

Abstract
Patients with systemic sclerosis (SSc) who express autoantibodies to centromeric proteins (CENPs) are at risk of developing pulmonary vascular disease and pulmonary arterial hypertension without fibrosis. Currently no biomarkers are available to predict these complications. We previously characterized the fine specificity of anti-CENP-A antibodies in SSc by screening a phage display library (expressing random 12-mer peptides), and identified phage clones whose peptides were differentially recognized by patients’ autoantibodies. Here, we examined if subgroups of SSc patients with different anti-CENP-A antibody subspecificities also differ clinically, and if serum reactivity to phage-displayed peptides can predict pulmonary vascular disease.
Clinical data and serum samples were collected from 84 anti-CENP-A-positive SSc patients. Indirect ELISAs were used to test serum reactivity. Pulmonary vascular disease was defined as high systolic pulmonary arterial pressure (sPAP) and low diffusing lung capacity for carbon monoxide (DLCO; percent of predicted values).
Sera were screened for reactivity to peptides expressed by phage clones pc4.2 and pc14.1, confirming our earlier observation of differential specificities. Linear regression showed that the levels of antibodies specific for the 2 phage clones were associated with clinical features of pulmonary vascular disease, but in opposite ways: anti-pc4.2 antibodies were positively associated with sPAP and inversely associated with DLCO, whereas anti-pc14.1 antibodies were inversely associated with sPAP and positively associated with DLCO. Anti-pc4.2 and anti-pc14.1 antibody levels predicted sPAP independently of DLCO. These associations were confirmed by logistic regression using antibodies as predictors and dichotomized sPAP (cutoff, 45 mm Hg) as outcome. The ratio of the 2 antibody levels was a useful marker in predicting high sPAP.
This study demonstrates that some SSc clinical features associate with subspecificities of anti-CENP-A antibodies. Moreover, it shows that a simple, inexpensive phage-based assay can predict which SSc patients have high sPAP and low DLCO, hence who are at greater risk of developing pulmonary arterial hypertension. The ability to identify these at-risk patients can contribute to clinical efficiency and effectiveness. Further research into the peptides expressed by the phage clones may reveal the molecular mechanisms that put some anti-CENP-A-positive patients at greater risk than others for pulmonary vascular disease.
Keywords: anticentromere antibodies, phage display peptide library, pulmonary arterial hypertension, systemic sclerosis
1. Introduction
Systemic sclerosis (SSc) is a disabling and incurable autoimmune disease of the connective tissue.[1,2] One of the clinical hallmarks of SSc is the production of autoantibodies to nuclear proteins.[3] Different subsets of SSc patients express antinuclear antibodies, in an almost exclusive manner, to DNA topoisomerase I, RNA polymerase III, Th/To antigen (RNase mitochondrial RNA processing and RNase P complexes), or centromeric proteins (CENPs).[3–5] The antibody specificity tends to associate with distinct clinical manifestations and with the overall severity of SSc.[5–8] For instance, patients with anti-DNA topoisomerase I antibodies are more likely to have interstitial lung disease (ILD) and musculoskeletal and myocardial involvement; anti-RNA polymerase III positivity is associated with scleroderma renal crisis; and anti-CENP-positive patients are at greater risk of precapillary pulmonary arterial hypertension (PAH) without fibrosis.[3,9–12] The reason for these clinical associations is still largely unknown, just as it is unknown why some patients progress to more severe stages of the disease. In particular, it is not known why only about 10% to 20% of anti-CENP-positive patients develop PAH, one of the major causes of death from SSc.[3] Identification of biomarkers able to define subgroups of anti-CENP-positive patients according to their risk for PAH is urgently needed.
By the time a definitive diagnosis of PAH is made, the disease is not responsive to therapy because the histopathological changes in the vascular and perivascular tissues are established and irreversible. Instead, early treatment has beneficial effects,[13] and early detection via a screening program results in patients having better prognosis than those who are preliminarily diagnosed in routine practice.[14] As a consequence, and in order to define a window of opportunity in which therapy is more effective, several prospective studies have searched for clinical and laboratories markers that, alone or combined in a evidence-based screening algorithm (e.g., the DETECT algorithm),[15] can predict PAH at its early onset.[10,15–17] So far these studies have found that low values of diffusing lung capacity for carbon monoxide (DLCO; <55% of predicted)[16,17] and high values of systolic pulmonary arterial pressure (sPAP; >40 mm Hg)[10,17] are predictive of PAH and can define a group of patients who have high probability of developing PAH in the following 5 years (pre-PAH period).[17] Nonetheless, right heart catheterization is always necessary for a definitive diagnosis.[17] In addition, a value of sPAP > 40 mm Hg is associated with a higher risk of death.[18] Nevertheless, there is still interest in identifying other easily measured variables that can predict subsets of SSc patients with high probability of developing PAH before irreversible vascular remodeling takes place.
As PAH (especially the form without fibrosis) presents mostly in SSc patients who express anti-CENP antibodies, it is possible that the exact fine specificity of these antibodies may predict PAH onset. Among the different CENPs expressed in humans, those most frequently targeted by autoantibodies in SSc are CENP-A and CENP-B.[19] Although these chromatin-binding proteins are unrelated, patients often have antibodies to both.[3] As differences in disease features were not found between CENP-A-positive and CENP-B-positive patients,[12,20] it is important to examine the fine specificities within these antibody populations.
We previously investigated the fine specificities of anti-CENP-A antibodies, focusing on those directed against the protein's 2 immunodominant epitopes located at the amino terminus. These epitopes are found in the regions spanning amino acids 1 to 17 (Ap1–17)[21] and 17 to 30 (Ap17–30).[22,23] In the former study,[21] IgG against Ap1–17 were affinity-purified from sera of 2 patients (pt4 and pt14) and used separately to pan a 12-mer phage display library, resulting in the isolation of 2 sets of phage clones specifically bound by pt4 and pt14 Ig, respectively. When anti-Ap1–17 Ig purified from 6 additional SSc patients was tested for binding to these phage clones, we observed a differential reactivity, or no binding at all, suggesting that this phage-based approach can distinguish different subsets of patients with different anti-CENP-A antibody subspecificities.
This translational study was therefore designed to determine if subsets of anti-CENP-positive SSc patients that differ in antibody subspecificity also differ in terms of clinical features. In particular, we tested if serological variables could predict the severity of pulmonary vascular disease or the likelihood of having PAH, thereby providing complementary prognostic information to routinely tested variables. We focused on the lung item of the SSc disease severity scale[24] and its 4 components, namely DLCO, forced vital capacity (FVC), fibrosis on radiographs (an indication of ILD), and sPAP estimated with Doppler ultrasonography. Here, we show that a phage-based serological assay identifies 2 subgroups of anti-CENP-positive SSc patients that differ in the severity of pulmonary vascular disease, as indicated by values of DLCO and estimated sPAP.
