

Abstract
Mastocytosis is a heterogeneous group of diseases with a young median age at diagnosis. Usually indolent and self-limited in childhood, the disease can exhibit aggressive progression in mid-adulthood. Our objectives were to describe the characteristics of the disease when diagnosed among elderly patients, for which rare data are available.
The French Reference Center conducted a retrospective multicenter study on 53 patients with mastocytosis >69 years of age, to describe their clinical, biological, and genetic features.
The median age of our cohort of patients was 75 years. Mastocytosis variants included were cutaneous (n = 1), indolent systemic (n = 5), aggressive systemic (n = 11), associated with a hematological non-mast cell disease (n = 34), and mast cell leukemia (n = 2). Clinical manifestations were predominantly mast cell activation symptoms (75.5%), poor performance status (50.9%), hepatosplenomegaly (50.9%), skin involvement (49.1%), osteoporosis (47.2%), and portal hypertension and ascites (26.4%). The main biological features were anemia (79.2%), thrombocytopenia (50.9%), leucopenia (20.8%), and liver enzyme abnormalities (32.1%). Of the 40 patients tested, 34 (85%), 2 (5%), and 4 (10%) exhibited the KIT D816V mutant, other KIT mutations and the wild-type form of the KIT gene, respectively. Additional sequencing detected significant genetic defects in 17 of 26 (65.3%) of the patients with associated hematological non-mast cell disease, including TET2, SRSF2, IDH2, and ASLX1 mutations. Death occurred in 19 (35.8%) patients, within a median delay of 9 months, despite the different treatment options available.
Mastocytosis among elderly patients has a challenging early detection, rare skin involvement, and/or limited skin disease; it is heterogeneous and has often an aggressive presentation with nonfortuitous associated myeloid lineage malignant clones, and thus a poor overall prognosis.
Keywords: ASLX1, elderly, KIT, mast cell, systemic mastocytosis, TET2
1. Introduction
Mastocytosis is a heterogeneous group of disorders characterized by abnormal growth and accumulation of mast cells (MCs) in 1 or more organ systems.[1] The most commonly affected tissues are the skin, bone marrow (BM), and gastrointestinal (GI) tract, whereas the liver, spleen, and lymph nodes are less commonly involved.[1,2] The WHO 2008 classification of hematopoietic malignancies has defined several variants including cutaneous mastocytosis (CM), extracutaneous mastocytoma, indolent systemic mastocytosis (ISM), systemic mastocytosis with associated clonal hematological non-mast cell-lineage disease (SM-AHNMD), aggressive SM (ASM), mast-cell leukemia (MCL), and mast cell sarcoma (MCS).[1] The clonal proliferation of MCs in different variants of matocytosis is usually related to an acquired gain-of-function mutation in the KIT gene, which encodes for a transmembrane tyrosine kinase receptor for stem cell factor, namely KIT/CD117.[3] KIT is expressed on maturing MC progenitors and on mature MCs. Most adults with mastocytosis present with the KIT D816V mutation (>90%), but additional genetic lesions involving epigenetic regulation, RNA splicing, and/or transcription have been recently described and are thought to contribute at least in part to the aggressiveness of the disease.[1,4–7] Of note, clinical manifestations of mastocytosis are variable and are either related to the release of MC mediators or to pathological MC infiltration, or both.[1] It has previously been described that the disease among children is usually benign, but the clinical and genetic presentations among elderly patients have not often been studied.[4,5,8]
Little data are available on mastocytosis in the elderly, in which age is also the background for additional mutations and clonal features for both mast-cell and other myeloid cell lineages. Indeed Lim et al and Brockow et al found that the late onset of the disease and an age at diagnosis >60 years is one of the most significant risk factors for death in systemic mastocytosis (SM) with a shorter overall survival (OS), as reported in a multivariate analysis; indeed, the cumulative probability of death in patients with ISM was 2.2% at 5 years and 11% at 25 years; and Lim et al showed that patients with SM-AHNMD and ASM display a life expectancy between 41 and 24 months compared with ISM patients that have the same life expectancy as healthy people.[9,10] Most other publications on mastocytosis patients >70 years of age are case reports showing a broad spectrum of clinical presentations in elderly patients, and underlining the challenge represented by the establishment of a positive diagnosis in such cases. Only 1 report has focused on a cohort of elderly patients, aged 70 years and older at the time of diagnosis.[11] In this study, 42 patients displayed a high rate of associated hematological disorders: KIT D816V mutation was present in 14 patients, negative in 3, and not tested in 25, and no report was available on additional mutations.[11]
Thus, given the limited amount of data available in the literature for such patients, the aims of our present study were therefore to systematically analyze clinical, biological, and genetic features encountered in mastocytosis in our nationwide cohort of 53 elderly patients (>69 years of age), and especially to report on the different complex genetic backgrounds and clinical presentations of the disease at diagnosis at this age.
