

Abstract
In the late 1980s, reports emerged describing experimental antibacterial quinolones having significant potency against eukaryotic Type II topoisomerases (topo II) and showing cytotoxic activity against tumor cell lines. As a result, several pharmaceutical companies initiated quinolone anticancer programs to explore the potential of this class in comparison to conventional human topo II inhibiting antitumor drugs such as doxorubicin and etoposide. In this review, we present a modern re-evaluation of the anticancer potential of the quinolone class in the context of today’s predominantly pathway-based (rather than cytotoxicity-based) oncology drug R&D environment. The quinolone eukaryotic SAR is comprehensively discussed, contrasted with the corresponding prokaryotic data, and merged with recent structural biology information which is now beginning to help explain the basis for that SAR. Quinolone topo II inhibitors appear to be much less susceptible to efflux-mediated resistance, a current limitation of therapy with conventional agents. Recent advances in the biological understanding of human topo II isoforms suggest that significant progress might now be made in overcoming two other treatment-limiting disadvantages of conventional topo II inhibitors, namely cardiotoxicity and drug-induced secondary leukemias. We propose that quinolone class topo II inhibitors could have a useful future therapeutic role due to the continued need for effective topo II drugs in many cancer treatment settings, and due to the recent biological and structural advances which can now provide, for the first time, specific guidance for the design of a new class of inhibitors potentially superior to existing agents.
Keywords: Quinolone, fluoroquinolone, topoisomerase, gyrase, topo II, topo IV, antitumor, anticancer, cytotoxic, antibacterial, selectivity, Paul Ehrlich, magic bullet, doxorubicin, etoposide, vosaroxin, quarfloxin, G quadruplex
INTRODUCTION: EUKARYOTIC AND PROKARYOTIC SELECTIVITY
In 1906 Paul Ehrlich described his vision of selective therapeutic agents by stating, in part,
“Such substances would then be able to exert their full action exclusively on the parasite harbored within the organism, and would represent, so to speak, magic bullets which seek their target of their own accord.” [1].
Ehrlich was undoubtedly alluding to the so-called “free” bullets of popular German folklore, the term “free” used in the sense of “free will” [2, 3] According to the legend, these specially crafted bullets need not be aimed precisely by the shooter, because once fired, they were free to seek out their targets on their own.
Those bullets always hit their target, never causing “collateral damage”. Antimicrobial therapies had been identified in the late 19th century which largely acted in this manner, i.e., the antibody mixtures (antisera) which Emil von Behring, with Erhlich’s help, had developed against diphtheria and tetanus toxins (1890) [4-7] It should be recalled that until the invention of antisera therapy all antimicrobial agents were essentially external antiseptics which were too unselective between pathogen and host to be used parenterally. With the anti-syphilis agent salvarsan, Ehrlich was to realize, albeit only partially, his magic bullet concept in the realm of small molecules as well. However, both the antisera of that time, as well as salvarsan occasionally did harm the host. Primarily due to the carryover of impurities, those polyclonal antibody serum treatments could cause serious immune reactions (serum sickness) [8, 9] while the therapeutic margin of salvarsan, an organoarsenic agent, was extremely narrow requiring careful management of the proper dosage [10, 11].
Selective therapeutics were sought not only as antimicrobial agents but also for use as antineoplastics, in the latter case selectivity being defined as discrimination between normal and aberrant proliferating eukaryotic cells. Following the model established by von Behring’s diphtheria toxin antibody preparation, Hericourt and Richet in 1895 immunized dogs with a human sarcoma and then transferred the serum to patients [12, 13]. The therapeutic effect achieved from this early antibody preparation was however poor. For the successful application of the concept of passive immunization to cancer therapy, further technical advances were required, only realized in the last few decades [5].
In the 1930s and 1940s, the synthetic sulfa drugs and the natural product penicillin more closely approached the ideal degree of selectivity that Ehrlich envisioned for antimicrobials. Application of synthetic chemicals or natural products to anticancer therapy however lagged antibacterial therapeutic applications. Although an extensive antitumor screening program was established in 1934 by the United States Government, by 1952 a decision was made at the National Cancer Institute (NCI) not to increase funding for cancer chemotherapy due to concerns that the effort was not sufficiently promising (that decision was reversed by 1955) [14-17]. Nevertheless during the 1940s and 1950s other anticancer screening programs were established by pharmaceutical companies and academic groups. Screening microbial fermentations for anticancer natural products in a manner analogous to that used to discover new antibiotics was a major strategy in this effort. However, many early microbial-based cytotoxic natural products discovered during this time were found to be too unselective for use as anticancer agents [18-22].
A key early achievement in antineoplastic selectivity arose from observations in the 1940s that folic acid given to patients diagnosed with chronic myeloid leukemia appeared to accelerate the disease while diets deficient in that nutrient caused a decrease in leukemia cell count [23, 24]. As a result of this knowledge, Lederle Labs, now part of Pfizer, applied the “antimetabolite” concept (first conceived in the early 1940s to explain the mechanism of sulfa antibacterials [25]) to design the therapeutically effective folic acid antagonists aminopterin and methotrexate (1, Fig. 1). Employing aminopterin, Sidney Farber at Harvard Medical School in 1948 reported for the first time that temporary remission of acute lymphoblastic leukemia in pediatric patients was achievable [26]. Later, scientists at Burroughs Wellcome developed trimethoprim (2, Fig. 1), a folic acid antagonist against bacteria. Mechanistically, all three of these agents were later shown to inhibit dihydrofolate reductase (DHFR) [23, 27-29]. Whereas the affinity of trimethoprim for bacterial DHFR was 1000-fold that of the human enzyme, [30]. the anticancer DHFR inhibitors exhibited substantial potency also against bacterial DHFR which translated into microbiological activity against a number of prokaryotes (Streptococcus faecalis, for example)[29, 31-34]. The prokaryotic activity present in the anticancer DHFR inhibitors is perhaps not unexpected insofar as measurement of DHFR-based bacterial inhibition was a component of Lederle’s anticancer compound screening process [35, 36]. In any case, concomitant antibacterial activity was not at that time considered to be a negative attribute for antitumor agents because the goal was selectivity against rapidly proliferating neoplasms compared to the relatively static cell populations of most other host tissue [37-40]. (Today, it could be a subject of debate whether antibacterial activity in an antitumor agent should be ignored [41]). The cellular “polypharmacology” of many early anticancer substances was summarized by Selman Waksman, discoverer of the anti-tuberculosis agent streptomycin, as follows:
“the antitumor agents vary greatly in chemical nature and biological activity, some being active against neoplasms and bacteria, others against neoplasms and fungi, and still others against neoplasms alone” [21].
Over the following decades, other synthetic and natural product agents were discovered acting through broad or specific mechanisms common to both eukaryotic and prokaryotic cells and having application (or potential application) as antibacterial or anticancer agents depending on the degree of selectivity that could be attained. An interesting illustration of prokaryotic vs eukaryotic selectivity in the domain of natural products is provided by a set of four molecules which at first glance appear quite dissimilar from one another: novobiocin (3, first reported 1956), geldanamycin (4, 1970), cyclothialidine (5, 1987), and radicicol (6, 1962) (Fig. 2). In fact all four compounds competitively bind to a unique ATP-binding fold--the Bergerat fold[42]--thereby inhibiting the ATPase activity of either bacterial Type II topoisomerase (novobiocin and cyclothialidine) or the eukaryotic anticancer chaperone target Hsp90 (geldanamycin and radicicol) [43-45]. A specific functional group motif plays a key role in the binding event for each prokaryotic/eukaryotic targeted pair of these molecules: a primary carbamate group for novobiocin and geldanamycin, and a phenol hydroxy group for cyclothialidine and radicicol (Fig. 2). These two functional groups are key anchoring points for the binding of these molecules to the Bergerat fold and involve an interaction with a critical aspartic acid - water motif in the enzyme ATP binding pocket: Asp73 (E. coli numbering, shown) or Asp79 (yeast numbering, shown) and Asp93 (human numbering). ATP itself binds to these aspartate-water motifs in the Bergerat fold via its purine 1-amine and 6-amino groups (Fig. 2; co-crystal structures have been obtained for the ATP analog ATPNP in bacterial topoisomerase, and for ADP in Hsp90). The anchoring interactions for all the compounds are highlighted in red in Fig. (2). In bacteria, the Asp73 binding interaction is so critical that no resistant mutants to competitive ATPase inhibitors have been found with a change in this amino acid. Even though the Bergerat fold is similar for both Type II bacterial topoisomerase and eukaryotic Hsp90, certain structural differences surrounding these N-terminal ATP binding pockets are sufficient to alter the general binding mode of the inhibitors outside the critical Asp-water motif interaction. Thus novobiocin and cyclothialidine both largely orient away from the remainder of the ATP binding site, while geldanamycin and radicicol generally overlap with the ATP binding site (Fig. 2). Novobiocin was employed for several decades as an antibacterial agent especially for therapy against penicillin-resistant Staphylococcus aureus infections, while cyclothialidine served as the starting point for a significant preclinical antibacterial optimization program at Roche [45]. Both geldanamycin and radicicol serve currently as starting points for the preparation of more optimized anticancer analogs, several of which have been investigated in clinical trials [46-51]. Unlike the anticancer DHFR inhibitors methotrexate and aminopterin which are also antibacterial by a DHFR mechanism, neither geldanamycin or radicicol exhibit appreciable cross inhibitory activity for bacteria, and do not inhibit prokaryotic topoisomerase [52-54]. Conversely neither novobiocin nor cyclothialidine significantly inhibit the N-terminal ATPase domain of Hsp90. This relatively compartmentalized selectivity profile for these four natural products is nevertheless subtly nuanced by recent discoveries that novobiocin can slightly inhibit Hsp90 activity by weakly binding to a C-terminal (apparently non-catalytic) ATP binding site, while radicicol has been shown to inhibit (weakly) a second human target, topo II, presumably via interaction with the enzyme’s ATPase Bergerat fold [55, 56].
Fig. (2).
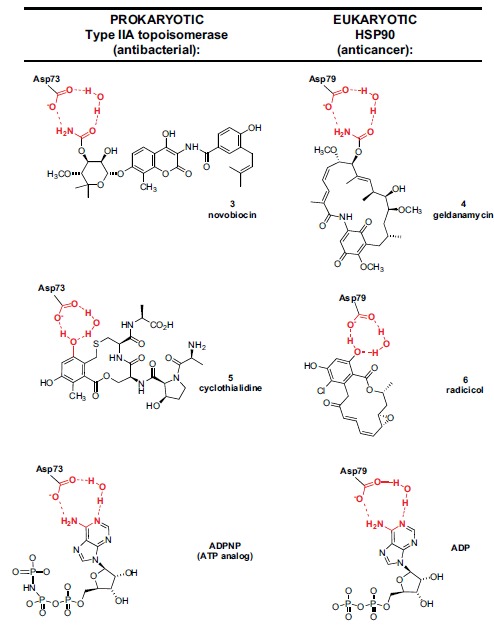
Bacterial Type II topoisomerase inhibitors novobiocin (3) and cyclothialidine (5) compared to eukaryotic Hsp90 inhibitors geldanamycin (4) and radicicol (6). All four natural products bind to a common ATPase Bergerat fold motif. Based on X-ray co-crystal structures, both novobiocin and geldanamycin “anchor” to the key Asp-water motif in the ATPase Bergerat fold via a primary carbamate moiety while cyclothialidine and radicicol analogously bind via a phenol hydroxyl group. The Asp-water motif interaction of both enzymes with ATP substrate is deduced from the crystallographic binding of ADPNP (adenosine 5′-(β,γ-imido)triphosphate, a stable ATP mimetic, shown) in bacterial topoisomerase and from the binding of ADP (shown) in Hsp90. The Asp-water motif binding interactions for all six compounds are highlighted in red. Novobiocin and cyclothialidine largely occupy a binding pocket adjacent to the ATP binding site, while geldanamycin and radicicol largely overlap with the ATP binding site.
As the above summary reveals, the term “selectivity” has evolved over the last century into a highly multifaceted descriptor, signifying not only “lack of harm to the host” but also embodying the concepts of molecular target selectivity among groups of related targets, the overall predilection of a scaffold series towards one therapeutic focus vs another (antibacterial vs anticancer, for example), as well as blends of all three concepts.
During the late 1950s and early 1960s Sterling Drug and Imperial Chemical Industries (ICI) had discovered--and in the case of Sterling, marketed--a new class of synthetic antibacterial agents: the quinolones [57]. Nalidixic acid (7, Fig. 3) was the first marketed agent in this class, launched in the US in 1964 [58]. Like novobiocin and cyclothialidine, the quinolone class was shown to inhibit bacterial Type II topoisomerases, specifically DNA gyrase as well as its bacterial “twin”, topoisomerase IV (topo IV). The interaction of quinolones with bacterial topoisomerases and the mechanism leading to bacterial cell death is different compared to the ATPase inhibitors novobiocin and cyclothialidine (see detailed discussion below). It has been shown that clinically relevant antibacterial quinolones do not substantially interact with eukaryotic Type II topoisomerase (topo II) and therefore display low mammalian cytotoxicity. However, by the late 1980s, several experimental quinolones prepared for
antibacterial programs were found to potently inhibit topo II and, in contrast to the clinically used quinolones, demonstrated eukaryotic cytotoxicity. As a consequence, several pharmaceutical companies (e.g. Abbott, Dainippon, Sterling) opportunistically investigated these compounds for application as “cytotoxic” anticancer drugs, while other companies (e.g. Pfizer and Parke Davis) studied the eukaryotic SAR only in order inform and “de-risk” their ongoing antibacterial programs. As of late 2014, the quinolone class is still under active investigation for both new antibacterial and anticancer therapies as evidenced by Phase III trials of the antibacterial delafloxacin 10 and the anticancer vosaroxin 11. The lines of research leading to both of these compounds can be traced through earlier analogs such as norfloxacin 8 and tosufloxacin 9 (Fig. 3). Technically, nalidixic acid 7, tosufloxacin 9, and vosaroxin 11 are 8-azaquinolones, also called 1,8-naphthyridones, while norfloxacin 8 and delafloxacin 10 are “pure” quinolones. However, the term quinolone (or fluoroquinolone) is often used informally to encompass both these core variations. The ring numbering for both quinolones and naphthyridones is depicted in Fig. (3) (nalidixic acid used an example).
Fig. (3).
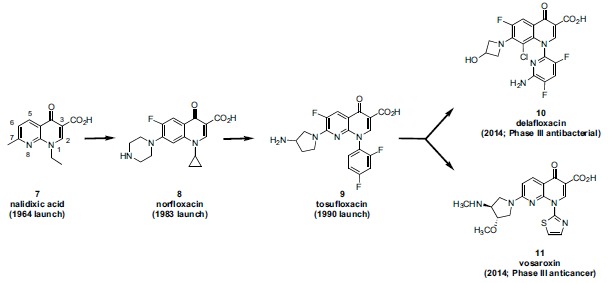
Antibacterial delafloxacin (10) and anticancer vosaroxin (11), inhibitors of bacterial and human Type II topoisomerases, respectively, were evaluated in Phase III studies during 2014. Inhibitors of bacterial Type II topoisomerase (DNA gyrase and topoisomerase IV) have been a significant class of antiinfectives since the launch of nalidixic acid (7) in 1964. The antibacterials norfloxacin (8) and tosufloxacin (9) can be viewed as intermediate agents on the evolutionary path toward both 10 and 11. The ring numbering for both quinolones and naphthyridones is represented by nalidixic acid (a naphthyridone).
The intent of this review is severalfold. The authors wish to convey how research on the quinolone class of antibacterials evolved during the 1980s and thereafter to provide potential novel options for anticancer treatment. The SAR for eukaryotic compared to prokaryotic activity within the quinolone class is subtle and a number of previous authors have made important observations in review articles regarding molecular features that predispose the scaffold toward one type of activity or the other [41, 59-68]. In this review, we would like to gather together in one place the existing threads regarding eukaryotic SAR for the class and provide additional observations based on current structural biology which may prove useful for our collective understanding of the molecular functionality that governs activities against human vs bacterial Type II topoisomerases. Such an understanding should prove useful for more effective design of both antibacterial as well as anticancer quinolones. We recognize that the majority of efforts during the 1990s at several pharmaceutical companies to develop quinolone-based topoisomerase anticancer agents were terminated prior to full optimization of any inhibitor series. Nevertheless, our analysis of the collective published data for these, and related, quinolone programs leads us to conclude that from a scientific perspective, the feasibility of success in developing therapeutically efficacious anticancer agents within this class should be high. Moreover, quinolone-based topo II inhibitors appear to be much less susceptible to efflux-mediated resistance which can sharply limit the therapeutic utility of conventional topo II drugs such as doxorubicin and etoposide. Further, recent advances in the biological understanding of human topo II isoforms suggest that significant progress might now be made in overcoming two other treatment-limiting disadvantages of therapy with conventional topo II inhibitors, namely cardiotoxicity and drug-induced secondary leukemias. Doxorubicin and etoposide and their analogs continue to be mainstays (despite their limitations) of cancer therapy alongside modern targeted therapies. New class topo II inhibitors having significant advantages in safety and efficacy over conventional drugs could occupy a viable place in therapeutic practice. Thus in this review, we present an argument that development of quinolone-based topo II inhibitors for the treatment of cancer represents an opportunity that is worthy of pursuit.
We limit the scope of chemical structures covered in this review largely to scaffolds which collectively would be viewed as “conventional” (antibacterial) quinolone structures. The reason for this limit on chemical scope is significant: the conventional quinolone scaffold is the outcome of several decades of optimization [67, 69-76] of key drug-like characteristics which resulted in the achievement of good physical properties, excellent pharmacokinetics, overall safety, good eukaryotic intracellular accumulation,[77-81] and simplicity of analog synthesis. Quinolone topo II anticancer programs which largely maintain this conventional structural starting point would have a clear medicinal chemistry advantage compared to programs employing more heavily modified scaffolds or which start from novel scaffolds [82, 83], since the drug-like properties for the latter cases will not be known, and are likely to be less ideal. The only exceptions to the stated scope in this review are the inclusion of Abbott’s isothiazoloquinolones and the agent quarfloxin whose structures deviate from conventional quinolones in a few regards. The authors chose to include the isothiazoloquinolones, a series representing a rare success of bioisosteric replacement of the quinolone 3-carboxy group, due to the similar antibacterial SAR of the series compared to standard quinolones, and the still acceptable drug-like properties of the scaffold. Quarfloxin was included due to its chemical evolution from an Abbott cytotoxic quinolone. That evolution however resulted in a substantial deviation from the classical quinolone structure, shifting its anticancer target from topo II to a G quadruplex motif and perhaps causing physical property issues that could not be overcome in the clinic.
1976-1980: PROKARYOTIC AND EUKARYOTIC TYPE II TOPOISOMERASES CHARACTERIZED
Topoisomerases are essential enzymes which resolve topological problems caused during DNA replication. The reader is referred to several excellent reviews for more details regarding the nomenclature, structure, and mechanism of this broad enzyme class [84-91]. Most relevant for this review is the Type II class of topoisomerases which transiently break double-stranded DNA, pass an intact DNA strand through the opening, and then reseal the double-strand nicks. The hydrolysis of ATP to ADP is employed to fuel the catalytic cycle. The other broad category of topoisomerases is Type I, a family of enzymes that transiently break (and then reseal) a single strand of double stranded DNA. Gyrase, a prokaryotic Type II topoisomerase, was first characterized in E. coli beginning in 1976. More than ten years later, evidence for the existence of a physiologically relevant “twin” to gyrase, namely topoisomerase IV (topo IV), began to accumulate, and by 1990-1992, E. coli topo IV had been characterized biochemically. Gyrase and topo IV from other prokaryotic species (e.g. Staphylococcus aureus) have also been characterized. Gyrase is primarily involved in negative supercoiling of DNA during replication while topo IV is involved primarily in DNA decatenation. Both of these prokaryotic topoisomerases are heterotetramers: the gyrase tetramer is composed of two subunits of GyrB and two of GyrA [(GyrB)2(GyrA)2] and the topo IV tetramer is composed of two subunits of ParE and two of ParC [(ParE)2(ParC)2] (Fig. 4). The GyrB and ParE subunits contain the ATP binding site and are thus the site of action for novobiocin and cyclothialidines (see above). Members of the quinolone class of antibacterials by contrast interact at the interface of the GyrA and GyrB subunits (for gyrase; ParC and ParE for topo IV), binding in a ternary manner together with the covalently bound (and cleaved) DNA strand [92, 93]. This inhibitory ternary complex is referred to as the “cleavable complex”, “interfacial complex”, or DNA covalent complex. (Fig. 5, sketch of ternary complex with the fluoroquinolone moxifloxacin 12). This complex generates “toxic” DNA fragments which initiate a cascade of events leading to cell death. Due to the generation of toxic DNA fragments, this particular mode of inhibition is also referred to as topoisomerase inhibitor induced “poisoning”, although this term is used more often in the context of eukaryotic rather than prokaryotic cell killing. The different mechanism of inhibition of gyrase and topo IV displayed by quinolones compared to ATP site inhibitors leads to some differences in the rate and/or manner by which bacterial cells are affected. Quinolones are generally regarded as rapidly bactericidal agents due to the generation of potently toxic action of DNA double strand breaks, whereas depending on the type of test, GyrB/ParE ATP site inhibitors are viewed as either bacteriostatic or more slowly bactericidal [94-96].
Eukaryotic topoisomerase II (topo II), another Type II enzyme, is a homodimer, each monomer corresponding to the conceptual fusion of the bacterial GyrB + GyrA (or ParE + ParC) domains (Fig. 6). This topoisomerase decatenates DNA and can relax positive supercoils produced during replication. In vertebrates, there are two isoforms: topo IIα which is essential for DNA replication during cell division, and topo IIβ which is not essential although it has been linked to proper neural development. The first evidence suggesting the existence of a eukaryotic topoisomerase counterpart to bacterial gyrase began to accumulate in the late 1970s and by 1980 characterization of Type II topoisomerase (topo II) was reported [97-99]. Several anticancer drugs inhibit eukaryotic topo II in a manner analogous to the mode of inhibition of gyrase and topo IV by antibacterial quinolones i.e. by stabilizing the “cleavable complex” thereby generating toxic DNA fragments leading to apoptosis. As alluded to above, eukaryotic-active agents operating through this mechanism are commonly referred to as DNA topo II poisons. However some eukaryotic topo II inhibiting agents, including some quinolones, seem to act via blended mechanisms wherein the cleavable complex mechanism may either be the predominant or minor pathway (see below).
1989-1990: TWO SEPARATE THERAPY WORLDS FINALLY COLLIDE
The field of antibacterial quinolone research and development, about 20 years old by 1980, was significantly re-energized in that year by reports of the vastly improved potency and microbiological spectrum of norfloxacin (8, Fig. 3; Kyorin Pharmaceutical), the first entry of the fluoroquinolone subclass [103, 104]. Fluoroquinolones are structurally characterized by substitution at the quinolone ring C-6 position with fluorine and at the C-7 position with a basic amino heterocyclic group. Physical properties and pharmacokinetics of fluoroquinolones were also superior to the first generation quinolones. The fluoroquinolones allowed for the first time effective treatment of infections caused by significant gram negative pathogens such as Pseudomonas aeruginosa; they also began to display enhanced potency against gram positive pathogens such as Staphylococcus aureus. Prior to norfloxacin, the quinolone class antibacterials occupied a fairly narrow therapeutic niche, used primarily for treatment of urinary tract infections caused by Escherichia coli and a few other gram negative pathogens. Whereas only a few pharmaceutical companies (Sterling Drug, Parke Davis (now Pfizer), Lilly, Riker and Dainippon) were involved significantly in the quinolone antibacterial field during its first two decades, after 1980 many more companies joined the effort, among them Pfizer, Abbott, Bayer, Daiichi, and Toyama. Consequently the number and types of additional structural variations synthesized and studied in the quinolone field vastly increased. Starting in 1981, not long after the discovery of norfloxacin, it was reported that nalidixic acid and oxolinic acid (another early quinolone) inhibited eukaryotic topo II, although at levels significantly higher than those required to inhibit E. coli DNA gyrase [105, 106]. Similar measurements made using other marketed quinolones confirmed that on the enzyme/ molecular level, the selectivity margin was concordant with the overall good safety profile of these agents used for treatment of bacterial infections in patients. A few years later, in 1984 the mechanism governing the therapeutic cytotoxicity of certain anticancer agents such as doxorubicin (adriamycin) 13 and etoposide 14 (Fig. 7) was discovered to be inhibition of human topo II (in tandem with the associated DNA damage caused by the formation of the cleavable complex) [107, 108].
Against this backdrop, W. E. Ross, who was one of the key early investigators in the anticancer topo II field, proclaimed presciently in 1985:
“Indeed, it may be interesting to re-evaluate a number of nalidixic acid congeners which were developed for antibacterial use but proved too toxic to mammalian cells. Some of these may prove to be inhibitors of mammalian topoisomerase II.” [109].
During the following several years, numerous reports continued to appear in the literature demonstrating the generally favorable biochemical selectivity margins between bacterial gyrase and eukaryotic topo II for marketed antibacterial quinolones and fluoroquinolones [110, 111]. Additionally several fluoroquinolones (including ciprofloxacin) were reported to inhibit, at high dose, eukaryotic cell growth via a topo II mechanism [110-114]. Finally during 1989-1990 the concept suggested by Ross was brought to clear experimental fruition. A paper from the Duke Cancer Center titled “Evidence for a common mechanism of action for antitumor and antibacterial agents that inhibit Type II DNA topoisomerases” provided data that “strongly suggest that diverse inhibitors of Type II topoisomerases share a common binding site”. Moreover, Pfizer scientists essentially “broke the ice” for the rest of the pharmaceutical industry by reporting an experimental quinolone CP-67015 26 (Fig. 8) having strong antibacterial activity that was clearly more potent in two eukaryotic DNA cleavage assays than any of the marketed antibacterial quinolones tested by other groups [115-117]. Although 26 was not as potent in these assays as the antitumor topo II inhibitor etoposide (e.g. 33 μg/mL vs 4.5 μg/mL for 26 and etoposide 14 respectively in a radiolabelled DNA cleavage assay), the Pfizer scientists associated that level of inhibition with positive in vivo cytogenetic results observed with 26, and cautioned that present and future antibacterial quinolones should be similarly screened for safety. Pfizer used its eukaryotic-based assays to develop extensive eukaryotic/prokaryotic inhibitory SAR for the quinolone scaffold in order to inform and “de-risk” its ongoing antibacterial program. Although eukaryotic topo II inhibitors were identified at Pfizer that equalled the potency of etoposide (see below), the company decided not to pursue an anticancer program based on those agents. Pfizer’s decision stands in contrast to that of several other companies (e.g. Sterling, Abbott, Banyu-Merck, and Dainippon) that did invest in dedicated anticancer quinolone programs over several years, typically while separate antibacterial quinolone programs ran concurrently (Chart 1).
Why would any company be interested in developing an anticancer quinolone in the early 1990s since there was already a selection of topo II cleavable com-
plex class drugs in wide use during that time, among them doxorubicin, etoposide, tenoposide, amascrine, and mitoxantrone? There are several answers. The early 1990s was still within an era in which cytotoxics dominated both the marketplace as well as the cancer drug research agendas of many pharmaceutical companies [118]. Research on “targeted” anticancer agents, such as small molecule kinase pathway inhibitors or monoclonal antibodies was just getting underway and was not yet dominant. Therefore development of a novel class of cytotoxic would be viewed as strategically desirable, especially a class that might circumvent several serious issues with the existing topo II inhibiting cytotoxics. The principle issues with the existing drugs and drug classes were as follows: 1) the anthracycline class--of which doxorubicin was a member--caused significant cardiotoxicity by an unknown mechanism; 2) the epipodophyllotoxins--etoposide and tenoposide--were poorly soluble and therefore difficult to formulate; in particular an oral formulation of etoposide showed an unacceptably wide range of bioavailability; 3) the anthracyclines and epipodophyllotoxins showed a significant incidence of secondary (drug-induced) leukemias due to an unknown mechanism; 4) resistance, primarily due to P-glycoprotein (P-gp) mediated efflux, developed frequently, with cross-resistance observed to all of the topo II classes; 5) anthracycline and epipodophyllotoxin synthetic chemistry was laborious so that the capacity to generate new analogs to solve specific issues was sharply limited [119-126]. By contrast, the differentiated, and highly pre-optimized, quinolone scaffold held promise to solve at least some of these issues: 1) the quinolone class was typically regarded as quite safe overall, without any significant cardiotoxicity (although occasional tendonitis was seen as a class effect), 2) quinolones have good physicochemical properties, are straightforward to formulate for both oral and parenteral use and display excellent and predictable bioavailability and pharmacokinetics, 3) quinolones easily cross cell membranes (both eukaryotic and prokaryotic) with good accumulation and therefore efflux-based resistance might not emerge as a major issue; and finally 5) because quinolone chemistry is extremely straightforward, new analogs can be synthesized quickly during problem-solving and optimization phases, offering some confidence that a preclinical program might proceed rapidly [73, 75, 127-132].
