

Abstract
Cardiac hypertrophy is characterized by maladaptive tissue remodeling that may lead to heart failure or sudden death. MicroRNAs (miRs) are negative regulators of angiotensin II and the angiotensin II receptor subtype 1 (AGTR1), which are two components involved in cardiac hypertrophy. In the present study, the interaction between angiotensin II receptor subtype 1 (AGTR1) signaling and miR-155 was investigated. Rat H9C2 (2–1) cardiomyocytes were transfected with miR-155 analogues or inhibitors, then stimulated with angiotensin II to induce cardiac hypertrophy. miR-155 expression was revealed to be altered following transfection with chemically-modified miR-155 analogues and inhibitors in rat cardiomyocytes. In cell cardiac hypertrophy models, the cell surface area, AGTR1, atrial natriuretic peptide and myosin heavy chain-β mRNA expression levels were revealed to be lower in cells stimulated with miR-155 analogue-transfected cells treated with angiotensin II compared with cells stimulated with angiotensin alone (P<0.05), as determined using reverse transcription-polymerase chain reaction (PCR), quantitative PCR and western blot analyses. Furthermore, calcineurin mRNA and protein, intracellular free calcium and nuclear factor of activated T-cells-4 proteins were downregulated in miR-155 analogue-transfected cells treated with angiotensin II, as compared with cells stimulated with angiotensin II alone (P<0.05). In conclusion, the current study indicates that miR-155 may improve cardiac hypertrophy by downregulating AGTR1 and suppressing the calcium signaling pathways activated by AGTR1.
Keywords: microRNA-155, cardiac hypertrophy, angiotensin II receptor type 1
Introduction
Cardiac hypertrophy, the thickening of heart muscle, is a compensatory response to physical stimuli or pathological insults. These pathological remodeling responses are often accompanied by fibrosis, pump failure, myocyte degeneration and apoptosis, which may culminate in heart failure and sudden death (1). Heart failure occurs when the heart is unable to pump blood at a rate proportional to the body's requirement for oxygen, or when this function leads to cardiogenic pulmonary edema. With >1 million hospitalizations annually (up 175% in the past 25 years), and costs of ~$15.4 billion, acute heart failure is a critical health concern. Furthermore, half of the patients discharged from the hospital are readmitted within 6 months. In-hospital mortality rates remain high, between 4 and 7% (2,3). Heart failure is a significant problem as the population ages. The prevalence in the US is 2.5% of the population, or 5 million patients (3) Cardiac hypertrophy is a major determinant of congestive heart failure. Angiotensin II is known to participate in cardiac hypertrophy by binding to angiotensin II receptor subtype 1 (AGTR1). In addition, AGTR1 overexpression has been closely associated with cardiac hypertrophy (4).
MicroRNAs (miRs) are endogenous, highly conserved, small non-coding RNAs that negatively regulate the expression of target genes at the post-transcriptional level (5). miR-155 is encoded by the human miR-155 host gene and is abundantly expressed in the lungs, heart and kidneys (6). Seok et al (7) and Heymans et al (8) have revealed that gain-of-function mutations in miR-155 exacerbate myocardial hypertrophy, whereas loss-of-function mutations ameliorate cardiac hypertrophy. Sethupathy et al (9) experimentally investigated the target sites for hsa-miR-155 within the 3′-untranslated region of the human AGTR1 gene and demonstrated that hsa-miR-155 downregulated the expression of AGTR1. These previous findings suggest that miR-155 ameliorates hypertension by modulating AGTR1 expression, as AGTR1 signaling occurs upstream of cardiac hypertrophy (10). The present study therefore aimed to test the hypothesis that miR-155 promotes cardiac hypertrophy by targeting AGTR1, and to determine the underlying molecular behavior of miR-155 in cardiac hypertrophy.