2. Patients and methods
2.1. Patients
The diagnosis of mastocytosis was established in accordance with the current WHO criteria for mastocytosis.[1,12] Fifty-three consecutive patients referred to the National Reference Center for Mastocytosis, Paris, France, and to regional centers of excellence in France were retrospectively enrolled in this multicenter study on the criteria of diagnosis over 69 years old. All patients gave their written informed consent. The study was approved by the local Ethics Committee of the Necker Enfants-Malades Hospital, and was carried out in accordance with ordinances of the Helsinki convention. This cohort consisted of 32 men and 21 women, with a median age of 75 years of age (range 70–90). The mastocytosis categories were as defined by the WHO criteria. In addition to BM biopsy establishing the diagnosis of systemic disease in all patients, liver, gastrointestinal, and cutaneous biopsies were performed for specific organic involvement confirmation respectively in 10 (18.9%), 12 (22.6%), and 27 (27.9%) patients. The treatments were retrospectively collected.
In our French mastocytosis reference center, 565 patients are followed, including 448 who are between 18 and 69 years old. In this cohort of young adults including 61% women, the median age at diagnosis is 38 years old.
3. Methods
3.1. Serum tryptase measurement
Total serum tryptase level (protryptase + β-tryptase) was determined using a fluorescent enzyme-linked immunoassay (Unicap, Pharmacia).[13] The limit of detection of this assay is 1 ng/mL, and in healthy controls, serum tryptase levels ranged from <1 to 15 ng/mL, with a median of 5 ng/mL.
3.2. KIT mutational status and analysis of additional genetic defects
The KIT mutational status was analyzed as previously described using a highly sensitive technique.[14,15] In addition, mutation analyses were performed to detect TET2, SRSF2, ASLX1, CBL, IDH2, JAK2, NRAS, and U2AF1 as already described.[13–17]
4. Results
4.1. Clinical presentation
Among the 53 elderly patients, the variants of mastocytosis were indolent SM (ISM, n = 5; 9.4%), aggressive SM (ASM, n = 11; 20.8%), ASM with associated hematologic non-MC lineage disease (ASM-AHNMD, n = 34; 64.2%), cutaneous mastocytosis (CM, n = 1; 1.9%), and mast cell leukemia (MCL, n = 2; 3.8%) (Fig. 1). The main clinical and biological features of the elderly patients are depicted in Table 1. The clinical presentation was very heterogeneous. A total of 50.9% (n = 27) of the patients displayed poor performance status, 75.5% (n = 40) presented with mast cell activation symptoms (flush n = 17, pruritus n = 19, diarrhea n = 12, abdominal pain n = 14); 24.5% (n = 13) had lymphadenopathy, 50.9% (n = 27) had hepatosplenomegaly, including 4 patients with only hepatomegaly and 6 patients with only splenomegaly. Ascites or other signs of portal hypertension-related disturbances were observed in 26.4% of patients (n = 14). Skin involvement was observed among 49% (n = 26) patients, mostly urticaria pigmentosa (UP) (n = 24) or less frequently telangiectasia macularis eruptiva persistans (TMEP) (n = 2). Forty-seven percent (n = 25) of patients displayed osteoporosis, including 12 of 25 (48%) patients with fractures (mostly at vertebral sites). Few patients (11.3%) displayed B findings as defined by the WHO classification, whereas C findings were documented in almost all patients (88.7%). Two patients affected by chronic eosinophilia (>1.7 × 109/L) exhibited specific patterns of cardiac and pulmonary involvement, respectively. Considering the clinical history and the onset of characteristic signs displayed by patients, the median time to diagnosis was estimated at 9 months, with extremes ranging from 3 to 12 months from the beginning of systemic signs, because some patients displayed cutaneous lesions for many years that were not linked to mastocytosis.