Chart 1 . Timeline depicting pharmaceutical companies having quinolone drug discovery programs for both antibacterial and anticancer, or anticancer only, application. Also depicted are companies that published quinolone eukaryotic SAR as part of their antibacterial programs (Pfizer, Parke Davis, Achillion). Other companies having antibacterial-only quinolone programs that did not contribute substantially to eukaryotic SAR are not shown. Key relevant scientific and clinical events are depicted on black vertical bars. With several exceptions, most project start and termination dates are only approximate as such estimates are frequently inferred from dates of the company’s patent applications and/or published articles. Project leaders from a few of these companies were contacted for this information. Dainippon licensed its lead anticancer quinolone (now called vosaroxin) to Sunesis. One of Abbott’s anticancer scaffolds was further evolved in academia and then at Cylene Pharmaceuticals, although the structure of the resultant anticancer clinical candidate (quarfloxin) deviates somewhat from the classical quinolone structure; also the target of quarfloxin is G-quadruplex DNA rather than topo II-DNA. Nevertheless, the authors chose to include quarfloxin due to its heritage and its achievement of a relatively advanced clinical status (Phase II).
Thus, having both the motivation and the technical means, a number of pharmaceutical companies initiated in-depth exploration of quinolone eukaryotic SAR starting in the early 1990s (see Chart 1).
MECHANISMS OF TOPO II INHIBITORS, INCLUDING QUINOLONES
There are a number of structurally diverse anticancer drugs (as well as experimental agents) which fall under the general heading “topo II inhibitors”: epipodophyllotoxins (etoposide and tenoposide), anthracyclines (doxorubicin and idarubicin), amsacrine (m-AMSA), ellipticines, quinolones, and others. No two classes appear to act by precisely the same mechanism or mechanisms, and even agents within the same scaffold series may display diversity in their types of interactions with the topo II DNA complex, topo II (without DNA), and/or DNA (prior to complex formation with topo II). Moreover, many details of the assigned mechanism(s) for most agents are not known with precision, and mechanistic studies continue today, even on well-established drugs [137, 138].
Nevertheless, some general observations regarding mechanism can be made. Etoposide and the other epipodophyllotoxins are often referred to as “non-intercalating” topo II poisons while doxorubicin and other anthracyclines are characterized as “DNA-intercalating” topo II poisons [139, 140]. Intercalation of agents into DNA does not necessarily lead by itself to topo II inhibition, although it may contribute to a cytotoxic effect independent of topo II [138, 141, 142]. Among the topo II poisons, some agents appear to stimulate DNA break formation (quinolones, ellipticines) whereas others slow DNA relegation (etoposide, amsacrine); both processes result in toxic (cell “poisoning”) DNA strand breaks leading ultimately to apoptosis [139, 143-145]. In the formation of the ternary cleavable complex, the molecular order of assembly has been debated: does the inhibitor first bind to DNA or to topo II, or does the inhibitor bind the preformed DNA-topo II complex? [139]. There are other agents, typically referred to as “catalytic topo II inhibitors”, which bind to the DNA topo II complex, inhibiting formation of the cleavable complex so that no “poisoning” effect (i.e. DNA fragmentation) occurs. Catalytic inhibitors can thereby antagonize topo II poisons by arresting the enzyme catalytic cycle prior to formation of the cleavable complex. Catalytic inhibition can occur at any of several points within the catalytic cycle and typically leads to mitotic failure [146, 147]. Further complicating the mechanistic landscape, some topo II inhibitors are metabolically converted to reactive species which covalently react with topo II or the topo II DNA complex, either before or after DNA cleavage. Alternatively, a reactive species may disrupt other cellular processes which also contribute to cytotoxicity. For example doxorubicin, in addition to its intercalating and topo II poisoning mechanisms, is believed to generate reactive oxygen species which may contribute to its antitumor effects as well as to cardiotoxicity [148, 149]. Ellipticine, as another example, intercalates DNA, acts as a topo II poison, and forms covalent DNA adducts mediated by cytochrome P450 [150, 151]. Yet another mechanistic element relevant to a fuller understanding of the therapeutic action of topoisomerase inhibitors is the degree of selectivity of an agent toward the two isoforms of topo II (α and β). In particular, it has recently been hypothesized that selective inhibition of the α topo II isoform in a clinical setting may potentially reduce or eliminate the cardiotoxicity and/or drug-induced secondary leukemias associated with current topo II drugs (fuller discussion below). Current clinical topoisomerase inhibitors appear to be isoform non-selective, yet certain new-class experimental inhibitors have been identified that demonstrate isoform selectivity [152-154].
A similarly complex mechanistic landscape is associated with quinolone topo II inhibitors and many details pertaining to proposed mechanisms have not been fully elucidated. For example, quinolones of the Pfizer series appear to act as topo II poisons, whereas analogs from the Abbott quinobenoxazine and Dainippon N-1 2-thiazole series appear to both intercalate DNA and show some degree of either topo II catalytic inhibition or covalent complex poisoning (described below). Specific sets of assay tools exist for parsing molecular mechanisms having to do with DNA and/or topo II interactions[142], but not all investigators have applied them comprehensively leading to some gaps in knowledge. Moreover, different labs may employ different conditions for the same type of assay (e.g. DNA cleavage assay) further complicating any broader endeavor to directly compare mechanistic results across companies. Due to these differences in assay use and technique, conflicting mechanistic assessments are occasionally reported between labs for the same compound. Moreover, in some cases, only cellular data is available so that a mechanistic assessment cannot be made. The discussion below will try to provide mechanistic information for each quinolone series to the extent that it is available. In general, as indicated earlier, topo II DNA cleavage assay data provides information on extent of topo II poisoning, while potency in the topo II DNA relaxation assay suggests non-poisoning catalytic enzyme inhibition. In some instances, DNA intercalation (e.g. DNA unwinding assay) data has also been reported. It is unknown at this time which mechanism or blend of mechanisms within the DNA topo II manifold may best match to positive outcomes for certain types of tumors in a clinical setting. Correlation of specific mechanism(s) with any future clinical results could, in theory, provide such information which could then be employed to guide subsequent inhibitor design.
Regarding selectivity of inhibition of the isoforms of topo II by any quinolone series, there unfortunately exists very little data reported at the present time. A range of topo II active quinolones in many subclasses will need to be profiled to gain a good understanding of that SAR. Based on the previously discussed potential clinical advantages of selectively targeting the α isoform, any medicinal chemistry program should first focus on structural modification favoring inhibition of (or interaction with) that isoform.
A further aspect of topo II inhibition which should be noted is the potential for concomitant inhibition of Type I topoisomerases. As previously mentioned, Type I enzymes, like their Type II counterparts, also relax supercoiled DNA, although by cutting and resealing a single strand, rather than both strands, of the DNA duplex [147, 155]. Type I topoisomerases differ structurally from Type II enzymes by existing as monomers (rather than dimers or tetramers) and by not requiring the hydrolysis of ATP as an energy source. Topotecan and irinotecan, synthetic analogs of the prototype natural product camptothecin (an alkaloid discovered in 1966) are anticancer drugs whose action appears to be mediated solely by interaction with topo I via the ternary (cleavage) complex with DNA, a complex analogous to the covalent complexes described for topo II poisons [147, 155-157]. Therefore, it may not be entirely surprising that there exist “dual” agents which interact with both topo I and topo II. Experimental dual-acting agents have been described in the literature, although none have yet been commercialized [158, 159]. In 1990, during the early profiling of clinical antibacterial quinolones against human topoisomerases, topo I inhibition by these drugs was measured but was found to be weak [117, 160, 161]. Subsequently, measurement of topo I activity was apparently no longer routinely reported for other quinolone analogs synthesized for either antibacterial or anticancer programs. More recently however, it was reported that a panel of quinobenoxazines, the 1,8-bridged quinolone series studied by Abbott in the early 1990s (see below), do potently inhibit both topo I and topo II [162]. There is unfortunately no published measurement of topo I inhibition for any other eukaryotic-active quinolone series discussed in this review.
While the mechanistic landscape of topo II inhibitors in general and quinolone topo II inhibitors in particular seems complex, the mechanistic landscape of “targeted” anticancer agents is no less challenging to navigate. Antitumor kinase inhibitors for example may target predominantly one, or more often, multiple kinases. While drug discovery scientists typically endeavor to focus on selective inhibition of one or two kinase targets, often that ideal is not achieved and agents having mixed target profiles—resulting less from design than from opportunism--are advanced. Interestingly, multi-kinase inhibitors having concomitant topoisomerase inhibitory activity have also been described [163].
PARKE DAVIS SAR
In 1989, Daiichi published E. coli gyrase vs mammalian topo II enzyme selectivity data for a panel of primarily commercialized quinolones, although CI-934 (23, Table 1), an advanced experimental antibacterial 6,8-difluoroquinolone from Parke Davis (now part of Pfizer) was also profiled [164]. Nalidixic acid aside, CI-934 showed the narrowest margin of bacterial selectivity among that group of drugs (only 18-fold selective vs gyrase compared to 1,192-fold in the case of ciprofloxacin; data not shown). From 1992 to 1995 Parke Davis published its own selectivity data on large panels of experimental antibacterial quinolones [165-167]. That company was attempting to understand the SAR for cytotoxicity of its antibacterial agents only for the purpose of designing safer quinolones, not to optimize for cytotoxicity as part of an anticancer program. Table 1 highlights a representative sample of their findings relating to cytotoxicity; Parke Davis did not report topo II biochemical data however. Nevertheless, if the assumption is made (albeit cautiously) that cell penetration and accumulation is generally equivalent for all the Parke Davis analogs, then the cell inhibition SAR could be used to suggest the corresponding SAR against the intracellular target(s). This assumption is not unreasonable as it has been well established that a broad range of antibacterial quinolones and fluoroquinolones demonstrate good accumulation in eukaryotic cells [77-81].
Several structural factors influencing greater cytotoxicity are the following:
At C-7, 3’-aminopyrrolidine, and to an even greater extent, 3’-aminomethylpyrrolidine increase cytotoxicity compared to piperazine (17 vs norfloxacin 8; 18 vs ciprofloxacin 15). Alkyl substitution of the amino group or sterically encumbered amino reduces cytotoxicity however (data not shown).
C-8 fluoro substitution increases cytotoxicity compared to C-8 H or N in place of CH at the 8-position (8-F ciprofloxacin 16 vs ciprofloxacin 15; 19 vs 18).
methyl or amino at C-5 increases cytotoxicity (20 vs 19; 21 vs 22).
N-1 cyclopropyl substituted analogs are more cytotoxic than the corresponding N-ethyl analogs (18 vs 17; ciprofloxacin 15 vs norfloxacin 8). N-difluorophenyl substitution is about the same as cyclopropyl. (tosufloxacin 9 vs 22).
Combinations of the above structural characteristics which potentiate cytotoxicity afford quite potent analogs, hundreds of times more cytotoxic than marketed agents such as norfloxacin and ciprofloxacin (for example, analogs 20 and 24: IC50 values < 7.8 μg/ml).
Since Parke Davis was not interested in pursuing an anticancer quinolone program, the company did no work to optimize eukaryotic activity in this series [168].
PFIZER SAR
Although the antibacterial fluoroquinolones of the early 1980s were seen as a significant improvement in potency and spectrum over the earlier generation of quinolones, certain gaps in microbial spectrum persisted, most notably against anerobic and gram positive pathogens. Two Pfizer patent applications published during 1986-1987 encompassed compounds which held promise to fill in some of those gaps [169, 170]. However, several of those compounds instead became the basis of Pfizer’s exploration of SAR against eukaryotic topo II and mammalian cytotoxicity [145, 171-175]. 7-Aryl substituted compounds, for example 26 and 27, were likely inspired by earlier quinolones such as Sterling’s rosoxacin 25, which had been marketed since the early 1980s (Fig. 8). In a non-radiolabelled DNA cleavage assay (measuring the generation of cell-toxic DNA fragments), ciprofloxacin showed no negligible cleavage (CC50 >1000 μg/ml) while pyridyl analog 26 and etoposide showed CC50 values at 73 and 7.5 μg/ml, respectively. For a broader SAR survey, Pfizer used a biochemical assay to measure relaxation of negatively supercoiled DNA, a gauge for inhibition of the overall catalytic activity of topo II. Cytotoxicity in CHO cells was found however to correlate best with the data from the DNA cleavage assay. Table 2 shows SAR from systematic modification at the N-1, C-7 and C-8 positions. Etoposide (14) ciprofloxacin (15), and norfloxacin (8) were included as controls. Several conclusions were drawn from this study:
1. A p-hydroxyphenyl substituent at C-7 was by far a preferred group for eukaryotic enzyme and cell potency (27, 32, 34). p-Methoxyphenyl (29, 33), m-hydroxyphenyl (28), or unsubstituted phenyl (31, 35) were all much less potent than the corresponding p-hydroxyphenyl analogs. It was speculated that the cytotoxicity shown by p-methoxyphenyl derivative 29 might derive from partial intracellular demethylation.
2. N-1 cyclopropyl substitution was superior to N-1 ethyl in the enzymatic assays (27, 31, 15 vs 34, 35, 8, respectively). (SAR for gyrase and antimicrobial activity have historically also favored cyclopropyl for ethyl at N-1).
3. 8-Fluoro substitution was superior to unsubstituted C-8 for eukaryotic enzyme activity (27 vs 32). This observation was concordant with the Parke Davis SAR, above. (Depending on the N-1 substituent, antibacterial activity can be slightly enhanced with an 8-fluoro substituent).
Among all the quinolones in Table 2, compound 27 (CP-115,953) is the most potent in both eukaryotic enzyme assays. In the cytotoxicity assay, 27 is the most potent quinolone and is equipotent compared to etoposide (3 μg/ml vs 5 μg/ml, respectively). Based on the cleavage assay however, one might have expected greater cell potency for 27 compared to etoposide since 27 showed an EC2 of 0.1 μg/ml compared to 10 μg/ml for etoposide (EC2 defined as the effective concentration of drug required to enhance double-stranded DNA cleavage two fold). The Pfizer authors speculated that factors such as uptake, efflux, or cellular metabolism might be factors influencing this discordance, or that alternatively, differences in the mechanisms for the two drugs may play a role. Etoposide acts primarily by slowing the rate of re-ligation of cleaved DNA while the Pfizer quinolones act primarily by stimulating the rate of DNA cleavage. In both cases, toxic DNA fragments would be accumulated intracellularly, yet other factors may play a role contributing to an overall cytotoxic effect at a given concentration of drug.
It is of interest to mention a related series of 7-aryl quinolones reported by Bristol Myers Squibb, which highlight garenoxacin (36), a recently commercialized non-6-fluoro antibacterial quinolone, compared to a few of its close analogs (Table 3). Addition of a 6-fluoro group (analog 37) in the presence of the 8-difluoromethoxy group increases inhibition of human topoisomerase and cytotoxicity while deletion of both 6 and 8 substituents (analog 38) further increases eukaryotic potency both biochemically and cellularly. Biochemical activity against bacterial topoisomerases are relatively little affected. One might speculate based on the Parke Davis and Pfizer SAR previously de-
scribed that the 6,8-difluoro analog of garenoxacin would be more potent still compared to 38 against eukaryotic topo II and cells.
Pfizer also studied the effect of stereochemistry in a 7-quinolyl-1,8-bridged quinolone (Fig. 9). In the topo II mediated DNA relaxation assay, the S-isomer (41) was slightly more potent than the R-isomer (40) (IC50 = 7 μg/ml and 16 μg/ml, respectively) while the racemate (39) showed an intermediate level of inhibition, as expected. However in the more clinically relevant DNA cleavage assay, the S-isomer (41) demonstrated stimulation of DNA cleavage (IC50 = 37 μg/ml) while surprisingly the R-isomer (40) was an antagonist of cleavage. Due to this antagonism, the corresponding racemate did not stimulate cleavage because 40 was able to effectively neutralize the stimulatory activity of 41. Based on an earlier discussion, this antagonism could reasonably be explained by the hypothesis that 30 is, mechanistically, a potent catalytic topo II inhibitor which would then have the ability to antagonize a topo II poison (41). For the antibacterial racemate ofloxacin 42, the corresponding S-isomer (44; levofloxacin) shows greater potency in a gyrase supercoiling relaxation assay than the R-isomer (43), a result analogous to the results in the eukaryotic DNA relaxation assay for the Pfizer isomers 40 and 41. Bacterial gyrase (or topo IV) mediated DNA cleavage data seems not to have been reported for ofloxacin and its enantiomers, so it is unknown whether one of the ofloxacin enantiomers might be antagonizing the other to any extent; ofloxacin has been a clinically useful antibacterial and so if there was any antagonism, it did not significantly impact its therapeutic use. Although Pfizer did not report corresponding eukaryotic topo II inhibitor data for ofloxacin or it enantiomers, a related investigation by Daiichi (Table 4) does provide such data. Daiichi’s bacterial data for ofloxacin (42) and its enantiomers (43 and 44) are analogous to Pfizer’s. The eukaryotic topo II relaxation assay data and low level of cytotoxicity are similar among these three analogs. Further, Daiichi examined two other analogs, 45 and 46, unremarkable in their prokaryotic potency, but showing greater potency in the eukaryotic assays, especially in the relaxation assay compared to 42-44 (Table 4). Analog 45, lacked a methyl group on the 1,8-bridge while the even more potent 46 incorporated an sp2 methylene exocyc-
lic to the bridge. Although there is no data to support a mechanistic basis for this increased eukaryotic (but not prokaryotic) potency, one might speculate that these flatter molecules might more easily intercalate DNA and express greater potencies selectively against topo II either as catalytic inhibitors or cleavable complex poisons. In this regard, analog 46 might be viewed structurally as on the “evolutionary path” toward the even more eukaryotic potent Abbott quinobenoxazines (see below) which incorporated sp2 character to a greater extent on the 1,8-bridge and which were shown to be both DNA intercalators and inhibitors of topo II catalytic function.
Finally, Pfizer also published eukaryotic and prokaryotic enzyme SAR on a series of sparfloxacin analogs having variations of the sparfloxacin 7-dimethylpiperazine heterocycle (Table 5). Sparfloxacin (47), a 5-amino-6,8-difluoro quinolone and etoposide (14) served as controls. All analogs in this survey contained the 6,8-difluoro motif which has previously been established in other series as potentiating eukaryotic potency. There was little variation in the prokaryotic DNA cleavage assay among these quinolone analogs, while the eukaryotic assay showed a wide range of activities dependent on the steric environment of the heterocycle. Thus more sterically compact C-7 groups (51-54) showed the greatest eukaryotic biochemical potency while the more sterically encumbered groups (47-50, including sparfloxacin itself) showed much weaker potency. C-5 substitution (NH2, H or F) in this series had little effect.
Pfizer, like Parke Davis, was not interested in pursuing an anticancer quinolone program and only used the eukaryotic SAR for the purpose of guiding further optimization of its antibacterial quinolone series toward safer analogs. Therefore the company made no effort to optimize eukaryotic activity in their series [176].
ABBOTT SAR (I): QUINOBENOXAZINE CLASS
Abbott established a fluoroquinolone antibacterial program in the early 1980s and within a few years had defined several promising scaffold variations for further optimization. One variation was an N-1-phenyl substituted core, while another—named quinobenoxazines (sometimes quinobenzoxazines)--bridged the N-1-phenyl to the quinolone C-8 via an ether oxygen. Each of these variations can be viewed as logical extensions of prior quinolone derivatization, extending an evolution that began in the late 1960s (Fig. 10).
Abbott recognized that analogs from the quinobenoxazine series possessed potent cytotoxicity (Table 6) [177-179]. By contrast, the non-bridged N-1 aryl analogs (Fig. 10) did not show a corresponding level of activity against eukaryotic cells. To assess the biochemical mechanism of cellular activity for the quinobenoxazines, analogs 55-57 were profiled for DNA intercalation (DNA unwinding assay), catalytic topo II inhibition (DNA decatenation assay), and topo II poisoning (topo II mediated duplex DNA cleavage assay) (Table 6). Analogs 55 and 56, both of which contain a basic amino H-bond donor on the C-7 heterocycle, demonstrated intercalation as well as topo II inhibition but in contrast to doxorubicin, neither of these analogs caused significant topo II dependent strand breaks (data not shown) indicating that they did not act as topo II poisons. Analog 57 having a neutral C-7 group was poorly active in all three biochemical assays. Correspondingly, 55 and 56 were significantly more potent than 57 in cytotoxicity assays (A549 and P388 lines, Table 6). These findings suggest that the stabilization of the topo II-DNA cleavable complex is not necessary for the cytotoxic activity of this class of quinolones but rather the catalytic inhibition of topo II drives cellular activity (DNA intercalation may additionally play a role). Abbott additionally demonstrated that 55 antagonized the DNA topo II poisoning activity of etoposide, further confirming the catalytic inhibition mechanism for 55. The remainder of the data discussed in Table 6 is cytotoxicity data. The enantiomers of 55 (55R and 55S) are similarly potent compared to each other and to the parent racemate in the cell assays. Substitution is tolerated on the 1,8-bridged phenyl ring (58-61). Although biochemical data is not available for 5-amino substituted analog 62, its cytotoxicity in two cell lines is greater than the corresponding 5-unsubstituted compound 55. This potentiating effect of a 5-amino substituent had been noted previously for the Parke Davis quinolone series (see above). Piperazine (rather than 3-aminopyrrolidine) at C-7 is tolerated as shown by analogs having the 5-amino substitution (65 vs 62). Monomethylation and dimethylation of the primary 5-amino group results in successively less potent analogs (62 > 64 and 65 > 66 > 67). Highly significant is the observation by Abbott that members of this quinolone series are much less susceptible to efflux (P-gp) mediated resistance. This is demonstrated for quinolones 55 (and its individual enantiomers), 56, and 58 for which cytotoxicity data was generated in the P388 ADR (adriamycin resistant) cell line having a multidrug re-
sistant (MDR) phenotype. Compared to the parent P388 line, those analogs are only 5-10-fold less active in the ADR line compared to doxorubicin (adriamycin) which shows a 1700-fold decrease. Resistance (and cross-resistance) among existing topo II drugs is a severe clinical problem limiting the usefulness of these otherwise highly effective agents in the treatment of some types of cancer. Alternative topo II drugs which are much less susceptible to efflux mediated resistance would represent a significant advance, providing more flexibility in treatment options.
In vivo, quinobenoxazine analog 56 produced a significant increased life span (ILS) and cures in three lines of i.p. implanted murine tumors, and was active against seven of nine solid tumors including s.c. murine tumors and human tumor xenografts [180]. Table 7 shows ILS and cure data along with tumor weight inhi-
bition (TWI) measurements for 56 dosed by single or multiple schedules against systemic murine tumors. Table 8 shows TWI and cure for 56 against human tumor xenographs. Collectively the data suggest that this quinolone analog is broadly active and can be interpreted as in vivo proof of concept for the action of this new scaffold topo II inhibitor. In spite of these promising preclinical results, Abbott chose not to continue its work on quinolone-based topo II inhibitors. Reasons include lack of a sufficiently robust cancer drug development organization within Abbott at that time to advance such agents, as well as a perception at Abbott, also growing within the wider industry since the early 1990s, that the future of cancer therapy lies with targeted agents rather than cytotoxics [181].
More recently, a different research group demonstrated that Abbott’s quinolone analog 55 functioned as
a dual inhibitor of topo II and HER2 (HER2 expression is down-regulated) [182]. Potential application in an oncology setting is of interest as HER2 overexpression is observed in ca. 6-35% of all gastric cancers; co-amplification of topo IIα occurs in ca. 32-90% of all cancers with HER2 amplification.
ABBOTT SAR II: ISOTHIAZOLOQUINOLONES (AND ACHILLION FOLLOWUP)
Another quinolone variation that Abbott identified during the mid-1980s was the isothiazoloquinolone scaffold [183-185]. Compounds from series possessed superior antibacterial potencies, particularly against gram positive pathogens, compared to other fluoroquinolones at the time. The unique feature of this series was incorporation of an isothiazolo ring in place of the 3-carboxy group, a very rare example of a successful bioisosteric replacement at this position. As with the quinobenoxazines, Abbott quickly identified potent cytotoxic members of this series and began a separate focused effort to develop this series as anticancer agents [186, 187].
In contrast to the quinobenoxazines, the isothiazoloquinolones act as topo II mediated DNA poisons (qualitative DNA cleavage assay, Table 9) as well as topo II inhibitors (DNA unknotting assay, Table 9). The commercial antibacterials ciprofloxacin 15 and norfloxacin 8 showed no activity in the topo II DNA cleavage assay. Isothiazoloquinolone analogs demonstrated cytotoxic activity in relevant cell lines comparable to or better than etoposide (68 and 70, Table 9). Of particular interest, SAR for cytotoxicity that had been established in prior quinolone investigations from Parke Davis seemed to map onto this modified scaffold. For example, cyclopropyl was superior to ethyl at N-1 (72 vs 73) for DNA cleavage, and 3’aminoethyl-pyrrolidine at C-7 was superior to substituted piperazines in the cytotoxicity assay (68 vs 69 and 70). Ultimately however, in spite of promising in vitro potency, this series was not progressed by Abbott as it “did not exhibit good activity against subcutaneously implanted murine solid tumours”; in particular, the development of analog 71 (A-65282) was terminated [62, 180]. The possible reasons for this poor translation of in vitro activity to the in vivo setting were not explained. As mentioned earlier, for strategic reasons, Abbott decided during the mid-1990s to abandon development of all quinolone scaffold topo II-targeted anticancer agents.