Materials and methods
Cell culture and reagents
Rat H9C2 (2–1) cardiomyocytes were purchased from the China Center for Type Culture Collection, Wuhan University (Wuhan, China). Lipofectamine 2000, TRIzol® and Platinum SYBR® Green qPCR SuperMix-UDG were purchased from Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA), angiotensin II was purchased from Sigma-Aldrich (St. Louis, MO, USA), and the mirVana PARIS RNA and Native Protein Purification kit was from Ambion (Thermo Fisher Scientific, Inc.). Rabbit anti-AGTR1 polyclonal antibody (cat. no. ab9391) was purchased from Abcam (Cambridge, UK). Rabbit anti-calcineurin-β (CaN-β; cat. no. BS6114), nuclear factor of activated T-cells (NFAT-4; cat. no. BS1762) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; cat. no. BS60630) polyclonal antibodies were purchased from Bioworld Technology, Inc. (St. Louis Park, MN, USA). Goat anti-rabbit polyclonal antibody labeled with horseradish peroxidase (HRP) (cat. no. BA1125) were purchased from Boster Biological Technology, Co., Ltd., (Wuhan, China). miR-155 analogue and inhibitor, with or without fluorescein (FAM) conjugation, were synthesized by GenePharma (Shanghai, China), in accordance with the miR-155 sequence provided in the miRBase database (www.mirbase.org/; accession no. MIMAT0030409; 5′-UUAAUGCUAAUUGUGAUAGGGGU-3′). The sequences were as follows: miR-155 analogue forward, 5′-UUAAUGCUAAUUGUGAUAGGGGU-3′ and reverse, 5′-CCCUAUCACAAUUAGCAUUAAUU-3′; and miR-155, 5′-ACCCCUAUCACAAUUAGCAUUAA-3′. The analogues and inhibitor were diluted with sterile water to a final concentration of 20 µmol/l and stored at −80°C until use.
miR-155 transfection
Rat cardiomyocytes were seeded into 6-well plates at a density of 1×108 cells/ml in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.). Cells were incubated at 37°C and 5% CO2 until 30–50% confluency was reached, after which the cardiomyocytes were transfected with either 8µl miR-155 analogue or inhibitors using lipofectamine, followed by incubation for 24 h in serum-free medium. Cells were then stimulated with 1×10−7 mmol/l angiotensin II for 48 h in various groups. FAM fluorescence was assessed at an excitation wavelength of 480 nm and an emission wavelength of 520 nm using the LAS X Widefield Systems (Leica Microsystems GmbH, Wetzlar, Germany).
Experimental groups
The cardiomyocytes were divided into six groups, as follows: i) Untreated control cells, subjected to no transfection or chemical regents; ii) treated with 1×10−7 mmol/l angiotensin (Ang II) only; iii) transfected with 80 nmol/l miR-155 analogue; iv) transfected with 80 nmol/l miR-155 inhibitors; v) 80 transfected with nmol/l miR-155 analogue and treated with 1×10−7 mmol/l AngII; and vi) transfected with 80 nmol/l miR-155 inhibitors and treated with 1×10−7 mmol/l AngII. The concentration of AngII was determined in accordance with a previous study by Zheng et al (11). A previous study by Cheng et al (12) was used to determine the concentration of miR-155 analogue and miR-155 inhibitor used.
Cell area measurement
Cardiomyocytes were fixed with 4% paraformaldehyde for 15 min and imaged using phase contrast microscopy. The LAS X Widefield Systems was used to count cardiomyocytes and measure the diameter of single cells. For each group, 10 images were captured under different perimeters, and 20 cardiomyocyetes were counted within each perimeter. ImageJ 1.45 software (National Institutes of Health, Bethesda, MA, USA) was used to calculate the number of cardiomyocetes and measure the average surface area. Three replicates were used in each experimental group.
Intracellular calcium ([Ca2+]i) measurements
Calcineurin and intracellular Ca2+ concentration are focal to the development of angiotensin II-induced cardiac hypertrophy (13,14). To determine intracellular calcium levels, 5 µmol/l Fura-2/AM (Biotium, Inc., Hayward, CA, USA) solution was added to the cell suspension in all groups for 30 min at 37°C. Cells were then washed twice with Hank's buffered salt solution supplemented with 0.2% bovine serum albumin (Roche Diagnostics-Basel, Switzerland). Measurements were then obtained with an excitation wavelength of 340 nm and an emission wavelength of 510 nm using a fluorescent spectrophotometer (Lengguang, Shanghai, China).
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative (PCR)
To detect atrial natriuretic peptide (ANP), myosin heavy chain-β (β-MHC), calcineurin-β (CaN-β), AGTR1, miR-155 and Rnu6 expression, cDNA was synthesized using Platinum SYBR Green qPCR SuperMix-UDG, according to the manufacturer's protocol. The RT-PCR primer sets are reported in Table I. Quantitative Stem-Loop RT-qPCR was used to detect the expression levels of miR-155, according to a previous study (15). These primers are listed in Table II. The RT-PCR cycling conditions were as follows: 94°C for 3 min; 35 cycles at 94°C for 30 sec; and 72°C for 1 min. Amplifications were performed in an ABI 7300 Real Time PCR System, under the following thermal cycling conditions: 50°C for 2 min, 95°C for 2 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 30 sec, and finally 20°C for 2 min. miR-155 expression levels were normalized to Rnu6 expression levels. Expression ratios were calculated by the 2−ΔΔCq method (16).
