuchel M., Bruckert E., Ginsberg H.N., et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014;35(32):2146–2157. doi: 10.1093/eurheartj/ehu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nordestgaard B.G., Chapman M.J., Humphries S.E., et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013;34(45):3478–90a. doi: 10.1093/eurheartj/eht273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjouke B., Kusters D.M., Kindt I., et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 2015;36(3):560–565. doi: 10.1093/eurheartj/ehu058. [DOI] [PubMed] [Google Scholar]
- 11.el-Hazmi M.A., al-Swailem A.R., Warsy A.S., al-Swailem A.M., Sulaimani R., al-Meshari A.A. Consanguinity among the Saudi Arabian population. J. Med. Genet. 1995;32(8):623–626. doi: 10.1136/jmg.32.8.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Hinai A.T., Al-Abri A., Al-Dhuhli H., et al. First case report of familial hypercholesterolaemia in an Omani family due to novel mutation in the low-density lipoprotein receptor gene. Angiology. 2013;64(4):287–292. doi: 10.1177/0003319712465171. [DOI] [PubMed] [Google Scholar]
- 13.Al-Waili K., Al-Zidi W.A., Al-Abri A.R., et al. Mutation in the PCSK9 Gene in Omani Arab Subjects with Autosomal Dominant Hypercholesterolaemia and its Effect on PCSK9 Protein Structure. Oman Med. J. 2013;28(1):48–52. doi: 10.5001/omj.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klautzer L., Becker J., Mattke S. The curse of wealth - Middle Eastern countries need to address the rapidly rising burden of diabetes. Int. J. Health Policy Manag. 2014;2(3):109–114. doi: 10.15171/ijhpm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sniderman A., Durrington P.N. Hyperlipidemia. 5th ed. Abingdon: Health Press; 2010. [Google Scholar]
- 16.Langslet G., Ose L. Screening methods in the diagnosis and assessment of children and adolescents with familial hypercholesterolaemia. Expert Rev. Cardiovasc. Ther. 2013;11(8):1061–1066. doi: 10.1586/14779072.2013.814851. [DOI] [PubMed] [Google Scholar]
- 17.Al-Rasadi K., Al-Waili K., Al-Sabti H.A., et al. Criteria for Diagnosis of Familial Hypercholesterolaemia: A Comprehensive Analysis of the Different Guidelines, Appraising their Suitability in the Omani Arab Population. Oman Med. J. 2014;29(2):85–91. doi: 10.5001/omj.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Risk of fatal coronary heart disease in familial hypercholes- terolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303(6807):893–896. doi: 10.1136/bmj.303.6807.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams R.R., Hunt S.C., Schumacher M.C., et al. Diagnosing heterozygous familial hypercholesterolaemia using new practical criteria validated by molecular genetics. Am. J. Cardiol. 1993;72(2):171–176. doi: 10.1016/0002-9149(93)90155-6. [DOI] [PubMed] [Google Scholar]
- 20.Kusters D.M., Huijgen R., Defesche J.C., et al. Founder mutations in the Netherlands: geographical distribution of the most prevalent mutations in the low-density lipoprotein receptor and apolipoprotein B genes. Neth. Heart J. 2011;19(4):175–182. doi: 10.1007/s12471-011-0076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seftel H.C., Baker S.G., Sandler M.P., et al. A host of hypercholesterolaemic homozygotes in South Africa. BMJ. 1980;281(6241):633–636. doi: 10.1136/bmj.281.6241.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Defesche J.C. Defining the challenges of FH screening for familial hypercholesterolaemia. J. Clin. Lipidol. 2010;4(5):338–341. doi: 10.1016/j.jacl.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 23.Bamimore M.A., Zaid A., Banerjee A., et al. Familial hypercholesterolaemia mutations in the Middle Eastern and North African region: A need for a national registry. J. Clin. Lipidol. 