Figure 1.
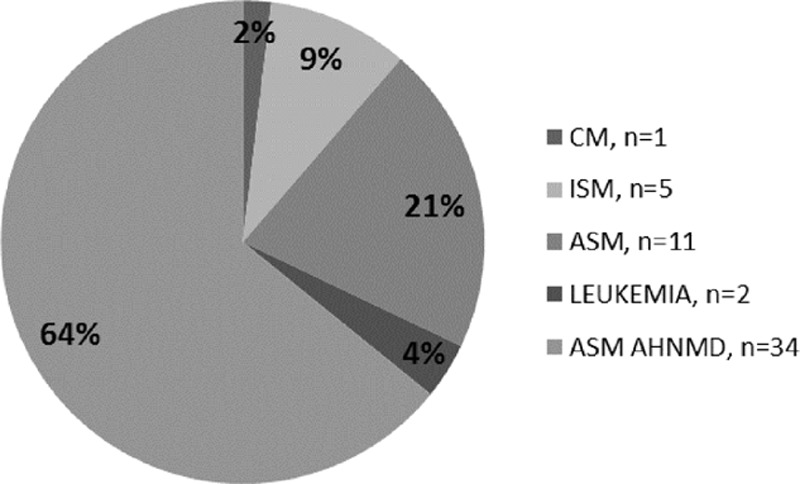
WHO classification of the cohort of 53 elderly mastocytosis patients.
Table 1.
Main features of the overall study population of 53 elderly mastocytosis patients subdivided into 3 groups: indolent forms (CM and ISM), aggressive forms (ASM, MCL, MCS), and ASM-AHNMD.
Thirty-four (64.2%) patients displayed SM-AHNMD. The distribution of associated non-mast cell hematological diseases is shown in Figure 2. The most frequent associated non-MC malignancies were myeloproliferative neoplasms in 19 patients (55.9%), followed by myelodysplastic syndrome (MDS) in 9 patients (26.5%) and acute leukemia in 3 patients (8.8%). Moreover, monoclonal gammopathy of undetermined significance or indolent myeloma were found in 3 (8.8%) patients.
Figure 2.
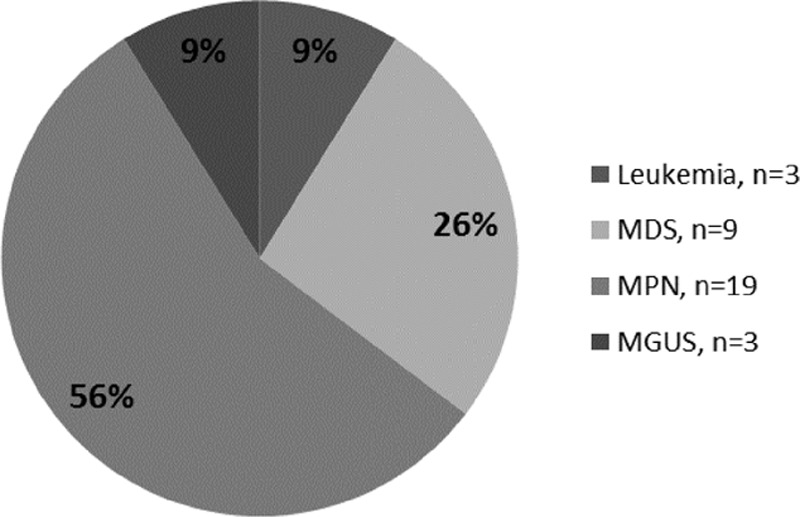
Details of the 34 patients with ASM-AHNMD according to type of associated clonal non-mast cell lineage disease. ASM = aggressive systemic mastocytosis, ASM-AHNMD = ASM with associated hematologic non-mast cell lineage disease.
When comparing the 3 subgroups of patients (Table 1): indolent (CM/ISM) versus aggressive (ASM, MCL), versus AHNMD, we observed the following features:
Indolent forms displayed more often mast cell activation symptoms and cutaneous localization and no deaths.
Aggressive forms displayed the worst performance status and more often had hepatosplenomegaly and C findings.