Table I.
Reverse transcription-polymerase chain reaction primers.
| Gene | Primers (5′-3′) | Sequence length (bp) | Melting temperature (°C) |
|---|---|---|---|
| β-MHC | |||
| F | AGGAAGAACCTACTGCGACTG | 264 | 53 |
| R | CATCCTTAGGGTTGGGTAGCAC | ||
| ANP | |||
| F | GGCTCCTTCTCCATCACC | 412 | 53 |
| R | CTCCAATCCTGTCAATCCTAC | ||
| CaN-β | |||
| F | CCATCTGGTAAAAGAAGGTCG | 198 | 53 |
| R | AGGTATCGTGTATTAGCAGGTGA | ||
| GAPDH | |||
| F | CAAGGTCATCCATGACAACTTTG | 496 | 58 |
| R | GTCCACCACCCTGTTGCTGTAG |
bp, base pairs; F, forward; R, reverse; β-MHC, myosin heavy chain-β; ANP, atrial natriuretic peptide; CaN-β, calcineurin-β; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Table II.
Rnu 6 and miR-155 primers.
| Gene | Reverse transcription-PCR primers (5′-3′) | Quantitative PCR primers (5′-3′) |
|---|---|---|
| Rnu 6 | CGCTTCACGAATTTCCGTGTCAT | F: CTCGCTTCGGCAGCACA |
| R: AACGCTTCACGAATTTGCGT | ||
| M-miR-155-5p | CTCAACTGGTGTCGTGGAGTCG | F: ACACTCCAGCTGGGTTAATGCTAATTGTGAT |
| GCAATTCAGTTGAGACCCCTAT | R: TGGTGTCGTGGAGTCG |
PCR, polymerase chain reaction; F, forward; R, reverse; miR, microRNA.
Western blot analysis
Total protein was obtained from the rat cardiomyocytes using radioimmunoprecipitation assay lysis buffer (Boster Biological Technology, Co., Ltd.). Protein samples (30 µg total protein) were separated by 10% SDS-PAGE, transferred to polyvinylidene fluoride membranes, and blocked with 5% skimmed milk for 1 h. The membranes were then incubated overnight at 4°C with rabbit polyclonal antibodies against AGTR1, GAPDH, CaN-β, and NFAT-4 (1:500 dilution). After washing the membranes with Tris-buffered saline supplemented with 0.05% Tween-20 (Boster Biological Technology, Co., Ltd.), the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (1:15,000 dilution) at room temperature for 1 h. The protein bands were visualized using enhanced chemiluminescence, after which images of the gels were captured and protein band intensities were quantified using Quantity One software, version 4.4 (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data were analyzed with SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). One-way analysis of variance and Tukey's post-hoc test were used to compare data among the various groups. P<0.05 was considered to represent a statistically significant difference. Data are presented as the mean ± standard error of the mean.
Results
miR-155 transfection increases miR-155 expression levels
To confirm that the miR-155 analogues had been successfully transfected into the cardiomyocytes, the fluorescence of FAM-labeled miR-155 analogues was observed and miR-155 mRNA expression levels were detected by RT-PCR (Fig. 1). miR-155 analogues labeled with FAM were successfully transfected into H9C2 (2–1) cells using lipofectamine (Fig. 1A-C), and miR-155 expression was significantly increased in cells transfected with FAM-labeled analogues, as compared with the control cells (P<0.05; Fig. 1D). Furthermore, downregulated miR-155 expression was demonstrated in the miR-155 inhibitor and angiotensin II + miR-155 inhibitor groups, as compared with the control group (P<0.05) In addition, the expression levels of miR-155 were significantly increased in the angiotensin II + miR-155 analogues group, as compared with the cells transfected with miR-155 analogues alone (P<0.05; Fig. 1D).
Figure 1.