2015;9(2):187–194. doi: 10.1016/j.jacl.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Goldberg A.C., Hopkins P.N., Toth P.P., et al. Familial hypercholesterolaemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolaemia. J. Clin. Lipidol. 2011;5(3):133–140. doi: 10.1016/j.jacl.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Soutar A.K., Naoumova R.P. Mechanisms of disease: genetic causes of familial hypercholesterolaemia. Nat. Clin. Pract. Cardiovasc. Med. 2007;4(4):214–225. doi: 10.1038/ncpcardio0836. [DOI] [PubMed] [Google Scholar]
- 26.Khlat M., Khoury M. Inbreeding and diseases: demographic, genetic, and epidemiologic perspectives. Epidemiol. Rev. 1991;13:28–41. doi: 10.1093/oxfordjournals.epirev.a036072. [DOI] [PubMed] [Google Scholar]
- 27.Rallidis L., Nihoyannopoulos P., Thompson G.R. Aortic stenosis in homozygous familial hypercholesterolaemia. Heart. 1996;76(1):84–85. doi: 10.1136/hrt.76.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mietus-Snyder M., Malloy M.J. Endothelial dysfunction occurs in children with two genetic hyperlipidemias: improvement with antioxidant vitamin therapy. J. Pediatr. 1998;133(1):35–40. doi: 10.1016/s0022-3476(98)70174-x. [DOI] [PubMed] [Google Scholar]
- 29.de Jongh S., Lilien M.R., Bakker H.D., Hutten B.A., Kastelein J.J., Stroes E.S. Family history of cardiovascular events and endothelial dysfunction in children with familial hypercholesterolaemia. Atherosclerosis. 2002;163(1):193–197. doi: 10.1016/s0021-9150(02)00003-5. [DOI] [PubMed] [Google Scholar]
- 30.Stroes E.S., Koomans H.A., de Bruin T.W., Rabelink T.J. Vascular function in the forearm of hypercholesterolaemic patients off and on lipid-lowering medication. Lancet. 1995;346(8973):467–471. doi: 10.1016/s0140-6736(95)91322-x. [DOI] [PubMed] [Google Scholar]
- 31.de Jongh S., Lilien M.R. op't Roodt J, Stroes ES, Bakker HD, Kastelein JJ. Early statin therapy restores endothelial function in children with familial hypercholesterolaemia. J. Am. Coll. Cardiol. 2002;40(12):2117–2121. doi: 10.1016/s0735-1097(02)02593-7. [DOI] [PubMed] [Google Scholar]
- 32.Marchesi S., Lupattelli G., Siepi D., et al. Short-term atorvastatin treatment improves endothelial function in hypercholesterolemic women. J. Cardiovasc. Pharmacol. 2000;36(5):617–621. doi: 10.1097/00005344-200011000-00011. [DOI] [PubMed] [Google Scholar]
- 33.Civeira F. International Panel on Management of Familial H. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolaemia. Atherosclerosis. 2004;173(1):55–68. doi: 10.1016/j.atherosclerosis.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Watts G.F., Sullivan D.R., Poplawski N., et al. Familial hypercholesterolaemia: a model of care for Australasia. Atheroscler. Suppl. 2011;12(2):221–263. doi: 10.1016/j.atherosclerosissup.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Marais A.D., Firth J.C., Rose A.G., Berger G.M. Fatal outcome of homozygous familial hypercholesterolaemia in a black patient. A case report. S. Afr. Med. J. 1990;77(11):588–590. [PubMed] [Google Scholar]
- 36.Cholesterol Treatment Trialists Collaboration Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raal F.J., Pilcher G.J., Panz V.R., et al. Reduction in mortality in subjects with homozygous familial hypercholesterolaemia associated with advances in lipid-lowering therapy. Circulation. 2011;124(20):2202–2207. doi: 10.1161/CIRCULATIONAHA.111.042523. [DOI] [PubMed] [Google Scholar]