AHNMD also displayed C findings, but more often portal hypertension and mast cell activation symptoms.
4.2. Biological features
Seventy-nine percent (n = 42) of the patients displayed anemia with a median hemoglobin level of 10.5 g/dL (range 7.7–14.8); 50.9% (n = 27) had thrombocytopenia with a median platelet count of 91 × 109/L (range 20–528 × 109/L), and 20.8% (n = 11) had leucopenia with a median PolyNuclear Neutrophils (PNN) count of 3.5 × 109/L (range 0.74–58 × 109/L) and a median total white blood cells count of 6 × 109/L (range 1.9–70 × 109/L) for the 53 patients. Abnormal liver function tests were present in 32.1% of the patients (n = 17). The median serum tryptase level was 176 ng/mL (range 5.45–1077, normal <15 ng/mL). The median blood albumin level was 37.5 g/L (range 23.8–45).
When comparing the different subgroups (Table 1), patients with aggressive forms more often had cytopenia and had higher tryptase levels.
4.3. KIT mutations and other genetic defects
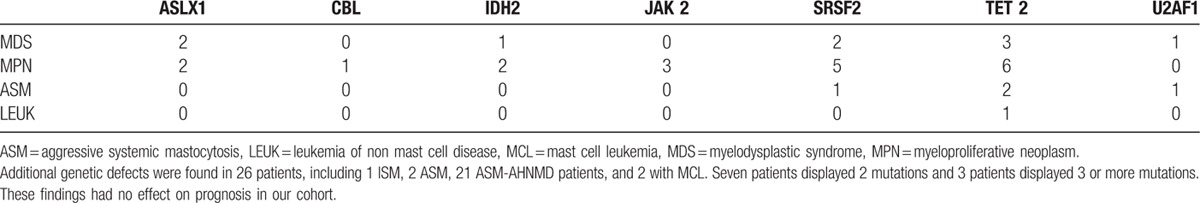
The entire coding regions of KIT were sequenced in 40 of the 53 patients (75.5%). When a mutation was detected, it was mostly the KIT D816V. Indeed, this mutation was retrieved in 34 of the 40 tested patients (85%), whereas 2 patients (5%) presented another KIT mutation, (p.(Phe504_Asn505delinsLeuLysPheLysThr) and p.Ser501_Ala502dup), and 4 patients (10%) displayed wild-type KIT. For 26 patients, including 1 ISM, 2 ASM, 21 SM-AHNMD patients, and the 2 patients with MCL, sequencing data for additional genetic defects were available and are shown in Table 2. Seventeen (65.3%) of them (including 14 SM-AHNMD, 2 ASM, and 1 MCL patient) exhibited TET2 (n = 12), SRSF2 (n = 8), IDH2 (n = 3), ASLX1 (n = 4), CBL (n = 1), U2AF1 (n = 2), and/or NRAS (n = 1) mutations. Ten patients had at least 2 mutations, mostly patients with SM-AHNMD variant (n = 9). No patient, even those with eosinophilia, exhibited the FIP1L1/PDGFRA mutation.
Table 2.
Types of additional mutation found among 26 late-onset systemic mastocytosis patients.
4.4. Treatment
All patients received at least symptomatic treatments with antihistamines (anti-H1R and anti-H2R) or proton pump inhibitors, low doses of corticosteroids (oral or local; n = 11), or best supportive care in some cases (bisphosphonate and/or transfusion regimen). For most patients, specific treatment was not proposed due to the patients’ poor general condition and/or associated malignant hematological disease with no available treatment options due to the patients’ advanced age. The type of chemotherapy regimen proposed was available and well documented in 17 patients with the systemic and aggressive forms of the disease. Nineteen patients received either cladribine (n = 11), velcade (n = 1), interferon α (n = 3), or thalidomide (n = 5). Six other patients, for whom chemotherapy regimens containing ARA-C and fludarabine failed, received midostaurin (PKC 412) in a context of high-risk advanced-stage myeloproliferative or myelodysplastic syndromes (n = 4), an aggressive form of mastocytosis (n = 1) or a mast cell leukemia (n = 1).