2. Patients and methods
2.1. Serum samples and clinical data
This retrospective study analyzed serum samples and clinical data from 85 SSc patients recruited at the Rheumatology Units of the Universities of Naples, Bari and Foggia from 2010 to 2014. The patients satisfied both the 1980 ACR and the 2013 ACR/EULAR criteria for the classification of SSc.[25,26] At the time when blood was drawn, patients underwent an extensive medical evaluation, including a routine medical history report, physical examination, and laboratory tests. For this study, patients were considered only if they were found, on routine hospital testing, to have antibodies to full-length recombinant CENP-B (determined using the Anti-Centromere B ELISA kit, Orgentec Diagnostika, Germany). Moreover, patients were included only if they were negative for anti-DNA topoisomerase I antibodies (tested with the Anti-Scl-70 ELISA kit, Orgentec) and without other overlapping connective tissue diseases or vasculitis. The set of 85 patients included in this study comprises 50 of the 75 patients (including patients pt4 and pt14) included in our previous study[23] and 35 additional patients with anti-CENP-B antibodies.
For each patient in this study, we collected data regarding sex, age at the time of enrolment, age at the onset of the first Raynaud phenomenon, and SSc subset (limited or diffuse according to LeRoy et al).[27] Disease duration was determined from the onset of Raynaud phenomenon. Existing clinical data were used to score patients on the 9 domains of Medsger et al disease severity scale,[24] where a score of 0 indicates normal findings while 4 indicates end-stage organ damage. In addition, we analyzed data pertinent to disease severity scale subitems for lung and heart, namely FVC, DLCO, estimated sPAP, presence of ILD (ILD; diagnosed with high-resolution CT and using Steele at al clinical decision rule),[12,28] left ventricular ejection fraction, and presence of electrocardiogram abnormalities. Finally, we obtained information on whether they had undergone right heart catheterization resulting in a diagnosis of precapillary pulmonary arterial hypertension (PAH) (mean ≥25 mm Hg and pulmonary artery wedge pressure ≤15 mm Hg).[29] Borderline PAH was considered when pulmonary arterial pressure values were between 21 and 24 mm Hg.[29]
2.2. Ethical issues
Approval for the collection of sera from patients and for the use of their clinical data for research purposes was obtained from the Ethics Committees of the Universities of Naples, Bari and Foggia. All subjects provided written informed consent to the use of clinical samples and data for research purposes in accordance with the ethical principles stated in the Declaration of Helsinki.
2.3. Reagents and immunoglobulin preparations
Chemicals were purchased from BDH Merck (Poole Dorset, UK) or Sigma–Aldrich (St. Louis, MO) unless otherwise indicated. A monoclonal antibody (mAb) to the bacteriophage M13 major coat protein, product of gene VIII (M13), was purchased from GE Healthcare Life Sciences (Milan, Italy). Horseradish-peroxidase (HRP)-conjugated goat anti-human IgG (Fc portion) was purchased from Jackson Immunoresearch Laboratories (West Grove, PA). CENP-A-derived peptides Ap1–17 and Ap17–30 were synthesized as described previously.[21,22] Their purity ranged between 92.4% and 98% as assessed by analytical reverse phase chromatography and mass spectroscopy. For serological assays, they were coupled to BSA using glutaraldehyde as previously described.[30]
Anti-Ap1–17 and anti-Ap17–30 Ig from 8 SSc patients’ sera had previously been prepared by affinity chromatography on Ap1–17- and Ap17–30-conjugated AffiGel columns.[21,22] For the purposes of this study, we purified these Ig fractions from sera of an additional 4 patients using identical methods. Antibody concentration was determined by UV absorption with 1.35 extinction coefficient at 280 nm for 1 mg/mL.
2.4. Phage preparation
This study used 4 phage clones previously isolated by panning a phage display library with affinity-purified anti-Ap1–17 Ig from 2 patients.[21] Specifically, these were 2 phage clones obtained with Ig from patient pt4 (pc4.1 and pc4.2) and 2 phage clones corresponding to patient pt14 (pc14.1 and pc14.2). These clones had been stored at −20 °C in 50% glycerol. For this study, the clones were amplified by inoculating into Escherichia coli cultures and incubating at 37 °C with vigorous shaking for 4 to 5 hours. Phage particle-enriched supernatants were used in serological assays.
2.5. Serological assays
First, SSc patients’ sera were screened for the presence of anti-Ap1–17 and anti-Ap17–30 antibodies in indirect ELISAs using synthetic peptides as described.[23] Briefly, 96-well polyvinylchloride microtiter plates were coated with BSA-conjugated peptide. Serum was diluted 100 times in phosphate-buffered saline (PBS) containing 0.1% BSA, and added to the wells. After 4 hours at 25 °C, wells were washed 3 times with PBS containing 0.05% Tween-20 (PBS-T20), and bound IgG was detected by sequential incubation with HRP-conjugated goat anti-human IgG (Fc portion) and o-phenylenediamine (0.5 mg/mL; 100 μL/well). Color development was stopped by adding 100 μL 2 N H2SO4 and the absorbance at 490 nm was read with the Benchmark microplate reader (Bio-Rad Laboratories, Hercules, CA). The cutoffs to discriminate patients who were positive and negative were obtained by receiver operating characteristics (ROC) analysis comparing the reactivity of the 85 anti-CENP-positive SSc sera to that of sera from 54 healthy blood donors, as previously described.[3] The cutoffs obtained during this study were 1.7 μg/mL for anti-Ap1–17 and 0.3 μg/mL for anti-Ap17–30 Ig.
Then, the reactivity of the 12 affinity-purified preparations of anti-Ap1–17 Ig toward the 4 peptide-expressing phage clones was tested in 96-well plates with indirect ELISAs as previously reported.[21,22] Briefly, wells were coated with 5 μg/mL purified Ig in an overnight incubation at 4 °C, and then incubated with phage-enriched supernatant diluted 16 times (100 μL/well) for 4 hours at room temperature. After 4 washings with PBS-T20, bound phage particles were detected with HRP-conjugated anti-M13 mAb and o-phenylenediamine, as above. Background binding was determined by incubating wells with supernatant from bacterial cultures not infected with phage. Specific binding was determined by subtracting the background binding from the binding in experimental wells. Samples were tested in duplicate, and the experiment was performed at least 3 times.
