

Abstract
Calorie restriction (CR) slows aging in numerous species. In the yeast Saccharomyces cerevisiae, this effect requires Sir2, a conserved NAD+-dependent deacetylase. We report that CR reduces nuclear NAD+ levels in vivo. Moreover, the activity of Sir2 and its human homologue SIRT1 are not affected by physiological alterations in the NAD+:NADH ratio. These data implicate alternate mechanisms of Sir2 regulation by CR.
Mild environmental stresses extend yeast life-span, including heat, osmotic stress, amino acid restriction, and glucose restriction (1–4). The latter two conditions are considered mimics of calorie restriction (CR), a diet that extends life-span in a broad range of species. The yeast NAD+-dependent histone deacetylase Sir2 (5–7) is required for life-span extension by glucose restriction (1, 3, 8) and low-intensity stress (8, 9). The function of Sir2 enzymes in longevity and cell survival appears to be conserved in higher organisms as well (10). Two models for how CR stimulates Sir2 activity are currently debated: by depleting nicotinamide, an inhibitory product of Sir2 itself, or by increasing either NAD+ or the NAD+:NADH ratio (1, 10). Although it is possible to affect Sir2 activity by genetically manipulating NAD+ metabolic pathways, it is not known whether NAD+ is a bona fide regulator of Sir2 in normal cells. A major obstacle has been determining the effective concentrations of these metabolites in living cells, as various levels of free NAD+ and NADH have been reported (11, 12).
To compare the levels of freely available NAD+ in the nuclei of wild-type and long-lived cells in which Sir2 is activated, a reporter assay was developed based on the Salmonella typhimurium NadR protein, a transcriptional repressor whose affinity for its recognition sequence, the NAD box, is specifically NAD+-dependent (13). NadR was converted to a yeast transcriptional activator by fusing its C terminus to a nuclear localization signal and the activation domain (AD) of the yeast Gal4 transcription factor (fig. S1) (14). Expression of the fusion protein was driven from a high-level constitutive promoter. A series of NAD boxes identical to their arrangement in the native S. typhimurium NAD regulon (fig. S2) were cloned upstream of a yeast HIS3 reporter (NADp-HIS3) and integrated as a single copy into the yeast genome. HIS3 expression is required for growth on media lacking histidine, and leaky expression of HIS3 can be suppressed by the inhibitor 3-aminotriazole (3-AT).
Only the strain carrying both the NadR-AD activator and the NADp-HIS3 reporter grew on plates lacking histidine with 3-AT (10 mM) (Fig. 1A). No growth was observed for control strains lacking the NadR-AD activator or with mutant NAD boxes (Fig. 1A), or for strains carrying other AD fusions (fig. S3). Loss-of-function mutations in the ribonucleotide kinase or nicotin-amide mononucleotide adenylyltransferase domains of NadR-AD also did not alter HIS3 expression (15). The NAD+ reporter system detected variations in nuclear NAD+ levels in vivo as a bna6Δ strain with ~30% lower steady-state NAD+ levels displayed retarded growth. In contrast, acetaldehyde, which regenerates NAD+ when reduced to ethanol, stimulated growth (Fig. 1B). Overexpression of HAP4, which increases respiration, also increased growth, whereas a mitochondrial mutant incapable of respiring grew very slowly (fig. S4).
Fig. 1.

A yeast reporter assay that detects freely available nuclear NAD+. (A) The reporter strain with both the nadR-AD plasmid and NAD-HIS3 construct was spotted in serial 10-fold serial dilutions onto reporter assay media: 2% glucose (w/v) synthetic complete (SC) medium with or without histidine + 3-aminotriazole (3-AT). The extent of growth on the test media is indicative of the availability of NAD+. Also shown are assays of negative control strains: empty vector (no NadR-AD), mutant NAD boxes (mut) (TGTgTA and its inverted repeat). Spots were incubated for ~48 hours at 30°C. (B) The NAD+ reporter system detects changes in availability of nuclear NAD+. NAD+ may be generated from nicotinic acid via the NAD+ pathway or from tryptophan via the de novo pathway, which is catalyzed by Bna1-6 (17, 18). Deletion of BNA6 decreases NAD+ levels, whereas exogenously supplied acetaldehyde increases NAD+ by oxidizing the NADH pool (24). BNA6 was deleted in the NAD+ reporter by replacing the entire coding region with a KanMX cassette and spotted on reporter assay media. The wild-type reporter strain was pregrown in liquid SC medium with or without acetaldehyde (10 mM) for 2 hours and then spotted on reporter assay media containing acetaldehyde (10 mM). (C) Life-span extending manipulations do not correlate with increased nuclear NAD+. To assay calorie-restricted cells, the NAD+ reporter strain was grown for 2 hours in defined SC medium with either 2.0% glucose (standard concentration) or 0.5% glucose (CR). Cultures were washed and spotted in serial 10-fold dilutions on assay media with the same glucose concentrations. The retarded growth of the strain on restricted medium was rescued by acetaldehyde (10 mM). (D) The NAD+ reporter strain was spotted on assay media and grown under two low-intensity stresses known to extend replicative life-span: high osmolarity (4% glucose) (2) and heat stress (37°C) (8, 9).
