

Abstract
Extracellular signal regulated kinase½ (ERK1/2) signaling is critical to endothelin-1 (ET-1)-induced cardiomyocyte hypertrophy. This study was to investigate ERK1/2 signaling and hypertrophic response to ET-1 stimulation in cardiomyocytes (CMs) from spontaneous hypertensive rats (SHR) and normotensive Wistar-Kyoto rats (WKY). Primary neonatal SHR and WKY CMs were exposed to ET-1 for up to 24 hrs. Minimal basal ERK1/2 phosphorylation was present in WKY CMs, while a significant baseline ERK1/2 phosphorylation was observed in SHR CMs. ET-1 induced a time- and dose-dependent increase in ERK1/2 phosphorylation in both SHR and WKY CMs. However, ET-1-induced ERK1/2 activation occurred much earlier with significantly higher peak phosphorylation level, and stayed elevated for longer duration in SHR CMs than that in WKY CMs. ET-1-induced hypertrophic response was more prominent in SHR CMs than that in WKY CMs as reflected by increased cell surface area, intracellular actin density, and protein synthesis. Pre-treatment with ERK1/2 phosphorylation inhibitor PD98059 completely prevented ET-1-induced ERK1/2 phosphorylation and increases in cell surface area and protein synthesis in SHR and WKY CMs. The specific PI3 kinase inhibitor LY294002 blocked ET-1-induced Akt and ERK1/2 phosphorylation, and protein synthesis in CMs. These data indicated that ERK1/2 signaling was differentially enhanced in CMs, and was associated with increased cardiac hypertrophic response to ET-1 in SHR. ET-1-induced ERK1/2 activation and cardiac hypertrophy appeared to be mediated via PI3 kinase/Akt signaling in SHR and WKY. The differential ERK1/2 activation in SHR CMs by ET-1 might represent a potential target for combination therapy of hypertension.
Keywords: Akt, cardiomyocyte hypertrophy, endothelin, ERK1/2, hypertension, spontaneous hypertensive rat
Introduction
Endothelin-1 (ET-1) is a multifunctional hormone that plays an important role in the control of peripheral vascular tone and the development of hypertension, as well as cardiac hypertrophy [1, 2]. Cardiac hypertrophy is a compensatory response of the heart to stresses such as mechanical stretch and sustained neurohormonal stimulation or growth factor release. It is characterized by excessive enlargement of cardiomyocytes (CMs) and progressive interstitial fibrosis [3]. Maladaptive cardiac hypertrophy could progress to congestive heart failure, a leading cause of morbidity and mortality in the world. Under normal situation, although CMs only account for 30% of the total cell number in the heart ventricle, they represent over 70% of the total ventricular mass [4]. Davis Heart & Lung Research Institute, the Ohio State University Medical Center, DHLRI Suite 200; 473 West 12th Ave, Columbus, OH 43210, USA; Tel: (614)247-7760; Fax: 614-293-5614; E-mail: zhenguo.liu@osumc.edu
Therefore, ventricular CMs play a critical role in ventricular remodeling during the progression of cardiac hypertrophy [5]. ET-1 is one of important factors involved in myocardial hypertrophy. It has been demonstrated that ET-1 induces significant hypertrophic changes of CMs in cultured neonatal rat CMs [6, 7]. In vivo studies have shown that ET-1 level is significantly elevated in plasma and myocardium in patients with hypertension and in spontaneously hypertensive rats (SHR); while prolonged suppression of endothelin prevents the development of hypertension and cardiac hypertrophy in SHR [8, 9].
Extracellular signal regulated kinase ½ (ERK1/2) belongs to the subfamily of mitogen-activated protein kinase (MAPK). ERK1/2 is activated by a variety of extracellular stimuli including ET-1, angiotensin II (AngII), and phenylephrine (PE), and is involved in the regulation of various biological functions like cell proliferation and differentiation [2, 10]. Recent observations indicate that ERK1/2 also plays a key role in the development of cardiac hypertrophy in both in vitro and in vivo experiments [11-13]. It is reported that suppression of MAPK signaling attenuates PE-induced hypertrophic response in cultured rat CMs [14]. ERK activities in cardiac muscle are found to be chronically increased in hypertensive rats from the onset of hypertension to the establishment of cardiac hypertrophy, although the role of ERK1/2 signaling in cardiac hypertrophy in vivo still remains to be established [15, 16].
The regulatory mechanisms for ERK1/2 signaling in hypertrophic myocardium have not been fully understood. A number of factors are involved in the activation of ERK1/2 including intracellular Ca2+, protein kinase C (PKC), and phosphoinositide 3-kinases (PI3 kinase) [17]. ET-1 is shown to initiate hypertrophic changes in CMs from Wistar-Kyoto rats (WKY) and Sprague-Dawley rats (SD) through ERK signaling pathway [18]. SHR rats develop sustained hypertension at their early age with massive left ventricular hypertrophy that is similar to the changes in patients with hypertensive heart diseases [19]. However, the mechanisms for cardiac hypertrophy remain largely unclear in SHR rats. In the present study, we evaluated ERK1/2 signaling and hypertrophic response to ET-1 stimulation in cultured neonatal CMs from SHR in comparison with its progenitor strain, normotensive WKY. PI3 kinase/Akt signaling was also examined in this study.