SSc patients’ sera were also tested for their reactivity to the specific peptides expressed by 2 of the selected phage clones in indirect ELISAs as follows. Polyvinylchloride 96-well plates were coated with mouse anti-M13 mAb by incubation with 50 μL of a 5 μg/mL solution in PBS for 12 hours at 4 °C. Wells were washed once with PBS-T20, and any free protein-binding sites were blocked with PBS containing 0.5% BSA. Then, 100 μL aliquots of undiluted phage-enriched supernatant were added and incubated for 2 hours at 25 °C to allow the mAb to capture the phages. After 3 washes with PBS-T20, serum samples (50 μL/well diluted 100 times in PBS containing 0.5% BSA) were added to the wells and incubated for 2 hours at 25 °C. As a negative control, some wells received diluted serum pooled from 5 healthy donors. As positive controls, some wells received diluted serum from pt4 or pt14. All samples were tested in duplicate. After incubation, wells were washed 4 times with PBS-T20, and bound IgG was detected with HRP-conjugated goat-antihuman IgG (Fc portion) and o-phenylenediamine, as above. Background binding was determined from the absorbance in the negative-control wells and was subtracted from the absorbance in other wells to determine the specific binding. Specific binding of experimental samples were expressed as percentage of the binding observed with the positive controls. The tests’ executers were blind to clinical data.
2.6. Statistical analyses
Multivariate forward (stepwise) linear regression was used to identify predictive associations between serological variables (anti-Ap1–17, anti-Ap17–30, and anti-Ap1–17 subspecificities determined with the phage-based assay) and clinical parameters describing pulmonary vascular changes that indicate a likelihood of having PAH (lung item of the SSc disease severity scale and its subitems). Age at diagnosis and disease duration were included as possible confounding predictors. Variables with a nonparametric distribution (i.e., antibody levels, sPAP, and DLCO) were log-transformed prior to inclusion in the models. The automatic linear modeling tool of SPSS v21 was used for the regression analyses. The sample size for the regression analysis was estimated taking into the account an expected partial R2 of 0.15, an α-error = 0.05, and power = 0.8 (1-β error probability), with a total number of predictors of 6 and 2 tested predictors. This analysis was performed with G∗power software v3.1.9.2 for windows.
Decision tree analysis was used to identify a cutoff in an independent variable (DLCO; antibody ratio) that partitioned the patients into clusters with widely different values of a dependent variable (sPAP). Logistic regression analysis was done to analyze the ability of antiphage antibodies to predict if patients had an estimated sPAP value above (or below) 45 mm Hg; this cutoff is more restrictive than the 40 mm Hg generally taken as suggestive of PAH.[10,17] Fisher exact test was used to assess the significance of the association among dichotomized variables. These statistical tests were performed using SPSS v21 for Windows. For all tests, a P value < 0.05 indicated statistical significance.
3. Results
To determine if the subspecificity of SSc patients’ autoantibodies is associated with clinical characteristics, we studied a group of 85 SSc patients who tested positive for anti-CENP-B antibodies and for whom we had clinical data and serum samples. As patients with antibodies to CENP-B usually also have antibodies to CENP-A, we expected our patients to be a good population for studying subspecificities to CENP-A. When tested for reactivity to synthetic CENP-A-derived peptides, 74 of the patients (87.0%) scored positive for anti-Ap1–17 antibodies and 77 (90.5%) for anti-Ap17–30 antibodies; 67 patients (78.8%) had both antibodies, while only 1 patient had neither and was excluded from subsequent analyses. The remaining 84 patients were studied for clinical associations with autoantibody fine specificities.
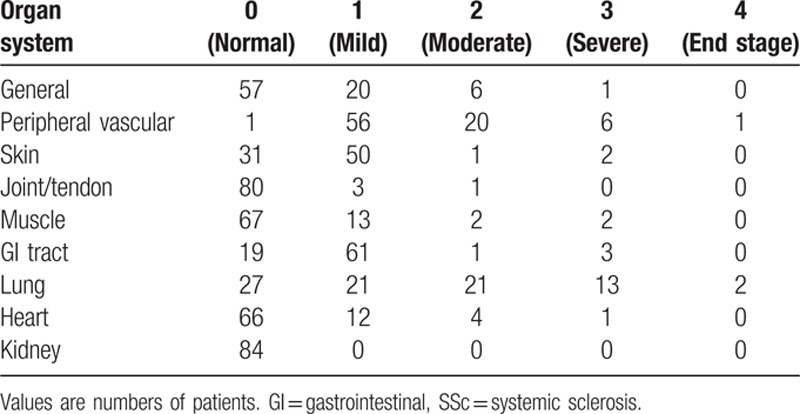
The SSc patients included in the study were predominantly female and had a mean disease duration of over 15 years (Table 1). Most patients had limited disease, with only 6 had diffuse SSc. On the disease severity scale, the most severely affected organ system was the lung (mean score, 1.31), but the most frequently affected was the gastrointestinal system (65 patients had some form of involvement), no patient had kidney involvement. The exact distribution of scores by organ system is given in Table 2, which shows, for example, that all patients but 1 scored > 0 for peripheral vascular involvement (having Raynaud phenomenon requiring vasodilators and possibly digital lesions). On the subscale items (Table 1), values of FVC were generally within the norm, with only 4 patients (4.8%) having values < 70% of predicted. Moreover, only 17 patients (20.3%) had evidence of ILD, whereas 37 patients (44.0%) had abnormally low DLCO. These findings indicate that the pulmonary disease in this study group was predominantly vascular rather than interstitial. Estimated sPAP was abnormally high in 20 cases, and 4 of the 10 patients who had right heart catheterization were diagnosed with PAH.
Table 1.
Characteristics of 84 patients with systemic sclerosis and anti-CENP-A antibodies.