Growth of the NAD+ reporter strain on histidine-3-AT assay medium was slower in CR cells compared with the control strain (0.5% versus 2.0% glucose) (Fig. 1C) but im proved with exogenous acetaldehyde, confirming that low NAD+ was responsible for slow growth. Moreover, mild osmotic stress (4% glucose medium) (2) and heat stress (37°C) (8, 9) did not increase reporter expression (Fig. 1D). Thus NAD+ levels did not correlate with yeast life-span, indicating that Sir2 is not regulated by the availability of NAD+ under these conditions.
To quantify the levels of total free NAD+ in CR cells, we used in vivo 13C NMR (carbon 13 nuclear magnetic resonance) spectroscopy, a highly accurate, noninvasive methodology. Only free, non–protein-bound NAD+ and/or free NAD+/NADH (16) pools are available for interactions with Sir2 and could potentially play a regulatory role. Therefore, a major advantage of the NMR technique over previous studies (17, 18) is that it detects only unbound metabolites; those bound to macromolecules are unobservable as a result of severe line broadening (11, 19). Rapid small molecule equilibration across the nuclear envelope (20) suggests that measurements of free cellular NAD are likely indicative of the nuclear and cytoplasmic concentrations. Although NMR cannot distinguish NADP+ from NAD+ in living cells, the pool of NADP+ is negligible and below the threshold of detection (21) (fig. S5).
In media with 2.0% or 0.5% glucose, intracellular NAD+ concentrations were stable and NADH was below the threshold of detection (<200 μM) (figs. S6 and S7). Consistent with the NAD+ reporter, NAD+ concentrations were lower in CR cells than in controls (3.97 ± 0.02 versus 4.28 ± 0.14 mM, respectively) (Fig. 2). Addition of glucose did not affect NAD+ levels, indicating that cells were not glucose-starved. This confirms that increased NAD+ is not the explanation for increased Sir2 activity in CR cells.
Fig. 2.
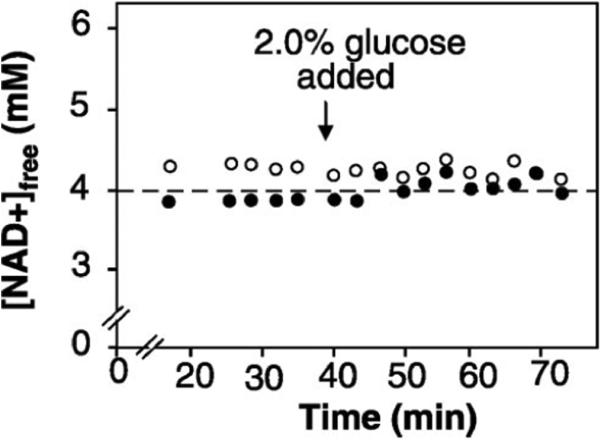
In vivo 13C NMR confirms that calorie restricted cells have lower NAD+. Intracellular concentrations of NAD+ as a function of time for cells grown in 2.0% glucose or 0.5% glucose as calculated by reference to a labeled lactate capillary standard. Additional glucose (2%) was added at time shown by arrow.
We estimate the NAD+:NADH ratio in live aerobic yeast cells to be at least 20, which agrees with classical estimates of the ratio (22, 23). Using a fluorescence-based deacetylation assay and two NAD+ concentrations (4.0 mM, as estimated by in vivo NMR, and 0.05 mM, a value in the range of previous Sir2 studies), we observed no significant effect on deacetylase activity of Sir2 or its human homologue SIRT1 within the physiological range of NAD+:NADH (Fig. 3, A to C). Furthermore, depletion of intracellular NADH with high concentrations of acetaldehyde did not apparently stimulate Sir2 in vivo, as determined by the extent to which two rDNA reporter genes, ADE2 and MET15, were silenced (Fig. 3D). These data indicate that physiological variations in NADH are unlikely to affect the activity of Sir2 or SIRT1.
Fig. 3.