Methods
Isolation and Culture of Cardiac Myocytes
The experimental protocol in this study followed the “Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health”, and was approved by the Experimental Animal Usage Ethics Committee of Shanghai Jiao-Tong University School of Medicine (SCXK2007-0005). Primary cultures of ventricular CMs were prepared from 1-2 day old neonatal SHR and WKY rats using the method previously described with minor modifications [20, 21]. Briefly, CMs were dispersed from the neonatal rat ventricles by a series of incubation at 37 ºC in D-hanks solution containing 1.0 mg/ml trypsin (1:250, Difco Laboratories). The dispersed cells were then cultured at a density of 3×105 cells per cm2 for 90 min (95% air / 5% CO2 at 37 ºC) to eliminate the fibroblasts. The unattached cells were transferred to, and cultured on collagen-coated plates at a density of 1.25×105 cells per cm2 in maintenance media consisting of 79% Modified Eagle’s Medium (MEM), 20% fetal bovine serum (Hyclone), and 1% penicillin/streptomycin solution (Gibco). After 48 hrs of culture, over 99% of the cells were CMs as estimated by immunocytochemical staining for the sarcomeric protein alpha-actin. The cells were then maintained in serum-free MEM for another 24 hrs, and divided into four treatment groups: control, cells treated with ET-1 (10 nM) alone, cells treated with ET-1 in the presence of ERK1/2 inhibitor PD98059 (50 µM), and cells treated with ET-1 in the presence of PI3 kinase inhibitor LY294002 (10 µM).
Immunocytochemistry and Microscopic Measurement of Cell Surface Area
The cells seeded in 6-well plates were stimulated with ET-1 for 24 hrs in the absence or presence of PD98059 or LY294009, and then fixed for 20 min at room temperature with 4% formaldehyde after rinsing with PBS. The cells were then permeabilized with 0.3% Triton X-100 in PBS, and exposed to the primary antibody (mouse anti-alpha actin monoclonal antibody, Calbiochem) with dilution factor of 1:200 in 1% BSA/PBS at 37 ºC for 1 hr, followed by incubation with peroxidase-conjugated secondary antibody (diluted 1:100 in 1% BSA/PBS). The nuclei of the cells were stained with hematoxylin. The microphotographs were scanned by the computerized-image analyzing system (Zeiss). The sizes of CMs were determined by measuring the areas of actin-positive cells. For each slide, the mean cell area was determined by dividing the total actin-positive area in three microphotographs (×10) by the number of independent myocyte nuclei (bi-nucleated cells counted as one cell).
Actin Staining
To visualize myofibrillar organization, fluorescent phalloidin staining was performed as described previously [18, 22]. After 24 hrs of culture in serum-free media, CMs were stimulated with ET-1 for 24 hrs. The cells on chamber slides were then prepared for staining by incubation with rhodamine-conjugated phalloidin for 1 hr in the dark. The preparations were readily examined and photographed using a confocal fluorescent microscope (Zeiss LSM 510) to determine the cytoskeleton actin organization.
[3H]-leucine Incorporation Assay
Protein synthesis was assessed using [3H]-leucine incorporation into CMs as described [23]. Briefly, the cells in 24-well plates were pretreated with PD98059 or LY294009 for 1 hr, and then exposed to ET-1 for 24 hrs. [3H]-leucine (1 µCi/ml or 3.7×104 Bq, Beijing Atom High Tech Co, Ltd) was added into the culture media at the same time as ET-1. The cells were then collected for the assay as described. The radioactivity incorporated into proteins of the cells in each group was determined by beta-liquid scintillation counting (Beckman; LS 6500).
Western Blot Analysis for ERK1/2 and Akt
CMs were stimulated with ET-1 (10 nM) for different time (0, 5, 10, 15, 30, 60 min) or with different doses (0, 5, 10, 20 nM) for 10 min with or without the treatment of PD98059 or LY294009, and then harvested for Western blot analysis as described previously [24]. The samples (12 µg) were separated on 10% SDS–PAGE, and transferred to a polyvinylidene difluoride membrane. The preparations were then incubated with primary antibodies (1:1000) against phosphor-ERK1/2 and Akt (Cell Signaling) for 2 hrs at room temperature. Subsequently, the preparations were exposed to horseradish peroxidase-conjugated IgG (1:2000 dilution, Protein Tech Group, Inc) for 1 hr at room temperature. The phosphorylated- and total- ERK1/2 or Akt were visualized by Western Lighting Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Inc). The intensity of individual band was analyzed using Image Qwin Software (Leica).
Statistical Analysis
All the data were presented as means ± SEM of “n” independent experiments. The data on cell surface measurement and [3H]-leucine incorporation assay were analyzed using ANOVA followed by Tukey post hoc multiple comparison tests. Western blot data were analyzed with Student’s t-test. A p value of 0.05 or less was considered statistically significant.