Changes to cellular expression levels of miR-155 following transfection with miR-155 analogue and/or inhibitor. (A-C) Immunofluorescence demonstrating cellular distribution of miR-155 analogues labeled with fluorescein (magnification, ×400). (D) Graph illustrating relative miRNA expression levels following transfection with miR-155 analogue or inhibitor and/or treatment with AngII. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. miR-155, microRNA-155; AngII, angiotensin II.
AGTR1 mRNA and protein expression
Angiotensin II-AGTR1 signaling was shown to be markedly activated in cardiac hypertrophy (17). Therefore the effects of miR-155 on the mRNA and protein expression levels of AGTR1 were evaluated in the presence of angiotensin II (Fig. 2). Transfection with miR-155 analogues or inhibitors alone had no effect on the AGTR1 mRNA and protein expression levels (P>0.05), as detected by RT-PCR and immunoblotting, respectively. Angiotensin II treatment significantly increased the mRNA and protein expression leves of AGTR1, as compared with the control cells (P<0.05). Notably, miR-155 analogues significantly reduced the effects of angiotensin II on miR-155 expression in cardiomyocytes (P<0.05), but these were not restored to control levels. The mRNA and protein expression levels of AGTR1 were significantly increased in the angiotensin II + miR-155 inhibitor group, as compared with the angiotensin II alone group (P<0.05).
Figure 2.

(A) Effects of miR-155 analogue or inhibitor on AGTR1 mRNA and protein expression in cardiomyocytes. (B) Relative density between these groups. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. AGTR1, angiotensin II receptor subtype 1; AngII, angiotensin II; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; bp, base pairs.
Myocardiocyte surface area and the mRNA expression of β-MHC and ANP
Cardiac hypertrophy is characterized by an increase in the size of cardiomyocytes, and in the synthesis of ANP and β-MHC (18,19). Cardiomyocytes incubated with angiotensin II, or transfected with miR-155 analogues or inhibitors and then treated with angiotensin II, had greater surface areas, as compared with control cells (Fig. 3A and B). The mRNA expression levels of β-MHC (Fig. 4A-C) and ANP (Fig. 4B-D) were significantly increased in the angiotensin II-treated group, as compared with the control cells (P<0.05), whereas the mRNA expression levels of ANP and β-MHC in cells transfected with miR-155 inhibitors or analogues alone were not significantly different from the control cells (P>0.05). Furthermore, β-MHC and ANP levels were significantly decreased in the angiotensin II-treated, miR-155 analogue-transfected group, as compared with the angiotensin II only group (P<0.05; Fig. 4B).
Figure 3.

Changes to myocardial cell surface area observed following transfection with miR-155 analogue or inhibitor and/or treatment with AngII. (A) Micrographs of the cells (magnification, ×200); (B) Cell surface area among the groups. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. AngII, angiotensin II.
Figure 4.

(A) β-MHC and (B) ANP mRNA expression levels following transfection with miR-155 analogue or inhibitor and/or treatment with AngII. (C and D) Relative density of β-MHC and ANP mRNA expression. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. AngII, angiotensin II; β-MHC, myosin heavy chain-β; ANP, atrial natriuretic peptide; bp, base pairs.
[Ca2 +]i, NFAT-4 and CaN-β expression
In previous studies, angiotensin II was shown to promote CaN-β-mediated calcium influx and NFAT-4 translocation into the nucleus by activation of CaN-β in cardiomyocytes, resulting in upregulation of the expression of ANP and β-MHC (20). Upon angiotensin II treatment, the miR-155 analogue-transfected cells had lower intracellular calcium levels, and NFAT-4 and CaN-β expression levels (P<0.05; Figs. 5–7). However, treatment of miR-155 inhibitor-transfected cells with angiotensin II caused increased intracellular calcium, NFAT-4 and CaN-β expression (P<0.05; Fig. 5–7).
Figure 5.
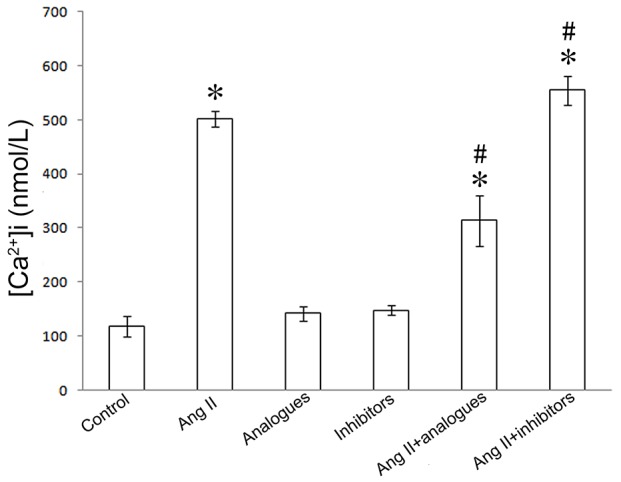
Intracellular calcium levels following transfection with miR-155 analogue or inhibitor and/or treatment with AngII. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. AngII, angiotensin II; [Ca2+]i, intracellular calcium levels.