During the early 2000s, the problem of infections due to MRSA (methicillin resistant S. aureus) motivated some companies to continue searching for new antibacterial agents with greater potency against this pathogen. Achillion Pharmaceuticals determined that isothiazoloquinolone analogs had good potency against MRSA as well as against other quinolone-resistant pathogens, and therefore established a program to search for a clinical candidate from this scaffold series [188-194]. Mindful of the inherent risk of unwanted eukaryotic cytotoxicity in this class, Achillion counterscreened their antibacterial analogs in a Hep2 (human laryngeal carcinoma) cytotoxicity assay. In vitro data for a number of analogs from their program, selected to highlight the eukaryotic SAR, are shown in Tables 10 and 11. Beyond cytotoxicity CC50 values, Achillion reported biochemical data on a few analogs to confirm the topo II DNA poisoning mode of action, although insufficient data was published to discern broad biochemical SAR. Therefore we again make the reasonable assumption that cellular SAR likely mimics the biochemical SAR. At first, Achillion concentrated on N7 aryl substituted analogs--the clinical quinolone garenoxacin (36; Table 3) serving as a model compound--due to the belief that such analogs would demonstrate enhanced antibacterial and reduced cytotoxic activity. As is generally the case with the quinolone scaffold however, expression of either eukaryotic or prokaryotic potency (or both) depends on a subtle simultaneous interplay among multiple substituents. Among the N7 aryl groups studied by Achillion were several that Pfizer and Sterling had previously investigated, such as p-hydroxyphenyl and 3,5-dimethyl-4-pyridyl (76 and 86, respectively, Table 10). The Achillion eukaryotic cellular SAR overlapped substantially with that of Pfizer (compare Table 2 to Table 10). In particular, 4-hydroxyphenyl (76) as well as 3-methoxy-4-hydroxyphenyl (78) were among the most potent in the isothiazoloquinolone series (CC50 values = 3 µg/ml and 2 µg/ml, respectively) demonstrating the preferred status of hydrogen-bond donating 4-hydroxy group in these analogs. Likewise other close analogs deviating from the preferred 4-hydroxy motif were generally less active or inactive (75, 77, 80) as Pfizer had noted in their series. A topo II poisoning mechanism was confirmed for 76 in a topo II DNA cleavage assay (EC2 = 6 µg/ml; EC2 defined as the concentration of drug required to enhance topo II mediated double strand DNA cleavage by 2-fold). Achillion moreover extended Pfizer’s limited eukaryotic SAR around the potent p-hydroxy analogs 27 and 32 (Table 2) with p-hydroxyphenyl, p-anilino, and aminoalkylphenyl substituted analogs (79 and 81-85; Table 10). In particular, both the 4-aminomethylphenyl (83) and 4-aminoethylphenyl (85) groups at the C-7 were similarly potent compared to the 4-hydroxyphenyl analog and again suggest that a H-bond donating group at C-7 defined within a narrow region of space is making a critical interaction with topo II in its covalent complex with DNA. An understanding of the SAR for a number of heteroaromatic groups at C-7 is less straightforward (e.g. 84 and 86-90). In particular it seems unusual that 84, the 3-pyridyl analog of potent aminomethyl compound 83, should be inactive in this assay. A fuller understanding of this SAR may need to await a more detailed structural understanding of the interactions of bound inhibitor in the covalent complex (see section below). Achillion additionally explored isoindoline substitutions related to the 7-isoindoline group of garenoxacin (Table 11). The potent aminomethyl inhibitor 83 can be viewed as a “ring-open” analog of the garenoxacin side chain. A number of analogs prepared for this survey (91-96) displayed a range of cytotoxicity values (1-40 µg/ml) which could be attenuated to varying degrees by an 8-methoxy group along with a hydrogen, rather than fluorine, at C-6 (the commercial antibacterial garanoxacin 36 contains an 8-difluoro-methyoxy group and a hydrogen at the C-6) which is concordant with some of the SAR that Bristol-Myers published for their own garenoxacin analogs (Table 3). Ultimately Achillion reverted to substituted pyrrolidine (i.e. non-aryl) groups at the isothiazoloquinolone C-7 to identify a “short list” of antibacterial candidates culminating in selection of ACH702 for development (100, Table 11). Ironically however, ACH702 displayed relatively potent cytotoxicity (CC50 = 4 µg/ml). The aminomethylpyrrolidine motif in 100 is similar to the C-7 groups identified earlier by both Parke Davis and Abbott (among others) as potentiators for eukaryotic activity (24, Table 1). Yet Achillion did achieve in 100 a level of potency against MRSA far superior to its other analogs, and therefore an argument could be made that despite its relatively potent level of cytotoxicity, a sufficient margin exists relative to the antibacterial potency to justify its selection (MRSA MIC = 0.06 µg/ml). Of interest from a eukaryotic potency perspective is the pyrrolidine SAR shown in Table 11 which is concordant with other historical Parke Davis SAR and our current understanding of the eukaryotic SAR as it pertains to the C-7 substituent. For example, removing the hydrogen bond donating potential from the potent aminomethyl analog 98 (CC50 = 4 µg/ml) by dimethylation affords a poorly active analog (99; CC50 >43 µg/ml), further re-inforcing the concept of a key H-bond accepting motif in that region of the topo II DNA covalent complex.
Even though Abbott ultimately found the isothiazoloquinolone series disappointing in animal models of implanted solid tumors (for reasons that were not apparently understood), this newer Achillion eukaryotic SAR (e.g. high potency of p-aminomethyl- and p-aminoethylphenyl analogs 83 and 85) is of potential future importance since it should map back onto a conventional quinolone scaffold for the purpose of design of more robust human topo II inhibitors.
STERLING SAR
Sterling Drug was a co-discoverer of the quinolone class of antibacterials in the late 1950s and was the first to commercialize an agent from the class, nalidixic acid, in 1964 in the US [57]. Sterling later investigated aryl substitution at the quinolone C-7 position and commercialized one such agent, rosoxacin (25 Fig. 8), which was used primarily as a treatment for gonorrhea. Searching for greater potency against Gram positive and anerobic pathogens within the quinolone core, Sterling identified additional 7-aryl substituted analogs, several of which were found to possess activity in eukaryotic topo II mediated DNA cleavage assays, although those analogs were less potent than etoposide. Sterling’s eukaryotic SAR for this 7-dimethylpyridyl quinolone series, along with several related analogs and reference agents is shown in Tables 12 and 13 [195, 196]. Much of the SAR confirms what was published by other companies from the same time, for example: 1. at N-1, the order of increasing potency is p-F-phenyl < ethyl < cyclopropyl (103-105); 2. the 5-NH2 group is tolerated (or slightly potentiates) activity (106 vs 103); 3. the (S)-methyl enantiomer of 1,8-bridged systems is the active enantiomer (both for prokaryotic and eukaryotic activity; 109 vs 111). Sterling additionally found that sulphur is superior to oxygen in 1,8-bridged compounds (110 vs 109).
Due to its relatively high level of biochemical potency and its promising level of cytotoxicity against P388 cells (IC50 = 8.7 µg/ml) Sterling chose compound 110 (WIN58161; Table 13) for further studies related to mechanism of action, in vivo antitumor profile, and susceptibility to cellular resistance mechanisms [197]. Analog 110 was shown not to intercalate DNA, and therefore, due to its topo II mediated DNA cleavage mechanism, it displays a mechanistic profile similar to etoposide. Table 14 shows that analog 110 exhibited a significant antitumor effect in five in vivo antitumor models. Table 15 demonstrates that 110, unlike reference agents etoposide, m-AMSA (amsacrine), and vincristine is not susceptible to either efflux- or topo II-mediated resistance mechanisms.
Additionally, Sterling evolved the traditional quinolone scaffold in more radical ways, exploring replacement of the C-3 carboxy group with other functionality and creating novel ring fusions [195, 198-200] For example, Sterling reported that the C-3 des-carboxy analog of 103 was similarly potent to parent 103 in the topo II DNA cleavage assay (IC50 5.6 µg/ml vs 2.8 µg/ml, respectively). The 3-carboxy group that is so essential for antibacterial activity can thus be omitted for eukaryotic-directed analogs largely due to the fact that the eukaryotic topo II DNA cleavage complex does not form the water-magnesium bridge present in gyrase cleavage complexes (Figs. 5 and 7); the topo II active quinolones utilize alternative interactions within the DNA-topo II cleaved (covalent) complex manifold. As stated previously however, the primary focus of this review is on the conventional quinolone scaffold (e.g. containing a 3-carboxy group) due to its highly pre-optimized—and predictable-- status with respect to physical properties, safety, PK and so forth. Analogs having more radical structural modification will not be covered here with the exception of the isothiazoloquinolones and quarfloxin, as already mentioned.
Based on their publication and patent record, Sterling appears to have essentially terminated its anticancer quinolone programs by the mid 1990s. There was no official reason communicated, but one might surmise that the acquisition of Sterling by Sanofi in 1994 could have played a role. Following such transitions, the loss of project champions and/or re-prioritization of projects by new management often result in project terminations, often for non-scientific reasons.
BANYU/MERCK SAR
Following the realization that prokaryotic and eu- karyotic topoisomerases acted through similar mecha- nisms, Banyu (in collaboration with Merck) screened a set of its experimental antibacterial quinolones against mammalian topo II [201, 202]. In so doing, Banyu identified (-)-BO-2367 (116) as a potent topo II inhibi- tor of DNA relaxation more active than etoposide in that biochemical assay (3.8 μM vs 7.4μM, respectively) and which prevented tumor growth in mice (Tables 16 and 17). The enantiomer of this quinolone, (+)-BO- 2367 (125) having the opposite stereochemistry in the C-7 side chain, retained activity against bacterial gy- rase, similar to ciprofloxacin, but was 100-fold less potent against eukaryotic topo II DNA relaxation, thus indicating that molecular details of the C-7 quinolone substituent were especially critical for eukaryotic topoi- somerase/DNA binding interactions (Table 17). (-)- BO-2376 (116) was about twice as potent as etoposide in enzymatic induction assays of DNA cleavage and cell-based assays of induction of formation of DNA cleavable complex (Table 16). Whereas (-)-BO-2376 increased survival vs control in two models (P388 and L1210) of murine leukemia compared to control, the survival times were inferior compared to etoposide. (-)- BO-2376 did show superiority to etoposide however as measured by extent of solid tumor growth in a murine s.c. colon tumor implant murine model (100% inhibi- tion vs 63% inhibition at 1.25 mg/kg/day for (-)-BO- 2376 and etoposide respectively; Table 17).
Banyu reported expanded SAR for this series with an analysis of the contribution of functionality at the quinolone N-1, C-5, C-7, and C-8 positions to induction of formation of DNA cleavable complex and to cytotoxicity (Table 16) [203]. At C-7, the SAR for both activities is strikingly sensitive to the position and environment of the primary distal amine. Saturation of the cyclohexene ring (which would alter the position in space of the attached amino group) and N-alkylation both reduced activity significantly (118, 119, 121). More substantial modifications at C-7 further reduced activity in both assays (123). Amino substitution at C-5, fluoro substitution at C-8 and cyclopropyl substitution at N-1 (compared to N-1 ethyl) each contributed to increases in potency for each assay. Methoxy was slightly less potent than fluoro at C-8 (117). Much of this SAR was concordant with that previously discussed for other companies investigations (e.g. Parke Davis, Pfizer, Achillion) thus further re-inforcing confidence in the collective trends. Neither Banyu or Merck seems to have followed up this interesting antitumor quinolone series. Based on the published record, Banyu and Merck had a vigorous ongoing portfolio of other anticancer discovery programs in the early 1990s, several of which were focused on “targeted agents” (for example, angiogenesis and ras oncogene). Therefore we speculate that other anticancer programs received higher priority than the topo II quinolones.
KYOWA SAR
Similar to Banyu/Merck, Kyowa (Japan) screened a large panel of clinical and experimental antibacterial quinolones in its collection for mammalian topo II and cytotoxic activity [204, 205]. Several analogs were found to be significantly active in the biochemical topo II mediated DNA cleavage assay (i.e., they produced linear DNA > 10% of substrate at a drug concentration of 250 μM) although were “generally less potent than etoposide.” The mechanism of DNA cleavage was determined to be through formation of the ternary cleavable complex. The entire panel of compounds was screened for antitumor activity in a murine P388 leukemia model, and it was found that all of the topo II active analogs were also active in this antitumor model. Among this set of topo II active analogs showing antitumor activity, S-116 was selected for further in vivo profiling. S-116 happens to be identical to the analog 24 (PD117579) also identified (but not further profiled) by Parke Davis (see Fig. 11). Compared to mitomycin in the murine P388 model, 24 demonstrated a 34% increase in life span (ILS) at a 25mg/kg dose compared to untreated controls, vs a 68% ILS shown by mitomycin at a 4 mg/kg dose. In the B16 melanoma cell murine model, a 100 mg/kg dose of 24 demonstrated a 128% ILS, comparable to mitomycin (118% ILS) at a 4 mg/kg dose.
It is important to note that although the in vitro and in vivo results of S-116 (24) seem modest by comparison to standard anticancer drugs, S-116 was merely the most active compound taken from a pre-existing antibacterial panel and was not further optimized for anti-
tumor activity. Thus S-116 should be viewed as an early “hit” (or “lead”) compound. By that standard, its in vitro and in vivo profile are impressive. In terms of SAR and design, it worth pointing out that S-166/PD117579 (24) and Banyu’s (-)-BO2376 (116) both contain an N-1 cyclopropyl group, an 8-fluoro, and a C-7 pyrrolidine bearing an amino group attached to the pyrrolidine 3’-position via a 1-carbon spacer. (Fig. 11) Thus, the primary amino groups for both compounds can occupy the same position in space to engage in binding interactions to topo II as part of the ternary complex. Kyowa, like Banyu, seems not to have pursued a larger drug discovery program based on their initial survey. According to the published record, Kyowa was involved in a number of natural product based anticancer programs (primarily cytotoxics) during the early 1990s, and perhaps the company considered those other projects to be of higher priority.
BAYER SAR
Bayer played a significant role in the field of antibacterial quinolone R&D during the 1980s and 1990s, having developed and commercialized both ciprofloxacin and moxifloxacin. During 2004-5 Bayer published two patent applications on the topic of anticancer quinolones [206, 207]. Analogs 126 and 127 (Fig. 12) are representative structures from these two applications. One of the applications lists semiquantitative data for nearly 200 examples showing that the majority of the analogs (such as 126) have CC50 values of less than 0.5µM in a Colo205 human colon carcinoma cell line proliferation assay. Bayer has not published this work in the journal literature and no further research in this area appears to be ongoing at Bayer. No information suggesting biochemical mechanism is reported. The particular substitution pattern of the Bayer quinolone series does not resemble that of any other quinolone series discussed in this review sufficiently closely to allow conjecture as to possible mechanism. While topo II may be involved, these Bayer quinolones may also be acting through a topo II independent mechanism.
DAINIPPON SAR: THE JOURNEY TO VOSA-ROXIN
In 1986 patent applications, both Parke Davis and Dainippon described N-1 2-thiazole quinolones or naphthyridones as antibacterial compounds (Fig. 13, 128 and 129) [208, 209]. The antibacterial activity for 128, later published by Parke Davis, was modest and no eukaryotic cytotoxicity was reported [210]. By 1995, Dainippon reported the related compound 11 (what was later to be named vosaroxin) in a patent application, featuring it as an antitumor agent [211]. During 2002-2004 Dainippon published detailed eukaryotic cytotoxicity SAR for their naphthyridone series, and additionally reported biochemical data for 11 [212-214]. In spite of potent cellular activity, Dainippon reported that 11 did not induce topo II mediated DNA cleavage at reasonable concentrations (IC50 = >1000 μg/mL) but did inhibit topo II DNA relaxation (IC50 = 3.2 μg/mL). As a control, Dainippon reported an IC50 of 1 μg/mL for etoposide in the topo II mediated DNA cleavage assay and an IC50 of >1000 μg/mL in the topo II DNA relaxation assay, consistent with etoposide’s historical characterization as a topo II DNA poison. This mechanistic profile for 11 is different from the typical topo II poisoning mechanism displayed by the majority of other cytotoxic quinolone series discussed in this review, yet is similar to the profile deduced for Abbott’s quinobenoxazine series. Abbott had determined that the cytotoxicity for 55 was driven predominantly by catalytic topo II inhibition along with a DNA unwinding (intercalation) activity. Although Dainippon did not assay for DNA intercalation, later studies by Hawtin et al. determined that intercalation for 11 was a component of its mechanistic profile [215]. Somewhat in contrast to the Dainippon mechanistic results, Hawtin determined that 11 can, in fact, be a topo II DNA poison at low micromolar levels of drug, but changes to a catalytic mechanism at higher drug concentration. For all of its other cytotoxic quinolone analogs, Dainippon reported only cellular data, so that wider assessments regarding either mechanism or target SAR cannot be confidently made for other members of the series. Nevertheless, as was suggested previously (above sections on Parke Davis and Achillion), one might reasonably (but cautiously) infer biochemical SAR from the available cellular SAR since it has been well-established historically that quinolone and fluoroquinolone penetration into eukaryotic cells is a facile process for a broad range of analogs.
Of particular note, the N-1 2-thiazole group and the 8-nitrogen of the 1,8-naphthyridone together appear to be a highly unique functional group combination imparting potent cytotoxicity (Tables 18 and 19). N-1 groups on the 1,8-naphthyridone scaffold other than a 2-thiazole resulted in compounds having distinctly inferior eukaryotic activity (135-144). All of the substituted thiazole N-1 containing compounds (131-134) shown in the first row of Table 18 display potent cytotoxicity (CC50 values from 0.05 to 0.26 μg / mL), with the unsubstituted 2-thiazole analog 130 being the most potent (CC50 = 0.02 μg/mL). For comparison, reference compounds etoposide 14 and doxorubicin 13 display CC50 values of 0.01 and 0.004 μg/mL, respectively. The 2-(3,4-thiadiazole) 135 as well as the 2-thiophene 136 N-1 analogs were inferior as were the isoxazole and pyrazole analogs (137-139) shown on the second row of Table 18. Six-membered aromatic groups (phenyl, 4-F-phenyl, 2-pyridyl) as well as 2-methylene thiazole and cyclopropyl were likewise sharply inferior (140-144). The N-1 2-thiazole group on scaffolds which did not have a nitrogen at the 8-position were also distinctly inferior as shown in Table 19. Thus, both 1,8-naphthyridone 130 and 6,8-diazaquinolone 145 are potent compounds, yet the corresponding 8-H and 8-F quinolones 146 and 147 show distinctly inferior potency. The effect of substitution at the C-5 and C-6 positions of the N-1 2-thiazole-1,8-naphthyridone scaffold is shown in Table 20. Apart from the negligible effect of fluorine vs hydrogen at C-6 (CC50 = 0.02 and 0.01 μg/mL for 130 and 148 respectively), both electron donating or withdrawing groups have a detrimental effect on activity (149-152). Substitution at the C-5 with both electron withdrawing and donating groups seems to be more tolerated (148, 153-155; Table 20). At C-7, N-pyrrolidines and piperazines substituted by (or fused with) small non-aromatic groups are widely tolerated (148, 11, 156-160), while aryl subsitution or fusion to the heterocycle is not tolerated (164, 165, Table 21). Alicycic N-substitution or direct substitution by aryl at C-7 is also not tolerated (161, 164, 165). Overall this SAR looks quite different from that for quinolone scaffolds acting through a predominant topo II mediated DNA cleavage mechanism (Parke Davis, Pfizer, Sterling, Kyowa, Banyu-Merck, and Abbott isothiazoloquinolones series).
A significant portion of the above SAR can be explained by assuming that an overall co-planar structure of the N-1 2-thiazole group with the bicyclic 1,8-naphthyridone core is favored for eukaryotic potency. Hawtin et al. first proposed a coplanarity hypothesis following an analysis of the cellular and biochemical activities of the “matched triplet” series comprised of vosaroxin (11), an Abbott-like quinobenoxazine analog which has forced co-planarity (166), and an unconstrained N-phenyl analog (167) which presumably lacks the ability to be co-planar (or at least ring co-planarity is not its preferred conformation) (Fig. 14) [215]. The forced coplanar analog 166 was the most potent (CC50 = 0.02 μM) in the cytotoxicity assay, followed by vosaroxin 11 which was about 10-fold less potent (CC50 = 0.19 μM), and finally the N-1 phenyl analog 167 was the least potent (CC50 of > 10 μM) (Fig. 14).
The co-planar preference of vosaroxin might be explained by a nitrogen-to-sulfur non-covalent interaction between the N-1 2-thiazole sulphur and the naphthyridone 8-nitrogen which could enforce the planar conformation depicted in Fig. (14) (partial structure 168). According to this argument, the N-1 2-thiazole C-S σ* orbital interacts with the lone pair of electrons from the 8-position naphthyridone ring nitrogen resulting in co-planarity between the scaffold aromatic rings [216]. Alternative non-sulfur containing heterocycles would not allow for this type of conformation constraint. The relatively high levels of potencies displayed by all of the thiazole analogs in the top row of Table 18 is consistent with this view. The thiadiazole and thiophene analogs (135, 136) are significantly less potent how-
Fig. (14).
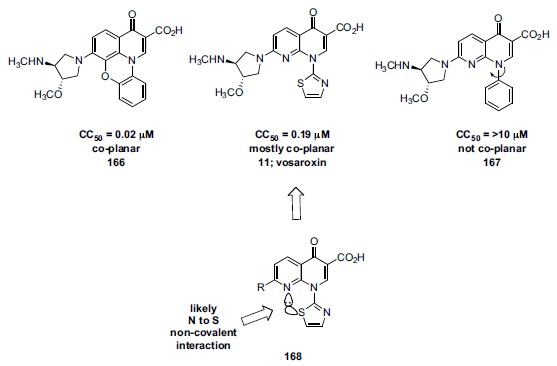
Effect of degree of planarity of the N-aryl substituent on cytotoxic potency. The N-thiazole napthyridone vosaroxin (11) likely achieves a planar bias due to constraint imposed by the C-8 nitrogen lone-pair donation to the thiazole sulfur. CC50 values represent the inhibition of proliferation of A549 cells.
ever, even though a sulphur is available for the type of interaction just described. This loss in potency could be explained by assuming that the 2-thiazole, rather than the thiophene or thiadiazole, is electronically optimal for this N to S non-covalent interaction. The 3-methyl-5-isoxazole analog 138 is moderately potent for reasons that are not entirely clear based on this argument. All of the remaining N-1 substituents in Table 18 are much less potent than the 2-thiazole series, consistent with this argument of non-covalent induced co-planarity. In Table 19, quinolones 150 and 151 lack the naphthyridone 8-nitrogen, and are poorly active, whereas the 6,8-diazaquinolone 149 allows the putative N to S interaction and is quite potent. At the naphthyridone C-5 position, substitution by electron withdrawing substituents leads to a 5-10 fold dropoff in potency, while electron donating substituents result in equipotent analogs (Table 20). This SAR might be explained by the requirement for suitable availability of the lone pair electrons on the naphthyridone 8-nitrogen; electron withdrawing substituents would disfavour availability of the lone pair (153, 154), while electron donating substituents (155) would maintain that availability. At the naphthyridone C-6 position, hydrogen and fluorine
substitution provides compounds roughly equivalent in potency whereas substitution with other (larger) groups, either electron donating or withdrawing, leads to moderate to severe reduction in cell potency (149-152). This SAR might be explained by a steric perturbation caused by those larger 6-substitutents on the orientation of the adjacent 7-pyrrolidine or 7-piperazine heterocycles. The precise orientation of the C-7 heterocycle may be an important component to overall potency. The acyclic 7-aminoethylamine 161 is much less potent, perhaps due to this requirement of a specifically oriented 7-heterocycle. The poor potency of the phenyl and 3,5-dimethyl-4-pyridyl substituents at C-7 might be explained by a poor fit into the required binding space for aryl groups in general in the context of this unique scaffold. In addition (or as an alternative) to this latter argument, an electron-donating amine may be required at C-7 (as part of a pyrrolidine or piperazine ring, for example) to fulfill the electronic demands (via electron donation) to the critical 8-nitrogen lone pair.
Dainippon generated additional cellular eukaryotic and prokaryotic SAR by modification of the quinolone C-3 group (Table 22) [213]. For prokaryotic activity, the C-3 carboxy substitution on the quinolone or 1,8-naphthyridone ring has for many decades served as the gold standard group for achieving excellent potency. At the target level, the structural basis for the importance of the carboxy group in antibacterial activity has been established to be the consequence of a key bridging interaction through a magnesium ion-water network to aspartate and serine residues in the GyrA (or ParC) gyrase (or topo IV) subunits (see Fig. 5). Although a few non-carboxy bioisosteric variants have been reported having good potency, (e.g. Abbott’s isothiazoloquinolone and more recently Pfizer’s aminodione series [217, 218]), all commercialized quinolone or naphthyridone drugs contain the canonical 3-carboxy group. As shown in Table 22, replacement of the 3-carboxy with hydrogen, carboxamide, or hydroxymethyl results in profound loss of antibacterial potency (169, 170, 171). By contrast, much less variation is seen in the eukaryotic cell activity (although the carboxy substitution is still preferred). Sterling had reported similar observations (see above). Structurally this relative lack of variation in eukaryotic potency by modification of the C-3 substitutent is due to the absence of the magnesium-water network which exists in prokaryotic topoisomerase-drug complexes, as previously discussed (compare Fig. 7 to Fig. 5). Eukaryotic active quinolones derive the bulk of their binding affinity in the cleavable complex from alternative interactions. As stated previously however, the primary focus of this review is on the conventional quinolone scaffold (i.e. containing a 3-carboxy group) due to its highly pre-optimized-and predictable- status with respect to physical properties, safety, PK and so forth.
Dainippon further characterized its most potent cell inhibitors for aqueous solubility at pH 7.2 and for in
vivo antitumor activity using mice implanted with P388 leukemia cells dosed i.p. at 3.13, 12.5, and 50 mg/kg [214]. A number of otherwise potent analogs possessed less than ideal aqueous solubility, whereas the analog which became vosaroxin (11) stood out with a superior solubility of 20.1 mg/kg. In vivo efficacy values, expressed as (median survival time of treated mice) / (median survival time of controls) X 100 of several analogs including 11 were comparable to that of etoposide. Analog 11 (vosaroxin) was chosen for development based on the combination of good in vivo efficacy and good aqueous solubility. Preclinical and clinical development of vosaroxin is described further below.
It is of interest to briefly explore a few additional divergences as well as similarities in quinolone/ naphthyridone SAR with respect to prokaryotic vs eukaryotic systems in the context of the Dainippon work. 6,8-Difluoro substitution on quinolone scaffolds tends to increase both eukaryotic and gram positive prokaryotic cellular activity. Whereas the N-1 2-thiazole group is uncommon in antibacterial quinolone scaffolds (and is not represented by any launched drug), depending on substitution elsewhere, N-1 2-thiazole quinolones can be antibacterially potent, as demonstrated by Dainippon example 130 (Table 22). Vosaroxin 11, which results from removal of the 6-fluorine from 171 as well as subtle decoration on the C-7 aminopyrrolidine is substantially less potent in gram positive and gram negative antibacterial assays compared to 130 (Table 22). N-1 2-Thiazolyl quinolone 128 (Parke Davis, Fig. 13) with a differently substituted C-7 pyrrolidine shows further loss of antibacterial potency, especially against S. aureus [219]. Finally, N-1 cyclopropyl and 4-fluoro- (or 2,4-difluoro-) phenyl have traditionally shown high antibacterial potency when matched with either quinolone of 1,8-napthyridone scaffolds, whereas as discussed, these N-1 groups were poor choices for eukaryotic potency within the napthyridone scaffold. Thus, in general, eukaryotic and prokaryotic activity, although at times overlapping can be directed by proper choices in substitution to predominate at one type of activity vs the other.
OVERALL SUMMARY OF QUINOLONE SAR FOR EUKARYOTIC TOPO IIA
Based on the quinolone scaffolds discussed in this review, there appear to be two distinct sets of SAR governing quinolone topo II inhibitors. One SAR set encompasses the quinolone series that mechanistically operate predominantly through the topo II DNA covalent complex (cleavable complex). The scaffolds that fall into this group (Group A, Fig. 15) are those from Parke Davis (and Kyowa), Pfizer, Banyu-Merck, Sterling, as well as Abbott’s isothiazoloquinolone series. The other SAR set encompasses the quinolones that act topo II catalytic inhibitors (operating at a step in the topo II catalytic cycle prior to formation of the cleavable complex) with possibly an intercalation component. The Abbott quinobenoxazine and the Dainippon/Sunesis N-1 2-thiazole naphthyridone scaffolds fall into this category (Group B, Fig. 15). It should be recalled however that reports describing the mechanism of vosaroxin 11 conflict to some extent, pointing perhaps to a more nuanced blend of mechanisms.
Thus, as detailed in this review, for Group A scaffolds:
6,8-difluoro substitution is strongly preferred.
H-bond donor at C-7 is preferred (e.g. HO or H2N; substitution on amino or steric congestion near the donor usually is detrimental to potency); proper spacer (length and orientation) is required.
N-1 cyclopropyl is preferred if there is not a 1,8-bridge.
If there is a 1,8-bridge, sulfur is preferred in the bridge, and (S)-stereochemistry is preferred for an appended methyl group.
Small C-5 groups such as primary amino or methyl usually increases potency.
For Group B scaffolds:
Enforced planarity of N-1 aryl group is required.
Substitution at other positions is widely tolerated, but requires some customization.
It is important to recognize that the guidances above are based on relatively small datasets involving panels of compounds which in many cases were not subjected to extensive (or any) medicinal chemistry optimization or additional exploration for the discovery of alternative structural elements that might further enhance DNA/topo II interactions and desired cytotoxicity. Yet, as has been amply demonstrated, much of the existing SAR across various quinolone scaffolds is concordant, re-inforcing confidence in a number of key SAR trends. Also, as previously stated, it is also important to recognize that quinolone SAR, either for eukaryotic or prokaryotic targets, is subtle and is the result of a blend of effects from multiple interacting substituents. Whereas the simple listing of guidances above may be helpful as a start, a fuller understanding of SAR typically requires more extensive analysis of the effects of interrelated functionality in individual compounds. This subtlety is illustrated in Fig. (16) which pairs three cytotoxic quinolones with corresponding structurally related commercialized quinolone antibiotics. Each of the commercialized (non-cytotoxic) antibacterials incorporates structural features which we have identified as potentiating cytotoxicity. Garenoxacin 36 contains a C-7 aryl group terminating in a H-bond donor analogous to the Pfizer cytotoxic analog 27 (Fig. 16). In this case, the difluoromethoxy group at C-8 as well as the fact that the C-7 amino group is secondary (and not primary) may counteract the target interactions that would otherwise express greater cytotoxicity. Sparfloxacin 47 contains the cytotoxicity-potentiating 6,8 difluoro motif as well as a 5-amino group, similar to the cell-potent Parke Davis analog 20. However, as was seen in Table 5 (Pfizer study), the C-7 dimethylpiperazine substituent of sparfloxacin is able to uniquely counteract the cytotoxicity that would otherwise be expressed with other C-7 substituents. Finally gemifloxacin 102 contains the cytotoxicity potentiating C-7 aminomethylpyrrolidine group that was a feature of the Kyowa/Parke Davis analog 24 (among other cell-potent analogs). Gemifloxacin however has a naphthyridone core and therefore lacks a C-8 fluorine which would otherwise contribute to cytotoxicity. The oxime substituent on pyrrolidine may additionally contribute to counteracting some of the cytotoxic effect of the C-7 aminomethylpyrrolidine group.