Results
ERK1/2 Signaling was Enhanced at Baseline and in Response to ET-1 in CMs from SHR
There was a low basal level of ERK1/2 phosphorylation in CMs from WKY rats as determined with Western blot (Fig. 1A). When exposed to ET-1, a time- and dose-dependent increase in ERK1/2 phosphorylation was observed in these cells. ERK1/2 phosphorylation reached its maximal level after 10 min incubation with ET-1. Exposure to ET-1 beyond 10 min or ET-1 concentration over 10 nM resulted in a rapid decline in ERK1/2 phosphorylation, and returned to baseline by 60 min (Fig. 1A & B). In contrast, a significant basal ERK1/2 phosphorylation was observed in CMs from SHR. When treated with ET-1, ERK1/2 phosphorylation increased rapidly, and reached its peak level by 5 min of ET-1 exposure. The elevated level of ERK1/2 activation dwindled slowly after 10 min, and remained at a high level after 60 min. The dose-response curve revealed that ERK1/2 activation started to occur at lower ET-1 dose in CMs from SHR as compared with the cells from WKY (Fig. 1B). The maximal level of ET-1-induced ERK1/2 activation in CMs from SHR was almost twice that in CMs from WKY. These results indicated that ET-1 induced an enhanced and sustained ERK1/2 phosphorylation in CMs from SHR compared to the ones from WKY.
Fig. (1).
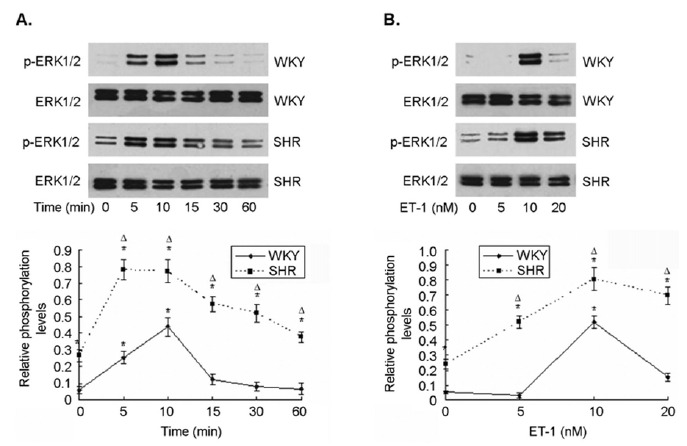
ERK1/2 activation was elevated in CMs from SHR at both baseline and in response to ET-1 stimulation. Primary neonatal SHR and WKY CMs were prepared, and exposed to different concentrations of ET-1 for up to 24 hrs. There was minimal basal ERK1/2 phosphorylation in CMs from WKY. In contrast, a significant level of basal ERK1/2 phosphorylation was observed in CMs from SHR. ET-1 induced a time- and dose-dependent increase in ERK1/2 phosphorylation in CMs from both SHR and WKY. However, ET-1-induced ERK1/2 phosphorylation occurred significantly earlier with higher maximal level of activation, and stayed elevated for longer time in CMs from SHR as compared with WKY. A representative Western blotting was shown in the top panel. The relative ERK1/2 phosphorylation in the cells from three independent experiments was summarized in the bottom panel.
ET-1 Induced an Increased Hypertrophic Response in CMs from SHR
As expected, when CMs were exposed to ET-1, the cells exhibited a symmetric growth with a significant increase in cell surface area (Fig. 2A & B). The cell surface area was increased by 40% and 30% for CMs from SHR and WKY rats, respectively, after ET-1 treatment (n=3, p < 0.01). The ET-1-induced increase in cell surface area was accompanied by an enhanced density of intracellular actin in the cells as evaluated by fluorescent phalloidin staining (Fig. 2C). The hypertrophic response to ET-1 was supported by increased protein synthesis as evaluated using [3H]-leucine incorporation assay. There was a low level of basal protein synthesis in the cells from both SHR and WKY rats without ET-1 stimulation that was significantly increased by 80% and 62% for CMs from SHR and WKY rats (p < 0.05, n=3, Fig. 4), respectively. These data demonstrated that ET-1 induced a greater hypertrophic response with increased cell surface area and protein synthesis in CMs from SHR than that in the cells from WKY rats.
Fig. (2).
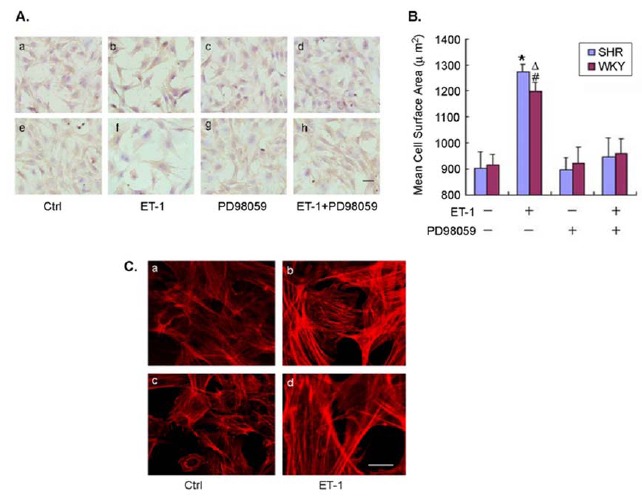
Enhanced hypertrophic response of CMs from SHR to ET-1 stimulation.