4.5. Prognosis and outcome
After a median follow-up of 17 months (range 7–50 months), death occurred in 35.8% (n = 19) of the patients. Considering the time to diagnosis, death occurred after a median time of 26 months from disease onset (range 10–62 months). Approximately one-third (n = 6) of the 19 deaths were related to the rapid progression of the AHNMD. Two-thirds (n = 12) of the deaths were related to the progression of mastocytosis and/or to the toxicity of the chemotherapy regimen, including both patients with respectively MCS or MCL. Fatal toxicities were as follows: BM failure with severe neutropenia and septic shock (n = 1), refractory thrombocytopenia and hemorrhage (n = 3) and refractory anemia (n = 2); specific organ involvement with liver (n = 3), heart (n = 1) or pulmonary (n = 2) failures. The remaining death was related to sudden heart failure of unknown origin.
When comparing the 3 different subgroups (Table 1), patients with aggressive forms of the disease displayed the worst prognosis.
5. Discussion
To our knowledge, this study reporting 53 elderly mastocytosis patients with a focus on additional mutations besides KIT is the largest published on elderly adults in the literature so far. The Table 3 summarizes the principal features of the 53 patients. Of note, data of our study point to several differences in the initial presentation between our elderly patients and patients with the “classical” disease mainly described in infancy and young adulthood. Indeed, elderly patients usually present with extremely poor performance status, symptoms of mast cell activation, hepatosplenomegaly, and sometimes ascites or osteoporosis with fractures, associated with cytopenia. Diagnosis of this rare disease in elderly patients is often long delayed, probably because gerontologists are less aware of the typical signs of the disease than the cutaneous symptoms, which is all the more frequent ad evocative when occurring at a younger age, as confirmed by our data. Indeed we found a relatively low frequency (less than half of cases) of urticaria pigmentosa (UP) in this cohort, contrasting with the high frequency of UP (up to 85%) reported for younger patients with systemic mastocytosis, and for whom UP constitutes a major diagnostic sign.[20,21] On the other hand, cutaneous involvement is found in about 90 percent of patients in the general mastocytosis population without an AHNMD, and about one-third of this population first develop characteristic lesions in adulthood; this form of the disease represents only 1.9% of our elderly population, while 88.7% of our patients exhibited C findings and almost 1 or more hematologic organ impairment.[8,18,19]
Table 3.
Main features of the 53 patients.
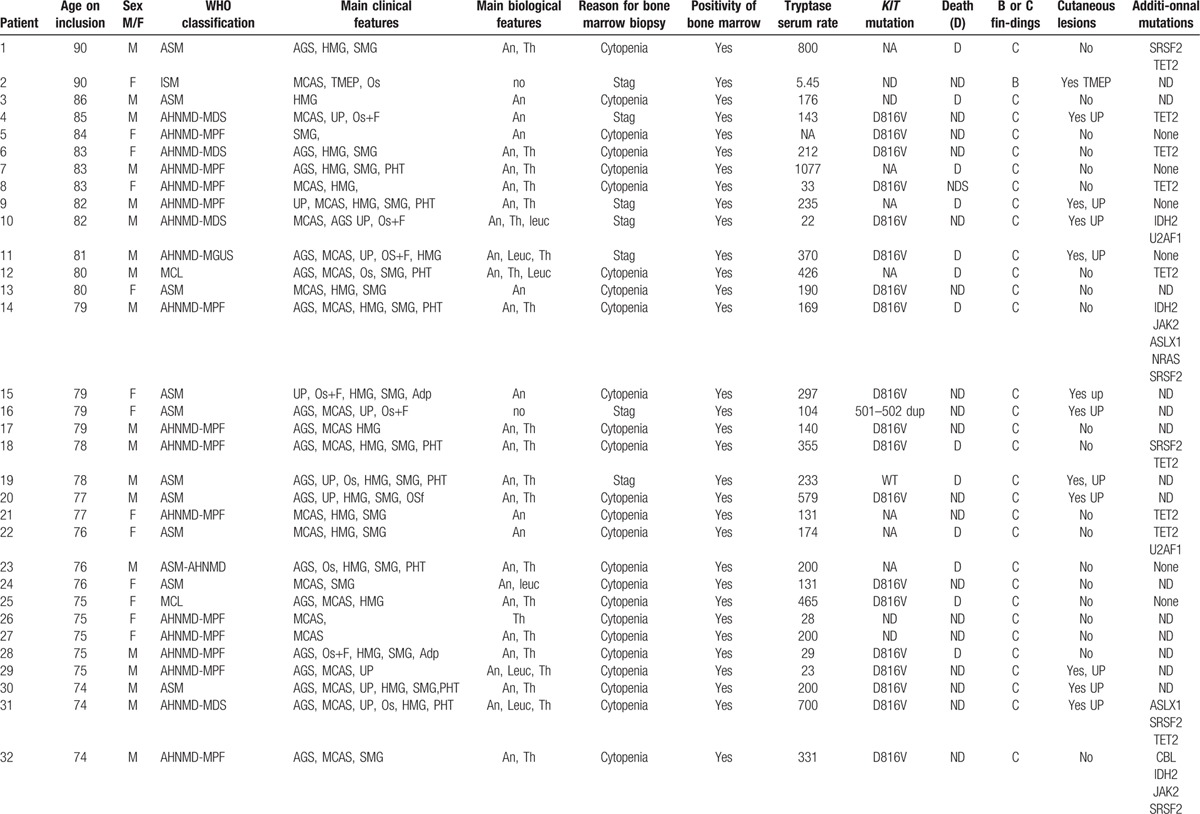
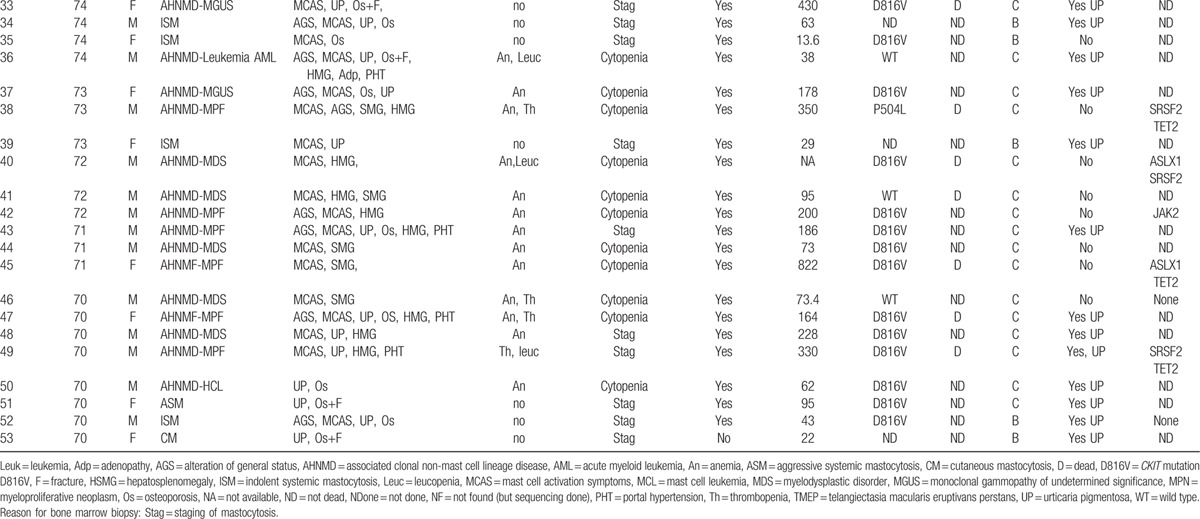
Table 3 (Continued).
Main features of the 53 patients.