Effect of NADH on yeast Sir2 and human SIRT1 activity. Error bars represent standard error of the mean. (A) Sir2 and SIRT1 deacetylation assays with 4 mM NAD+ and increasing concentrations of NADH. Activity values shown are normalized against the activities of the recombinant proteins in the absence of NADH. Activities were 23 and 100 pmol hour−1 μg−1 for Sir2 at 0.05 mM and 4 mM NAD+ respectively and 160 and 500 pmol hour −1 μg−1 for SIRT1. (B) Sir2 and SIRT1 deacetylation assays with 0.05 mM NAD+ and increasing concentrations of NADH. For clarity, data is shown on two different scales, the upper panel for 0 to 1 μM NADH and (C) the lower panel for higher NADH concentrations. (D) Ribosomal DNA locus (RDN1) silencing assays with or without acetaldehyde (10 mM). Cells were pretreated for 2 to 3 hours and then spotted to assay plates in 10-fold dilutions or single spots for the ADE2 and MET15 assay, respectively. Silencing of the RDN1::ADE2 reporter results in growth retardation on plates lacking adenine, whereas silencing of the RDN1::MET15 reporter leads to a brown coloration on Pb2+-containing medium.
Our measurements of intracellular NAD+ demonstrate that, under aerobic conditions, the steady-state levels of NAD+ do not fluctuate greatly; moreover, its fluctuations during CR negatively correlate with Sir2 activity. The redox state of NAD is also unlikely to regulate Sir2. Nicotinamide is a negative regulator of Sir2 activity in vivo, but whether this is the main mechanism of Sir2 regulation, or whether there are other regulatory mechanisms, remains to be determined.
Supplementary Material
Acknowledgments
We are grateful to D. Moazed, P. Frey, M. Agosto, R. Veech, J. Foster (pFW38–46), L. Guarente (pADH-HAP4), and B. Forrester (NSY64) for advice and/or reagents. Synthesis of labeled nicotinic acid was carried out by R. Ventura and C. Maycock. This work was supported by the National Institute on Aging and the Harvard-Armenise Foundation. D.S. is an Ellison Medical Research Foundation Special Fellow. R.A. is supported by a John Taplan Postdoctoral Fellowship, O.M. by the American Federation of Aging Research.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/1088697/DC1
Materials and Methods
SOM Text
Figs. S1 to S7
References and Notes
References and Notes
- 1.Lin SJ, Defossez PA, Guarente L. Science. 2000;289:2126. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 2.Kaeberlein M, Andalis AA, Fink GR, Guarente L. Mol. Cell. Biol. 2002;22:8056. doi: 10.1128/MCB.22.22.8056-8066.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang JC, Jaruga E, Repnevskaya MV, Jazwinski SM. FASEB J. 2000;14:2135. doi: 10.1096/fj.00-0242fje. [DOI] [PubMed] [Google Scholar]
- 4.Swiecilo A, Krawiec Z, Wawryn J, Bartosz G, Bilinski T. Acta Biochim. Polym. 2000;47:355. [PubMed] [Google Scholar]
- 5.Smith JS, et al. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6658. [Google Scholar]
- 6.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 7.Landry J, et al. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5807. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson RM, Bitterman KJ, Wood JG, Med vedik O, Sinclair DA. Nature. 2003;423:181. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shama S, Lai CY, Antoniazzi JM, Jiang JC, Jazwinski SM. Exp. Cell. Res. 1998;245:379. doi: 10.1006/excr.1998.4279. [DOI] [PubMed] [Google Scholar]
- 10.Hekimi S, Guarente L. Science. 2003;299:1351. doi: 10.1126/science.1082358. [DOI] [PubMed] [Google Scholar]
- 11.Neves AR, et al. J. Biol. Chem. 2002;13:28088. doi: 10.1074/jbc.M202573200. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Q, Piston DW, Goodman RH. Science. 2002;295:1895. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 13.Penfound T, Foster JW, Bacteriol J. 1999;181:648. doi: 10.1128/jb.181.2.648-655.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Materials and methods are available as supporting material on Science Online.
- 15.Anderson RM, et al. unpublished data.
- 16.Lin SJ, Guarente L. Curr. Opin. Cell Biol. 2003;15:241. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 17.Anderson RM, et al. J. Biol. Chem. 2002;277:18881. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- 18.Sandmeier JJ, Celic I, Boeke JD, Smith JS. Genetics. 2002;160:877. doi: 10.1093/genetics/160.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei Y, Lin J, Frey PA. Biochemistry. 2001;40:11279. doi: 10.1021/bi011085z. [DOI] [PubMed] [Google Scholar]
- 20.Paine PL, Cell Biol J. 1975;66:652. doi: 10.1083/jcb.66.3.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Unkefer CJ, London RE. J. Biol. Chem. 1984;259:2311. [PubMed] [Google Scholar]
- 22.Sies H. Metabolic Compartmentation. Academic Press; London: 1982. [Google Scholar]
- 23.Fulco M, et al. Mol. Cell. 2003;12:51. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 24.Pahlman AK, Granath K, Ansell R, Hohmann S, Adler L. J. Biol. Chem. 2001;276:3555. doi: 10.1074/jbc.M007164200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.