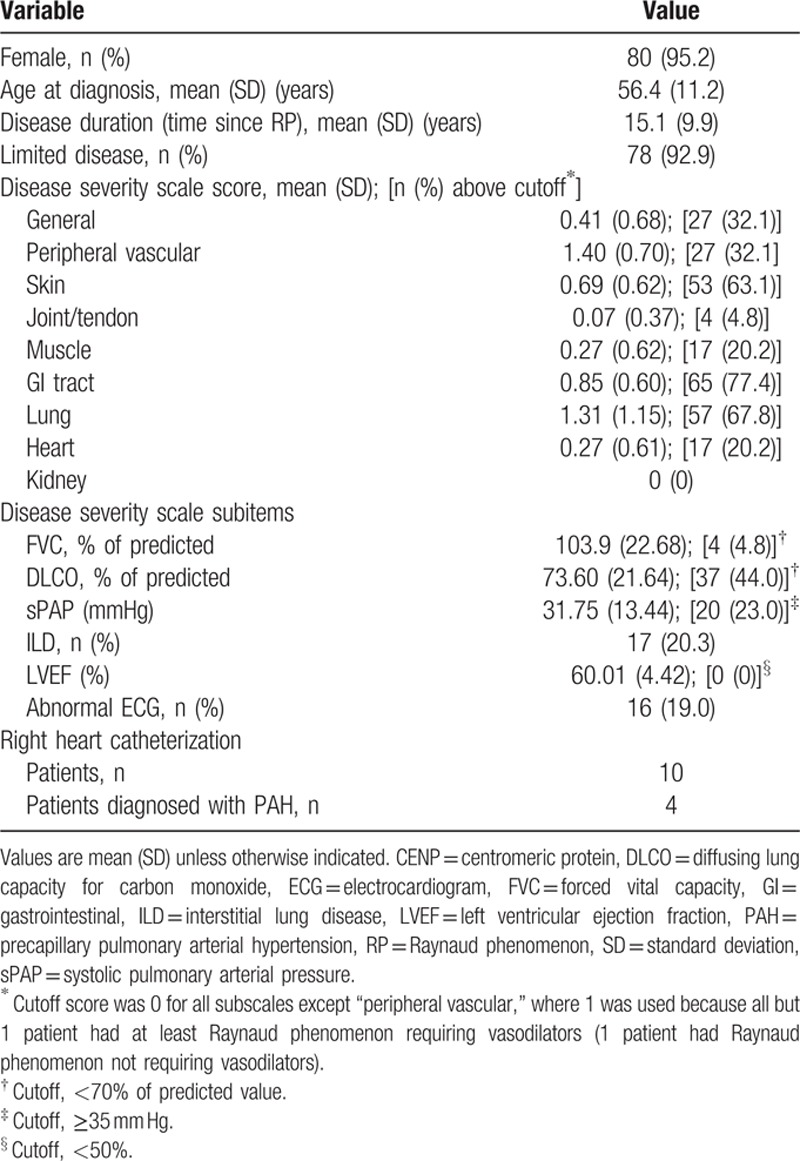
Table 2.
Scores on the disease severity scale[24] for the 84 SSc patients.
3.1. Phage-expressed peptides define subsets of anti-CENP-A-positive SSc patients
Affinity-purified anti-Ap1–17 Ig from 12 of the 84 SSc patients was tested for reactivity to peptides expressed by 2 pairs of phage clones previously isolated from a phage display library using similar Ig from patients pt4 and pt14. In indirect ELISAs using the 12 preparations of purified Ig as capture antibodies and 1 of the 2 pt4 phage clones as antigen (Fig. 1A, B), the reactivity of pt4 Ig to both clones was confirmed by strong absorbance; moreover, 6 Ig preparations gave negligible absorbance in both tests, while 5 Ig preparations (pt3, pt5, pt7, pt9, and pt21) gave absorbance values >0.5 for at least 1 phage clone. When the experiment was repeated using the pt14 phage clones (Fig. 1C, D), no reactivity was seen at all, with the exception of pt14 Ig (the exact Ig used to isolate the phage clones). Hence, 12-mer phage-expressed peptides that are specifically bound by pt4 and pt14 Ig are recognized in a differential manner by anti-Ap1–17 Ig purified (on the basis of their affinity to the 17-mer synthetic peptide) from 10 other SSc patients.
Figure 1.
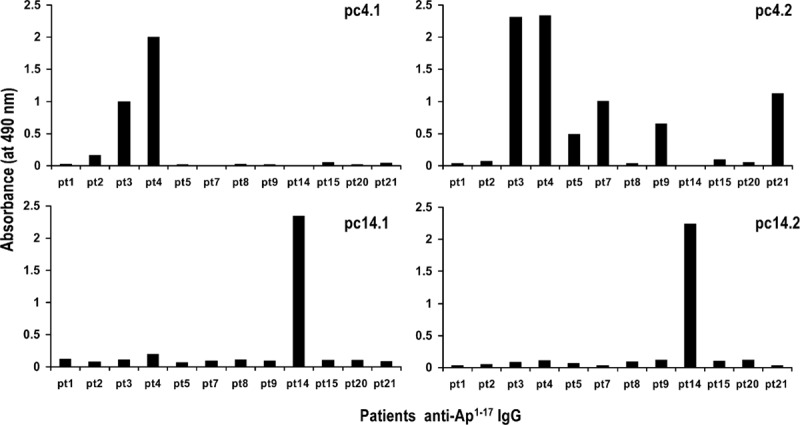
Differential recognition of affinity-purified anti-Ap1–17 Ig from 12 SSc patients for peptides expressed by 4 phage clones, previously isolated from a phage display library using Ig from 2 of the patients. (A, B) Phage clones isolated using patient pt4 Ig. (C, D) Phage clones isolated using patient pt14 Ig. Each data point is the mean of duplicate wells. The data are representative of 2 experiments.
Next, we choose 2 phage clones (pc4.2 and pc14.1) to use in screening sera from the entire SSc study group, in an indirect ELISA with antiphage mAb as capture antibody and phage as antigen. This analysis showed that 49 sera samples recognized both peptides, while 17 bound neither (Fig. 2A). The median binding to pc4.2 was 7.3% of that obtained with pt4 Ig, while the median binding to pc14.1 was 3.8% of that obtained with pt14 Ig, although in both cases there was a broad right skew in the distribution (Fig. 2B). These results therefore confirm, in a larger group of SSc patients, that there are wide subspecificities among the anti-CENP-A antibodies expressed by different patients.
Figure 2.
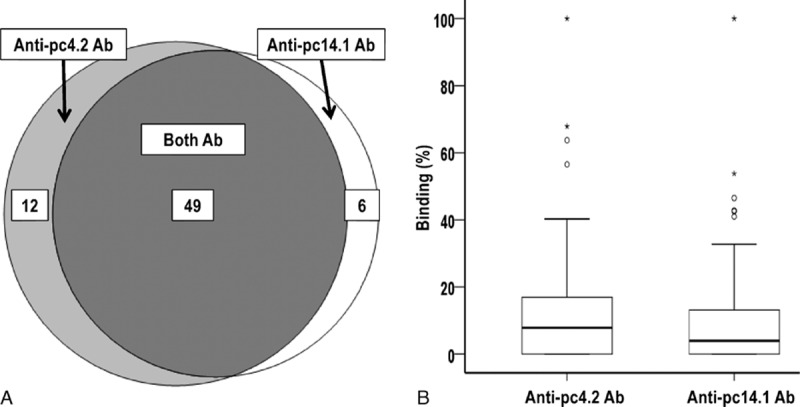
Subspecificities of the 84 SSc patients’ sera for 2 phage-expressed centromeric protein (CENP)-A-derived peptides. (A) Venn diagram showing overlapping sets of patients with antibodies recognizing peptides expressed by 2 phages; sera from 17 patients did not react with either phage clone. (B) Binding of anti-pc4.2 and anti-pc14.1 antibodies expressed as a percentage of the binding obtained with positive control sera from pt4 or pt14. The horizontal bar marks the median and the box indicates the interquartile range; outlier values (more than 1.5 times the interquartile range) are marked with a circle, while extreme outliers values (more than 3 times the interquartile range) are marked with an asterix.