Fig. (4).
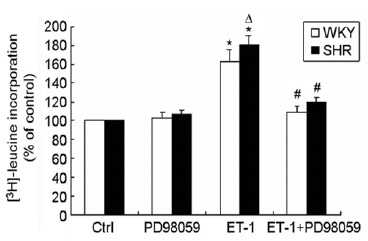
The specific ERK1/2 kinase inhibitor PD98059 suppressed ET-1-induced protein synthesis in CMs. Enhanced protein synthesis by ET-1 was observed in CMs in SHR and WKY rats, especially in the cells from SHR. Pre-treatment of the cells with PD98059 completely blocked ET-1-induced protein synthesis as evaluated with [3H]-leucine incorporation assay. Data were presented as means ± SEM of three different experiments (each in triplicate).
Inhibition of ERK1/2 Signaling Prevented ET-1-Induced Cardiomyocyte Hypertrophy
To further investigate the role of ERK1/2 signaling in ET-1-induced cardiomyocyte hypertrophy, the cells were pre-treated with the specific ERK1/2 phosphorylation inhibitor PD98059. As expected, treatment with PD98059 completely prevented the ET-1-induced ERK1/2 phosphorylation in CMs from both SHR and WKY rats as analyzed with Western blot (Fig. 3). PD98059 treatment had no effect on the cell surface area in the control cells (no exposure to ET-1) (Fig. 2B). However, pre-treatment with PD98059 totally blocked the ET-1-induced increase in cell surface area in CMs from both SHR and WKY rats (Fig. 2B). The surface area of the cells treated with ET-1 in the presence of PD98059 was the same as that of control cells. Similarly, when the cells were pre-treated with PD98059, the ET-1-induced increase in [3H]-leucine incorporation was completely reversed in CMs from both SHR and WKY rats (Fig. 4). Of note, treatment with PD98059 had no impact on the basal [3H]-leucine incorporation in CMs from either SHR or WKY rats. These results further suggested that ET-1-induced hypertrophy was mediated through ERK1/2 signaling in both SHR and WKY rats.
Fig. (3).
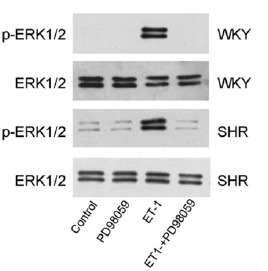
ET-1-induced phosphorylation of ERK1/2 was blocked by PD98059.
ERK1/2 phosphorylation was significantly increased by ET-1 in the neonatal CMs from both SHR and WKY rats. Pretreatment of the cells with PD98059 (50 µM) for 30 min completely prevented ET-1-induced increase in ERK1/2 phosphorylation as evaluated with Western blotting.
Inhibition of PI3 kinase/Akt Signaling Attenuated ET-1-induced ERK1/2 Activation and Protein Synthesis
To determine the involvement of PI3 kinase/Akt signaling in ET-1-induced ERK1/2 phosphorylation and protein synthesis, CMs from both SHR and WKY rats were pre-treated with specific PI3 kinase inhibitor LY294002 for 30 min prior to exposure to ET-1. There was detectable level of Akt phosphorylation in CMs from both SHR and WKY rats at baseline that was significantly increased when exposed to ET-1 in a time-dependent manner (Fig. 5). The maximal
Fig. (5).
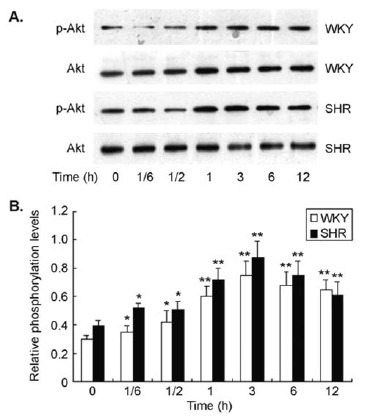
ET-1 stimulated Akt phosphorylation in cultured CMs. There was a low level of Akt phosphorylation in CMs that was significantly increased by ET-1 stimulation in a time-dependent manner in both SHR and WKY rats. ET-1-induced Akt phosphorylation reached maximal level at 3 hrs after stimulation in the cells.
ET-1-induced Akt activation occurred after 3 hrs of stimulation, and slowly declined afterwards. The pattern of ET-1-induced Akt activation was similar in the cells from both SHR and WKY rats. When the cells were pre-treated with LY294002, the baseline Akt phosphorylation was not affected. However, the ET-1-induced increase in Akt phosphorylation was completely reversed to the baseline level (Fig. 6A). Interestingly, pre-treatment of CMs with LY294002 also completely prevented ET-1-induced ERK1/2 phosphorylation in both SHR and WKY rats (Fig. 6B), suggesting that PI3 kinase / Akt signaling is crucial to ET-1-induced ERK1/2 activation in these cells. In parallel to ERK1/2 activation, pre-treatment of CMs with LY294002 also inhibited ET-1-induced protein synthesis as evaluated with [3H]-leucine incorporation assay (Fig. 6c. These data indicate that PI3 kinase / Akt /ERK1/2 signaling plays an important role in ET-1-induced hypertrophy of CMs in both SHR and WKY rats.