- 38.Wiegman A., Gidding S.S., Watts G.F., et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur. Heart J. 2015;•••:ehv157. doi: 10.1093/eurheartj/ehv157. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Endo A. The discovery and development of HMG-CoA reductase inhibitors. J. Lipid Res. 1992;33(11):1569–1582. [PubMed] [Google Scholar]
- 40.Bitzur R., Cohen H., Kamari Y., Harats D. Intolerance to statins: mechanisms and management. Diabetes Care. 2013;36(Suppl. 2):S325–S330. doi: 10.2337/dcS13-2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banach M., Rizzo M., Toth P.P., et al. Statin intolerance - an attempt at a unified definition. Position paper from an International Lipid Expert Panel. Arch. Med. Sci. 2015;11(1):1–23. doi: 10.5114/aoms.2015.49807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waters D.D., Ho J.E., DeMicco D.A., et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J. Am. Coll. Cardiol. 2011;57(14):1535–1545. doi: 10.1016/j.jacc.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 43.Bruckert E., Gagne C., Gaudet D. Homozygous familial hypercho- lesterolemia: a novel therapy with ezetimibe. Atherosclerosis. 2002;3(2):81. [Google Scholar]
- 44.Gagné C., Gaudet D., Bruckert E. Ezetimibe Study G. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolaemia. Circulation. 2002;105(21):2469–2475. doi: 10.1161/01.cir.0000018744.58460.62. [DOI] [PubMed] [Google Scholar]
- 45.Gagne C., Gaudet D., Bruckert E., et al. Ezetimibe significantly reduces low-density lipoprotein cholesterol in homozygous familial hypercholesterolaemia. J. Am. Coll. Cardiol. 2002;39(S1):227. [Google Scholar]
- 46.Kassner U., Thomas H., Steinhagen-Thiessen E., Vogt A. Intensified lipid lowering treatment with lipid-apheresis and atorvastatin coadministered with ezetimibe in two children with homozygous familial hypercholesterolaemia. Atheroscler. Suppl. 2004;5(Suppl. 1):120. [Google Scholar]
- 47.Kolovou G.D., Dedoussis G.V., Anagnostopoulou K.K., et al. Management of a patient with a null low-density lipoprotein receptor mutation: a case report. Angiology. 2006;57(6):729–732. doi: 10.1177/0003319706294421. [DOI] [PubMed] [Google Scholar]
- 48.Schmidt H.H., Tietge U.J., Buettner J., et al. Liver transplantation in a subject with familial hypercholesterolaemia carrying the homozygous p.W577R LDL-receptor gene mutation. Clin. Transplant. 2008;22(2):180–184. doi: 10.1111/j.1399-0012.2007.00764.x. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto A., Harada-Shiba M., Endo M., et al. The effect of ezetimibe on serum lipids and lipoproteins in patients with homozygous familial hypercholesterolaemia undergoing LDL-apheresis therapy. Atherosclerosis. 2006;186(1):126–131. doi: 10.1016/j.atherosclerosis.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 50.Cannon C.P., Blazing M.A., Giugliano R.P., et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015;372(25):2387–2397. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 51.Schuff-Werner P., Fenger S., Kohlschein P. Role of lipid apheresis in changing times. Clin. Res. Cardiol. Suppl. 2012;7(Suppl. 1):7–14. doi: 10.1007/s11789-012-0049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Dughaishi T., Al-Waili K., Banerjee Y., et al. Successful Direct Adsorption of Lipoproteins (DALI) apheresis during pregnancy in an Omani woman with homozygous familial hypercholesterolaemia. Open Cardiovasc Med. doi: 10.2174/1874192401509010114. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefanutti C., Julius U. Lipoprotein apheresis: state of the art and novelties. Atheroscler. Suppl. 2013;14(1):19–27. doi: 10.1016/j.atherosclerosissup.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 54.Thompson G.R., Miller J.P., Breslow J.L. Improved survival of patients with homozygous familial hypercholesterolaemia treated with plasma exchange. Br. Med. J. (Clin. Res. Ed.) 1985;291(6510):1671–1673. doi: 10.1136/bmj.291.6510.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kroon A.A., van't Hof M.A., Demacker P.N., Stalenhoef A.F. The rebound of lipoproteins after LDL-apheresis. Kinetics and estimation of mean lipoprotein levels. Atherosclerosis. 2000;152(2):519–526. doi: 10.1016/s0021-9150(00)00371-3. [DOI] [PubMed] [Google Scholar]