Figure 7.
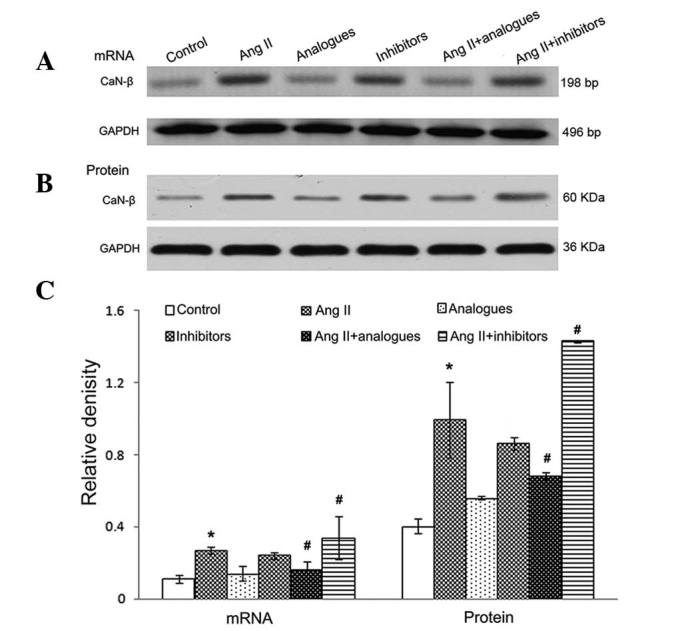
CaN-β (A) mRNA and (B) protein expression levels following transfection with miR-155 analogue or inhibitor and/or treatment with AngII. (C) Relative density of between these groups. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. AngII, angiotensin II; CaN-β, calcineurin-β; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; bp, base pairs.
Discussion
The angiotensin II-AGTR1 and calcineurin signal transduction pathways have vital roles in the development of myocardial hypertrophy (21). Angiotensin II has previously been reported to regulate the expression of numerous miRNAs, including miR-29b, miR-129-3p, miR-132, miR-132 and miR-212 (22). Sayed et al (23) reported in an in vivo model that numerous miRNAs are involved in the regulation of calcineurin signaling, and miR-155 was central to this cardiac hypertrophy model. Cheng et al (12) revealed that miR-155 inhibited AGTR1 expression, possibly through binding to the 3′ non-coding region of the AGTR1 gene, resulting in AGTR1 gene silencing.
The present study demonstrated that, compared with normally growing cardiomyocytes, miR-155 mRNA expression increased 3–4 fold upon transfection with miR-155 analogues when combined with angiotensin II treatment. However, transfection with miR-155 inhibitors had the opposite effect in cells treated with angiotensin II. Overexpression of miR-155 was able to effectively inhibit AGTR1 expression at the mRNA and protein level in hypertrophic cardiomyocytes. When hypertrophic H9C2 (2–1) cells were incubated with miR-155 analogues, intracellular calcium concentrations were reduced, and those of CaN-β and NFAT-4 were upregulated. Furthermore, cell area was increased in hypertrophic rat cardiomyocytes, and β-MHC and ANP levels, which are indicative of the degree of hypertrophy, were reduced. It was therefore speculated that miR-155 may inhibit angiotensin II-induced cardiac hypertrophy by silencing the AGTR1 and the calcineurin signal transduction pathways. However, transfection with miR-155 inhibitors did not alter ANP or β-MHC levels and cell area in hypertrophic H9C2 (2–1) cells, despite an increase in [Ca2+]i, CaN-β and NFAT-4 levels.