Fig. (16).
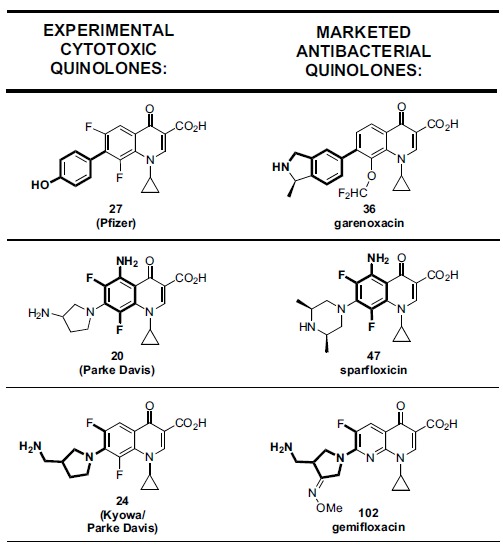
Comparison of three experimental cytotoxic quinolones with corresponding marketed antibacterial quinolones (27 vs 39; 116 vs 47; 24 vs 102) showing structural elements (in bold) common between the pairs which are considered to potentiate cytotoxicity.
SUNESIS: VOSAROXIN (QUINPREZOTM) – CLIN-ICAL DEVELOPMENT OF THE DAINIPPON EQUITY
In 2003, Sunesis entered into an agreement with Dainippon Pharmaceuticals (now Sumitomo Dainippon), obtaining a worldwide exclusive license to vosaroxin (11) and related compounds [220]. Preclinical studies demonstrated that vosaroxin, in comparison to a variety of standard agents such as doxorubicin, cisplatin, paclitaxel, etc, was highly potent in vitro against a panel of human hematologic and solid tumor cell lines and also demonstrated strong dose-dependent growth inhibition against a number of tumor types in rodent xenograph models [221, 222]. Vosaroxin was minimally metabolized in vitro, and in animals demonstrated favourable pharmacokinetic properties (dose-proportional exposure, low variability and moderate clearance) [223-226]. Due to its mechanism leading to replication-dependent DNA damage, vosaroxin induces irreversible cell cycle arrest in the G2 phase followed by apoptosis [227].
Notably, vosaroxin differentiates from standard chemotherapeutic agents in several important ways. First vosaroxin maintains potent cytotoxicity in cell lines resistant to other agents where the resistance is due to overexpression of P-glycoprotein (P-gp, an ATP driven efflux pump). P-gp overexpression is a well characterized tumor resistance mechanism, and vosaroxin was shown not to be susceptible to that mechanism. By contrast, other topo II anticancer agents (anthracyclines, epipodophyllotoxins, etc.) are susceptible to resistance development by this mechanism. Treatment failure due to resistance is a major issue with those conventional agents [228]. Moreover, once resistance develops to one agent, others within the traditional topo II class are less effective due to cross-resistance. As discussed in this review, Abbott and Sterling found examples of their quinolone series to be either non-susceptible or much less susceptible to efflux-based resistance mechanisms in relevant tumor cell lines compared to standard agents. Although there seems to be no additional data to support the hypothesis that quinolones are in general minimally susceptible to mammalian efflux, one can look to the antibacterial quinolone literature for further clues. Although reports describe variable susceptibility of quinolones to P-gp and other ABC (ATP Binding Cassette) efflux pumps in caco-2, MDCK (Madlin Darby canine kidney), and
macrophage cell lines [229-234]. the majority of commercialized (as well as non-commercialized) antibacterial quinolones are known to be well-absorbed orally. Indeed good oral bioavailability is a characteristic of the class and demonstrates that intestinal P-gp mediated efflux is not a general issue for the class. Based on this argument, we might speculate that anticancer quinolones beyond the several examples discussed in this review might also not be severely susceptible to ABC pumps in cancer cell lines. A second point of differentiation is that vosaroxin avoids the p53 resistance pathway by activation of caspase-3, a key mediator of apoptosis independent of p53 [235]. A third point of differentiation of vosaroxin compared to the anthracycline class agents is the absence of generation of reactive oxygen species (ROS) by vosaroxin. It has been suggested that ROS generation by the anthracyclines is in part responsible for the cardiotoxicity displayed by the class (in addition to those agents lack of target selectivity between of the two isoforms of topoisomerase) [215, 236, 237]. Therefore vosaroxin might not be expected to display cardiotoxicity, at least by the ROS mechanism. On the other hand, vosaroxin apparently does inhibit both isoforms of topoisomerase II (α and β)[215, 238]; selectivity for the α form, as previously discussed, is hypothesized to confer (in part) protection from cardiotoxicity. In any case, there are no clinical reports of cardiotoxicity associated with vosaroxin. Perhaps due to the fact that a major fraction of vosaroxin’s cell killing mechanism is related to topoisomerase-independent DNA intercalation compared to inhibition of topo II in the catalytic mode and/or in the cleavable complex, cardiotoxicity might therefore not be seen in spite of lack of isoform selectivity. It should be noted however that (cardiotoxic) doxorubicin also has been shown to act by the dual mechanisms of DNA intercalation and topo II cleavable complex stabilization, and also does not does not display isoform selectivity. Yet doxorubicin displays biochemical differences compared to vosaroxin. Whereas the levels of cleavable complex induced by both vosaroxin and doxorubicin are similar at a fixed drug concentration (1 μM), similar levels of DNA fragments are achieved by doxorubicin at 10-fold lower concentration (0.1 μM) compared to vosaroxin (1 μM) [215]. Also doxorubicin favors DNA cleavage sites different from those of vosaroxin. Obviously, for DNA intercalating topo II inhibitors, the precise mechanistic details associated with cardiac toxicity remain to be fully elucidated.
Vosaroxin was shown in vitro to be synergistic with cytarabine, a standard chemotherapeutic agent often used in combination with conventional topo II inhibitors [235]. In the clinic, vosaroxin, alone or in combination with cytarabine, has been studied most thoroughly in patients with acute myeloid leukaemia (AML). AML is very difficult to treat, and there have been essentially no major treatment advances in several decades. A number of new agents, including targeted signalling pathway modulators have recently failed in the clinic for AML, or registered only modest results vs comparator. The current standard of care for AML is cytarabine + an anthracycline (or anthracyclinedione) [239, 240]. In AML patients, Phase I and II trial results for vosaroxin were reported as encouraging [221, 241-244]. For example, in a Phase II study in newly diagnosed and previously untreated AML patients over 60 years old and having additional risk factors that precluded conventional induction therapy, treatment with vosaroxin alone resulted in an overall remission rate of 32%; the dose-limiting toxicity was stomatitis (oral mucositis). A recent Phase III trial studied the vosaroxin/cytarabine combination vs placebo/cytara-bine in patients with relapsed or refractory AML. Results were modest however. The combination did not meet the primary endpoint (overall survival, OS) but did register significant effect vs the comparator in complete remission (CR) rates, more so in patients over 60 years of age [220, 245]. Sunesis is currently studying the use of vosaroxin in combination with decitabine in patients with myelodysplastic syndrome (MDS), although these trials are at an earlier stage.
CYLENE PHARMA: QUARFLOXIN - EVOLUTION OF ABBOTT QUINOBENOXAZINE ANTICANCER EQUITY
Following Abbott’s decision in the mid 1990s to terminate its quinolone anticancer programs, Laurence Hurley, a professor at the University of Texas, Austin (later at the University of Arizona, Tuscon) continued investigations on Abbott’s quinobenoxazine series [246-249].} During that time, Hurley was simultaneously interested in G-quadruplex biology and the implications of modulating quadruplex structure by small molecules [250]. G-quadruplexes, first identified in 1962, are four-stranded DNA structures comprised of stacked planar sets of four guanine bases. They are dynamic structures which arise from, and can revert back to, G-rich DNA duplexes or single strands. These structures can be found in gene promoter regions of DNA and therefore can be viewed as potential therapeutic targets [251, 252]. For example, stabilization of certain G-quadruplexes by small molecules in promoter regions could influence gene expression or other potentially relevant biological processes. Evidence indicating that quadruplex-containing telomeres cannot be extended by telomerase (and therefore cannot contribute to the immortalization of cancer cells) launched a wide search for G-quadruplex stabilizers which continues today [253-255].
Focusing on Abbott’s A-62176 (55) quinobenoxazine, Hurley’s lab prepared analogs modified by fusion of additional planar rings to the N-1, 8-O linked phenyl group [256, 257]. It was reasoned that due to the wide planar surfaces within G-quadruplexes, the extended aromatic conjugated systems of such quinolones might serve to effectively stack with and stabilize those nucleotide structures. Quinobenoxazine analogs were indeed identified possessing G-quadruplex interactive properties and having residual topo II poisoning activity, i.e. dual mechanism agents. Interestingly two such structures, 172 and 173 (Fig. 17), differing only by the stereochemistry of the pendant amino group displayed mixed mode activity, one having a bias toward topo II poisoning, the other toward G-quadruplex interaction [258]. Further evolution of the series led to 174, named quarfloxin which possessed still greater selectivity for G-quadruplex over duplex DNA and did not have any topo II poisoning effect (Fig. 17) [259, 260]. Interestingly therefore, during the evolution of A-62176 (55) to quarfloxin, a cytotoxic effect was retained, yet the mechanism of action completely changed. This modern illustration of a switch of mechanism within a chemical series with retention of phenotypic biologic effect (i.e. antitumor action) mirrors to some extent the historical chemical evolution of podophyllotoxin, an antitumor tubulin stabilizer, into the epipodophyllotoxin class (etoposide and tenoposide as examples), which are antitumor topo II poisons (Fig. 17, lower panel) [261].
It is believed that quarfloxin’s G-quadruplex interactions do not involve telomerase, but rather trigger a cascade of events leading to silencing of MYC gene expression. According to this view, quarfloxin is concentrated in the cell nucleolus where it binds to ribosomal DNA template G-quadruplexes, displacing nucleolin. Nucleolin in turn migrates to the nucleoplasm and binds to the MYC G-quadruplex inhibiting MYC expression and inducing apoptosis. Cylene Pharma conducted Phase I early Phase II clinical studies of quarfloxin during 2005-2009 in patients with carcinoid/neuroendocrine tumors [262]. In these studies, quarfloxin was described as “well tolerated” with “signs of biological benefit”. However, development was terminated “largely because of tissue distribution problems” [263]. More recently another company, Tetragene, has licensed quarfloxin.
The termination of the development of quarfloxin due to tissue distribution issues strongly suggests that quarfloxin no longer retained the optimal drug-like characteristics of the classic quinolone scaffold. Antibacterial quinolones typically have outstanding tissue distribution properties. In particular, the hydrophobic and highly planar naphthylene group fused to the 1,8-bridge and the 3-amide group (rather than 3-carboxy) in quarfloxin represent extensive structural modifications which may have resulted in sub-optimal physical properties and DMPK characteristics. Significant structural modifications to an otherwise highly optimized scaffold invite equally significant risks of incurring inferior drug-like characteristics including unknown safety profiles. Therefore, a potential lesson is this: if a new therapeutic activity (e.g. antitumor activity) is discovered in the quinolone scaffold, structural modifications should be kept to an absolute minimum consistent with the expression of that new activity. The modifications that were required to evolve the DNA/topo II inhibitor A-62176 (55) toward analogs specifically targeting G-quadruplexes suggest that the G-quadruplex may be a biological target significantly more challenging to “drug” than the topo II DNA target manifold discussed in the review. Beyond quarfloxin, other known G-quadruplex-interacting molecules tend to be large and highly planar, suggesting that physical properties could be an issue for the entire class [264].
RECENT BIOLOGICAL and STRUCTURE-BASED ADVANCES RELEVANT TO MODERN TOPO II INHIBITOR DESIGN
Whereas the existence of two isoforms of eukaryotic topo II had been recognized since the late 1980s, only following cloning and expression of human recombinant topo IIα and IIβ in the mid 1990s could reliable differential measurements of inhibition be achieved for drugs or experimental agents [265, 266]. By about 2010, a hypothesis had been developed stating that selective inhibition of the topo IIα isoform (leaving the IIβ isoform unaffected) would translate into certain therapeutic advantages, in particular enhanced safety, in comparison to the relatively unselective inhibition effected by existing topo II anticancer agents (doxorubicin and etoposide, as examples). Specifically the cardiotoxicity of anthracycline chemotherapy and the incidence of secondary (drug-induced) leukemias caused by most existing topo II drugs had been linked to inhibition of topo IIβ while the desired antiproliferative therapeutic properties had been linked to inhibition of topo IIα [149, 267-272]. This hypothesis, still somewhat controversial[273, 274], prompted several investigators to advocate clinical evaluation of topo IIα selective agents for both safety and efficacy [269, 271, 272]. At least one such topo IIα selective agent, the benzo[c]phenanthridine NK314, had recently undergone early clinical evaluation (NK314 does not have a quinolone structure) [275-277].
During the quinolone anticancer investigations of the 1990s and early 2000s there was not yet a scientific basis for any expectation that new topo II agents should fundamentally improve upon established drugs in terms of reducing cardiotoxicity and/or secondary leukemias. Additionally during that time, there was no consensus yet suggesting that quinolone-based topo II antitumor agents might overcome the issue of efflux-based resistance. In the absence of such incentives, one might reasonably speculate that pharmaceutical companies would be left with few reasons to advocate for quinolone topo II programs especially as pathway-targeted small molecule and biologic drug programs began to take shape simultaneously in the early 1990s (see below). Today those science-based incentives clearly exist.
Over the last 5 or so years, detailed structural information relating to both prokaryotic and eukaryotic Type II topoisomerases in complex with inhibitors has been reported. Crystal structures of ternary (cleavable) complexes of several fluoroquinolones (e.g. moxifloxacin 11) in bacterial topo IV have been solved [92, 278]. Crystal structures of human topo IIα bound to DNA and of human topo IIβ ternary (cleavable) complexes with several small molecule inhibitors including doxorubicin and etoposide have been solved [90, 134-136, 279]. Based on this structural information, insight is now accumulating to allow, for the first time, reasonable structure-based discussions of SAR for quinolones in the context of both prokaryotic and eukaryotic targets. For example, Berger and Osheroff have highlighted the importance to antibacterial activity and resistance development of a number of key quinolone-enzyme interactions, most notably the magnesium-water network which bridges to the quinolone 3-carboxy group (Fig. 5). The binding of the new class of quinazolinediones, a quinolone-like scaffold lacking the “critical” 3-carboxy group, can now be modelled and the degree of potency and the relative lack of bacterial cross-resistance to this scaffold can be rationalized [217, 280]. For quinolone and non-quinolone inhibitors of eukaryotic topo II, similar explanations for the structural basis of binding and SAR for those agents is emerging. Osheroff has begun to rationalize the basis for the eukaryotic potency of the topo II active quinolone CP-115955 (16, Pfizer) and the differences between eukaryotic and prokaryotic structures which influence the degree of selectivity between the two [281]. Considering that many decades of quinolone research has already been performed, and is ongoing to this day, this structural information has the potential to be transformative by permitting rational design for this scaffold for the first time [279, 281-283].
CONCLUSIONS: CRITICAL PERSPECTIVE ON TODAY’S POTENTIAL OF QUINOLONES AS ANTICANCER DRUGS
In 1991 J. Michael Bishop published the review “Molecular Themes in Oncogenesis” which summarized for investigators in the field of anticancer research entirely new opportunities to explore as the basis for drug discovery [284]. In contrast to conventional cytotoxic research and development, Bishop and others set a new vision that argued for therapeutic corrections to the specific biomolecular pathway aberrations arising from the genetic mutations that ultimately lead to cancer. In review articles on cancer therapy during this time, companies such as Merck, Parke Davis, and Pfizer signalled that the pharmaceutical industry was indeed embracing this new vision for cancer therapy [285-288]. Patent applications encompassing what would become imatinib, as well as many other “targeted” therapies, both small molecule and biologic were already being filed in the early 1990s, concurrent with publication of the topo II-based anticancer quinolone studies by Abbott, Sterling, Banyu-Merck and others described in this review. Over the intervening 25 years, dozens of targeted anticancer drugs have been commercialized and have proven to be significant medical successes. Nevertheless, the following recent statement provides a degree of counterbalance:
“We shouldn’t delude ourselves, or our patients, in thinking that standard chemotherapy is a thing of the past”[289].
This statement made in March 2015 by Mikkael L. Sekeres, an oncologist and director of the leukaemia program at the Cleveland Clinic, highlights the reality of today, and likely the foreseeable future, in which relatively non-specific cell division inhibitors--including topo II agents--will still retain a significant role in cancer therapy.
Therefore, a parallel vision for the pharmaceutical industry could be to now also develop significantly improved versions of “standard chemotherapeutic agents”, specifically versions whose use is not limited by off-target effects, susceptibility to efflux-mediated resistance development, and/or poor pharmacokinetics. In light of the discussions presented in this review, a worthy goal would be to replace doxorubicin, etoposide, and their current analogs, by a safer and more effective class of topo II inhibitors [290, 291]. Such a goal is scientifically not unrealistic insofar as strong hypotheses have been recently framed (discussed above) offering explanations for the biological basis underlying the treatment-limiting side effects of both the anthracycline and epipodophyllotoxin classes. Moreover, based on the data analyzed in this review and taking into account the well-known optimized physical and metabolic properties of the quinolone class, the quinolone scaffold possesses a unique capacity to deliver on this goal by not only potentially addressing all the major issues of the conventional topo II classes but also retaining a highly desirable profile in the key parameters of drug-likeness. More specifically, quinolone scaffold topo II antitumor agents could, in principle, incorporate all of the following eleven attributes which, if met, would represent a significant advance in comparison to the current conventional topo II drugs:
reduced (or no) susceptibility to MDR (P-gp-based) resistance development, or cross-resistance to standard topo II agents.
reduced (or no) potential for development of drug-induced secondary cancers.
reduced (or no) cardiotoxicity.
oral and i.v. formulations with highly predictable pharmacokinetics.
The reduced susceptibility of quinolones to efflux-based resistance (including lack of cross-resistance to established topo II inhibitors) in clinically relevant cancer cell lines has already been demonstrated by vosaroxin, by several analogs from Abbott’s quinobenoxazine series, and by Sterling’s 7-pyridyl quinolone. Additional quinolone topo II inhibitors will need to be profiled to assess the wider generality and SAR of this phenomenon within the quinolone class. The potential of the quinolone class to ameliorate drug-induced secondary cancers and cardiotoxicity of conventional topo II agents is based on the current hypothesis that selectively targeting the α topo II isoform could circumvent both of these key issues. Only recently have experimental topo II inhibitors (non-quinolone classes) been shown to possess isoform selectivity [152-154]. Isoform selectivity data for the quinolone class would need to be broadly generated, and resultant SAR exploited to identify and pursue α-isoform selective quinolone agents.
Additional advantages of pursuing quinolone scaffold topo II inhibitors are based on the following well-known qualities of the corresponding antibacterial class:
good class safety (increased risk of tendonitis is one safety issue of the class).
typically excellent physical properties, including good aqueous solubility and low protein binding.
good tissue distribution and good penetration into human cells.
-
good metabolic and clearance profiles allowing once or twice a day dosing.
Another potential advantage of quinolone topo II inhibitors is anticipated to be:
the prospect of achieving greater antitumor potency compared to current topo II drugs.
The potential for excellent antitumor potency in the quinolone class compared to established topo II classes was demonstrated by Pfizer, Banyu, Abbott, and Dainippon insofar as those companies discovered analogs at least equivalent in potency compared to etoposide. It should be emphasized that, in the case of Pfizer and Banyu, these companies did not expend great effort to find potent antitumor quinolones, but rather those compounds merely happened to be selected from a pre-existing sets of antibacterial quinolones. Abbott terminated its anticancer quinolone program after just a few years of focused work in the area and did not have an opportunity to continue optimization of their series while Dainippon ended their in-house effort after a limited optimization program, selecting a single compound (vosaroxin) to out-license. All the quinolone topo II drug discovery projects discussed in this review (Chart 1) were terminated prior to standard sustained and iterative lead optimization efforts. One factor that played a role in at least some of these termination decisions programs was, as alluded to above, the ascendancy of the targeted-based approaches during that time. Therefore, based on historical medicinal chemistry experience, it can be reasonably anticipated that proper lead optimization efforts encompassing those series (or other eukaryotic-active quinolone series) could result in the generation of drug candidates having superior potency compared to both etoposide and doxorubicin. Moreover, the structural biology of topo II is now gaining momentum and could, in principle, have a synergistically positive impact on the design of more potent quinolones.
An additional characteristic of the quinolone class, viewed by the authors as an advantage, is the demonstrated
ability of the quinolone scaffold to be tuned to single or multiple mechanisms within the DNA-topo II manifold, potentially allowing adjustment of mechanism(s) to optimally match different therapeutic needs.
As we have seen, the quinolone scaffold can encompass single mechanisms or blended mechanisms within the DNA-topo II interacting manifold. The Pfizer series, typified by CP-155955 (16), appears to exert cytotoxic potency primarily via the cleavable complex (“topo II poisoning”) mechanism while the Abbott quinobenoxazine series typified, by A-62176 (55), appears to act by a combination of intercalation and topo II catalytic (non-poisoning) inhibition. Thus the quinolone structural framework affords flexibility to optimize to a specific mechanism or blend of mechanisms, any of which might ultimately afford therapeutic advantages in certain tumor settings or in combination with other agents. Vosaroxin, which acts primarily as an intercalator, did not achieve as robust of a response in the AML setting as was hoped. This single example should not necessarily be taken as representative of the anticancer potential for the quinolone class. Would a quinolone having primarily a cleavable complex mechanism been more efficacious in that therapeutic setting? Would greater cellular potency, regardless of mechanism, have resulted in a more robust outcome? As with any class of antitumor agents (e.g. tyrosine kinase inhibitors), a number of iterative cycles need to be examined in order to assess the potential of new class agents in various therapeutic settings. Translational feedback from the clinic to the discovery setting to fine-tune follow-on drug candidates is a historically critical element to eventual clinical success. For the class of quinolone topo II inhibitors, that standard drug development strategy did not have an opportunity to play out. Moreover there are several recently proposed scientific strategies which might be utilized in conjunction with new class topo II inhibitors to further enhance their safety and/or efficacy. For example: 1) chemical hybridization methods exist which allow cytotoxics to more specifically target tumor cells; 2) novel agents have been reported that induce mitosis in otherwise quiescent cells within solid tumors, potentially sensitizing those otherwise resistant cells to topo II inhibitors; 3) novel DNA repair inhibitors have been described which potentiate topo II poisons; 4) modern kinase inhibitor agents have has been reported to be synergistic with topo II agents [290, 292-294].
Finally, a key but highly underappreciated advantage of the quinolone class is the
ease of synthesis of new analogs.
Historically, ease of synthesis of diverse analogs of any class of drugs provides that class with a strategic advantage by facilitating rapid cycle time in troubleshooting any biological, physical property, or toxicological issue by interrogating successive waves of new analogs. One of the reasons the quinolone class has been so successful both medically and commercially over the decades is the simple fact that many analogs, diversified simultaneously at multiple scaffold positions, could be easily synthesized, allowing stepwise improvements in essentially all critical drug-related categories (potency, antimicrobial spectrum, evolving resistance issues, pharmacokinetic profile, etc.).
OTHER POTENTIAL ANTICANCER MECANISMS OF CLASSICAL QUINOLONES
The classical 3-carboxy quinolone scaffold has also demonstrated interactions with other potential anti-cancer targets and pathways beyond topo II and the G quadruplex. Although many of these reports are preliminary, the list of targets and pathways include PI3K, CK2, MAPK, migration/invasion pathways, and novel cell cycle arrest pathways [295-302].
The authors hope that this review has provided a balanced and insightful overview of the potential of the quinolone scaffold to be developed into a new class of anticancer topo II inhibitors having significant advantages in the clinic over currently employed topo II classes.
Fig. (1).

Dihydrofolate reductase (DHFR) inhibitors aminopterin and methotrexate (1) and trimethoprim (2) developed for anticancer and antibacterial therapy, respectively. Aminopterin and methotrexate also possess DHFR-based antibacterial activity, while trimethoprim is selective for bacterial DHFR.
Fig. (4).
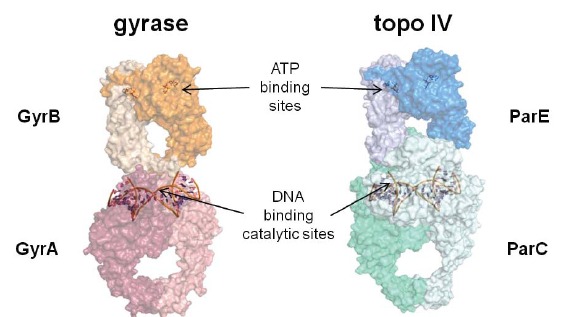
Models for bacterial Type II topoisomerase tetramers: DNA gyrase (left) and topoisomerase IV (right). The arrows show the relative locations of the ATP binding sites in GyrB and ParE and the DNA cleavage/ligation catalytic sites in GyrA and ParC which are shown binding the so-called “gateway” segment of DNA. Each monomer within the two tetramers is defined by a different color or shade of color. The models were constructed with S. pneumoniae ParC (PDB code 4I3H)[91]. E. coli ParE (1S16)[100], C. psychrerythraea 34H GyrA (3LPX), and E. coli GyrB (1EI1)[101] using the X-ray crystal structure of the complete tetramer from S. cerevisiae (4GFH)[102] as a template.
Fig. (5).
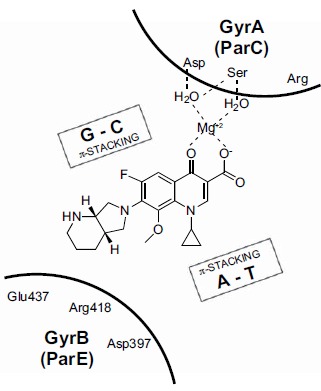
Schematic representation of antibacterial fluoroquinolone moxifloxacin 12 as part of the ternary complex (“cleavable complex”) with bacterial gyrase (GyrA/GyrB) or topo IV (ParC/ParE) and DNA. Moxifloxacin and other fluoroquinolones make interactions with both enzyme subunits, as well as p-stacking interactions with the DNA base pairs. A strand break between two base pairs, mediated by a topoisomerase catalytic tyrosine, is not shown.
Fig. (6).
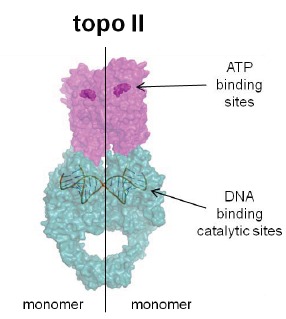
Model for the eukaryotic Type II topoisomerase, topo II, in covalent (“cleavable”) complex with DNA. This representation is a composite of the two PDB entries 1ZXN (N terminal human topo IIα with ATP analog ADPNP in the ATP binding site) from Wei et al. [133] and 3QX3 (C-terminal catalytic domain of human topo IIβ with bound and cleaved DNA and bound etoposide) from Wu et al. [134]. The pink represents the ATP binding domains, analogous to bacterial GyrB (or ParE) and the green represents the DNA binding domains, analogous to bacterial GyrA (or ParC). The vertical line indicates the enzyme dimeric composition as two symmetrical monomers (right and left).
Fig. (7).