Fig. (6).
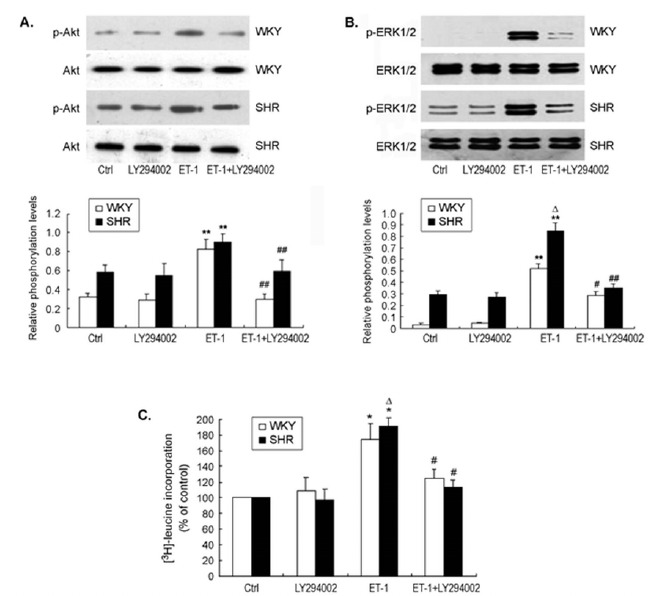
Inhibition of PI3 kinase / Akt signaling prevented ET-1-induced ERK1/2 phosphorylation and protein synthesis in CMs from SHR and WKY rats.
Discussion
ET-1 is a 21-amino acid peptide that is abundantly expressed in CMs, vascular endothelial cells, and vascular smooth muscle cells primarily as a local paracrine/autocrine hormone [25]. ET-1 plays an important role in the pathogenesis of a variety of diseases such as hypertension, atherosclerosis, and acute myocardial infarction [26-28]. ET-1 also contributes to the development of cardiac hypertrophy [6]. This was confirmed in the present study as evidenced by increased cell surface area, enhanced intracellular actin density, and protein synthesis in the neonatal CMs from both SHR and WKY rats. We also demonstrated that ET-1-induced myocardial cell hypertrophy was mediated through PI3 kinase / Akt / ERK1/2 signaling pathway in neonatal cardiac myocyte preparations.
It is well established that ERK1/2 signaling is important in ET-1-induced cardiac hypertrophy [13, 17, 29]. However, there is no previous report that showed ERK1/2 signaling was involved in cardiac hypertrophy induced by ET-1 in SHR. For the first time, we observed that the level of ERK1/2 phosphorylation was significantly enhanced in CMs from SHR at baseline, and remained at high level after ET-1 stimulation compared to the cells from WKY rats. The enhanced ERK1/2 activation in response to ET-1 stimulation was associated with augmented ET-induced hypertrophy as reflected with increased cell surface area and protein synthesis in CMs from SHR as compared with WKY. This may provide a new mechanism for the development of cardiac hypertrophy in SHR and possibly in patients with essential hypertension. Of note, the blood pressure in the neonatal SHR is not elevated until a few weeks after birth in these animals [30, 31]. The changes in ERK1/2 signaling occurred much earlier than the development of left ventricle hypertrophy and hypertension. This may provide a window for the prevention of ventricular hypertrophy in SHR and hypertensive patients by modifying ERK1/2 signaling pathway. Interestingly, increased activation of ERK1/2 by AngII stimulation was also observed in vascular smooth muscle cells from SHR as compared with WKY [32, 33]. Inhibition of ERK1/2 activity with PD98059 improved endothelial function and reduced arterial reactivity to Ang II in SHR in vivo, and blocked vascular smooth muscle cell growth responses to AngII in vitro [32, 34].
The role of Akt signaling in ET-1-induced cardiac hypertrophy is controversial although it is important to physiological cardiac growth and cardiac hypertrophy induced by other growth factors like connective tissue growth factor [35, 36]. It has been recently shown that isolated CMs exhibit an enhanced protein synthesis in response to ET-1 stimulation with increased incorporation of [3H]-leucine into CMs from the Akt1 knock-out mice. Interestingly, the Akt1 knock-out animals develop an exacerbated form of cardiac hypertrophy in the setting of aortic constriction [35], indicating that Akt1 inhibits cardiac hypertrophy in this animal model. The level of phosphorylated Akt was not increased in isolated neonatal CMs from WKY rats [37]. In our study, however, the data clearly showed that ET-1 stimulated Akt activation in CMs from both SHR and WKY rats. Our results also indicated that PI3 kinase / Akt signaling was involved in ET-induced cardiac hypertrophy in vitro as evidenced by the fact that the specific PI3 kinase inhibitor LY294002 prevented ET-induced protein synthesis. The reason(s) for the apparent opposite roles of Akt signaling in cardiac hypertrophy is very likely due to the different models used. Further studies are needed to clarify the difference in the role of Akt signaling in ET-1-induced cardiac hypertrophy.