Besides KIT mutations found recurrently in SM, we were able to detect mutations in TET2, JAK2, or SRSF2 genes, which are usually found in myelodysplastic/proliferative disorders. In the study by Schwaab et al,[5] 27 of 39 patients (69.2%) with different forms of systemic mastocytosis also exhibited some of these additional defects. In our study, we found almost similar rates. Indeed, we observed at least one of these additional mutations in 17 of 26 (65.3%) of our patients tested who had associated hematological malignancies. The finding of common molecular neoplastic mechanisms and/or a common hematopoietic progenitor cell origin for both mastocytosis and myelodysplastic/myeloproliferative disorders should be therefore of great interest. We have recently reported that TET2 deficiency has the ability to affect KIT D816V+ MCs, and therefore promote more severe forms of the disease.[15] In our cohort of elderly patients, we found a frequency of 46.2% of TET2 mutations, which is higher than that (20%) reported in the literature.[6,7,9,15,17] Patients with TET2 mutations harbored the following variants of mastocytosis: ASM (n = 2), ASM-AHNMD (n = 8), ISM (n = 1), and MCL (n = 1). By contrast, SRSF2 mutations were observed in 8 patients (30.8% of our cohort), including 7 affected by a severe AHNMD form, a frequency that is similar to that reported in the literature (24%–37%).[6,14,17]
The mortality rate is high in the cohort (35.8%), but this is somewhat expected since most of the patients are classified as SM-AHNMD and because elderly patients usually display more comorbidities than young patients. According to the literature and to our results, the OS was worst among patients with additional genetic defects compared with those where the KIT D816V mutation was the sole defect identified.[17] Taken together, these results underline the critical need to sequence not only KIT but also additional genes to better characterize mastocytosis and the frequently associated AHNMDs, these latter malignancies playing a major role in the poor prognosis of the elderly. Indeed, the KIT mutant D816V may disappear in some advanced forms of the disease which is best explained by selection and transformation of more malignant subclones derived from a more primitive neoplastic stem cell compartment where KIT D816V is not expressed.[7] Therefore, new treatments such as kinase inhibitors targeting both mast cell and other myeloid cell malignant proliferation could open up interesting therapeutic prospects in the context of elderly patients displaying mastocytosis.[22,23]
In conclusion, the clinical and prognostic profiles described herein confirmed the variable presentation of mastocytosis in elderly patients. Indeed, besides the dominant patterns of limited and/or self-limited benign cutaneous disease in infancy, and mostly indolent systemic disease with frequent neuropsychological features in young adulthood, elderly patients in our cohort exhibit a more severe aggressive outcome, less frequent skin involvement and/or limited disease, but frequent nonfortuitous association with other myeloid malignancies, probably due to the high frequency of aggressive and AHNMD forms of mastocytosis in the ageing population.[8,24–28] A better knowledge of KIT and other additional myeloid lineage mutations should help improve therapeutic differential targeting of this heterogeneous disease.[5–7,9,14,15,17,29]
Footnotes
Abbreviations: AGS = alteration of general status, ASM = aggressive systemic mastocytosis, CEREMAST = Centre de Reference des Mastocytoses ( = mastocytosis reference center), CM = cutaneous mastocytosis, HMG = hepatomegaly, HSMG = hepatosplenomegaly, ISM = indolent systemic mastocytosis, MCAS = mast cell activation symptoms, MCL = mast cell leukemia, MCS = mast cell sarcoma, MDS = myelodysplastic syndrome, MGUS = monoclonal gammopathy of undetermined significance, MPN = myeloproliferative neoplasm, PHT = portal hypertension, SM-AHNMD = systemic mastocytosis with associated clonal nonmast cell lineage disease, SMG = splenomegaly, TMEP = telangiectasia macularis eruptivans perstans, UP = urticaria pigmentosa, WT = wild type.
AR and AA contributed equally to the study. OH and SG-L codirected the study.
The authors report no conflicts of interest.
References
- [1].Akin C, Valent P. Diagnostic criteria and classification of mastocytosis in 2014. Immunol Allergy Clin North Am 2014;34:207–18. [DOI] [PubMed] [Google Scholar]