Two online prediction tools used to determine the miRNA targets, TargetScan and PicTar, were previously reported (24). These revealed that AGTR1 may be a target of miR-155. In addition, AGTR1 was combined with angiotensin II to enhance CaN-β (21). Myogenic enhancer factor 2, which has been reported to be involved in the regulation of CaN-β, is a regulatory target of miR-155 (25). Suppressor of cytokine signaling 1 from macrophages may also be a target of miR-155 and participate in cardiac hypertrophy (8). Conversely, angiotensin II may also elevate intracellular calcium concentration via the phospholipase C-inositol trisphosphate signaling pathway (26), and increase mitogen-activated protein kinase levels to induce cardiac hypertrophy (27). These data indicate that multiple factors cause cardiac hypertrophy, including the effects of AGTR1. miR-155 may therefore represent a target at multiple sites during the development of cardiac hypertrophy induced by angiotensin II The present study solely investigated the role of miR-155 within hypertrophic cardiomyocytes, however. The calcium signaling pathway may have an important role in the development of cardiac hypertrophy, and in the apoptosis of myocardial cells (28). In the current study, miR-155 inhibitors did not attenuate hypertrophy of H9C2 (2–1) cells. It is possible that when miR-155 is inhibited, activation of the calcium signaling pathway may lead to the apoptosis of a proportion of myocardial cells, thereby reducing the overall levels of myocardial hypertrophy markers.
In conclusion, the results of the present study demonstrated that miR-155 overexpression effectively inhibits angiotensin II-induced myocardial hypertrophy by preventing the effects of AGTR1. As a result, intracellular free calcium and NFAT-4 nuclear translocation were reduced. Additional studies are therefore required to determine the role of miR-155 as an important target in the treatment of cardiac hypertrophy.
Figure 6.

NFAT-4 protein expression levels following transfection with miR-155 analogue or inhibitor and/or treatment with AngII. *P<0.05, vs. the control group; #P<0.05, vs. the AngII group. AngII, angiotensin II; NFAT-4, nuclear factor of activated T-cells-4.
Acknowledgements
The present study was supported by a grant from the Municipal Science and Technology Bureau (grant no. ZD2012014).
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2.Abraham WT, Fonarow GC, Albert NM, Stough WG, Gheorghiade M, Greenberg BH, O'Connor CM, Sun JL, Yancy CW, Young JB. OPTIMIZE-HF Investigators and Coordinators: Predictors of in-hospital mortality in patients hospitalized for heart failure: Insights from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) J Am Coll Cardiol. 2008;52:347–356. doi: 10.1016/j.jacc.2008.04.028. [DOI] [PubMed] [Google Scholar]
- 3.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. 2013 ACCF/AHA guideline for the management of heart failure: Executive summary: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128:1810–1852. doi: 10.1161/CIR.0b013e31829e8807. [DOI] [PubMed] [Google Scholar]
- 4.Savoia C, Burger D, Nishigaki N, Montezano A, Touyz RM. Angiotensin II and the vascular phenotype in hypertension. Expert Rev Mol Med. 2011;13:e11. doi: 10.1017/S1462399411001815. [DOI] [PubMed] [Google Scholar]
- 5.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: A typical multifunctional microRNA. Biochim Biophys Acta. 2009;1792:497–505. doi: 10.1016/j.bbadis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Seok HY, Chen J, Kataoka M, Huang ZP, Ding J, Yan J, Hu X, Wang DZ. Loss of microRNA-155 protects the heart from pathological cardiac hypertrophy. Circ Res. 2014;114:1585–1595. doi: 10.1161/CIRCRESAHA.114.303784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L, Wijnands E, et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation. 2013;128:1420–1432. doi: 10.1161/CIRCULATIONAHA.112.001357. [DOI] [PubMed] [Google Scholar]