One of the major findings in the present study is that inhibition of Akt signaling with LY294002 also led to the suppression of ET-1-induced ERK1/2 activation in CMs from both SHR and WKY rats. It is very likely that the hypertrophic response of CMs to ET-1 is through PI3 kinase / Akt / ERK1/2 signaling cascade in both SHR and WKY rats. It is known that PI3 kinase is involved in the regulation of a number of cell functions including membrane trafficking, actin rearrangement and adhesion, as well as cell survival [38]. PI3 kinase also plays a critical role in cardiac growth and contractility [39, 40]. Recent studies demonstrate that PI3 kinase is involved in maladaptive cardiac hypertrophy triggered by neurohormonal mediators and biomechanical stress [41]. The mechanisms for PI3 kinase-mediated cardiac growth and hypertrophy are believed to be associated with the activation of heterotrimeric G-protein-coupled receptors, growth factors or hormones [40, 41]. PI3 kinase / Akt signaling is another major pathway for PI3 kinase function including cell survival and proliferation [42, 43]. There are extensive cross-talking and interactions between PI3 kinase / Akt pathway and ERK pathways on the regulations of a variety of biological events [44-47]. ERK pathway is frequently co-activated along with PI3 kinase / Akt pathway, and functioning synergistically [44, 47]. In our study, the specific PI3 kinase inhibitor LY294002 blocked ET-1-induced Akt phosphorylation and ERK1/2 activation, as well as protein synthesis in CMs, suggesting that ET-1-induced cardiac hypertrophy is mediated through PI3 kinase Akt / ERK1/2 signaling pathway in SHR and WKY rats. However, the mechanism for ET-1-induced PI3 kinase activation is unclear. It is possible that ET-1 stimulates PI3 kinase directly [19].
In conclusion, this study demonstrated that ERK1/2 signaling was significantly enhanced both at baseline and in response to ET-1 stimulation in isolated neonatal CMs in SHR as compared with WKY rats. The elevated ET-1 signaling was associated with increased hypertrophic response to ET-1 stimulation in CMs in SHR. PI3 kinase / Akt signaling was involved in ET-1-induced ERK1/2 activation and cardiac hypertrophy in CMs from both SHR and WKY rats. The data from this study might provide a molecular explanation for the development of cardiac hypertrophy in SHR, and help to identify potential molecular targets for prevention and treatment of cardiac hypertrophy and combination therapy of hypertension.
ACKNOWLEDGEMENTS
This work was supported by a grant from the National Nature Science Foundation of China (grant number 30670830 to NF), and partially by NIH R01 HL094650 (ZGL).
LIST OF ABBREVIATIONS
- AngII
Angiotensin II
- BSA
Bovine Serum Albumin
- CMs
Cardiomyocytes
- ERK1/2
Extracellular Signal Regulated Kinase½
- ET-1
Endothelin-1
- MAPK
Mitogen-Activated Protein Kinase
- PE
Phenylephrine
- PI3
Phosphoinositide 3-Kinases
- PKC
Protein Kinase C
- SHR
Spontaneous Hypertensive Rat
- WKY
Normotensive Wistar-Kyoto Rat
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Meyers K.E., Sethna C. Endothelin antagonists in hypertension and kidney disease. Pediatr. Nephrol. 2013;28(5):711–720. doi: 10.1007/s00467-012-2316-4. [DOI] [PubMed] [Google Scholar]
- 2.Vignon-Zellweger N., Heiden S., Miyauchi T., Emoto N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci. 2012;91(13-14):490–500. doi: 10.1016/j.lfs.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 3.Wilkins B.J., Molkentin J.D. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2004;322(4):1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 4.Eghbali M., Czaja M.J., Zeydel M., et al. Collagen chain mRNAs in isolated heart cells from young and adult rats. J. Mol. Cell. Cardiol. 1988;20:267–276. doi: 10.1016/s0022-2828(88)80059-2. [DOI] [PubMed] [Google Scholar]
- 5.Harada M., Itoh H., Nakagawa O., et al. Significance of ventricular myocytes and nonmyocytes interaction during cardiocyte hypertrophy: evidence for endothelin-1 as a paracrine hypertrophic factor from cardiac nonmyocytes. Circulation. 1997;96:3737–3744. doi: 10.1161/01.cir.96.10.3737. [DOI] [PubMed] [Google Scholar]
- 6.Ito H., Hirata Y., Hiroe M., et al. Endothelin-1 induces hypertrophy with enhanced expression of muscle-specific genes in cultured neonatal rat cardiomyocytes. Circ. Res. 1991;69:209–215. doi: 10.1161/01.res.69.1.209. [DOI] [PubMed] [Google Scholar]