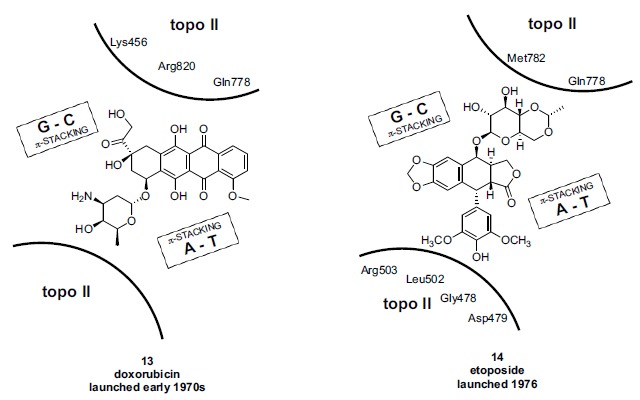
Schematic representation of anticancer agents doxorubicin 13 (left panel) and etoposide 14 (right panel) bound as the ternary complex (“cleavable complex”) with eukaryotic topo II and DNA. The binding modes of 13 and 14 are broadly similar to that of moxifloxacin 12 in the prokaryotic ternary complex (compare Fig. 5). Renderings adapted from crystal structure information by Wu et al. and Chan et al. [135, 136] employing topo IIβ. Topo II amino acids which form known interactions with the drugs based on crystallography are shown in each of the two panels, although the specific interactions are not depicted. A strand break between two base pairs, mediated by a topoisomerase catalytic tyrosine, is not shown.
Fig. (8).
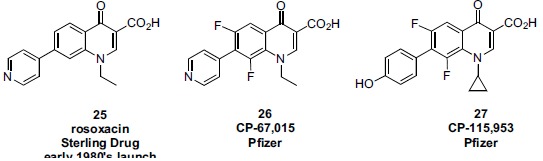
Structures of Pfizer’s experimental quinolones CP-67015 (26) and CP-115,953 (27) compared to Sterling’s earlier launched drug rosoxacin 25. The cytotoxicity displayed by 27 is equivalent to the marketed anticancer drug etoposide 14.
Fig. (9).
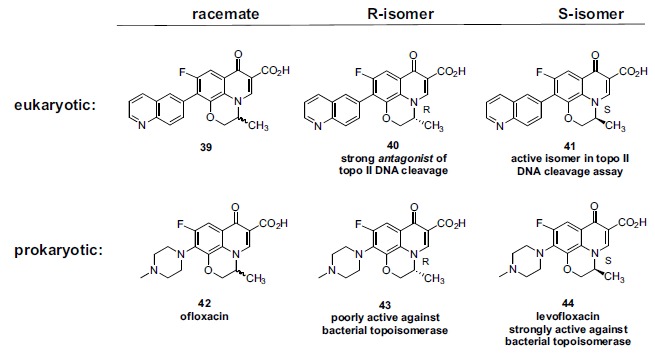
Pfizer C-7 quinolone substituted 1,8-bridged analogs; racemates and enantiomers.
Fig. (10).
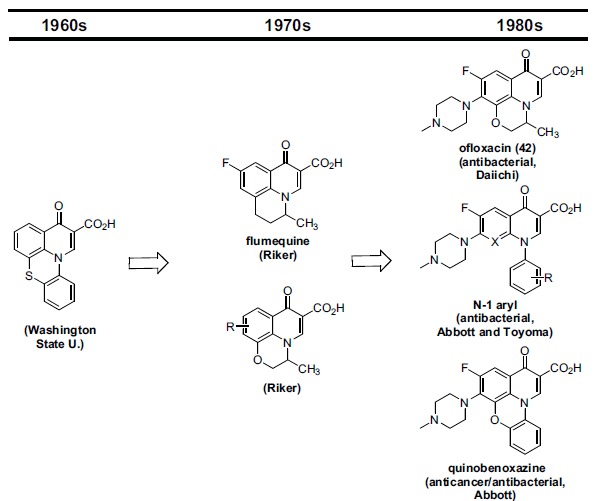
Evolution of early antibacterial 1,8-bridged quinolone scaffolds to ofloxacin (42) and the antibacterial N-1phenyl and eukaryotic/prokaryotic cell active quinobenoxazine variants of Abbott.
Fig. (11).
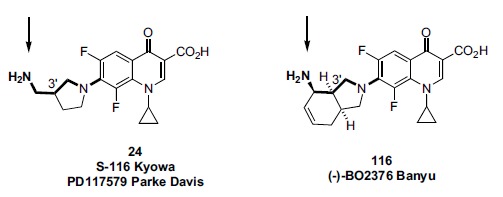
Similarity of position in space of the primary amino groups of S-116/PD117579 (24) and (-)-BO2376 (116). The common bonds for placement of the primary amino group at C-7 for the two compounds are shown in bold.
Fig. (12).
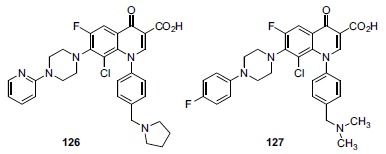
Representative quinolone structures from Bayer patent applications reporting antiproliferative/anticancer activities.
Fig. (13).
Comparison of the structures of N-thiazole quinolone analogs from Parke Davis and Dainippon.
Fig. (15).
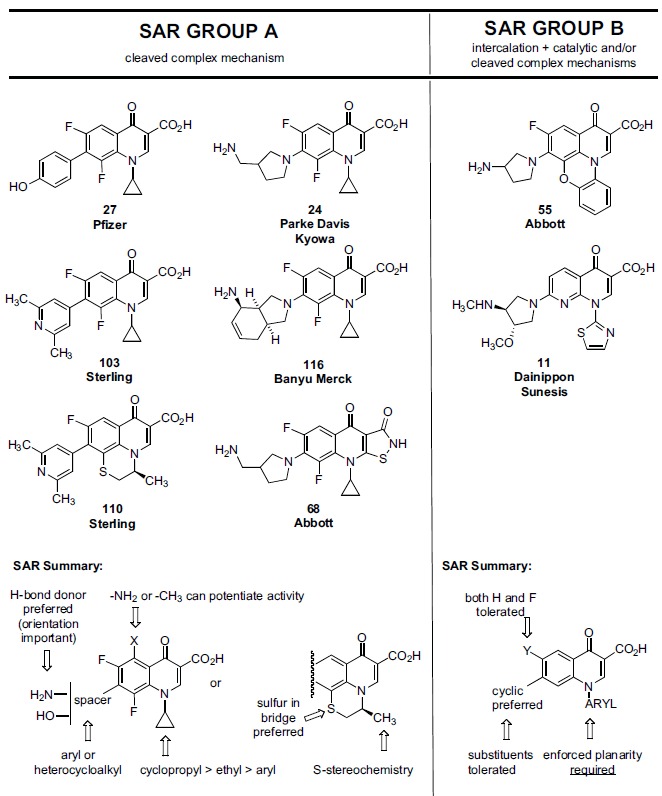
Eukaryotic-active quinolone scaffolds discussed in this review, grouped by overall mechanism and associated SAR (Group A and B). SAR summaries for each group is shown at the bottom of the Figure.
Fig. (17).
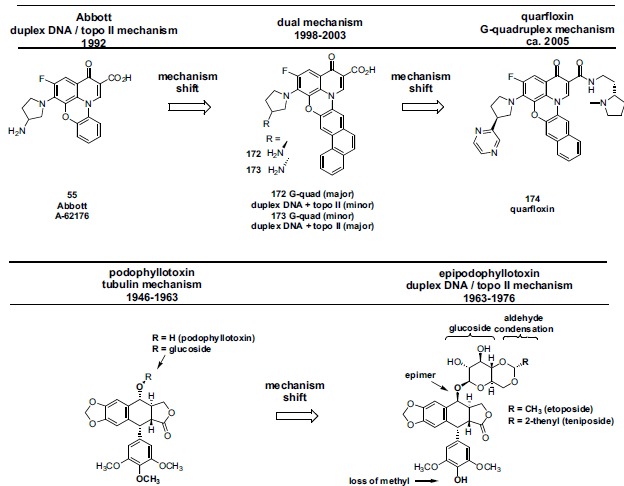
Evolution of Abbott quinobenoxazine 55 through 172 and 173 which partially display G-quaduplex activity in addition to DNA/topo II activity, to quarfloxin 174 having pure G-quadruplex activity (upper panel). An earlier example of a scaffold evolution of an anticancer agent resulting in a different mechanism of action is the transformation of podophyllotoxin (tubulin mechanism) to the epipodophyllotoxin class having a DNA/topo II-based mechanism (lower panel).
Table 1. Parke Davis eukaryotic cytotoxicity SAR at quinolone N-1, C-5, C-7 and C-8. All the analogs in the table were potent antibacterials (data not shown).
| Compound | R7 | X | R5 | R1 |
CH V79 cells a
CC50 (μg/ml)a |
|---|---|---|---|---|---|
|
8 norfloxacin |
CH | H | ≥500 | ||
|
15 ciprofloxacin |
CH | H | 330 | ||
|
16 8-F- ciprofloxacin |
CF | H | 47 | ||
| 17 | CH | H | 280 | ||
| 18 | CH | H | 92 | ||
| 19 | CF | H | 30 | ||
| 20 | CF | -NH2 | <7.8 | ||
| 21 | N | -CH3 | 13 | ||
| 22 | N | H | 98 | ||
|
9 tosufloxacin |
N | H | 120 | ||
|
23 CI-934 |
CF | H | 190 | ||
|
24 PD117579 |
CF | H | <7.8 |
aChinese hamster V79 cells
Table 2. Pfizer eukaryotic SAR at quinolone N-1, C-7, and C-8. CP-115,953 (27) is the most potent analog and displays a similar level of cytotoxicity compared to etoposide (14). Cytotoxicity correlated better with stimulation of DNA cleavage (generation of cell-toxic fragments) than with inhibition of DNA relaxation (measure of topo II catalytic activity).
| Compound | R7 | R8 | R1 |
Topo II DNA relaxation
IC50 (μg/ml)a,b |
Topo II DNA cleavage
EC2 (μg/ml)a,b, c |
CHO cellsd
CC50 (μg/ml)a |
|---|---|---|---|---|---|---|
|
14 etoposide |
- | - | - | * | 10 | 5 |
|
27 CP-115,953 |
F | 1.2 | 0.1 | 3 | ||
| 28 | F | 73 | 7 | 30 | ||
| 29 | F | >185 | >185 | 36 | ||
| 30 | F | 50 | 19 | 37 | ||
| 31 | F | 67 | 36 | >170 | ||
|
16 8-F- ciprofloxacin |
F | 46 | 22 | 82 | ||
| 32 | H | 11 | 1 | * | ||
| 33 | H | 120 | >175 | * | ||
|
15 ciprofloxacin |
H | 35 | 56 | >165 | ||
| 34 | F | 39 | 4 | * | ||
| 35 | F | >165 | >165 | * | ||
|
8 norfloxacin |
H | >165 | 162 | * |
aValues originally reported as μM were converted to μg/ml for this table; bDNA topoisomerase II purified from calf thymus; cEC2 is defined as the effective concentration of drug required to enhance double-stranded DNA cleavage twofold; dChinese hamster ovary cells; *no data reported.
Table 3. Garenoxacin (36) and two analogs differently substituted at C-6 and C-8. Ciprofloxacin 15 was used as control. Within this panel, 37 is most potent analog against eukaryotic topo II and against human cells.
| Compound | E. coli gyrase IC50 (μg/ml) |
S. aureus topo IV
IC50 (μg/ml) |
topo II DNA relax.
IC50 (μg/ml) |
HEp-2
CC50 (μg/ml) |
HeLa
CC50 (μg/ml) |
|
|---|---|---|---|---|---|---|
|
36 garenoxacin |
0.17 | 2.19 | 509.7 | >166 | >166 | |
| 37 | 0.16 | 4.6 | 128.1 | 41.1 | 203 | |
| 38 | 0.19 | 2.04 | 89.2 | 8.2 | 10.0 | |
|
15 ciprofloxacin |
0.18 | 2.34 | 873 | 115 | 94.2 | |
Table 4. Daiichi prokaryotic and eukaryotic SAR for ofloxacin isomers and related analogs.
| Compound | R |
gyrase
IC50 (μg/ml) |
E. coli
KL-16 MIC (μg/ml) |
topo II
DNA relaxation IC50 (μg/ml) |
CFU-GM cellsa
CC50 (μg/ml) |
|---|---|---|---|---|---|
|
42 ofloxacin |
0.76 | 0.05 | 1,870 | 103 | |
|
43 (R-isomer) |
4.7 | 0.78 | 2,550 | 289 | |
|
44 levofloxacin (S-isomer) |
0.38 | 0.025 | 1,380 | 138 | |
| 45 | 3.1 | 0.10 | 178 | 43 | |
| 46 | 0.70 | 0.05 | 64 | 22 |
aMurine granulocyte macrophage progenitor cells.
Table 5. Pfizer prokaryotic and eukaryotic biochemical SAR for quinolone C-5 and C-7 substitutions in an N-1-cyclopropyl, 6,8-difluoro quinolone scaffold. Etoposide 14 was used as control.
| Compound | R5 | R7 |
E. coli
gyrase cleavage endpoint (μg/ml)a,b |
Topo II DNA cleavage
IC50 (μg/ml)a,c |
|---|---|---|---|---|
|
47 sparfloxacin |
-NH2 | 0.18 | >780 | |
| 48 | -F | 0.19 | >800 | |
| 49 | -H | 0.39 | >760 | |
| 50 | -NH2 | 0.19 | 257 | |
| 51 | -NH2 | * | 62 | |
| 52 | -H | 0.19 | 55 | |
| 53 | -NH2 | 0.19 | 29 | |
| 54 | -NH2 | 0.19 | 30 | |
|
14 etoposide |
- | - | * | 8.3 |
aValues originally reported as μM were converted to μg/ml for this table; blowest drug concentration that stimulates cleavage above that for the drug-free control; cCC50 is the concentration of drug that produces half-maximal stimulation of cleavage; *no data reported.
Table 6. Abbott quinobenoxazine series SAR. Significantly, a much lower susceptibility to MDR efflux was displayed by quinolones in this series compared to doxorubicin (wild type P388 vs P388 (ADR) lines).
| Compound | R5 | R7 | a/b |
Min. DNA un-winding conc.a
(μg/mL) |
Decatenation inhib.b
(μg/mL) |
A549c
CC50 (μg/mL) |
P388d
CC50 (μg/mL) |
P388/
ADRe (MDR) CC50 (μg/mL) |
|---|---|---|---|---|---|---|---|---|
|
doxof 13 |
- | - | - | * | * | 0.31 | 0.0015 | 2.59 |
| 55g | H | A | H/H | 0.5 | 0.45 | 0.26 | 0.019 | 0.12 |
| 55(R)h | H | A | H/H | * | * | 0.42 | 0.021 | 0.10 |
| 55(S)i | H | A | H/H | * | * | 0.23 | 0.017 | 0.12 |
| Compound | R5 | R7 | a/b |
Min. DNA un-winding conc.a
(μg/mL) |
Decatanation inhib.b
(μg/mL) |
A549c
CC50 (μg/mL) |
P388d
CC50 (μg/mL) |
P388/
ADRe (MDR) CC50 (μg/mL) |
|---|---|---|---|---|---|---|---|---|
| 56j | H | B | H/H | 3 | 2 | 0.59 | 0.04 | 0.39 |
| 57 | H | C | H/H | 128 | >100 | 2 | 0.5 | * |
| 58 | H | A | H/Me | * | * | 0.12 | 0.038 | 0.24 |
| 59 | H | A | Me/Me | * | * | 0.034 | 0.012 | * |
| 60 | H | A | H/OMe | * | * | 0.29 | 0.05 | * |
| 61 | H | A | H/Ph | * | * | 0.16 | 0.07 | * |
| 62 | NH2 | A | H/H | * | * | 0.069 | 0.0053 | * |
| 63 | NH2 | B | H/H | * | * | 0.10 | 0.0047 | * |
| 64 | NHMe | A | H/H | * | * | 0.39 | 0.039 | * |
| 65 | NH2 | D | H/H | * | * | 0.06 | 0.030 | * |
| 66 | NHMe | D | H/H | * | * | 1.1 | 0.79 | * |
| 67 | NMe2 | D | H/H | * | * | 6.4 | 3.9 | * |
a Measured at 10mM MgCl2; b drug concentration that inhibits 50% of the conversion of catenated to decatenated k-DNA catalyzed by calf thymus topoisomerase II; c human breast cancer cell line; d murine leukemia cell line; e adriamycin resistant/multidrug resistant; f doxorubicin (adriamycin); g 55 = A-62176 (racemate); h R-enantiomer of 55; i S-enantiomer of 55; j 56 = A-74932 (diasteromeric mixture); *data not reported.
Table 7. In vivo activity of Abbott quinobenoxazine 56 against systemic murine tumors.
| Tumor type | Treatment route and schedule | Best % TWIa | Best % ILSb | Cures (%) | Dose (mg/kg) |
|---|---|---|---|---|---|
| P388 | i.p. BID D1-9 | * | 85 | 10 | 10.0 |
| P388/ADR (MDR) | i.p. BID D1-9 | * | 50 | * | 10.0 |
| M5076 | i.p. BID D1-9 | 68 | 95 | 30 | 10.0 |
| i.v. Q4Dx3 | 99 | * | * | 40.0 | |
| C26 | i.p. QD D1-9 | 62 | 92 | 40 | 10.0 |
| i.v. Q4Dx3 | 95 | * | * | 40.0 | |
| B16 | i.p. BID D1-9 | 67 | * | * | 5.0 |
| i.v. Q4Dx3 | * | 13 | 19.6 | * | |
| Lewis lung | i.p. QD D1-9 | 65 | * | * | 2.5 |
| i.v. Q4Dx3 | 59 | 31 | * | 40.0 |
aTumor weight inhibition; bincreased life span; *data not reported.
Table 8. In vivo activity of Abbott quinobenoxazine 56 against human tumor xenographs.
| Tumor | Treatment route and schedule | Dose (mg/kg) | Best % TWIa | Cures (%) |
|---|---|---|---|---|
| CX-1 colon |
i.p. QD D2-11 | 20 | 79 | * |
| MBA 9812 mammary | i.p. QD D2-11 | 20 | 76 | * |
| LX-1 lung |
i.p. QD D2-11 | 20 | 43 | * |
| MX-1 mammary | i.v. Q4Dx3 | 40 | 84 | 30 |
| HT29 colon |
i.v. Q4Dx3 | 40 | 21 | * |
aTumor weight inhibition; *data not reported.
Table 9. Abbott isothiazoloquinolone SAR for topo II catalytic inhibition (unknotting), topo II mediated DNA cleavage, and cytotoxicity in two cell lines. Etoposide serves as a reference compound.
| Compound | R7 | R8 | R1 |
topo II DNA unknotting IC50
(µg/ml) |
topo II DNA cleavage |
P388D1a
CC50 (µg/ml) |
A549b
CC50 (µg/ml) |
|---|---|---|---|---|---|---|---|
|
14 etopoc |
- | - | - | * | * | 0.05 | 1.0 |
| 68 | F | * | strong | <0.05 | 0.3 | ||
| 69 | F | * | strong | 3.1 | 8.3 | ||
| 70 | F | * | strong | 0.23 | 2.9 | ||
|
71 A-65282 |
F | 8 | strong | * | * | ||
| 72 | F | 8 | strong | * | * | ||
| 73 | F | 25-50 | weak | * | * | ||
| 74 | H | 16-25 | none | * | * | ||
|
15 ciprod |
H | >50 | none | * | * | ||
|
8 norfloxe |
H | >50 | none | * | * | ||
a Leukaemia cell line; bhuman breast cancer cell line; Ciprofloxacin 15, moxifloxacin 12, and gemifloxacin 102 serve as reference compounds; *data not reported.
Table 10. Achillion isothiazoloquinolone SAR for MRSA (MIC) and cytotoxicity in Hep2 cells.
| Compound | R7 |
MRSAa
MIC (µg/ml) |
Hep2b
CC50 (µg/ml)c |
Compound | R7 |
MRSAa
MIC (µg/ml) |
Hep2b
CC50 (µg/ml)c |
|---|---|---|---|---|---|---|---|
| 75 | 16 | >35 | 83 | 8.0 | 2 | ||
| 76 | 0.125 | 3 | 84 | 32 | >38 | ||
| 77 | 0.25 | >37 | 85 | >64 | 4 | ||
| 78 | 0.125 | 2 | 86 | 0.25 | 19 | ||
| 79 | 4.0 | 10 | 87 | 4.0 | 3 | ||
| 80 | 2.0 | 35 | 88 | 1.0 | >40 | ||
| 81 | 0.5 | 13 | 89 | 2.0 | >35 | ||
| 82 | 4.0 | 20 | 90 | 1.0 | 4 |
aMethicillin resistant S. aureus (methicillin- and quinolone-resistant, vancomycin intermediate-resistant; bHep2 = human laryngeal carcinoma cell line; CC50 = concentration of drug lethal to 50% of cells; 72h incubation with drug; cvalues originally reported as μM were converted to μg/ml for this table.
Table 11. Achillion isothiazoloquinolone SAR for MRSA (MIC) and cytotoxicity in Hep2 cells (CC50). Ciprofloxacin 15, moxifloxacin, and gemifloxacin 102 serve as reference compounds.
| Compound | R6 | R7 | X |
MRSAa
MIC (µg/ml) |
Hep2 cellsb
CC50 (µg/ml)c [topo II EC2 (µg/ml)]c,d |
|---|---|---|---|---|---|
| 91 | F | CH | 2 | 3 | |
| 92 | F | COCH3 | 0.5 | 8 [32] | |
| 93 | H | COCH3 | 2 | 40 | |
| 94 | F | CH | 1 | 1 | |
| 95 | F | COCH3 | 0.5 | 6 [15] | |
| 96 | H | COCH3 | 2 | 19 | |
| 97 | F | COCH3 | 2 | 10 | |
| 98 | F | COCH3 | 0.25 | 4 | |
| 99 | F | COCH3 | 4 | >43 | |
|
100 ACH702 |
F | COCH3 | 0.06 | 4 | |
| 101 | F | COCH3 | 2 | 28 | |
|
15 ciprofloxacin |
F | CH | 32 | >33 [>50] | |
|
12 moxifloxacin |
F | COCH3 | 2 | >40 [>60] |
| Compound | R6 | R7 | X |
MRSAa
MIC (µg/ml) |
Hep2 cellsb
CC50 (µg/ml)c [topo II EC2 (µg/ml)]c,d |
|---|---|---|---|---|---|
|
102 gemifloxacin |
F | N | 2 | 18 [>59] | |
aMethicillin resistant S. aureus (methicillin- and quinolone-resistant, vancomycin intermediate-resistant); bHep2 = human laryngeal carcinoma cell line; CC50 = concentration of drug lethal to 50% of cells; 72h incubation with drug; cvalues originally reported as μM were converted to μg/ml for this table; dEC2 = concentration of drug required to enhance enzyme-mediated cleavage of double-stranded DNA twofold.
Table 12. Sterling biochemical SAR of N-1, C-5 and C-7 substituted 6,8-difluoro analogs.
| Compound | R5 | R7 | R1 |
topo II DNA cleavage
EC50 (µg/ml)a,b |
|---|---|---|---|---|
|
103 WIN 57294 |
H | 2.8 | ||
| 104 | H | -CH2CH3 | 40 | |
| 105 | H | >181 | ||
| 106 | -NH2 | 2.4 |
| Compound | R5 | R7 | R1 |
topo II DNA cleavage
EC50 (µg/ml)a,b |
|---|---|---|---|---|
| 107 | H | 5.8 | ||
| 108 | H | 54 | ||
|
19 Parke Davisc |
H | 119 | ||
|
27 Pfizer CP-115953d |
H | 0.43 | ||
|
14 etoposide |
- | - | - | 0.48 |
aMeasurement of HeLa cell topo II covalently complexed; the EC50 is the concentration of compound with activity equal to 50% of the activity observed with the nearly saturating dose of the reference agent m-AMSA (EC50 = 0.72); bValues originally reported as μM were converted to μg/ml for this table; cParke Davis data in Table 1; dPfizer data in Table 2.
Table 13. Sterling biochemical SAR for 1,8 bridged analogs.
| Compound | R5 | R6 | R7 | X | R’ | R |
Topo II DNA
cleavage EC50 (µg/ml)a, b |
|---|---|---|---|---|---|---|---|
| 109 | H | F | O | -CH3 | H | 11 | |
|
110 WIN 58161 |
H | F | S | -CH3 | H | 3.6 | |
| 111 | H | F | O | H | -CH3 | >205 | |
| 112 | F | F | O | -CH3 | H | 20 |
| Compound | R5 | R6 | R7 | X | R’ | R |
Topo II DNA
cleavage EC50 (µg/ml)a, b |
|---|---|---|---|---|---|---|---|
| 113 | H | H | O | -CH3 | H | >102 | |
| 114 | H | F | O | -CH3 | H | 19 | |
|
42 ofloxc |
H | F | O | mono -CH3 racemate |
>101 | ||
|
14 etopod |
- | - | - | - | - | - | 0.48 |
aDefinition see Table 12; bValues originally reported as μM were converted to μg/ml for this table; cofloxacin; detoposide.
Table 14. Sterling in vivo antitumor data for analog 110 (WIN-58161).
| Tumor | Drug route/schedule | Max. tolerated total dosea (mg/kg) | %T/Cb | %ILSc |
|---|---|---|---|---|
| Panc03 | s.c., qd 3-9 | 781 | 17 | |
| Colo38 | s.c., qd 3-9 | 504 | 0 | |
| Mam16C | s.c., qd 1-4 | 263 | 15 | |
| B16 | i.p., qd 1,5,9 | 1500 | 66 | |
| P388 | i.p., qd 1,5,9 | 1014 | 90 |
aMaximum non-lethal dose; btumor growth inhibition where T and C are median tumor weights of the treatment and control groups respectively; cpercent increased life span.
Table 15. Sterling cell-based resistance study: lack of cross-resistance in two cell lines (VLB100 and VM-1) for analog 110 (WIN-58161).
| IC50 (µg/ml)a | |||
|---|---|---|---|
| Drug | CEM | VLB100b | VM-1c |
| 110 WIN58161 | 19 | 16 | 15 |
| 14 etoposide | 3.0 | 18 | 17 |
| m-AMSAd | 0.26 | 2.1 | 2.0 |
| vinblastine | 2.8 | 150 | 2.5 |
aValues originally reported as μM were converted to μg/ml for this table; bP-gp MDR cell line derived from the CEM human leukemic lymphoblast cell line, originally derived as resistant to vinblastine but also resistant to common topo II inhibitors; ctenoposide resistant cell line derived from CEM in which drug resistance to topo II inhibitors is a consequence of alteration in topo II; damsacrine (4'-[9-acridinylamino]-methanesulfon-m-anisidide).