- 9.Sethupathy P, Borel C, Gagnebin M, Grant GR, Deutsch S, Elton TS, Hatzigeorgiou AG, Antonarakis SE. Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3′ untranslated region: A mechanism for functional single-nucleotide polymorphisms related to phenotypes. Am J Hum Genet. 2007;81:405–413. doi: 10.1086/519979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marian AJ. Experimental Therapies in Hypertrophic Cardiomyopathy. J Cardiovasc Transl Res. 2009;2:483–492. doi: 10.1007/s12265-009-9132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng L, Xu CC, Chen WD, Shen WL, Ruan CC, Zhu LM, Zhu DL, Gao PJ, et al. MicroRNA-155 regulates angiotensin II type 1 receptor expression and phenotypic differentiation in vascular adventitial fibroblasts. Biochem Biophys Res Commun. 2010;400:483–488. doi: 10.1016/j.bbrc.2010.08.067. [DOI] [PubMed] [Google Scholar]
- 12.Cheng W, Liu T, Jiang F, Liu C, Zhao X, Gao Y, Wang H, Liu Z, et al. microRNA-155 regulates angiotensin II type 1 receptor expression in umbilical vein endothelial cells from severely pre-eclamptic pregnant women. Int J Mol Med. 2011;27:393–399. doi: 10.3892/ijmm.2011.598. [DOI] [PubMed] [Google Scholar]
- 13.Gómez AM, Ruiz-Hurtado G, Benitah JP, Domínguez-Rodríguez A. Ca(2+) fluxes involvement in gene expression during cardiac hypertrophy. Curr Vasc Pharmacol. 2013;11:497–506. doi: 10.2174/1570161111311040013. [DOI] [PubMed] [Google Scholar]
- 14.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 15.Varkonyi-Gasic E1, Wu R, Wood M, Walton EF, Hellens RP. Protocol: A highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods. 2007;3:12. doi: 10.1186/1746-4811-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Saris JJ, 't Hoen PA, Garrelds IM, Dekkers DH, den Dunnen JT, Lamers JM, Jan Danser AH. Prorenin induces intracellular signaling in cardiomyocytes independently of angiotensin II. Hypertension. 2006;48:564–571. doi: 10.1161/01.HYP.0000240064.19301.1b. [DOI] [PubMed] [Google Scholar]
- 18.Cavallero S, González GE, Puyó AM, Rosón MI, Pérez S, Morales C, Hertig CM, Gelpi RJ, Fernández BE. Atrial natriuretic peptide behaviour and myocyte hypertrophic profile in combined pressure and volume-induced cardiac hypertrophy. J Hypertens. 2007;25:1940–1950. doi: 10.1097/HJH.0b013e3282435b1e. [DOI] [PubMed] [Google Scholar]
- 19.Haddad F, Qin AX, Bodell PW, Zhang LY, Guo H, Giger JM, Baldwin KM. Regulation of antisense RNA expression during cardiac MHC gene switching in response to pressure overload. Am J Physiol Heart Circ Physiol. 2006;290:H2351–H2361. doi: 10.1152/ajpheart.01111.2005. [DOI] [PubMed] [Google Scholar]
- 20.Lunde IG, Kvaløy H, Austbø B, Christensen G, Carlson CR. Angiotensin II and norepinephrine activate specific calcineurin-dependent NFAT transcription factor isoforms in cardiomyocytes. J Appl Physiol. 2011;111:1278–1289. doi: 10.1152/japplphysiol.01383.2010. [DOI] [PubMed] [Google Scholar]
- 21.Marian AJ. Hypertrophic cardiomyopathy: From genetics to treatment. Eur J Clin Invest. 2010;40:360–369. doi: 10.1111/j.1365-2362.2010.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeppesen PL, Christensen GL, Schneider M, Nossent AY, Jensen HB, Andersen DC, Eskildsen T, Gammeltoft S, Hansen JL, Sheikh SP. Angiotensin II type 1 receptor signalling regulates microRNA differentially in cardiac fibroblasts and myocytes. Br J Pharmacol. 2011;164:394–404. doi: 10.1111/j.1476-5381.2011.01375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 24.Kozomara A, Griffiths-Jones S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seok HY, Tatsuguchi M, Callis TE, He A, Pu WT, Wang DZ. miR-155 inhibits expression of the MEF2A protein to repress skeletal muscle differentiation. J Biol Chem. 2011;286:35339–35346. doi: 10.1074/jbc.M111.273276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinne A, Blatter LA. Activation of NFATc1 is directly mediated by IP3 in adult cardiac myocytes. Am J Physiol Heart Circ Physiol. 2010;299:H1701–H1707. doi: 10.1152/ajpheart.00470.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izumi Y, Kim S, Zhan Y, Namba M, Yasumoto H, Iwao H. Important role of angiotensin II-mediated c-Jun NH(2)-terminal kinase activation in cardiac hypertrophy in hypertensive rats. Hypertension. 2000;36:511–516. doi: 10.1161/01.HYP.36.4.511. [DOI] [PubMed] [Google Scholar]
- 28.Liu Q, Wilkins BJ, Lee YJ, Ichijo H, Molkentin JD. Direct interaction and reciprocal regulation between ASK1 and calcineurin-NFAT control cardiomyocyte death and growth. Mol Cell Biol. 2006;26:3785–3797. doi: 10.1128/MCB.26.10.3785-3797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]