- 7.Shubeita H.E., Mcdonough P.M., Harris A.N., et al. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy. J. Biol. Chem. 1990;265:20555–20562. [PubMed] [Google Scholar]
- 8.Mckinsey T.A., Olson E.N. Cardiac hypertrophy: sorting out the circuitry. Curr. Opin. Genet. Dev. 1999;9:267–274. doi: 10.1016/s0959-437x(99)80040-9. [DOI] [PubMed] [Google Scholar]
- 9.Stasch J.P., Hirth-Dietrich C., Frobel K., et al. Prolonged endothelin blockade prevents hypertension and cardiac hypertrophy in stroke-prone spontaneously hypertensive rats. Am. J. Hypertens. 1995;8:1128–1134. doi: 10.1016/0895-7061(95)00224-D. [DOI] [PubMed] [Google Scholar]
- 10.Page C., Doubell A.F. Mitogen-activated protein kinase (MAPK) in cardiac tissues. Mol. Cell. Biochem. 1996;157:49–57. doi: 10.1007/BF00227880. [DOI] [PubMed] [Google Scholar]
- 11.Bueno O.F., Molkentin J.D. Involvement of Extracellular Signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ. Res. 2002;91:776–781. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y.M., Shioda N., Han F., et al. DY-9760e inhibits endothelin-1-induced cardiomyocyte hypertrophy through inhibition of CaMKII and ERK activities. Cardiovasc. Ther. 2009;27:17–27. doi: 10.1111/j.1755-5922.2008.00068.x. [DOI] [PubMed] [Google Scholar]
- 13.Yue T.L., Gu J.L., Wang C., et al. Extracellular signal-regulated kinase plays an essential role in hypertrophic agonists, endothelin-1 and phenylephrine-induced cardiomyocyte hypertrophy. J. Biol. Chem. 2000;275:37895–37901. doi: 10.1074/jbc.M007037200. [DOI] [PubMed] [Google Scholar]
- 14.Glennon P.E., Kaddoura S., Sale E.M., et al. Depletion of mitogen-activated protein kinase using an antisense oligodeoxynucleotide approach downregulates the phenylephrine-induced hypertrophic response in rat cardiac myocytes. Circ. Res. 1996;78:954–961. doi: 10.1161/01.res.78.6.954. [DOI] [PubMed] [Google Scholar]
- 15.Izumi Y., Kim S., Murakami T., et al. Cardiac mitogen - activated protein kinase activities are chronically increased in stroke-prone hypertensive rats. Hypertension. 1998;31:50–56. doi: 10.1161/01.hyp.31.1.50. [DOI] [PubMed] [Google Scholar]
- 16.Purcell N.H., Wilkins B.J., York A., et al. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. USA. 2007;104:14074–14079. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molkentin J.D., Dorn G.W. Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu. Rev. Physiol. 2001;63:391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 18.Ueyama T., Kawashima S., Sakoda T., et al. Requirement of activation of the extracellular signal-regulated kinase cascade in myocardial cell hypertrophy. J. Mol. Cell. Cardiol. 2000;32:947–960. doi: 10.1006/jmcc.2000.1135. [DOI] [PubMed] [Google Scholar]
- 19.Katada T., Kurosu H., Okada T., et al. Synergistic activation of a family of phosphoinositide 3-kinase via G-protein coupled and tyrosine kinase-related receptors. Chem. Phys. Lipids. 1999;98:79–86. doi: 10.1016/s0009-3084(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 20.Simpson P. Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells is an alpha 1 adrenergic response. J. Clin. Invest. 1983;72:732–738. doi: 10.1172/JCI111023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson P., Savion S. Differentiation of rat myocytes in single cell cultures with and without proliferating nonmyocardial cells. Circ. Res. 1982;50:101–116. doi: 10.1161/01.res.50.1.101. [DOI] [PubMed] [Google Scholar]
- 22.Torsoni A.S., Marin T.M., Velloso L.A., et al. RhoA/ROCK signaling is critical to FAK activation by cyclic stretch in cardiac myocytes. Am. J. Physiol. 2005;289:H1488–H1496. doi: 10.1152/ajpheart.00692.2004. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi N., Saito Y., Kuwahara K., et al. Hypertrophic responses to cardiotrophin-1 are not mediated by STAT3, but via a MEK5-ERK5 pathway in cultured cardiomyocytes. J. Mol. Cell. Cardiol. 2005;38:185–192. doi: 10.1016/j.yjmcc.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 24.Filipeanu C.M., Zhou F., Claycomb W.C., et al. Regulation of the cell surface expression and function of angiotension II type I receptor by Rab1-mediated endoplasmic reticulum-to Golgi transport in cardiac myocytes. J. Biol. Chem. 2004;279:41077–41084. doi: 10.1074/jbc.M405988200. [DOI] [PubMed] [Google Scholar]
- 25.Yanagisawa M. The endothelin system, a new target for therapeutic intervention. Circulation. 1994;89:1320–1322. doi: 10.1161/01.cir.89.3.1320. [DOI] [PubMed] [Google Scholar]