3.2. Antibody subspecificities predict severity of pulmonary vascular disease
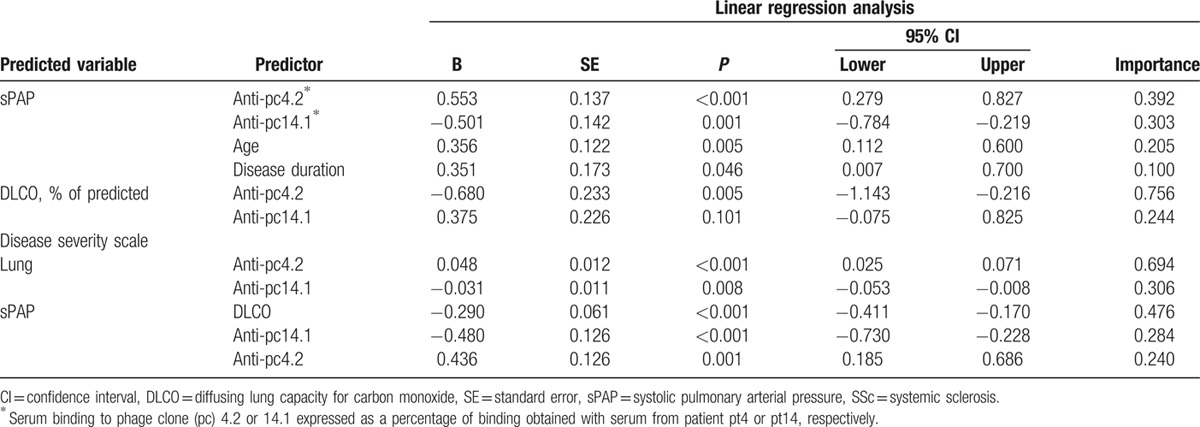
To search for associations between the SSc patients’ serological variables and the clinical variables describing pulmonary vascular disease, we used multivariate forward linear regression. In the model to predict sPAP (R2 = 0.26), both antiphage antibodies were retained, as were age and disease duration, while the antibodies to peptides Ap1–17 and Ap17–30 were excluded (Table 3). A striking finding was the contrast between the positive association observed for anti-pc4.2 levels (percentage binding) and the inverse association observed for anti-pc.14.1 antibody. In particular, an increase in anti-pc4.2 antibody levels was associated with an increase in sPAP (B = 0.553; P < 0.001), while an increase in anti-pc14.1 antibody levels was associated with a decrease in sPAP (B = −0.501; P = 0.001); age and disease duration were associated with sPAP in a positive manner. In the model for DLCO (R2 = 0.07), only the antiphage antibodies were retained, with an opposite pattern: anti-pc4.2 levels were inversely associated (B = −0.680; P = 0.005) while anti-pc14.1 were positively associated albeit in a not-significant manner (B = 0.375; P = 0.101). Finally, the antiphage antibodies were the only independent factors retained in the model to predict the lung item on the disease severity score (R2 = 0.15). In this case, anti-pc4.2 antibodies were positively associated (B = 0.048; P < 0.001), while anti-pc14.1 antibodies were inversely associated (B = −0.031; P = 0.008).
Table 3.
Multivariate forward linear regression to identify predictors of pulmonary function in SSc patients.
To better understand if the values of estimated sPAP in our study group were indicative of PAH, we examined the relationship between sPAP and DLCO, a strong predictor of PAH. We therefore used decision tree analysis to see if patients could be clustered into subgroups with distinct sPAP values (dependent variable) on the basis of DLCO (independent variable). This analysis showed that a cutoff of DLCO equal to 56% of predicted was the best discriminator, separating the cohort into a high sPAP group (n = 16; mean, 47.14 mm Hg) and a low sPAP group (n = 68; mean, 28.18 mm Hg) (P < 0.001). This result justifies the testing of DLCO as a predictor variable for sPAP in linear regression.
Therefore, forward multivariate linear regression was repeated by including DLCO among the predictor variables. As shown in Table 3, anti-pc4.2 and anti-pc14.1 antibody levels were retained in the model (R2 = 0.41), along with DLCO, while age and disease duration were not. This result indicates that the antiphage antibodies were independent of DLCO in predicting an increased sPAP, whereas age and disease duration did not add any information beyond that provided by DLCO and the antibodies.
3.3. Phage-based assay can predict suspected PAH
Given that high levels of anti-pc4.2 and low levels of anti-pc14.1 antibodies were predictive of high sPAP on linear regression, we tested the possibility that these variables could also predict if a patient was above or below a 45 mm Hg sPAP cutoff suggestive of PAH. Table 4 shows that anti-pc4.2 (P = 0.029) and anti-pc14.1 (P = 0.001) antibody levels were retained in the model: an estimated sPAP > 45 mm Hg was associated with high levels of anti-pc4.2 and low levels of anti-pc14.1 antibodies. This result confirms their ability to identify a subgroup of SSc patients with high sPAP and probable PAH.
Table 4.
Forward logistic regression analysis to identify whether anti-pc4.2 and anti-pc14.1 antibodies are predictive of sPAP above a cutoff indicative of PAH.

Because of the opposite associations of anti-pc4.2 and anti-pc14.1 antibodies with SSc patients’ clinical parameters, we used decision tree analysis to identify a ratio of antibody levels that partitioned the patients into 2 distinct groups according to values of sPAP. This analysis revealed that an anti-pc4.2/anti-pc14.1 ratio of 5.25 was the best cutoff (P = 0.008), forming 2 groups: a low sPAP group of 69 patients were identified by a ratio ≤5.25; these patients had mean sPAP of 28.4 mm Hg (SD = 8.6 mm Hg). Additionally, a high sPAP group of 15 patients were identified by a ratio >5.25; these patients had a mean sPAP of 44.8 mm Hg (SD = 21.5 mm Hg). To illustrate how this ratio could be used to predict the presence of PAH, we tested different sPAP cutoffs (from 35 to 50 mm Hg) and observed that patients with a high ratio were significantly more likely to rank above the sPAP cutoff than patients with a low ratio (Fig. 3).
Figure 3.