Table 16. Banyu eukaryotic biochemical and cellular SAR at quinolone N-1, C-5, C-7 and C-8 positions. Etoposide 14 was used as control.
| Compound | R7 | R8 | R5 | R1 |
Induction of DNA cleavable complex
(T/C)a |
L1210 cells CC50
(μg/ml) |
|---|---|---|---|---|---|---|
| 115 | F | -NH2 | 17.3 | 0.006 | ||
|
116 (-)-BO2367 |
F | H | 15.3 | 0.012 | ||
| 117 | -OCH3 | H | 14.3 | 0.026 | ||
| 118 | F | H | 10.0 | 0.17 | ||
| 119 | F | H | 8.6 | 0.30 | ||
| 120 | H | H | 6.7 | 0.50 | ||
| 121 | F | H | 6.7 | 1.1 | ||
| 122 | H | H | 1.8 | 1.7 | ||
| 123 | F | H | 1.5 | 4.0 | ||
|
16 8-F-ciprofloxacin |
F | H | 1.4 | 30 |
| Compound | R7 | R8 | R5 | R1 |
Induction of DNA cleavable complex
(T/C)a |
L1210 cells CC50
(μg/ml) |
|---|---|---|---|---|---|---|
| 124 | F | H | 1.0 | >50 | ||
|
14 etoposide |
- | - | - | - | 7.9 | 0.017 |
aRatio increase of complex formation, drug treated compared to control
Table 17. Banyu eukaryotic and prokaryotic biochemical and cellular data for enantiomers 116 and 125 and in vivo antitumor activities in 3 murine models. Etoposide and ciprofloxacin were used as controls.
| In vitro or in vivo assay |
14
etopo-side |
15
cipro-floxacin |
||
|---|---|---|---|---|
| E. coli gyrase DNA supercoiling IC50 (μg/ml)a | 0.2 | 1.0 | * | 1.0 |
| L1210 topo II DNA relaxation IC50 (μg/ml)a | 1.5 | 24 | 4.4 | > 50 |
| L1210 topo II induction of DNA cleavage IC50 (μg/ml)a |
1.2 | > 12 | ½ as active vs of (-)-BO | * |
| L1210 topo II induction of DNA cleavable complex formation in intact cells IC50 (μg/ml)a | 3-17 fold increase vs control over range 0.10 to 28 μg/ml |
minimal complex formation at 9 μg/ml | ½ as active vs of (-)-BO | * |
| P388 cytotoxicity CC50 (μg/ml)a |
0.004 | 0.50 | 0.004 | * |
| L1210 cytotoxicity CC50 (μg/ml)a |
0.017 | 1.7 | 0.020 | * |
| Antitumor activity i.p.-implanted murine leukaemia P388 0.313mg/kg/day i.p. | ||||
| Survival days mean: | 16.4b | * | 22.2b | * |
| T/C%c | 146 | * | 198 | * |
| Antitumor activity on i.p.-implanted murine leukaemia L1210 0.313mg/kg/day i.p. | ||||
| Survival days mean: | 21.2d | * | 31.4d | * |
| T/C%: | 155 | * | 229 | * |
| Antitumor activity on s.c.-implanted colon 26 solid tumor 1.25 mg/kg/day s.c. | ||||
| Tumor weight mean (g): | 0.00e | * | 1.28e | * |
| % inhibition | 100 | * | 63 | * |
aValues originally reported as μM were converted to μg/ml for this table; bcontrol = 11.2 days; cRatio (%) of survival time of drug treated animals to control; dcontrol = 13.7 days; econtrol tumor weight mean = 3.46g.
Table 18. Dainippon SAR at N-1: eukaryotic cytotoxicity (P388 cell line).
| R1 | ||||
|---|---|---|---|---|
|
P388 cells
CC50 (μg/ml)a | ||||
| Compound # | ||||
| 0.02 | 0.26 | 0.09 | 0.05 | 0.05 |
| 130 | 131 | 132 | 133 | 134 |
| 5.5 | 1.2 | 9.4 | 0.58 | >10 |
| 135 | 136 | 137 | 138 | 139 |
| 2.4 | 2.1 | >10 | >10 | 7.9 |
| 140 | 141 | 142 | 143 | 144 |
| Etoposide (14) = 0.01 (μg/ml) | ||||
| Doxorubicin (13) = 0.004 (μg/ml) | ||||
aMurine lymphocytic leukemia cell line
Table 19. Dainippon SAR at the quinolone 6- and 8-positions suggesting that co-planarity of the 2-thiazole ring is a determent of potency for this scaffold.
| Compound | R6 | R8 |
P388 cells
CC50 (μg/ml)a |
|---|---|---|---|
| 130 | CF | N | 0.02 |
| 145 | N | N | 0.05 |
| 146 | CF | CH | >10 |
| 147 | CF | CF | 55 |
aMurine lymphocytic leukemia cell line
Table 20. Dainippon cellular SAR showing the effect of electron donating and withdrawing substituents at the quinolone C-5 and C-6.
| Compound | R6 | P388 cells CC50 (μg/ml)a | Compound | R5 | P388 cells CC50 (μg/ml)a |
|---|---|---|---|---|---|
| 148 | -H | 0.01 | 148 | -H | 0.01 |
| 130 | -F | 0.02 | - | - | - |
| 149 | -Cl | 0.23 | 153 | -Cl | 0.05 |
| - | - | - | 154 | -CF3 | 0.10 |
| 150 | -NO2 | 2.29 | - | - | - |
| 151 | -NH2 | 1.01 | 155 | -NH2 | 0.01 |
| 152 | -OH | 4.31 | - | - | - |
aMurine lymphocytic leukemia cell line
Table 21. Dainippon SAR showing the effect of variation of the quinolone C-7 substituent on eukaryotic cytotoxicity.
| R7 | |||
|---|---|---|---|
|
P388 cells
CC50 (μg/ml)a | |||
| Compound # | |||
| 0.01 | 0.07 | 0.008 | 0.02 |
| 148 | 156 | vosaroxin; 11 | 157 |
| 0.06 | 0.08 | 0.02 | >10 |
| 158 | 159 | 160 | 161 |
| >10 | >1 | 6.33 | 7.37 |
| 162 | 163 | 164 | 165 |
| Etoposide (14) = 0.01 (μg/ml) | |||
| Doxorubicin (13) = 0.004 (μg/ml) | |||
aMurine lymphocytic leukemia cell line
Table 22. Dainippon SAR for the C-3 position of N-1 2-thiazole quinolones: biochemical prokaryotic and eukaryotic IC50 values and cytotoxicity in a P388 murine cell line.
| Scaffold | Compound | R3 |
Antibacterial activity
MIC (μg/ml) |
Cytotoxicity
CC50 (μg/ml)a |
|
|---|---|---|---|---|---|
|
E. coli
NIHJ JC-2 |
S. aureus
209 JC-1 |
murine P388b | |||
| 130 | -CO2H | 0.025 | 0.39 | 0.02 | |
| 169 | -H | 12.5 | 12.5 | 0.18 | |
| 170 | -CONH2 | >100 | >100 | 0.27 | |
| 11 | -CO2H vosaroxin |
1.56 | 3.13 | 0.01 | |
| 171 | -CH2OH | >100 | 25 | 0.06 | |
a Concentration that reduces cell viability by 50%; bmurine lymphocytic leukaemia cell line.
AcknowledgEments
Declared none.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Ehrlich P. Address Delivered at the Dedication of the Georg-Speyer-Haus. In: Himmelweit F., editor. The Collected Papers of Paul Ehrlich. Vol. III. UK: Pergamon Press, Ltd.; 1960. pp. 53–63. [Google Scholar]
- 2.Drews J. Paul Ehrlich: magister mundi. Nat. Rev. Drug Discov. 2004;3:797–801. doi: 10.1038/nrd1498. [DOI] [PubMed] [Google Scholar]
- 3.Weber C.M. Der Freischütz.
- 4.Grundmann K. Emil von Behring: the founder of serum therapy.
- 5.Strohl W.R. Therapeutic Monoclonal Antibodies: Past, Present, and Future. In: Zhiqiang A., editor. Therapeutic Monoclonal Antibodies. Hoboken: John Wiley & Sons, Inc.; 2009. pp. 3–50. [Google Scholar]
- 6.Casadevall A. Passive antibody administration (immediate immunity) as a specific defense against biological weapons. Emerg. Infect. Dis. 2002;8:833–841. doi: 10.3201/eid0808.010516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casadevall A., Scharff M.D. Return to the past: the case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 1995;21:150–161. doi: 10.1093/clinids/21.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casadevall A. Antibody-based therapies for emerging infectious diseases. Emerg. Infect. Dis. 1996;2:200–208. doi: 10.3201/eid0203.960306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strohl W.R., Strohl L.M. Multiple Antibody and Multi-specificity Approaches. In: Strohl W.R., Strohl L.M., editors. Therapeutic Antibody Engineering. Cambridge: Woodhead Publishing Limited; 2012. pp. 299–328. [Google Scholar]
- 10.Wadia M.D. Death after salvarsan. BMJ. 1917;1(2923):13–14. doi: 10.1136/bmj.1.2923.13-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmitt A. Cases of death due to salvarsan. Their causes with regard to injuries due to salvarsan. Munch. Med. Wochenschr. 1914;61:1337–1340. [Google Scholar]
- 12.Sharkey R.M., Goldenberg D.M. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J. Clin. 2006;56:226–243. doi: 10.3322/canjclin.56.4.226. [DOI] [PubMed] [Google Scholar]
- 13.Hericourt J., Richet C.H. Physologie pathologique - de la serotherapie dan la traitement du cancer. Comptes Rendus Hebd. Seanc. Acad. Sci. (Paris) 1895;120:567–569. [Google Scholar]
- 14.Goldin A., Schepartz S.A., Venditti J.M., Devita V.T. Historical Development and Current Strategy of the National Cancer Institute Drug Development Program. In: Busch H., editor. Methods in Cancer Research. Vol. 26. New York: Academic Press; 1979. pp. 165–245. [Google Scholar]
- 15.Zubrod C.G., Schepartz S., Leiter J., Endicott J.M., Carrese I.M., Baker C.G. The chemotherapy program of the National Cancer Institute: history, analysis, and plans. Cancer Chemother. Rep. 1966;50:349–540. [Google Scholar]
- 16.DeVita V.T., Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68:8643–8653. doi: 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- 17.National Cancer Institute Creation of CCNSC.
- 18.Kidd J.G. Effects of an antibiotic from Aspergillus fumigatus fresenius on tumor cells in vitro, and its possible identity with gliotoxin. Science. 1947;105(2733):511–513. doi: 10.1126/science.105.2733.511. [DOI] [PubMed] [Google Scholar]
- 19.Umezawa H., Takeuchi T., Nitta K., Maeda K. Studies on anti-tumor substances producted by microorgansims, I. On the anti-tumor substance No. 289. J. Antibiot. 1953;•••:45–51. [PubMed] [Google Scholar]
- 20.Umezawa H., Takeuchi T., Nitta K., Okami Y. Studies on anti-tumor substances produced by microorganisms, III. On sarkomycin producted by a strain resembling to Streptomyces erythrochromogenes. J. Antibiot. 1953;•••:147–152. [PubMed] [Google Scholar]
- 21.Waksman S.A. Search for Antitumor and Antiviral Agents. In: Waksman S.A., editor. The Antibiotic Era. Tokyo: The Waksman Foundation of Japan, Inc.; 1975. pp. 95–104. [Google Scholar]
- 22.Schepartz S.A. Historical overview of the National Cancer Institute fermentation program. Recent Res. Cancer. 1978;63:30–32. doi: 10.1007/978-3-642-81219-4_3. [DOI] [PubMed] [Google Scholar]
- 23.Bertino J.R. Methotrexate: Historical Aspects. In: Cronstein B.N., Bertino J.R., editors. Milestones in Drug Therapy: Methotrexate. Basel: Birkhaeuser; 2000. pp. 1–8. [Google Scholar]
- 24.Heinle R.W., Welch A.D. Experiments with pteroylglutamic acid and pteroylglutamic acid deficiency in human leukemia. J. Clin. Invest. 1948;27:539. [Abstract]. [PubMed] [Google Scholar]
- 25.Lesch J.E. A Mechanism Revealed. In: Lesch J.E., editor. The First Miracle Drugs: How the Sulfa Drugs Transformed Medicine. Oxford: Oxford University Press; 2007. pp. 251–265. [Google Scholar]
- 26.Farber S., Diamond L.K., Mercer R.D., Sylvester R.F., Wolf J.A. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin). N. Engl. J. Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 27.Kompis I.M., Islam K., Then R.L. DNA and RNA synthesis: antifolates. Chem. Rev. 2005;105:593–620. doi: 10.1021/cr0301144. [DOI] [PubMed] [Google Scholar]
- 28.Hitchings G.H. The Utilisation of Biochemical Differences Between Host and Parasite as a Basis for Chemotherapy.. In: Goodwin L.G., Nimmo-Smith R.H., editors. Symposium on the Relation Between Chemotherapeutic Drugs, Infecting Organisms and Hosts; 1962. pp. 196–210. [Google Scholar]
- 29.Hitchings G.H. 1988.
- 30.Hitchings G.H. 1966.
- 31.Bodet C.A., Jorgensen J.H., Drutz D.J. Antibacterial activities of antineoplastic agents. Antimicrob. Agents Chemother. 1985;28:437–439. doi: 10.1128/aac.28.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobs J.Y., Michel J., Sacks T. Bactericidal effect of combinations of antimicrobial drugs and antineoplastic antibiotics against Staphylococcus aureus. Antimicrob. Agents Chemother. 1979;15:580–586. doi: 10.1128/aac.15.4.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaykovsky M., Brown B.L., Modest E.J. Methotrexate analogs. 6. Replacement of glutamic acid by varous amino acid esters and amines. J. Med. Chem. 1975;18:909–912. doi: 10.1021/jm00243a010. [DOI] [PubMed] [Google Scholar]
- 34.Chaykovsky M., Rosowsky A., Papathanasopoulos N., Chen K.K., Modest E.J. Methotrexate analogs. 3. synthesis and biological properties of some side-chain altered analogs. J. Med. Chem. 1974;17:1212–1216. doi: 10.1021/jm00257a015. [DOI] [PubMed] [Google Scholar]
- 35.Oleson J.J., Hutchings B.L., Subbarow Y. Studies on the inhibitory nature of 4-aminopteroylglutamic acid. J. Biol. Chem. 1948;175:359–365. [PubMed] [Google Scholar]
- 36.Hutchings B.L., Mowat J.H., Oleson J.J., Stokstad E.L., Boothe J.H., Waller C.W., Angier R.B., Semb J., Subbarow Y. Pteroylaspartic acid, an antagonist for pteroylglutamic acid. J. Biol. Chem. 1947;170:323–328. [Google Scholar]
- 37.Takeuchi T. Bacteriological studies on actinomycetes products exhibiting antitumor activity, I. On antimicrobial effects of synthetic anti-tumor substances and antimicrobial spectra of actinomycetes screened for the anti-tumor activity. J. Antibiot. 1954;7:29–36. [PubMed] [Google Scholar]
- 38.Hamilton-Miller J.M. Antimicrobial activity of 21 anti-neoplastic agents. Br. J. Cancer. 1984;49:367–369. doi: 10.1038/bjc.1984.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Calame W., van der Waals R., Douwes-Idema N., Mattie H., van Furth R. Antibacterial effect of etoposide in vitro. Antimicrob. Agents Chemother. 1988;32:1456–1457. doi: 10.1128/aac.32.9.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pittillo R.F., Schabel F.M., Wilcox W.S., Skipper H.E. Experimental evaluation of potential anticancer agents. XVI. Basic study on effects of certain anticancer on kinetic behavior of model bacterial cell populations. Cancer Chemother. Rep. Part 1. 1965;47:1–26. [PubMed] [Google Scholar]
- 41.Barrett J.F. Quinolone antibacterials and derivatives as antineoplastic agents. Expert Opin. Investig. Drugs. 1996;5:1021–1031. [Google Scholar]
- 42.Dutta R., Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem. Sci. 2000;25:24–28. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 43.Lewis R.J., Singh O.M., Smith C.V., Skarzynski T., Maxwell A., Wonacott A.J., Wigley D.B. The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 44.Roe S.M., Prodromou C., O'Brian R., Ladbury J.E., Piper P.W., Pearl L.H. Structural basis for Inhibiton of the Hsp90 moelcular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 45.Bisacchi G.S., Manchester J.I. A new-class antibacterial--almost. Lessons in drug discovery and development: a critical analysis of more than 50 years of effort toward ATPase inhibitors of DNA gyrase and topoisomerase IV. ACS Inf. Dis. 2015;1:4–41. doi: 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- 46.Bhat R., Tummalapalli S.R., Rotella D.P. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J. Med. Chem. 2014;57:8718–9728. doi: 10.1021/jm500823a. [DOI] [PubMed] [Google Scholar]
- 47.Kim T., Keum G., Pae A.N. Discovery and development of heat shock protein 90 inhibitors as anticancer agents: a review of patented potent geldanamycin derivatives. Expert Opin. Ther. Pat. 2013;22:1–25. doi: 10.1517/13543776.2013.780597. [DOI] [PubMed] [Google Scholar]
- 48.Williams R. Discontinued in 2013: oncology drugs. Expert Opin. Investig. Drugs. 2015;24:95–110. doi: 10.1517/13543784.2015.971154. [DOI] [PubMed] [Google Scholar]
- 49.Jhaveri K., Taldone T., Modi S., Chiosis G. 2012. [DOI] [PMC free article] [PubMed]
- 50.Hong D.S., Banerji U., Tavana B., George G.C., Aaron J., Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (Hsp90): lessons learned and future directions. Cancer Treat. Rev. 2013;39:375–387. doi: 10.1016/j.ctrv.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 51.Ozgur A., Tutar Y. Heat shock protein 90 inhibitors in oncology. Curr. Proteomics. 2014;11:2–16. [Google Scholar]
- 52.Corbett K.D., Berger J.M. Structural basis for topoisomerase VI inhibition by the anti-Hsp90 drug radicicol. Nucleic Acids Res. 2006;34:4269–4277. doi: 10.1093/nar/gkl567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Evans G., White N.H. Radicicolin and radicicol, two new antibiotics produced by Cylindrocarpon radicicola. Trans. Br. Mycol. Soc. 1966;49:563–576. [Google Scholar]
- 54.DeBoer C., Meulman P.A., Wnuk R.J., Peterson D.H. Geldanamycin, a new antibiotic. J. Antibiot. 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- 55.Donnelly A., Blagg B.S. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide binding pocket. Curr. Med. Chem. 2008;22:311–316. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gadelle D., Graille M., Forterre P. The HSP90 and DNA topoisomerase VI inhibitor radicicol also inhibits human type II DNA topoisomerase. Biochem. Pharmacol. 2006;72:1207–1216. doi: 10.1016/j.bcp.2006.07.040. [DOI] [PubMed] [Google Scholar]
- 57.Bisacchi G.S. Origins of the quinolone class of antibacterials: an expanded “discovery story”. J. Med. Chem. 2015;••• doi: 10.1021/jm501881c. [DOI] [PubMed] [Google Scholar]
- 58.Lesher G.Y. Nalidixic Acid and Other Quinolone Carboxylic Acids. In: Grayson M., Eckroth D., editors. Kirk-Othmer Encyclopedia of Chemical Technology, 3rd. Vol. 2. New York: John Wiley; 1978. pp. 782–789. [Google Scholar]
- 59.Gootz T.D., Osheroff N. Quinolones and Eukaryotic Topoisomerases. In: Hooper D.C., Wolfson J.S., editors. Quinolone Antimicrobial Agents, 2nd. Washington, DC: American Society for Microbiology; 1993. pp. 139–160. [Google Scholar]
- 60.Radl S., Dax S. Quinolone congeners as mammalian topoisomerase II inhibitors. Curr. Med. Chem. 1994;1:262–270. [Google Scholar]
- 61.Radl S. From chloroquine to antineoplastic drugs? The story of antibacterial quinolones. Arch. Pharm. 1996;329:115–119. doi: 10.1002/ardp.19963290302. [DOI] [PubMed] [Google Scholar]
- 62.Chu D.T. The future role of quinolones. Expert Opin. Ther. Pat. 1996;6:711–737. [Google Scholar]
- 63.Xia Y., Yang Z.Y., Morris-Natschke S.L., Lee K.H. Recent advances in the discovery and development of quinolones and analogs as antitumor agents. Curr. Med. Chem. 1999;6:179–194. [PubMed] [Google Scholar]
- 64.Anderson V.E., Osheroff N. Type II toposisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 2001;7:339–355. doi: 10.2174/1381612013398013. [DOI] [PubMed] [Google Scholar]
- 65.Larsen A.K., Escargueil A.E., Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003;99:167–181. doi: 10.1016/s0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 66.Sissi C., Palumbo M. The quinolone family: from antibacterials to anticancer agents. Curr. Med. Chem. Anticancer Agents. 2003;3:439–450. doi: 10.2174/1568011033482279. [DOI] [PubMed] [Google Scholar]
- 67.Wagman A.S., Wentland M.P. 2007. Quinolone Antibacterial Agents. [Google Scholar]
- 68.Srivastava S.K., Jha A., Agarwal S.K., Mukherjee R., Burman A.C. Synthesis and structure-activity relationships of potent antitumor active quinoline and naphthyridine derivatives. Anticancer. Agents Med. Chem. 2007;7:685–709. doi: 10.2174/187152007784111313. [DOI] [PubMed] [Google Scholar]
- 69.Andriole V.T. 2000. [Google Scholar]
- 70.Scholar E.M., Pratt W.B. The Fluoroquinolones. In: Scholar E.M., Pratt W.B., editors. The Antimicrobial Drugs, 2nd. Oxford: Oxford University Press; 2000. pp. 257–279. [Google Scholar]
- 71.Domagala J.M., Hagen S.E. Structure-activity Relationships of the Quinolone Qntibacterials in the New Millennium: Some Things Change and Some Do Not. In: Hooper D.C., Rubenstein E., editors. Quinolone Antimicrobial Agents, 3rd. Washington, D.C.: ASM Press; 2003. pp. 3–18. [Google Scholar]
- 72.Sheehan G., Chew N.S. The History of Quinolones. In: Ronald A.R., Low D.E., editors. Fluoroquinolone Antibiotics. Basel: Birkhaeuser Verlag; 2003. pp. 1–10. [Google Scholar]
- 73.Bambeke F.V., Michot J.M., Eldere J.V., Tulkens P.M. Quinolones in 2005: an update. Clin. Microbiol. Infect. 2005;11:256–280. doi: 10.1111/j.1469-0691.2005.01131.x. [DOI] [PubMed] [Google Scholar]
- 74.Andriole V.T. The quinolones: past, present, and future. Clin. Infect. Dis. 2005;41(Suppl. 2):S113–S119. doi: 10.1086/428051. [DOI] [PubMed] [Google Scholar]
- 75.Mitscher L.A. Bacterial topoisomerase inhibitors: quinolone and pyridone antibacterial agents. Chem. Rev. 2005;105:559–592. doi: 10.1021/cr030101q. [DOI] [PubMed] [Google Scholar]
- 76.Wiles J.A., Bradbury B.J., Pucci M.J. New quinolone antibiotics: a survey of the literature from 2005 to 2010. Expert Opin. Ther. Pat. 2010;20:1295–1319. doi: 10.1517/13543776.2010.505922. [DOI] [PubMed] [Google Scholar]
- 77.Taira K., Koga H., Kohno S. Accumulation of a newly developed fluoroquinolone, OPC-17116, by human polymorphonuclear leukocytes. Antimicrob. Agents Chemother. 1993;37:1877–1881. doi: 10.1128/aac.37.9.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carryn S., Chanteus H., Seral C., Mingeot-Leclercq M.P., Van Bambeke F., Tulkens P.M. Intracellular pharmacodynamics of antibiotics. Infect. Dis. Clin.N. Am.; 2003. pp. 615–634. [DOI] [PubMed] [Google Scholar]
- 79.Seral C., Barcia-Macay M., Mingeot-Leclercq M.P., Tulkens P.M., Van Bambeke F. Comparative activity of quinolones (ciprofloxacin, levofloxacin, moxifloxacin and garenoxacin) against extracellular and intracellular infection by Listeria monocytogenes and Staphylococcus aureus in J774 macrophages. J. Antimicrob. Chemother. 2005;55:511–517. doi: 10.1093/jac/dki059. [DOI] [PubMed] [Google Scholar]
- 80.Baltch A.L., Bopp L.H., Smith R.P., Michelsen P.B., Ritz W.J. Antibacterial activities of gemifloxacin, levofloxacin, gatifloxacin, moxifloxacin, and erythromycin against intracellular Legionella pneumophila and Legionella micdadei in human monocytes. J. Antimicrob. Chemother. 2005;56:104–109. doi: 10.1093/jac/dki186. [DOI] [PubMed] [Google Scholar]
- 81.Van Bambeke F., Barcia-Macay M., Lemaire S., Tulkens P.M. Cellular pharmacodynamics and pharmacokinetics of antibiotics: current views and perspectives. Curr. Opin. Drug Discov. Devel. 2006;9:218–230. [PubMed] [Google Scholar]
- 82.Sui Z., Altom J., Nguyen V.N., Fernandez J., Bernstein J.I., Hiliard J.J., Barrett J.F., Podlogar B.L., Ohemeng K.A. Synthesis and inhibitory activity of novel tri-and tetracyclic quinolines against topoisomerases. Bioorg. Med. Chem. 1998;6:735–742. doi: 10.1016/s0968-0896(98)00030-3. [DOI] [PubMed] [Google Scholar]
- 83.Corbett A.H., Guerry P., Pflieger P., Osheroff N. A pyrimido[1,6a]benzimidazole that enhances DNA cleavage mediated by eukaryotic topoisomerase II: a novel class of topoisomerase II-targeted drugs with cytotoxic potential. Antimicrob. Agents Chemother. 1993;37:2599–2605. doi: 10.1128/aac.37.12.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 85.Forterre P., Gribaldo S., Gadelle D., Serre M.C. The origin and evolution of DNA topoisomerases. Biochimie. 2007;89:427–446. doi: 10.1016/j.biochi.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 86.Schoeffler A.J., Berger J.M. DNA topoisomerases: harnessing and constraining energy to govern chromosome topology. Q. Rev. Biophys. 2008;41:41–101. doi: 10.1017/S003358350800468X. [DOI] [PubMed] [Google Scholar]
- 87.Forterre P., Gadelle D. Phylogenomics of DNA topoisomerases: their origin and putative roles in the emergence of modern organisms. Nucleic Acids Res. 2009;37:679–692. doi: 10.1093/nar/gkp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sissi C., Palumbo M. In front of and behind the replication fork: bacterial type IIA topoisomerases. Cell. Mol. Life Sci. 2010;67:2001–2024. doi: 10.1007/s00018-010-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schvartzman J.B., Martinez-Robles M.L., Hernandez P., Krimer D.B. The benefit of DNA supercoiling during replication. Biochem. Soc. Trans. 2013;41:646–651. doi: 10.1042/BST20120281. [DOI] [PubMed] [Google Scholar]
- 90.Chang C.C., Wang Y.R., Chen S.F., Wu C.C., Chan N.L. New insights into DNA binding by type IIA topoisomerases. Curr. Opin. Struct. Biol. 2013;23:125–133. doi: 10.1016/j.sbi.2012.11.011. [DOI] [PubMed] [Google Scholar]
- 91.Laponogov I., Veselkov D.A., Crevel I.M., Pan X., Fisher L.M., Sanderson M.R. Structure of an ‘open’ clamp type II topoisomerase-DNA complex provides a mechanism for DNA capture and transport. Nucleic Acids Res. 2013;41:9911–9923. doi: 10.1093/nar/gkt749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Laponogov I., Sohi M.K., Veselkov D.A., Pan X.S., Sawhney R., Thompson A.W., McAuley K.E., Fisher L.M., Sanderson M.R. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 2009;16:667–669. doi: 10.1038/nsmb.1604. [DOI] [PubMed] [Google Scholar]
- 93.Bax B.D., Chan P.F., Eggleston D.S., Fosberry A., Gentry D.R., Gorrec F., Giordano I., Hann M.M., Hennessy A., Hibbs M., Huang J., Jones E., Jones J., Brown K.K., Lewis C.J., May E.W., Saunders M.R., Singh O., Spitzfaden C.E., Shen C., Shillings A., Theobald A.J., Wohlkonig A., Pearson N.D., Gwynn M.N. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature. 2010;466:935–940. doi: 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- 94.Mani N., Gross C.H., Parsons J.D., Hanzelka B., Muh U., Mullin S., Liao Y., Grillot A.L., Stamos D., Charifson P.S., Grossman T.H. In vitro characterization of the antibacterial spectrum of novel bacterial type II topoisomerase inhibitors of the aminobenzimidazole class. Antimicrob. Agents Chemother. 2006;50:1228–1237. doi: 10.1128/AAC.50.4.1228-1237.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stokes N.R., Thomaides-Brears H.B., Barker S., Bennett J.M., Berry J., Collins I., Czaplewski L.G., Gamble V., Lancett P., Logan A., Lunniss C.J., Peasley H., Pommier S., Price D., Smee C., Haydon D.J. Biological evaluation of benzothiazole ethyl urea inhibitors of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2013;57:5977–5986. doi: 10.1128/AAC.00719-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Drlica K., Malik M., Kerns R.J., Zhao X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008;•••:385–392. doi: 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee J.S., Morgan A.R. the topological trapping of circular DNAs on agarose: unexpected restrictions on DNA rotation. Can. J. Biochem. 1978;56:585–591. doi: 10.1139/o78-088. [DOI] [PubMed] [Google Scholar]