- 26.Little P.J., Ivey M.E., Osman N. Endothelin-1 actions on vascular smooth muscle cell functions as a target for the prevention of atherosclerosis. Curr. Vasc. Pharmacol. 2008;6:195–203. doi: 10.2174/157016108784911966. [DOI] [PubMed] [Google Scholar]
- 27.Niccoli G., Lanza G.A., Shaw S., et al. Endothelin-1 and acute myocardial infarction: a no-reflow mediator after successful percutaneous myocardial revascularization. Eur. Heart J. 2006;27:1793–1798. doi: 10.1093/eurheartj/ehl119. [DOI] [PubMed] [Google Scholar]
- 28.Sugden P.H. Signalling pathways in cardiac myocyte hypertrophy. Ann. Med. 2001;33:611–622. [PubMed] [Google Scholar]
- 29.Sugden P.H., Clerk A. Endothelin signalling in the cardiac myocyte and its pathophysiological relevance. Curr. Vasc. Pharmacol. 2005;3:343–351. doi: 10.2174/157016105774329390. [DOI] [PubMed] [Google Scholar]
- 30.Seubert J.M., Xu F., Graves J.P., et al. Differential renal gene expression in prehypertensive and hypertensive spontaneously hypertensive rats. Am. J. Physiol. Renal Physiol. 2005;289:F552–F561. doi: 10.1152/ajprenal.00354.2004. [DOI] [PubMed] [Google Scholar]
- 31.Trippodo N.C., Frohlich E.D. Similarities of genetis(spontaneous) hypertension. Man and rat. Circ. Res. 1981;48:309–319. doi: 10.1161/01.res.48.3.309. [DOI] [PubMed] [Google Scholar]
- 32.El Mabrouk M., Touyz R.M., Schiffrin E.L. Differential ANG II-induced growth activation pathways in mesenteric artery smooth muscle cells from SHR. Am. J. Physiol. Heart Circ. Physiol. 2001;281:H30–H39. doi: 10.1152/ajpheart.2001.281.1.H30. [DOI] [PubMed] [Google Scholar]
- 33.Touyz R.M., Deschepper C., Park J.B., et al. Inhibition of mitogen-activated protein/extracellular signal-regulated kinase improves endothelial function and attenuates Ang II-induced contractility of mesenteric resistance arteries from spontaneously hypertensive rats. J. Hypertens. 2002;20:1127–1134. doi: 10.1097/00004872-200206000-00024. [DOI] [PubMed] [Google Scholar]
- 34.Touyz R.M., He G., El Mabrouk M., et al. Differential activation of extracellular signal-regulated protein kinase 1/2 and p38 mitogen activated-protein kinase by AT1 receptors in vascular smooth muscle cells from Wistar-Kyoto rats and spontaneously hypertensive rats. J. Hypertens. 2001;19:553–559. doi: 10.1097/00004872-200103001-00006. [DOI] [PubMed] [Google Scholar]
- 35.DeBosch B., Treskov I., Lupus T.S., et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 36.Hayata N., Fujio Y., Yamamoto Y., et al. Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochem. Biophys. Res. Commun. 2008;370:274–278. doi: 10.1016/j.bbrc.2008.03.100. [DOI] [PubMed] [Google Scholar]
- 37.Arino T., Tanonaka K., Kawahara Y., et al. Effects of tanshinone VI on phosphorylation of ERK and Akt in isolated cardiomyocytes and cardiac fibroblasts. Eur. J. Pharmacol. 2008;580:298–305. doi: 10.1016/j.ejphar.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Toker A., Cantley L.C. Signaling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 39.Crackower M.A., Oudit G.Y., Kozieradzki I., et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 40.McMullen J.R., Shioi T., Zhang L., et al. Osphoinositide 3-kinase (p110 alpha) plays a critical role for the induction of physiological, but not pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patrucco E., Notte A., Barberis L., et al. PI3K gamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and –independent effects. Cell. 2004;118:375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 42.Cantley L.C. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 43.Katso R., Okkenhaug K., Ahmadi K., et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 44.Chambard JC, Lefloch R, Pouysségur J, et al. ERK implication in cell cycle regulation. 1773. [DOI] [PubMed]
- 45.Hayashi H., Matsuzaki O., Muramatsu S., et al. Centaurin-alpha1 is a phosphatidylinositol 3-kinase-dependent activator of ERK1/2 mitogen-activated protein kinase. J. Biol. Chem. 2006;281:1332–1337. doi: 10.1074/jbc.M505905200. [DOI] [PubMed] [Google Scholar]
- 46.Hayashi H., Tsuchiya Y., Nakayama K., et al. Down-regulation of the PI3-kinase/Akt pathway by ERK MAP kinase in growth factor signaling. Genes Cells. 2008;13:941–947. doi: 10.1111/j.1365-2443.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- 47.Laprise P., Langlois M.J., Boucher M.J., et al. Down-regulation of MEK/ERK signaling by E-cadherin-dependent PI3K/Akt pathway in differentiating intestinal epithelial cells. J. Cell. Physiol. 2004;199:32–39. doi: 10.1002/jcp.10432. [DOI] [PubMed] [Google Scholar]