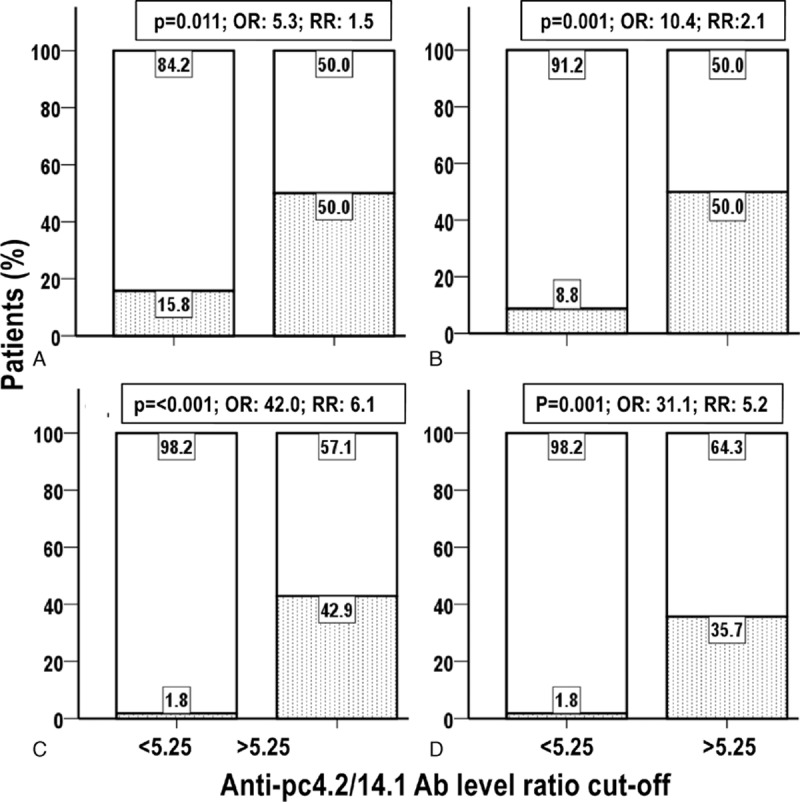
Association between high sPAP and high anti-pc4.2/anti-pc14.1 antibody ratio. Percentage distribution in the low and high anti-pc4.2/pc14.1 antibody groups of patients with low (open bar) and high (dotted bar) sPAP. Low and high sPAP were defined using the sPAP cutoffs of (A) 35 mm Hg, (B) 40 mm Hg, (C) 45 mm Hg, and (D) 50 mm Hg. P: Fisher exact test P; OR = odds ratio, sPAP = systolic pulmonary arterial pressure, RR = relative risk.
3.4. Anti-pc4.2/anti-pc14.1 ratio and PAH
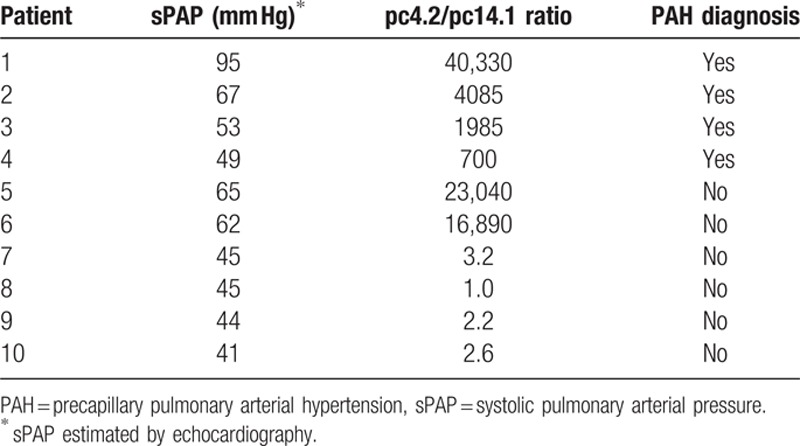
Of the 12 patients with estimated sPAP > 40 mm Hg, 10 underwent right heart catheterization and 4 were diagnosed with PAH (Table 5). The 4 patients with PAH all had anti-pc4.2/anti-pc14.1 ratios markedly higher than 5.25. Of the remaining 6 patients without PAH, only 2 had anti-pc4.2/anti-pc14.1 ratios higher than 5.25. One of these 2 patients had borderline PAH and received a definitive diagnosis of PAH within 1 year of follow-up. The other patient had a transient increase in sPAP due to an acute myocardial infarction 1 month earlier. In the subsequent follow-up sPAP returned to normal without clinical signs of PAH. Clinical follow-up of patients without PAH will clarify if patients with high ratios are more likely to develop PAH than those with low ratio.
Table 5.
Diagnosis of PAH at right heart catheterization in 10 patients with sPAP > 40 mm Hg, and relation to anti-pc4.2/pc14.1 ratio.
4. Discussion
The 2 most common autoantigens in SSc patients with autoantibodies to CENPs are CENP-A and CENP-B. They can be detected by indirect immunofluorescence assay and by a CENP ELISA.[4] The ELISA is more sensitive in that it can detect anti-CENP antibodies in sera that do not show the anti-CENP staining pattern typically seen with indirect immunofluorescence assays.[4] Previous investigations in anti-CENP-positive patients using the more sensitive CENP ELISA did not find differences in disease features between those with antibodies to CENP-A versus CENP-B.[12,20] Here, we looked at a still finer level of antibody specificity, by limiting our analysis to the first epitope, Ap1–17, and focusing on shorter peptide sequences within it. From our previous study,[21] we had phage clones expressing 12-mer peptides that were specifically recognized by sera from a small number of SSc patients. Using 2 of these phage clones and a larger group of 84 SSc patients, we confirmed our previous observation that patients’ anti-Ap1–17 antibodies have unique subspecificities, recognizing one, both or neither of the phage-expressed peptides. Through linear regression, we found that antibodies specific for the 2 peptides were associated with clinical features of pulmonary vascular disease, but in opposite ways: anti-pc4.2 antibodies were positively associated with sPAP and inversely associated with DLCO (percent of predicted values), whereas anti-pc14.1 antibodies were inversely associated with sPAP and positively associated with DLCO.
Our work with phage-based assays has several points of interest. First, the peptides displayed by phages are biosynthesized and displayed in a consistent manner, differently from synthetic peptides that must be cross-linked, in random manner, to a carrier protein. Second, phages are easily propagated without costly reagents or instruments. From the clinical viewpoint, the particular assay used here, with pc4.2 and pc14.1 phage clones, can readily distinguish SSc patients with high sPAP and low DLCO. Instead, immunoreactivity to the larger epitopes, Ap1–17 and Ap17–30, were not predictive of these clinical parameters.