- 98.Falaschi A., Cobianchi F., Riva S. DNA-binding proeins and DNA-unwinding enzymes in eukaryotes. Trends Biochem. Sci. 1980;5:154–157. [Google Scholar]
- 99.Tao-shih H., Brutlag D. ATP-dependent DNA topoisomerase from Drosophila melanogaster reversibly catenates duplex DNA rings. Cell. 1980;21:115–125. doi: 10.1016/0092-8674(80)90119-1. [DOI] [PubMed] [Google Scholar]
- 100.Bellon S., Parsons J.D., Wei Y., Hayakawa K., Swenson L.L., Charifson P.S., Lippke J.A., Aldape R., Gross C.H. Crystal structures of Escherichia coli topoisomerase IV ParE Subunit (24 and 43 Kilodaltons): a single residue dictates differences in novobiocin potency against topoisomerase IV and DNA gyrase. Antimicrob. Agents Chemother. 2004;48:1856–1864. doi: 10.1128/AAC.48.5.1856-1864.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brino L., Urzhumtsev A., Mousli M., Bronner C., Mitschler A., Oudet P., Moras D. Dimerization of Escherichia coli DNA-gyrase B provides a structural mechanism for activating the ATPase catalytic center. J. Biol. Chem. 2000;275:9468–9475. doi: 10.1074/jbc.275.13.9468. [DOI] [PubMed] [Google Scholar]
- 102.Schmidt B.H., Osheroff N., Berger J.M. Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 2012;19:1147–1154. doi: 10.1038/nsmb.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ito A., Hirai K., Inoue M., Koga H., Suzue S., Irikura T., Mitsuhashi S. In vitro antibacterial activity of AM-715, a new nalidixic acid analog. Antimicrob. Agents Chemother. 1980;17:103–108. doi: 10.1128/aac.17.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koga H., Itoh A., Murayama S., Suzue S., Irikura T. Structure-activity relationships of antibacterial 6,7- and 7,8-disubstituted 1-alkyl-1,4-dihydro-4-oxoquinoline-3-carbo-xylic acids. J. Med. Chem. 1980;23:1358–1363. doi: 10.1021/jm00186a014. [DOI] [PubMed] [Google Scholar]
- 105.Mattern M.R., Scudiero D.A. Dependence of mammalian DNA synthesis on DNA supercoiling III. Characterization of the inhibition of replicative and repair-type DNA synthesis by novobiocin and nalidixic acid. Biochim. Biophys. Acta. 1981;653:248–253. doi: 10.1016/0005-2787(81)90160-x. [DOI] [PubMed] [Google Scholar]
- 106.Miller K.G., Liu L.F., Englund P.T. A homogeneous Type II DNA topoisomerase from HeLa cell nuclei. J. Biol. Chem. 1981;256:9334–9339. [PubMed] [Google Scholar]
- 107.Chen G.L., Yang L., Rowe T.C., Halligan B.D., Tewey K.M., Liu L.F. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 1984;259:13560–13566. [PubMed] [Google Scholar]
- 108.Tewey K.M., Towe T.C., Yang L., Halligan B.D., Liu L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science. 1984;226:466–468. doi: 10.1126/science.6093249. [DOI] [PubMed] [Google Scholar]
- 109.Ross W.E. DNA topoisomerases as targets for cancer therapy. Biochem. Pharmacol. 1985;34:4191–4195. doi: 10.1016/0006-2952(85)90273-4. [DOI] [PubMed] [Google Scholar]
- 110.Hussy P., Maass G., Tuemmler B., Grosse F., Schomburg U. Effect of 4-quinolones and novobiocin on calf thymus DNA polymerase alpha primase complex, topoisomerases I and II, and growth of mammalian lymphoblasts. Antimicrob. Agents Chemother. 1986;29:1073–1078. doi: 10.1128/aac.29.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Oomori Y., Yasue T., Aoyama H., Hirai K., Suzue S., Yokota T. Effects of fleroxacin on HeLa cell functions and topoisomerase II. J. Antimicrob. Chemother. 1988;22(Suppl. D):91–97. doi: 10.1093/jac/22.supplement_d.91. [DOI] [PubMed] [Google Scholar]
- 112.Somekh E., Lev B., Schwartz E., Barzilai A., Rubenstein E. The effect of ciprofloxacin and pefloxacin on bone marrow engraftment in the spleen of mice. J. Antimicrob. Chemother. 1989;23:247–251. doi: 10.1093/jac/23.2.247. [DOI] [PubMed] [Google Scholar]
- 113.Somekh E., Douer D., Shaked N., Rubenstein E. In vitro effects of ciprofloxacin and fleroxacin on growth of normal human hematopoietic progenitor cells and on leukemic cell lines. J. Pharmacol. Exp. Ther. 1989;248:415–418. [PubMed] [Google Scholar]
- 114.Pessina A., Neri M.G., Nuschiato A., Mineo E., Cocuzza G. Effet of fluoroquinolones on the in-vitro proliferation of myeloid precursor cells. J. Antimicrob. Chemother. 1989;24:203–208. doi: 10.1093/jac/24.2.203. [DOI] [PubMed] [Google Scholar]
- 115.Holden H.E., Barrett J.F., Huntington C.M., Muehlbauer P.A., Wahrenburg M.G. Genetic profile of a nalidixic acid analog: a model for the mechansim of sister chromatid exchange induction. Environ. Mol. Mutagen. 1989;13:238–252. doi: 10.1002/em.2850130308. [DOI] [PubMed] [Google Scholar]
- 116.Barrett J.F., Gootz T.D., McGuirk P.R., Farrell C.A., Sokolowski S.A. Use of in vitro topoisomerase II assays for studying quinolone antibacterial agents. Antimicrob. Agents Chemother. 1989;33:1697–1703. doi: 10.1128/aac.33.10.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barrett J.F., Sutcliffe J.A., Gootz T.D. In vitro assays used to measure the activity of topoisomerases. Antimicrob. Agents Chemother. 1990;34:1–7. doi: 10.1128/aac.34.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hertzberg R.P., Johnson R.K. Antineoplastic Agents. In: Briston J.A., editor. Annual Reports in Medicinal Chemistry. Vol. 28. San Diego: Academic Press; 1993. pp. 167–176. [Google Scholar]
- 119.Chambers S.K., Chopyk R.L., Chambers J.T., Schwartz P.E., Duffy T.P. Development of leukemia after doxorubicin and cisplatin treatment for ovarian cancer. Cancer. 1989;64:2459–2461. doi: 10.1002/1097-0142(19891215)64:12<2459::aid-cncr2820641210>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 120.Albain K.S., Le Beau M.M., Ullirsch S.H. Implication of prior treatment with drug combinations including inhibitors of topisomerase II in therapy-related monocytic leukemia with a 9;11 translocation. Genes Chromosomes Cancer. 1990;2:53–58. doi: 10.1002/gcc.2870020110. [DOI] [PubMed] [Google Scholar]
- 121.Whitlock J.A., Greer J.P., Lukens J.N. Epipodophyllotoxin-related leukemia. Identification of a new subset of secondary leukemia. Cancer. 1991;68:600–604. doi: 10.1002/1097-0142(19910801)68:3<600::aid-cncr2820680326>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 122.Ratain M.J., Rowley J.D. Therapy-related acute myeloid leukemia secondary to inhibitors of topoisomerase II: from the bedside to the target genes. Ann. Oncol. 1992;3:107–111. doi: 10.1093/oxfordjournals.annonc.a058121. [DOI] [PubMed] [Google Scholar]
- 123.Fleming R.A., Miller A.A., Stewart C.F. Etoposide: an update. Clin. Pharm. 1989;8:274–293. [PubMed] [Google Scholar]
- 124.Beck W.T., Danks M.K. Mechanisms of resistance to drugs that inhibit DNA topoisomerases. Semin. Cancer Biol. 1991;2:235–234. [PubMed] [Google Scholar]
- 125.Takano H., Kohno K., Matsuo K., Matsuda T., Kuwano M. DNA topoisomerase-targeting antitumor agents and drug resistance. Anticancer Drugs. 1992;3:323–330. doi: 10.1097/00001813-199208000-00002. [DOI] [PubMed] [Google Scholar]
- 126.Hande K.R. Antitumor antibiotics, epipodophyllotoxins, and vinca alkaloids. Curr. Opin. Oncol. 1992;4:1080–1088. doi: 10.1097/00001622-199212000-00013. [DOI] [PubMed] [Google Scholar]
- 127.Reuman M., Lesher G.Y. Antibacterial Agents, Synthetic – Quinolones. In: Kroschwitz J.I., editor. Kirk-Othmer Encyclopedia of Chemical Technology 4th. Vol. 2. New York: John Wiley; 1992. pp. 854–869. [Google Scholar]
- 128.Fernandes P.B., Chu D.T. Qunolones. In: Bailey D.M., editor. Annual Reports in Medicinal Chemistry. Vol. 22. San Diego: Academic Press; 1987. pp. 117–126. [Google Scholar]
- 129.Fernandes P.B., Chu D.T. Quinolone Antibacterial Agents. In: Bailey D.M., editor. Annual Reports in Medicinal Chemistry. Vol. 23. San Diego: Academic Press; 1988. pp. 133–140. [Google Scholar]
- 130.Hooper D.C., Wolfson J.S. 1993. [Google Scholar]
- 131.Vila J., Fabrega A., Roca I. Efflux pumps as an important mechanism for quinolone resistance. Adv. Enzymol. RAMB. 2011;77:167–235. doi: 10.1002/9780470920541.ch5. [DOI] [PubMed] [Google Scholar]
- 132.Aldred K.J., Kerns R.J., Osheroff N. Mechanism of quinolone action and resistance. Biochemistry. 2014;53:1565–1574. doi: 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wei H., Ruthenburg A.J., Bechis S.K., Verdine G.L. Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J. Biol. Chem. 2005;280:37041–37047. doi: 10.1074/jbc.M506520200. [DOI] [PubMed] [Google Scholar]
- 134.Wu C.C., Li T.K., Farh L., Lin L.Y., Lin T.S., Yu Y.J., Yen T.J., Chiang C.W., Chan N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459–462. doi: 10.1126/science.1204117. [DOI] [PubMed] [Google Scholar]
- 135.Wu C.C., Li Y.C., Wang Y.R., Li T.K., Chan N.L. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res. 2013;41:10630–10640. doi: 10.1093/nar/gkt828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chan N.L., Li T.K., Wu C.C., Wang Y.R. 2014.
- 137.Yang F., Kemp C.J., Henikoff S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. Fund. Mol.M. 2015;773:9–15. doi: 10.1016/j.mrfmmm.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ketron A.C., Denny W.A., Graves D.E., Osheroff N. Amsacrine as a topoisomerase II poison: importance of drug-DNA interactions. Biochemistry. 2012;51:1730–1739. doi: 10.1021/bi201159b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fortune J.M., Osheroff N. Topoisomerase II as a Target for Anticancer Drugs: When Enzymes Stop Being Nice. In: Moldave K., editor. Progress in Nucleic Acid Research and Molecular Biology. Vol. 64. San Diego: Academic Press; 2000. pp. 221–253. [DOI] [PubMed] [Google Scholar]
- 140.Topcu Z. DNA topoisomerases as targets for anticancer drugs. J. Clin. Pharm. Ther. 2001;26:405–416. doi: 10.1046/j.1365-2710.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- 141.Liu L.F. DNA toposiomerase poisons as antitumor drugs. Annu. Rev. Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- 142.Palchaudhuri R., Hergenrother P.J. DNA as a target for anticancer comounds: methods to determine the mode of binding and mechanism of action. Curr. Opin. Biotechnol. 2007;18:497–503. doi: 10.1016/j.copbio.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 143.Osheroff N., Corbett A.H., Elsea S.H., Westergaard M. Defining functional drug-interaction domains on topoisomerase II by exploiting mechanistic differences between drug classes. Cancer Chemother. Pharmacol. 1994;34(Suppl.):S19–S25. doi: 10.1007/BF00684859. [DOI] [PubMed] [Google Scholar]
- 144.Corbett A.H., Hong D., Osheroff N. Exploiting mechanistic differences between drug classes to define functional drug interaction domains on topoisomerase II. J. Biol. Chem. 1993;268:14394–14398. [PubMed] [Google Scholar]
- 145.Robinson M.J., Martin B.A., Gootz T.D., McGuirk P.R., Osheroff N. Effects of novel fluoroquinolones on the catalytic activities of eukaryotic topoisomerase II: influence of the C-8 fluorine group. Antimicrob. Agents Chemother. 1992;36:751–756. doi: 10.1128/aac.36.4.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nitiss J.L. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim. Biophys. Acta. 1998;1400:63–81. doi: 10.1016/s0167-4781(98)00128-6. [DOI] [PubMed] [Google Scholar]
- 147.Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 148.Nitiss K.C., Nitiss J.L. Twisting and ironing: doxorubicin cardiotoxicity by mitochondrial DNA damage. Clin. Cancer Res. 2014;20:4737–4739. doi: 10.1158/1078-0432.CCR-14-0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vejpongsa P., Yeh E.T. Prevention of anthracycline-induced cardiotoxicity. Challenges and opportunities. J. Am. Coll. Cardiol. 2014;64:938–945. doi: 10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- 150.Stiborova M., Sejbal J., Dorek-Dohalska L., Aimova D., Poljakova J., Forsterova K., Rupertova M., Wiesner J., Hudecek J., Wiessler M., Frei E. The anticancer drug ellipticine forms covalent DNA adducts, mediated by human cytochromes P450, through metabolism to 13-hydroxyellipticine and ellipticine N2-oxide. Cancer Res. 2004;64:8374–8380. doi: 10.1158/0008-5472.CAN-04-2202. [DOI] [PubMed] [Google Scholar]
- 151.Stiborova M., Poljakova J., Ecksschlager T., Kizek R., Frei E. Analysis of covalent ellipticine- and doxorubicin-derived adducts in DNA of neuroblastoma cells by the 32P-postlabeling technique. Biomed. Pap. 2012;156:115–121. doi: 10.5507/bp.2012.043. [DOI] [PubMed] [Google Scholar]
- 152.Regal K.M., Mercer S.L., Deweese J.E. HU-331 is a catalytic inhibitor of topoisomerase IIα. Chem. Res. Toxicol. 2014;27:2044–2051. doi: 10.1021/tx500245m. [DOI] [PubMed] [Google Scholar]
- 153.Bau J.T., Kang Z., Austin C.A., Kurz E.U. Salicylate, a catalytic inhibitor of topoisomerase II, inhibits DNA cleavage and is selective for the α-isoform. Mol. Pharmacol. 2013;85:198–207. doi: 10.1124/mol.113.088963. [DOI] [PubMed] [Google Scholar]
- 154.Gao H., Huang K.C., Yamasaki E.F., Chan K.K., Chohan L., Snapka R.M. XK469, a selective topoisomerase IIβ poison. Proc. Natl. Acad. Sci. USA. 1999;96:12168–12173. doi: 10.1073/pnas.96.21.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 156.Bailly C. Topoisomerase I poisons and suppressors as anticancer drugs. Curr. Med. Chem. 2000;7:39–58. doi: 10.2174/0929867003375489. [DOI] [PubMed] [Google Scholar]
- 157.Choi I., Kim C., Choi S. Binding mode analysis of topoisomerase inhibitors, 6-arylamino-7-chloro-quinazoline-5,8-diones, within the cleavable complex of human topoisomerase I and DNA. Arch. Pharm. Res. 2007;30:1526–1535. doi: 10.1007/BF02977321. [DOI] [PubMed] [Google Scholar]
- 158.Denny W.A., Baguley B.C. Dual topoisomerase I/II inhibitors in cancer therapy. Curr. Top. Med. Chem. 2003;3:339–353. doi: 10.2174/1568026033452555. [DOI] [PubMed] [Google Scholar]
- 159.Salerno S., Settimo F.D., Taliani S., Simorini F., La Motta C., Fornaciari G., Marini A.M. Recent advances in the development of dual topoisomerase I and II inhibitors as anticncer drugs. Curr. Med. Chem. 2010;17:4270–4290. doi: 10.2174/092986710793361252. [DOI] [PubMed] [Google Scholar]
- 160.Moreau N.J., Robaus H., Baron L., Tabary X. Inhibitory effects of quinolones on pro- and eukaryotic DNA topoisomerases I and II. Antimicrob. Agents Chemother. 1990;34:1955–1960. doi: 10.1128/aac.34.10.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gootz T.D., Barrett J.F., Sutcliffe S.A. Inhibitory effects of quinolone antibacterial agents on eucaryotic topoisomerases and related test systems. Antimicrob. Agents Chemother. 1990;34:8–12. doi: 10.1128/aac.34.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kang D.H., Kim J.S., Jung M.J., Lee E.S., Jahng Y., Kwon Y., Na Y. New insight for fluoroquinophenoxazine derivatives as possibly new potent topoisomerase I inhibitor. Bioorg. Med. Chem. Lett. 2008;18:1520–1524. doi: 10.1016/j.bmcl.2007.12.053. [DOI] [PubMed] [Google Scholar]
- 163.Hasinoff B.B., Wu X., Nitiss J.L., Kanagasabai R., Yalowich J.C. The anticancer multi-kinase inhibitor dovitinib also targets topoisomerase I and topoisomerase II. Biochem. Pharmacol. 2012;84:1617–1626. doi: 10.1016/j.bcp.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hoshino K., Sato K., Une T., Osada Y. Inhibitory effects of quinolones on DNA gyrase of Escherichia coli and topoisomerase II of fetal calf thymus. Antimicrob. Agents Chemother. 1980;33:1816–1818. doi: 10.1128/aac.33.10.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Suto M.J., Domagala J.M., Roland G.E., Mailloux G.B., Cohen M.A. Fluoroquinolones: relationships between structural variations, mammalian cell cycotoxicity, and antimicrobial activity. J. Med. Chem. 1992;35:4745–4750. doi: 10.1021/jm00103a013. [DOI] [PubMed] [Google Scholar]
- 166.Domagala J.M. Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J. Antimicrob. Chemother. 1994;33:685–706. doi: 10.1093/jac/33.4.685. [DOI] [PubMed] [Google Scholar]
- 167.Sanchez J.P., Gogliotti R.D., Domagala J.M., Gracheck S.J., Huband M.D., Sesnie J.A., Cohen M.A., Shapiro M.A. The synthesis, structure-activity, and structure-side effect relationships of a series of 8-alkoxy- and 5-amino-8-alkoxyquinolone antibacterial agents. J. Med. Chem. 1995;38:4478–4487. doi: 10.1021/jm00022a013. [DOI] [PubMed] [Google Scholar]
- 168.Domagala J.M. 2015.
- 169.Gilligan P.J., McGuirk P.R., Witty M.J. 1987. [Google Scholar]
- 170.Gilligan P.J., Witty M.J., McGuirk P.R. Substituted dihydroquinolones carboxylic acids, anti-bacterial compositions containing them. E. 1986;•••:184384. [Google Scholar]
- 171.Elsea S.H., Osheroff N., Nitiss J.L. Cytotoxicity of quinolones toward eukaryotic cells. Identification of topoisomerase II as the primary cellular taret for the quinolone CP-115,953 in yeast. J. Biol. Chem. 1992;267:13150–13153. [PubMed] [Google Scholar]
- 172.Froelich-Ammon S.J., McGuirk P.R., Gootz T.D., Jefson M.R., Osheroff N. Novel 1-8-bridged chiral quinolones with activity against topoisomerase II: stereospecificity of the eukaryotic enzyme. Antimicrob. Agents Chemother. 1993;37:646–651. doi: 10.1128/aac.37.4.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Elsea S.H., McGuirk P.R., Gootz T.D., Moynihan M., Osheroff N. Drug features that contribute to the activity of quinolones against mammalian topoisomerase II and cultured cells: correlation between enhancement of enzyme-mediated DNA cleavage in vitro and cytotoxic potential. Antimicrob. Agents Chemother. 1993;37:2179–2186. doi: 10.1128/aac.37.10.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Robinson M.J., Elsea S.H., Martin B.A., Gootz T.D., McGuirk P.R., Sutcliff J.A., Osheroff N. A Novel Quinolone with Potent Activity Against Eukaryotic DNA Topoisomerase II.; 1993. [Google Scholar]
- 175.Gootz T.D., McGuirk P.R., Moynihan M.S., Haskell S.L. Placement of alkyl substituents on the C-7 piperazine ring of fluoroquinolones: dramatic differential effects on mammalian topoisomerase II and DNA gyrase. Antimicrob. Agents Chemother. 1994;38:130–133. doi: 10.1128/aac.38.1.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gootz T.D. 2015.
- 177.Chu D.T., Hallas R., Tanaka S.D., Alder J., Balli D., Plattner J.J. Synthesis and antitumor activities of tetracyclic quinolone antineoplastic agents. Drugs Exp. Clin. Res. 1994;20:177–183. [PubMed] [Google Scholar]
- 178.Permana P.A., Snapka R.M., Shen L.L., Chu D.T., Clement J.J., Plattner J.J. Quinobenoxazines: a class of novel antitumor quinolones and potent mammalian DNA topoisomerase Ii catalytic inhibitors. Biochemistry. 1994;33:11333–11339. doi: 10.1021/bi00203a031. [DOI] [PubMed] [Google Scholar]
- 179.Chu D.T., Hallas R., Clement J.J., Alder J., McDonald E., Plattner J.J. Synthesis and antitumour activities of quinolone antineoplastic agents. Drugs Exp. Clin. Res. 1992;18:275–282. [PubMed] [Google Scholar]
- 180.Clement J.J., Burres N., Jarvis K., Chu D.T., Swiniarski J., Alder J. Briological characterization of a novel antitumor quinolone. Cancer Res. 1995;55:830–835. [PubMed] [Google Scholar]
- 181.Chu D.T. 2015.
- 182.Kim H.L., Jeon K.H., Jun K.Y., Choi Y., Kim D.K., Na Y., Kwon Y. A-62176, a potent topoisomerase inhibitor, inhibits the expression of human epidermal growth factor receptor 2. Cancer Lett. 2012;325:72–79. doi: 10.1016/j.canlet.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 183.Chu D.T., Fernandes P.B., Claiborne A.K., Shen L., Pernet A.G. Structure-activity relationships in quinolone antibacterials: design, synthesis and biological activities of novel isothiazoloquinolones. Drugs Exp. Clin. Res. 1988;14:379–383. [PubMed] [Google Scholar]
- 184.Chu D.T., Fernandes P.B., Claiborne A.D., Shen L., Pernet A.G. 1989. [Google Scholar]
- 185.Chu D.T., Lico I.M., Claiborne A.K., Plattner J.J., Pernet A.G. Structure-activity relationship of quinolone antibacterial agents: the effects of C-2 substitution. Drugs Exp. Clin. Res. 1990;16:215–224. [PubMed] [Google Scholar]
- 186.Chu D.T., Plattner J.J., Klein L.L., Shen L.L. Tricyclic quinoline antineoplastic agents. E. 1990;•••:424802. [Google Scholar]
- 187.Kohlbrenner W.E., Wideburg N., Weigl D., Saldivar A., Chu D.T. Induction of calf thymus topoisomerase II-mediated DNA breakage by the antibacterial isothiazoloquinolones A-65281 and A-65282. Antimicrob. Agents Chemother. 1992;36:81–86. doi: 10.1128/aac.36.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wiles J.A., Hashimoto A., Thanassi J.A., Cheng J., Incarvito C.D., Deshpande M., Pucci M.J., Bradbury B.J. Isothiazolopyridones: synthesis, structure, and biological activity of a new class of antibacterial agents. J. Med. Chem. 2006;49:39–42. doi: 10.1021/jm051066d. [DOI] [PubMed] [Google Scholar]
- 189.Wiles J.A., Song Y., Wang Q., Lucien E., Hashimoto A., Cheng J., Marlor C.W., Ou Y., Podos S.D., Thanassi J.A., Thoma C.L., Deshpande M., Pucci M.J., Bradbury B.J. Biological evaluation of isothiazoloquinolones containing aromatic heterocycles at the 7 position: in vitro activity of a series of potent antibacterial agents that are effective against methicillin-resistant Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2006;16:1277–1281. doi: 10.1016/j.bmcl.2005.11.064. [DOI] [PubMed] [Google Scholar]
- 190.Wang Q., Lucien E., Hashimoto A., Pais G.C., Nelson D.M., Song Y., Thanassi J.A., Marlor C.W., Thoma C.L., Cheng J., Podos S.D., Ou Y., Deshpande M., Pucci M.J., Buechter D.D., Bradbury B.J., Wiles J.A. Isothazoloquinolones with enhanced antistaphylococcal activities against multidrug-resistant strains: effects of structural modifications at the 6-, 7-, and 8-positions. J. Med. Chem. 2007;50:199–210. doi: 10.1021/jm060844e. [DOI] [PubMed] [Google Scholar]
- 191.Pucci M.J., Cheng J., Podos S.D., Thoma C.L., Thanassi J.A., Buechter D.D., Mushtaq G., Vigliotti G.A., Bradbury B.J., Deshpande M. In vitro and in vivo antibacterial activities of heteroaryl isothiazolones against resistant gram-positive pathogens. Antimicrob. Agents Chemother. 2007;51:1259–1267. doi: 10.1128/AAC.01315-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng J., Thanassi J.A., Thoma C.L., Bradbury B.J., Deshpande M., Pucci M.J. Dual targeting of DNA gyrase and topoisomerasae IV: target interactions of heteroaryl isothiazolones in Staphylococcus aureus. Antimicrob. Agents Chemother. 2007;51:2445–2453. doi: 10.1128/AAC.00158-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pucci M.J., Podos S.D., Thanassi J.A., Leggio M.J., Bradbury B.J., Deshpande M. In vitro and in vivo profiles of ACH-702, an isothiazoloquinolone against bacterial pathogens. Antimicrob. Agents Chemother. 2011;55:2860–2871. doi: 10.1128/AAC.01666-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kim H.Y., Wiles J.A., Wang Q., Pais G.C., Lucien E., Hashimoto A., Nelson D.M., Thanassi J.A., Podos S.D., Deshpande M., Pucci M.J., Bradbury B.J. Exploration of the activity of 7-pyrrolidino-8-methoxyiso-thiazoloquinolones against methicillin-resistant Staphylococcus aureus (MRSA). J. Med. Chem. 2011;54:3268–3282. doi: 10.1021/jm101604v. [DOI] [PubMed] [Google Scholar]
- 195.Wentland M.P., Lesher G.Y., Reuman M., Gruett M.D., Singh B., Aldous S.C., Dorff P.H., Rake J.B., Coughlin S.A. Mammalian topoisomerase II inhibitory activity of 1-cyclopropyl-6,8-difluoro-1,4-dihydro-7-(2,6-dimethyl-4-pyridinyl)-4-oxo-3-quinolinecarboxylic acid and related derivatives. J. Med. Chem. 1993;36:2801–2809. doi: 10.1021/jm00071a010. [DOI] [PubMed] [Google Scholar]
- 196.Wentland M.P., Lesher G.Y., Reuman M., Pilling G.M., Saindane M.T., Perni R.B., Eissenstat M.A., Weaver J.D., Singh B., Rake J., Coughlin S.A. Relationship of structure of bridged (2,6-dimethyl-4-pyridinyl)-quinolones to mammalian topoisomerase II inhibition. Bioorg. Med. Chem. Lett. 1993;3:1711–1716. [Google Scholar]
- 197.Coughlin S.A., Danz D.W., Robinson R.G., Klingbeil K.M., Wentland M.P., Corbett T.H., Waud W.R., Zwelling L.A., Altschuler E., Bales E., Rake J.B. Mechanism of action and antitumor activity of (S)-10-(2,6-dimethyl-4-pyridinyl)-9-fluoro-3-methyl-7-oxo-2,3-dihydro-7H-pyridol[1,3,3-de]-[1,4]benzothiazine-6-carboxylic acid (WIN 58161). Biochem. Pharmacol. 1995;50:111–122. doi: 10.1016/0006-2952(95)00016-s. [DOI] [PubMed] [Google Scholar]
- 198.Wentland M.P., Aldous S.C., Gruett M.D., Perni R.B., Powles R.G., Kanz D.W., Klingbeil K.M., Peverly A.D., Robinson R.G., Corbett T.H., Rake J.B., Coughlin S.A. The antitumor activity of novel pyrazoloquinoline derivatives. Bioorg. Med. Chem. Lett. 1995;5:405–410. [Google Scholar]
- 199.Kuo G.H., Eissenstat M.A., Wentland M.P., Robinson R.G., Klingbeil K.M., Danz D.W., Coughlin S.A. Potent mammalian topoisomerase II inhibitors: 1-cyclopropyl-6,8-difluoro-1,4-dihydro-7-(2,6-dimethyl-4-pyridinyl)-4-substituted quinolones. Bioorg. Med. Chem. Lett. 1995;5:399–404. [Google Scholar]
- 200.Eissenstat M.A., Kuo G.H., Weaver J.D., Wentland M.P., Robinson R.G., Klingeil K.M., Danz D.W., Corbett T.H., Coughlin S.A. 3-Benzyl-quinolones: novel, potent inhibitors of mammalian topoisomerase II. Bioorg. Med. Chem. Lett. 1995;5:1021–1026. [Google Scholar]
- 201.Okura A., Yohinari T., Arakawa H., Nakagawa S., Mano E., Ushijima R. 1992.