- [2].Hermine O, Lortholary O, Leventhal PS, et al. Case-control cohort study of patients’ perceptions of disability in mastocytosis. PloS One 2008;3:e2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cruse G, Metcalfe DD, Olivera A. Functional deregulation of KIT: link to mast cell proliferative diseases and other neoplasms. Immunol Allergy Clin North Am 2014;34:219–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lanternier F, Cohen-Akenine A, Palmerini F, et al. Phenotypic and genotypic characteristics of mastocytosis according to the age of onset. PloS One 2008;3:e1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schwaab J, Schnittger S, Sotlar K, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013;122:2460–6. [DOI] [PubMed] [Google Scholar]
- [6].Soucie E, Brenet F, Dubreuil P. Molecular basis of mast cell disease. Mol Immunol 2015;63:55–60. [DOI] [PubMed] [Google Scholar]
- [7].Jawhar M, Schwaab J, Schnittger S, et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015;29:1115–22. [DOI] [PubMed] [Google Scholar]
- [8].Méni C, Bruneau J, Georgin-Lavialle S, et al. Paediatric mastocytosis: a systematic review of 1747 cases. Br J Dermatol 2015;172:642–51. [DOI] [PubMed] [Google Scholar]
- [9].Brockow K. Epidemiology, prognosis, and risk factors in mastocytosis. Immunol Allergy Clin North Am 2014;34:283–95. [DOI] [PubMed] [Google Scholar]
- [10].Lim K-H, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood 2009;113:5727–36. [DOI] [PubMed] [Google Scholar]
- [11].Butterfield JH, Weiler CR. Systemic mastocytosis in the elderly. Am J Hematol 2013;88:406–8. [DOI] [PubMed] [Google Scholar]
- [12].Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- [13].Georgin-Lavialle S, Lhermitte L, Baude C, et al. Blood CD34-c-Kit+ cell rate correlates with aggressive forms of systemic mastocytosis and behaves like a mast cell precursor. Blood 2011;118:5246–9. [DOI] [PubMed] [Google Scholar]
- [14].Hanssens K, Brenet F, Agopian J, et al. SRSF2-p95 hotspot mutation is highly associated with advanced forms of mastocytosis and mutations in epigenetic regulator genes. Haematologica 2014;99:830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Soucie E, Hanssens K, Mercher T, et al. In aggressive forms of mastocytosis, TET2 loss cooperates with c-KITD816V to transform mast cells. Blood 2012;120:4846–9. [DOI] [PubMed] [Google Scholar]
- [16].Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009;360:2289–301. [DOI] [PubMed] [Google Scholar]
- [17].Bibi S, Langenfeld F, Jeanningros S, et al. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol Allergy Clin North Am 2014;34:239–62. [DOI] [PubMed] [Google Scholar]
- [18].Horan RF, Austen KF. Systemic mastocytosis: retrospective review of a decade's clinical experience at the Brigham and Women's Hospital. J Invest Dermatol 1991;96(3 suppl):5S–13S. [PubMed] [Google Scholar]
- [19].Gruchalla RS. Southwestern Internal Medicine Conference: mastocytosis: developments during the past decade. Am J Med Sci 1995;309:328–38. [DOI] [PubMed] [Google Scholar]
- [20].Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. J Invest Dermatol 1991;96(3 suppl):15S–8S. [PubMed] [Google Scholar]
- [21].Azaña JM, Torrelo A, Mediero IG, et al. Urticaria pigmentosa: a review of 67 pediatric cases. Pediatr Dermatol 1994;11:102–6. [DOI] [PubMed] [Google Scholar]
- [22].Arock M, Akin C, Hermine O, et al. Current treatment options in patients with mastocytosis: status in 2015 and future perspectives. Eur J Haematol 2015;474–90. [DOI] [PubMed] [Google Scholar]
- [23].Siebenhaar F, Akin C, Bindslev-Jensen C, et al. Treatment strategies in mastocytosis. Immunol Allergy Clin North Am 2014;34:433–47. [DOI] [PubMed] [Google Scholar]
- [24].Azaña JM, Torrelo A, Mediero IG, et al. Urticaria pigmentosa: a review of 67 pediatric cases. Pediatr Dermatol 1994;11:102–6. [DOI] [PubMed] [Google Scholar]
- [25].Kettelhut BV, Metcalfe DD. Pediatric mastocytosis. J Invest Dermatol 1991;96(3 suppl):15S–8S. [PubMed] [Google Scholar]
- [26].Torrelo A, Alvarez-Twose I, Escribano L. Childhood mastocytosis. Curr Opin Pediatr 2012;24:480–6. [DOI] [PubMed] [Google Scholar]
- [27].Lange M, Niedoszytko M, Renke J, et al. Clinical aspects of paediatric mastocytosis: a review of 101 cases. J Eur Acad Dermatol Venereol JEADV 2013;27:97–102. [DOI] [PubMed] [Google Scholar]
- [28].Lange M, Nedoszytko B, Górska A, et al. Mastocytosis in children and adults: clinical disease heterogeneity. Arch Med Sci AMS 2012;8:533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bodemer C, Hermine O, Palmérini F, et al. Pediatric mastocytosis is a clonal disease associated with D816 V and other activating c-KIT mutations. J Invest Dermatol 2010;130:804–15. [DOI] [PubMed] [Google Scholar]