In our analysis, age remained included in the 1st model to predict sPAP. The relationship between age and increased sPAP was previously reported by Kollert et al[31] in a cohort of 76 patients with SSc. These investigators found that age and sCD90, the marker that was the object of their study, were independent variables predicting PAH. However, neither Kollert et al's study nor the present one addressed the role of age-related comorbidities,[32] or the very rare possibility of an increase in sPAP in otherwise healthy individual.[33]
In this study, sPAP was estimated on echocardiography because this is the recommended procedure for yearly screening of SSc patients for early detection of PAH.[34,35] Moreover, patients may refuse right heart catheterization, the gold standard method; in fact, only 10 of the 12 patients with estimated sPAP ≥40 mm Hg in this study underwent right heart catheterization. In this subgroup, patients with very high anti-pc4.2/anti-pc14.1 ratios were more likely to have PAH, although 2 patients with high ratios did not meet the diagnostic criteria for PAH. Although these data are suggestive, there are too few patients to permit a statistical analysis about the predictivity of ratio; this is a limitation of our study.
Three lines of evidence in our study indicate that high sPAP and low DLCO (expression of pulmonary vascular disease and predicted by anti-phage clone antibody levels) suggest the risk of PAH in our cohort: only 4 patients had FVC < 70% of predicted while 37 had DLCO < 70% of predicted, suggesting that hypoxia contributed only marginally to the sPAP increase (type III PAH); the 56% DLCO cutoff found here to distinguish patients with high versus low sPAP is similar to the 55% cutoff previously reported by Hsu et al[16] in a cohort in which PAH was confirmed by right heart catheterization; and our finding that DLCO was an independent predictor of sPAP is in agreement with work by Chang et al,[36] who reported that a progressive decline of DLCO was an independent predictor of PAH (assessed by catheterization).
PAH can also be predicted by endothelial cell proteins (e.g., CD90),[31] markers of natural immunity activation (e.g., CXCL4 and pentraxin 3),[37,38] or N-terminal pro-brain natriuretic peptide,[39] with a diagnosis of pulmonary hypertension being made in these studies with echocardiography,[31] right heart catheterization,[37,38] or both.[39] Although the mechanisms underlying the association of these markers and increased sPAP can be deduced, given the demonstrated role of endothelial cell activation or natural immunity in the early vasculopathy events leading to PAH,[40] the mechanisms underlying the ability of anti-pc4.2 and anti-pc14.1 antibody levels to predict an increased sPAP remain to be determined.
The peptides expressed by the 2 sets of phage clones permitted us to identify 2 motifs (consensus sequences) that were partially overlapping: 10KPXXP14 (pt4) and 5RXSXKP11 (pt14)[21] and the antiphage antibodies studied here recognize different residues (or different conformations) of the amino terminus of CENP-A. At present we can only speculate on why they associate with clinical variables in opposite directions. One possible explanation is that these antibodies cross-react with different antigens on endothelial cells or cells involved in the natural immune response, and thereby influence the progression of vascular damage, as previously demonstrated for antiendothelial cell antibodies[41,42] and antifibroblast antibodies[43] in SSc. If this is the case, we can speculate that anti-pc14.1 antibodies exert a protective effect by delaying or blocking the vascular occlusive changes that cause sPAP to increase in SSc,[44] while anti-pc4.2 antibodies would exacerbate these changes. Sequencing the peptides expressed by the phage clones pc4.2 and pc14.1 and identifying the vascular proteins containing those sequences will tell if this interpretation is correct.
The ability to identify a subset of anti-CENP-positive patients with a high likelihood of having worse vascular phenotypes (elevated sPAP and decreased DLCO), and therefore at higher risk of developing PAH with a consequently lower survival,[18] narrows the number of patients requiring close clinical surveillance while lifting the burden of surveillance (and the associated costs) from those at lower risk. However, caution must be exercised in interpreting our findings, because we studied a homogeneous group of Italian patients followed at 3 regional rheumatological centers. The results presented here must be confirmed prospectively in a larger multiethnic cohort including patients with PAH assessed by right heart catheterization. Finally, follow-up of these patients will reveal to what extent the early recognition of these subsets will affect their prognosis in the clinical settings of very early[45] and full-blown SSc.
Acknowledgements
The authors thank Silvestro Montrone, Professor of Statistic, Department of Management Sciences, University of Bari, for valuable discussion on the statistical analyses; and Mrs Maria Daniele and Mr Vito Iacovizzi for their excellent secretarial assistance; Valerie Matarese for providing editorial advice and scientific editing.
Footnotes
Authorship: FP and EF conceived and designed the study; FP, GV, SV, IEF, AC, MP, and FPC acquired the data; FP, GV, and EF analyzed and interpreted the data; FP, EF, IEF, SV, and AC drafted the manuscript, while GV, FP, FPC, and MP critically revised it for intellectual content; all authors read and approved the final version of the manuscript. All persons listed gave permission to be named.
Funding: This work was supported by a grant from the Italian Group Against Systemic Sclerosis (GILS).
The authors have no conflicts of interest to disclose.
References
- 1.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007; 117:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guy S, Kong J, Cheema GS, et al. The immunobiology of systemic sclerosis. Semin Arthritis Rheum 2008; 38:132–160. [DOI] [PubMed] [Google Scholar]
- 3.Perosa F, Prete M, Di LG, et al. Anti-centromere protein A antibodies in systemic sclerosis: significance and origin. Autoimmun Rev 2016; 15:102–109. [DOI] [PubMed] [Google Scholar]
- 4.Mahler M, You D, Baron M, et al. Anti-centromere antibodies in a large cohort of systemic sclerosis patients: comparison between immunofluorescence, CENP-A and CENP-B ELISA. Clin Chim Acta 2011; 412:1937–1943. [DOI] [PubMed] [Google Scholar]
- 5.Mehra S, Walker J, Patterson K, et al. Autoantibodies in systemic sclerosis. Autoimmun Rev 2013; 12:340–354. [DOI] [PubMed] [Google Scholar]
- 6.Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81:139–153. [DOI] [PubMed] [Google Scholar]
- 7.Senecal JL, Henault J, Raymond Y. The pathogenic role of autoantibodies to nuclear autoantigens in systemic sclerosis (scleroderma). J Rheumatol 2005; 32:1643–1649. [PubMed] [Google Scholar]
- 8.Meyer OC, Fertig N, Lucas M, et al. Disease subsets, antinuclear antibody profile, and clinical features in 127 French and 247 US adult patients with systemic sclerosis. J Rheumatol 2007; 34:104–109. [PubMed] [Google Scholar]