- 202.Yoshinari T., Mano E., Arakawa H., Kurama M., Iguchi T., Nakagawa S., Tanaka N., Okura A. Stereo(C7)-dependent topoisomerase II inhibition and tumor growth supression by a new quinolone, BO-2367. Jpn. J. Cancer Res. 1993;84:800–806. doi: 10.1111/j.1349-7006.1993.tb02047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Arakawa H., Mano E., Hakoda N., Yoshinari T., Nakagawa S., Okura A. Potent antitumor activity of quinolone compounds with an unsaturated aminoazabicyclo group at the C-7 position of the quinolone ring. Anticancer Drug Des. 1996;11:221–229. [PubMed] [Google Scholar]
- 204.Yamashita Y., Ashizawa T., Morimoto M., Hosomi J., Nakano H. Antitumor quinolones with mammalian topoisomerase II mediated DNA cleavage activity. Cancer Res. 1992;52:2818–2822. [PubMed] [Google Scholar]
- 205.Nakano H., Yamashita Y., Ashizawa T., Morimoto M., Komuro T., Hosomi J. Anticancer agents containing quinolonecarboxylic acids. J.P.: 1991. p. 03024013. [Google Scholar]
- 206.Dumas J., Khire U., Lasch S., Nagarathnam D., Scott W.J. 2004.
- 207.Khire U., Liu X.G., Nagarathnam D., Wood J., Wang L., Liu D., Zhao J., Guernon L., Zhang L. 2005.
- 208.Mich T.F., Domagala J.M. 1986. [Google Scholar]
- 209.Matsumoto J., Koshi M., Hiroshi E., Nakamura S. 1986. [Google Scholar]
- 210.Domagala J.M., Hanna L.D., Heifetz C.L., Hutt M.P., Mich T.F., Sanchez J.P., Solomon M. New structure-activity relationships of the quinolone antibacterials using the target enzyme. The development and application of a DNA gyrase assay. J. Med. Chem. 1986;29:394–404. doi: 10.1021/jm00153a015. [DOI] [PubMed] [Google Scholar]
- 211.Tomita K., Chiba K., Kashimoto S., Shibamon K.I., Tsuzuki Y. 1995.
- 212.Tomita K., Tsuzuki Y., Shibamori K.I., Tashima M., Kajikawa F., Sato Y., Kashimoto S., Chiba K., Hino K. Synthesis and structure-activity relationships of novel 7-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acids as antitumor agents. Part 1. J. Med. Chem. 2002;45:5564–5575. doi: 10.1021/jm010057b. [DOI] [PubMed] [Google Scholar]
- 213.Tsuzuki Y., Tomita K., Sato Y., Kashimoto S., Chiba K. Synthesis and structure-activity relationships of 3-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridines as novel antitumor agents. Bioorg. Med. Chem. Lett. 2004;14:3189–3193. doi: 10.1016/j.bmcl.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 214.Tsuzuki Y., Tomita K., Shibamori K.I., Sato Y., Kashimoto S., Chiba K. Synthesis and structure-activity relationships of novel 7-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acids as antitumor agents. Part 2. J. Med. Chem. 2004;47:2097–2109. doi: 10.1021/jm0304966. [DOI] [PubMed] [Google Scholar]
- 215.Hawtin R.E., Stockett D.E., Byl J.A., McDowell R.S., Tan N., Arkin M.R., Conroy A., Yang W., Osheroff N., Fox J.A. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLoS One. 2010;5:1–10. doi: 10.1371/journal.pone.0010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Beno B.R., Yeung K.S., Bartberger M.D., Pennington L.D., Meanwell N.A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015;58(11):4383–4438. doi: 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- 217.Drlica K., Mustaev A., Towle T.R., Luan G., Kerns R.J., Berger J.M. Bypassing fluoroquinolone resistance with quinazolinediones: studies of drug-gyrase-DNA complexes having implication for drug design. ACS Chem. Biol. 2014;9:2895–2904. doi: 10.1021/cb500629k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Huband M.D., Cohen M.A., Zurack M., Hanna D.L., Skerlos L.A., Sulavik M.C., Givson G.W., Gage J.W., Ellsworth E., Stier M.A., Gracheck S.J. In vitro and in vivo activities of PD 0305970 and PD 0326448, new bacterial gyrase/topoisomerase inhibitors with potent antibacterial activities versus multidrug-resistant Gram-positive and fastidious organism groups. Antimicrob. Agents Chemother. 2007;51:1191–1201. doi: 10.1128/AAC.01321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Domagala J.M., Heifetz C.L., Hutt M.P., Mich T.F., Nichols J.B., Solomon M., Worth D.F. 1-Substituted 7-[3-[(ethylamino)methyl]-1-pyrrolidinyl]-6,8difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids. New quantitative structure-activity relationships at N1 for the quinolone antibacterials. J. Med. Chem. 1988;31:991–1001. doi: 10.1021/jm00400a017. [DOI] [PubMed] [Google Scholar]
- 220.
- 221.Abbas J.A., Stuart R.K. Vosaroxin: a novel antineoplastic quinolone. Expert Opin. Investig. Drugs. 2012;21:1223–1233. doi: 10.1517/13543784.2012.699038. [DOI] [PubMed] [Google Scholar]
- 222.Hoch U., Lynch J., Sato Y., Kashimoto S., Kajikawa F., Furutani Y., Silerman J.A. Voreloxin, formerly SNS-595, has potent activity against a broad panel of cancer cell lines and in vivo tumor models. Cancer Chemother. Pharmacol. 2009;64:53–65. doi: 10.1007/s00280-008-0850-3. [DOI] [PubMed] [Google Scholar]
- 223.Evanchik M.J., Allen D., Yoburn J.C., Silverman J.A., Hoch U. Metabolism of (+)-1,4-dihydro-7-(trans-3-methoxy-4-methylamino-1-pyrrolidinyl)-4-oxo-1-(2-thiazolyl)-1,8-naphthyridine-3-carboxylic acid (voreloxin; formerly SNS-595), a novel replication-dependent DNA-damaging agent. Drug Metab. Dispos. 2009;37:594–601. doi: 10.1124/dmd.108.023432. [DOI] [PubMed] [Google Scholar]
- 224.Silverman J., Hoch U., Evanchick M., Furutani Y., Walker D. In vitro and in vivo activity of SPS-595, a novel cell cycle inhibitory cytotoxic in murine syngeneic and human xenograft tumor models. Proc. Am. Assoc. Cancer Res. 2004;•••:45. [Abstract 3840]. [Google Scholar]
- 225.Hoch U., Evanchick M., Silverman J. 2005. [Google Scholar]
- 226.Advani R.H., Hurwitz H.I., Gordon M.S., Ebbinghaus S.W., Mendelson D.S., Wakelee H.A., Hoch U., Silverman J.A., Havrilla N.A., Berman C.J., Fox J.A., Allen R.S., Adelman D.C. Voreloxin, a first-in-class anticancer quinolone derivative, in relapsed-refractory solid tumors: a report on two dosing schedules. Clin. Cancer Res. 2010;16:2167–2175. doi: 10.1158/1078-0432.CCR-09-2236. [DOI] [PubMed] [Google Scholar]
- 227.Mills D.A., Fekrazad H.M., Verschraegen C.F. SNS-595, a naphthyridine cell cycle inhibitor and stimular of apoptotis for the treatment of cancers. Curr. Opin. Invest. 2008;9:647–657. [PubMed] [Google Scholar]
- 228.Moscow J., Schneider E., Sikic B.I., Morrow C.S., Cowan K.H. 2006. Drug Resistance and its Clinical Circumvention. [Google Scholar]
- 229.Jin H.E., Song B., Kim S.B., Shim W.S., Kim D.D., Chong S., Chung S.J., Shim C.K. Transport of gemifloxacin, a 4th generation quinolone antibiotic, in the Caco-2 and engineered MDCKII cells, and potential involvement of efflux transporters in the intestinal absorption of the drug. Xenobiotica. 2013;43:355–367. doi: 10.3109/00498254.2012.720740. [DOI] [PubMed] [Google Scholar]
- 230.Vallet C.M., Marquez B., Nhiri N., Anantharajah A., Mingeot-Leclercq M.P., Tulkens P.M., Lallemand J.Y., Jacquet E., Van Bambeke F. Modulation of the expression of ABC transporters in murine (J774) macrophages exposed to large concentrations of the fluoroquinolone antibiotic moxifloxacin. Toxicology. 2011;290:178–186. doi: 10.1016/j.tox.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 231.Kwatra D., Vadlapatla R.K., Vadlapudi A.D., Pal D., Mitra A.K. Interaction of gatifloxacin with efflux tranporters: a possible mechanism for drug resistance. Int. J. Pharm. 2010;395:114–121. doi: 10.1016/j.ijpharm.2010.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Marquez B., Caceres N.E., Mingeot-Leclercq M.P., Tulkens P.M., Van Bambeke F. Identification of the efflux tranporter of the fluoroquinolone antibiotic ciprofloxacin in murine macrophages: studies with ciprofloxacin-resitant cells. Antimicrob. Agents Chemother. 2009;53:2410–2416. doi: 10.1128/AAC.01428-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Michot J.M., Seral C., Van Bambeke F., Mingeot-Leclercq M.P., Tulkens P.M. Influence of efflux transporters on the accumulation and efflux of four quinolones (ciprofloxacin, levofloxacin, garenoxacin, and moxifloxacin) in J774 macrophages. Antimicrob. Agents Chemother. 2005;49:2429–2437. doi: 10.1128/AAC.49.6.2429-2437.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lowes S., Simmons N.L. Multiple pathways for fluoroquinolone secretion by human intestinal epithelial (Caco-2) cells. Br. J. Pharmacol. 2002;135:1263–1275. doi: 10.1038/sj.bjp.0704560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Walsby E.J., Coles S.J., Knapper S., Burnett A.K. The topoisomerase II inhibitor voreloxin causes cell cycle arrest and apoptosis in myeloid leukemia cells and acts in synergy with cytarabine. Haematologica. 2011;96:393–399. doi: 10.3324/haematol.2010.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ichikawa Y., Ghanefar M., Bayeva M., Wu R., Khechaduri A., Prasad S.V., Mutharasan R.K., Naik T.J., Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Mjos K.D., Cawthray J.F., Jamieson G., Fox J.A., Orvig C. Iron (III)-binding of the anticancer agents doxorubicin and vosaroxin. Dalton T. 2015;44:2348–2358. doi: 10.1039/c4dt02934h. [DOI] [PubMed] [Google Scholar]
- 238.Moualla H., Mills D.A., Hromas R., Verschraegen C.F. Voreloxin. Drugs Future. 2009;34:363–374. [Google Scholar]
- 239.Sasine J.P., Schiller G.J. Emerging strategies for high-risk and relapsed/refractory acute myeloid leukemia: novel agents and approaches currently in clinical trials. Blood Rev. 2015;29:1–9. doi: 10.1016/j.blre.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 240.Lancet J.E. New agents: great expectations not realized. Best Pract. Res. Cl. Ha. 2013;26:269–274. doi: 10.1016/j.beha.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 241.Freeman C., Keane N., Swords R., Giles F. Vosaroxin: a new valuable tool with the potential to replace anthracyclines in the treatment of AML. Expert Opin. Pharmacother. 2013;14:1417–1427. doi: 10.1517/14656566.2013.799138. [DOI] [PubMed] [Google Scholar]
- 242.El-Amm J., Tabbara I.A. Vosaroxin for acute myeloid leukemia. Clin. Investig. (Lond.) 2014;4:147–152. [Google Scholar]
- 243.Lancet J.E.; Roboz, G.J.; Cripe, L.D.; Michelson, G.C.; Fox, J.A.; Leavitt, R.D.; Chen, T.; Hawtin, R.; Craig, A.R.; Ravandi, F.; Maris, M.B.; Stuart, R.K.; Karp, J.E. A phase 1b/2 study of vosaroxin in combination with cytarabine in patients with relapsed or refractory acute myeloid leukemia. Heamatologica. 2015;100:231–237. doi: 10.3324/haematol.2014.114769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Stuart R.K., Cripe L.D., Maris M.B., Cooper M.A., Stone R.M., Dakhil S.R., Turturro F., Stock W., Mason J., Shami P.J., Strickland S.A., Costa L.J., Borthakur G., Michelson G.C., Fox J.A., Leavitt R.D., Ravandi F. REVEAL-1, a phase 2 dose regimen optimization study of vosaroxin in older poor-risk patients with previously untreated acute myeloid leuaemia. Br. J. Haematol. 2015;168:796–805. doi: 10.1111/bjh.13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ravandi F., Ritchie E.K., Sayar S.H., Lancet J.E., Craig M.D., Vey N., Strickland S.A., Schiller G.J., Jabbour E., Erba H.P., Pigneux A., Horst H.A., Recher C., Klimek V.M., Cortes J., Feldman E.J., Craig A.R., Fox J.A., Ward R., Smith J., Acton G., Mehta C., Kantarjian H.M., Stuart R.K. 2014. [Google Scholar]
- 246.Fan J.Y., Sun D., Yu H., Kerwin S.M., Hurley L.H. Self-assembly of a quinobenzoxazine-Mg+2 complex on DNA: a new paradigm for the structure of a drug-DNA complex and implications for the structure of the quinolone bacterial gyrase-DNA complex. J. Med. Chem. 1995;38:408–424. doi: 10.1021/jm00003a003. [DOI] [PubMed] [Google Scholar]
- 247.Yu H., Hurley L.H., Kerwin S.M. Evidence for the formation of 2:2 drug-Mg+2 dimers in solution and for the formation of dimeric drug complexes on DNA from the DNA-accelerated photochemical reaction of antineoplastic quinobenzoxazines. J. Am. Chem. Soc. 1996;118:7040–7048. [Google Scholar]
- 248.Kwok Y., Zeng Q., Hurley L.H. Structural insight into a quinolone-topoisomerase II-DNA complex. J. Biol. Chem. 1999;274:17226–17235. doi: 10.1074/jbc.274.24.17226. [DOI] [PubMed] [Google Scholar]
- 249.Kwok Y., Sun D., Clement J.J., Hurley L.H. The quinobenzoxazines: relationship between DNA binding and biological activity. Anticancer Drug Des. 1999;14:443–450. [PubMed] [Google Scholar]
- 250.Sun D., Thompson B., Cathers B.E., Salazar M., Kerwin S.M., Trent J.O., Jenkins T.C., Neidle S., Hurley L.H. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 251.Hurley L.H., Wheelhouse R.T., Sun D., Kerwin S.M., Salazar M., Fedoroff O.Y., Han F.X., Han H., Izbicka E., Von Hoff D.D. G-quadruplexes as targets for drug design. Pharmacol. Ther. 2000;85:141–158. doi: 10.1016/s0163-7258(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 252.Wong H.M., Payet L., Huppert J.L. Function and targeting of G-quadruplexes. Curr. Opin. Mol. Ther. 2009;11:146–155. [PubMed] [Google Scholar]
- 253.Crees Z., Girard J., Rios Z., Botting G.M., Harrington K., Shearrow C., Wojdyla L., Stone A.L., Uppada S.B., Devito J.T., Puri N. Oligonucleotides and G-quadruplex stabilizers: targeting telomers and telomerase in cancer therapy. Curr. Pharm. Des. 2014;20:6422–6437. doi: 10.2174/1381612820666140630100702. [DOI] [PubMed] [Google Scholar]
- 254.Neidle S. Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J. 2010;277:1118–1125. doi: 10.1111/j.1742-4658.2009.07463.x. [DOI] [PubMed] [Google Scholar]
- 255.Tomlinson C.G., Cohen S.B., Bryan T.M. Inhibition of Telomerase: Promise, Progress, and Potential Pitfalls. In: Neidle S., editor. Cancer Drug Design and Discovery, 2nd. Vol. 7. Amsterdam: Elsevier, Inc.; 2014. pp. 491–527. [Google Scholar]
- 256.Zeng Q., Dwok Y., Kerwin S.M., Mangold G., Hurley L.H. Design of new topoisomerase Ii inhibitors based upon a quinobenzoxazine self-assembly model. J. Med. Chem. 1998;41:4273–4278. doi: 10.1021/jm980265c. [DOI] [PubMed] [Google Scholar]
- 257.Duan W., Rangan A., Vankayalapati H., Kim M.Y., Zeng Q., Sun D., Han H., Fedoroff O.Y., Nishioka D., Rha S.Y., Izbicka E., Von Hoff D.D., Hurley L.H. Design and synthesis of fluoroquinophenoxazines that interact with human telomeric G-quadruplexes and their biological effects. Mol. Cancer Ther. 2001;1:103–120. [PubMed] [Google Scholar]
- 258.Kim M.Y., Duan W., Guzman-Gleason M., Hurley L.H. Design, synthesis, and biological evaluation of a series of fluoroquinoanthroxazines with contrasting dual mechanisms of action against topoisomerase II and G-quadruplexes. J. Med. Chem. 2003;46:571–583. doi: 10.1021/jm0203377. [DOI] [PubMed] [Google Scholar]
- 259.Balasubramanian S., Hurley L.H., Neidle S. Targeting G-guadruplexes in gene promotors: a novel anticancer strategy? Nat. Rev. Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Brooks T.A., Hurley L.H. Targeting MYC expression through G-quadruplexes. Genes Cancer. 2010;1:641–649. doi: 10.1177/1947601910377493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Staehelin H.F., Wartburg A. The chemical and biological route from podophyllotoxin glucoside to etoposide: ninth Cain memborial award lecture. Cancer Res. 1991;51:5–15. [PubMed] [Google Scholar]
- 262.
- 263.College of Pharmacy University of Arizone COP scientists further anti-cancer drug development.
- 264.Alcaro S., Musetti C., Distinto S., Casatti M., Zagotto G., Artese A., Parrotta L., Moraca F., Costa G., Ortuso F., Maccioni E., Sissi C. Identification and characterization of new DNA G-quadruplex binders selected by a combination of ligand and structure-based virtual screening approaches. J. Med. Chem. 2013;56:843–855. doi: 10.1021/jm3013486. [DOI] [PubMed] [Google Scholar]
- 265.Gatto B., Leo E. Drugs acting on the beta isoform of human topoisomerase II (p180). Curr. Med. Chem. 2003;3:173–185. doi: 10.2174/1568011033482486. [DOI] [PubMed] [Google Scholar]
- 266.Austin C.A., Marsh K.L. Eukaryotic DNA topoisomerase IIβ. BioAssays. 1998;20:215–226. doi: 10.1002/(SICI)1521-1878(199803)20:3<215::AID-BIES5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 267.Azarova A.M., Lyu Y.L., Lin C.P., Tsai Y.C., Lau J.Y., Wang J.C., Liu L.F. Roles of DNA toposiomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA. 2007;104:11014–11019. doi: 10.1073/pnas.0704002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Cowell I.G., Austin C.A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health. 2012;9:2075–2091. doi: 10.3390/ijerph9062075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 270.Chen W., Qiu J., Shen Y. Topoisomerase IIα, rather than IIβ, is a promising taret in development of anti-cancer drugs. Drug Discov. Ther. 2012;6:230–237. [PubMed] [Google Scholar]
- 271.Vejpongsa P., Yeh E.T. Topoisomerase 2β: a promising molecular target for primary prevention of anthracycline-induced cardiotoxicity. Clin. Pharmacol. Ther. 2014;95:45–52. doi: 10.1038/clpt.2013.201. [DOI] [PubMed] [Google Scholar]
- 272.Pendleton M.J., Lindsey R.H., Felix C.A., Grimwade D., Osheroff N. Toposiomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014;1310:98–110. doi: 10.1111/nyas.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Song J.H., Kweon S.H., Kim H.J., Lee T.H., Min W.S., Kim H.J., Kim Y.K., Hwang S.Y., Kim T.S. High TOP2B/TOP2A expression ratio at diagnosis correlates with favourable outcome for standard chemotherapy in acute myeloid leukemia. Br. J. Cancer. 2012;107:108–115. doi: 10.1038/bjc.2012.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Mordente A., Meucci E., Silvestrini A., Martorana G.E., Giardina B. Anthracyclines and mitochondria. Adv. Exp. Med. Biol. 2012;942:385–419. doi: 10.1007/978-94-007-2869-1_18. [DOI] [PubMed] [Google Scholar]
- 275.Hisatomi T., Sueoka-Aragane N., Sato A., Tomimasu R., Ide M., Kurimasa A., Okamoto K., Kimura S., Sueoka E. NK314 potentiates antitumor activity with adult T-cell leukemia-lymphoma cells by inhibition of dual targets on topoisomerase IIα and DNA-dependent protein kinase. Blood. 2011;117:3575–3584. doi: 10.1182/blood-2010-02-270439. [DOI] [PubMed] [Google Scholar]
- 276.Toyoda E., Kagaya S., Cowell I.G., Kurosawa A., Kamoshita K., Nishikawa K., Iiizumi S., Koyama H., Austin C.A., Adachi N. NK314, a topoisomerase II inhibitor that specifically targets the α isoform. J. Biol. Chem. 2008;283:23711–23718. doi: 10.1074/jbc.M803936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Guo L., Liu X., Nishikawa K., Plunkett W. Inhibition of topisomerase IIα and G2 cell cycle arrest by NK314, a novel benzo[c]phenanthridine currently in clinical trials. Mol. Cancer Ther. 2007;6:1501–1508. doi: 10.1158/1535-7163.MCT-06-0780. [DOI] [PubMed] [Google Scholar]
- 278.Wohlkonig A., Chan P.F., Fosberry A.P., Homes P., Huang J., Kranz M., Leydon V.R., Miles T.J., Pearson N.D., Perera R.L., Shillings A.J., Gwynn M.N., Bax B.D. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 2010;17:1152–1153. doi: 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- 279.Wendorff T.J., Schmidt B.H., Heslop P., Austin C.A., Berger J.M. The structure of DNA-bound human topoisomerase II alpha: conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012;424:109–124. doi: 10.1016/j.jmb.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Aldred K.J., Schwanz H.A., Li G., McPherson S.A., Turnbough C.L., Kerns R.J., Osheroff N. Overcoming taret-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem. Biol. 2013;8:2660–2668. doi: 10.1021/cb400592n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Aldred K.J., Schwanz H.A., Li G., Williamson B.H., McPherson S.A., Turnbough C.L., Kerns R.J., Osheroff N. The activity of quinolone CP-115,955 against bacterial and human type II topoisomerases is mediated by different interactions. Biochemistry. 2015;54:1278–1286. doi: 10.1021/bi501073v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Perez J.J., Lupala C.S., Gomez-Gutierrez P. Designing type II topoisomerase inhibitors: a molecular modeling approach. Curr. Top. Med. Chem. 2014;14:40–50. doi: 10.2174/1568026613666131113150046. [DOI] [PubMed] [Google Scholar]
- 283.Chen S.H., Chan N.L., Hsieh T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013;82:139–170. doi: 10.1146/annurev-biochem-061809-100002. [DOI] [PubMed] [Google Scholar]
- 284.Bishop J.M. Molecular themes in oncogenesis. Cell. 1991;64:235–248. doi: 10.1016/0092-8674(91)90636-d. [DOI] [PubMed] [Google Scholar]
- 285.Larson E.R., Fisher P.H. New Approaches to Antitumor Therapy. In: Allen R.C., editor. Annual Reports in Medicinal Chemistry. Vol. 24. San Diego: Academic Press, Inc.; 1989. pp. 121–128. [Google Scholar]
- 286.Dobrusin E.M., Fry D.W. Protein Tyrosine Kinases and Cancer. In: Bristol J.A., editor. Annual Reports in Medicinal Chemistry. Vol. 27. San Diego: Academic Press, Inc.; 1992. pp. 169–176. [Google Scholar]
- 287.Bolton G.L., Sebolt-Leopold J.S., Hodges J.C. Ras Oncogene Directed Approaches in Cancer Chemotherapy. In: Bristol J.A., editor. Annual Reports in Medicinal Chemistry. Vol. 29. San Diego: Academic Press, Inc.; 1994. pp. 166–174. [Google Scholar]
- 288.Gibbs J.B., Oliff A. Pharmaceutical research in molecular oncology. Cell. 1994;79:193–198. doi: 10.1016/0092-8674(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 289.Sekeres M.L. Trying to Fool Cancer. 2015.
- 290.Bailly C. Cell-targeted cytotoxics: a new generation of cytotoxic agents for cancer treatment. Phytochem. Rev. 2014;13:171–181. [Google Scholar]
- 291.Nitiss J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Yano S., Zhang Y., Zhao M., Hiroshima Y., Miwa S., Uehara F., Kishimoto H., Tazawa H., Bouvet M., Fujiwara T., Hoffman R.M. Tumor-targeting Salmonella typhimurium A1-R decoys quiescent cancer cells to cycle as visualized by FUCCI imaging and become sensitive to chemotherapy. Cell Cycle. 2014;13:3958–3963. doi: 10.4161/15384101.2014.964115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Macy M.E., DeRyckere D., Gore L. Vandetanib mediates anti-leukemia activity by multiple mechanisms and interacts synergistically with DNA damaging agents. Invest. New Drugs. 2012;30:468–479. doi: 10.1007/s10637-010-9572-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Willmore E., De Caux S., Sunter N.J., Tilby M.J., Jackson G.H., Austin C.A., Durkacz B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103:4659–4665. doi: 10.1182/blood-2003-07-2527. [DOI] [PubMed] [Google Scholar]
- 295.Sha S., Han H.W., Gao F., Liu T.B., Li Z., Zu C., Zhong W.Q., Zhu H.L. Discovery of fluoroquinolone derivatives as potent, selective inhibitors of PI3K. MedChemComm. 2015;6:2029–2035. [Google Scholar]
- 296.Chen T.C., Hsu Y.L., Tsai Y.C., Chang Y.W., Kuo P.L., Chen Y.H. Gemifloxacin inhibits migration and invasion and induces mesenchymal-epithelial transition in human breast adenocarcinoma cells. J. Mol. Med. 2014;92:53–64. doi: 10.1007/s00109-013-1083-4. [DOI] [PubMed] [Google Scholar]
- 297.Yadav V., Sultana S., Yadav J., Saini N. Gatifloxacin induces S and G2-phase cell cycle arrest in pancreatic cancer cells via p21/p27/p53. PLoS One. 2012;7:e47796. doi: 10.1371/journal.pone.0047796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Seo K.W., Holt R., Jung Y.S., Rodriguez C.O., Chen X., Rebhun R.B. Fluoroquinolone-mediated inhibition of cell growth, S-G2/M cell cycle arrest, and apoptosis in canine osteosarcoma cell lines. PLoS One. 2012;7:e42960. doi: 10.1371/journal.pone.0042960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Niwa M., Hotta K., Kanamori Y., Hirota M., Kozawa O., Fujimoto S. Interaction of human neutrophils and grepafloxacin, fluoroquinolone antibiotics: involvement of p38MAPK activation. Ensho. Saisei. 2005;25:501–506. [Google Scholar]
- 300.Smart D.J., Halicka H.D., Traganos F., Darzynkiewicz Z., Williams G.M. Ciprofloxacin-induced G2 arrest and apoptosis in TK6 lymphoblastoid cells is not dependent on DNA double-strand break formation. Cancer Biol. Ther. 2008;7:113–119. doi: 10.4161/cbt.7.1.5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Aranha O., Grignon R., Fernandes N., McDonnell T.J., Wood D.P., Sarkar F.H. Suppression of human prostate cancer cell growth by ciprofloxacin is associated with cell cycle arrest and apoptosis. Int. J. Oncol. 2003;22:787–794. [PubMed] [Google Scholar]
- 302.Golub A.G., Yakovenko O.Y., Bdzhola V.G., Sapelkin V.M., Zien P., Yarmoluk S.M. Evaluation of 3-carboxy-4(1H)-quinolones as inhibitors of human protein kinase CK2. J. Med. Chem. 2006;49:6443–6450. doi: 10.1021/jm050048t. [DOI] [PubMed] [Google Scholar]