- 56.Stefanutti C., Thompson G.R. Lipoprotein apheresis in the management of familial hypercholesterolaemia: historical perspective and recent advances. Curr. Atheroscler. Rep. 2015;17(1):465. doi: 10.1007/s11883-014-0465-6. [DOI] [PubMed] [Google Scholar]
- 57.Daniels S.R., Gidding S.S., de Ferranti S.D. National Lipid Association Expert Panel on Familial H. Pediatric aspects of familial hypercholesterolaemias: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolaemia. J. Clin. Lipidol. 2011;5(3) Suppl.:S30–S37. doi: 10.1016/j.jacl.2011.03.453. [DOI] [PubMed] [Google Scholar]
- 58.Malatak J.J. Liver transplantation as treatment for familial homozygous hypercholesterolaemia: too early or too late. Pediatr. Transplant. 2011;2011(15):123–125. doi: 10.1111/j.1399-3046.2010.01458.x. [DOI] [PubMed] [Google Scholar]
- 59.Cannon C.P. on behalf of the IMPROVE IT investigators. IMPROVE-IT trial: a comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes after acute coronary syndromes. American Heart Association Scientific Sessions; Chicago: Illinois.; 2014. [Google Scholar]
- 60.Blazing MA, Giugliano RP, Cannon CP, et al. Evaluating cardiovascular event reduction with ezetimibe as an adjunct to simvastatin in 18,144 patients after acute coronary syndromes: final baseline characteristics of the IMPROVE-IT study population. Am Heart J. 2014;168(2):205–212. doi: 10.1016/j.ahj.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 61.Aegerion Pharmaceuticals Inc Juxtapid prescribing informatio. 2013.
- 62.Genzyme Corporation Kynamro prescribing informatio Juxtapid prescribing informatios. http://www.juxtapid.com/sites/default/files/downloads/ Prescribing_Information.pdf. 2013.
- 63.Santos R.D., Duell P.B., East C., et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur. Heart J. 2015;36(9):566–575. doi: 10.1093/eurheartj/eht549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sohrabpour A., Rezvan H., Amini-Kafiabad S., Dayhim M., Merat S., Pourshams A. Prevalence of nonalcoholic steatohepatitis in iran: a population based study. Middle East J. Dig. Dis. 2010;2(1):14–19. [PMC free article] [PubMed] [Google Scholar]
- 65.Horton J.D., Cohen J.C., Hobbs H.H. PCSK9: a convertase that coordinates LDL catabolism. J. Lipid Res. 2009;50(Suppl.):S172–S177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raal F.J., Honarpour N., Blom D.J., et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2014:384. doi: 10.1016/S0140-6736(14)61374-X. [ePub ahead of print.]. [DOI] [PubMed] [Google Scholar]
- 67.Cuchel M., Meagher E.A., du Toit Theron H., et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40–46. doi: 10.1016/S0140-6736(12)61731-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.France M., Schofield J., Kwok S., Soran H. Treatment of homozygous familial hypercholesterolaemia. Clin. Lipidol. 2014;9(1):101–118. [Google Scholar]
- 69.Al-Allaf F.A., Coutelle C., Waddington S.N., David A.L., Harbottle R., Themis M. LDLR-Gene therapy for familial hypercholesterolaemia: problems, progress, and perspectives. Int. Arch. Med. 2010;3:36. doi: 10.1186/1755-7682-3-36. [DOI] [PMC free article] [PubMed] [Google Scholar]