- 9.Mitri GM, Lucas M, Fertig N, et al. A comparison between anti-Th/To- and anticentromere antibody-positive systemic sclerosis patients with limited cutaneous involvement. Arthritis Rheum 2003; 48:203–209. [DOI] [PubMed] [Google Scholar]
- 10.Walker UA, Tyndall A, Czirjak L, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2007; 66:754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hesselstrand R, Scheja A, Wuttge DM. Scleroderma renal crisis in a Swedish systemic sclerosis cohort: survival, renal outcome, and RNA polymerase III antibodies as a risk factor. Scand J Rheumatol 2012; 41:39–43. [DOI] [PubMed] [Google Scholar]
- 12.Hudson M, Mahler M, Pope J, et al. Clinical correlates of CENP-A and CENP-B antibodies in a large cohort of patients with systemic sclerosis. J Rheumatol 2012; 39:787–794. [DOI] [PubMed] [Google Scholar]
- 13.Galie N, Rubin L, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet 2008; 371:2093–2100. [DOI] [PubMed] [Google Scholar]
- 14.Humbert M, Yaici A, de GP, et al. Screening for pulmonary arterial hypertension in patients with systemic sclerosis: clinical characteristics at diagnosis and long-term survival. Arthritis Rheum 2011; 63:3522–3530. [DOI] [PubMed] [Google Scholar]
- 15.Coghlan JG, Denton CP, Grunig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73:1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsu VM, Moreyra AE, Wilson AC, et al. Assessment of pulmonary arterial hypertension in patients with systemic sclerosis: comparison of noninvasive tests with results of right-heart catheterization. J Rheumatol 2008; 35:458–465. [PubMed] [Google Scholar]
- 17.Hsu VM, Chung L, Hummers LK, et al. Development of pulmonary hypertension in a high-risk population with systemic sclerosis in the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS) cohort study. Semin Arthritis Rheum 2014; 44:55–62. [DOI] [PubMed] [Google Scholar]
- 18.Hachulla E, Clerson P, Airo P, et al. Value of systolic pulmonary arterial pressure as a prognostic factor of death in the systemic sclerosis EUSTAR population. Rheumatology (Oxford) 2015; 54:1262–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fritzler MJ, Rattner JB, Luft LM, et al. Historical perspectives on the discovery and elucidation of autoantibodies to centromere proteins (CENP) and the emerging importance of antibodies to CENP-F. Autoimmun Rev 2011; 10:194–200. [DOI] [PubMed] [Google Scholar]
- 20.Hanke K, Becker MO, Brueckner CS, et al. Anticentromere-A and anticentromere-B antibodies show high concordance and similar clinical associations in patients with systemic sclerosis. J Rheumatol 2010; 37:2548–2552. [DOI] [PubMed] [Google Scholar]
- 21.Favoino E, Digiglio L, Cuomo G, et al. Autoantibodies recognizing the amino terminal 1-17 segment of CENP-A display unique specificities in systemic sclerosis. PLoS One 2013; 8:e61453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perosa F, Vicenti C, Racanelli V, et al. The immunodominant epitope of centromere-associated protein A displays homology with the transcription factor forkhead box E3 (FOXE3). Clin Immunol 2010; 137:60–73. [DOI] [PubMed] [Google Scholar]
- 23.Perosa F, Favoino E, Cuomo G, et al. Clinical correlates of a subset of anti-CENP-A antibodies cross-reacting with FOXE3p53-62 in systemic sclerosis. Arthritis Res Ther 2013; 15:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Medsger TA, Jr, Bombardieri S, Czirjak L, et al. Assessment of disease severity and prognosis. Clin Exp Rheumatol 2003; 21:S42–S46. [PubMed] [Google Scholar]
- 25.Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic, Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum 1980; 23:581–590. [DOI] [PubMed] [Google Scholar]
- 26.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013; 65:2737–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988; 15:202–205. [PubMed] [Google Scholar]
- 28.Steele R, Hudson M, Lo E, et al. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res (Hoboken) 2012; 64:519–524. [DOI] [PubMed] [Google Scholar]
- 29.Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37:67–119. [DOI] [PubMed] [Google Scholar]
- 30.Perosa F, Favoino E, Caragnano MA, et al. Generation of biologically active linear and cyclic peptides has revealed a unique fine specificity of rituximab and its possible cross-reactivity with acid sphingomyelinase-like phosphodiesterase 3b precursor. Blood 2006; 107:1070–1077. [DOI] [PubMed] [Google Scholar]
- 31.Kollert F, Christoph S, Probst C, et al. Soluble CD90 as a potential marker of pulmonary involvement in systemic sclerosis. Arthritis Care Res (Hoboken) 2013; 65:281–287. [DOI] [PubMed] [Google Scholar]
- 32.Fisher MR, Mathai SC, Champion HC, et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum 2006; 54:3043–3050. [DOI] [PubMed] [Google Scholar]
- 33.Ghamra ZW, Dweik RA. Primary pulmonary hypertension: an overview of epidemiology and pathogenesis. Cleve Clin J Med 2003; 70 suppl 1:S2–S8. [DOI] [PubMed] [Google Scholar]
- 34.Hachulla E, de Groote P, Gressin V, et al. The three-year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitudinal study in France. Arthritis Rheum 2009; 60:1831–1839. [DOI] [PubMed] [Google Scholar]
- 35.Vachiery JL, Coghlan G. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev 2009; 18:162–169. [DOI] [PubMed] [Google Scholar]
- 36.Chang B, Schachna L, White B, et al. Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma. J Rheumatol 2006; 33:269–274. [PubMed] [Google Scholar]
- 37.van Bon L, Affandi AJ, Broen J, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med 2014; 370:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shirai Y, Okazaki Y, Inoue Y, et al. Elevated levels of pentraxin 3 in systemic sclerosis: associations with vascular manifestations and defective vasculogenesis. Arthritis Rheumatol 2015; 67:498–507. [DOI] [PubMed] [Google Scholar]
- 39.Thakkar V, Stevens WM, Prior D, et al. N-terminal pro-brain natriuretic peptide in a novel screening algorithm for pulmonary arterial hypertension in systemic sclerosis: a case-control study. Arthritis Res Ther 2012; 14:R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Pavec J, Humbert M, Mouthon L, et al. Systemic sclerosis-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2010; 181:1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Negi VS, Tripathy NK, Misra R, et al. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol 1998; 25:462–466. [PubMed] [Google Scholar]
- 42.Mihai C, Tervaert JW. Anti-endothelial cell antibodies in systemic sclerosis. Ann Rheum Dis 2010; 69:319–324. [DOI] [PubMed] [Google Scholar]
- 43.Terrier B, Tamby MC, Camoin L, et al. Identification of target antigens of antifibroblast antibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med 2008; 177:1128–1134. [DOI] [PubMed] [Google Scholar]
- 44.al-Sabbagh MR, Steen VD, Zee BC, et al. Pulmonary arterial histology and morphometry in systemic sclerosis: a case-control autopsy study. J Rheumatol 1989; 16:1038–1042. [PubMed] [Google Scholar]
- 45.Valentini G, Marcoccia A, Cuomo G, et al. Early systemic sclerosis: analysis of the disease course in patients with marker autoantibody and/or capillaroscopic positivity. Arthritis Care Res (Hoboken) 2014; 66:1520–1527. [DOI] [PubMed] [Google Scholar]