

Abstract
Hyperactive Ras promotes proliferation and malignant phenotypic conversion of cells in cancer. Ras protein must be associated with cellular membranes for its oncogenic activities through post-translational modifications, including farnesylation. Farnesyltransferase (FTase) is essential for H-Ras membrane targeting, and H-Ras, but not N-Ras, has been demonstrated to cause an invasive phenotype in MCF10A breast epithelial cells. In the present study, it was observed that an FTase inhibitor (FTI), FTI-277, blocked epidermal growth factor (EGF)-induced H-Ras activation, but not N-Ras activation in MDA-MB-231 cells, which express wild-type H-Ras and N-Ras. FTI-277 exerted a more potent inhibitory effect on the proliferation of H-Ras-MCF10A cells and Hs578T breast cancer cells expressing an active mutant of H-Ras than that of MDA-MB-231 cells. The invasive/migratory phenotypes of the H-Ras-MCF10A and Hs578T cells were effectively inhibited by FTI-277 treatment. By contrast, FTI-277 did not affect the invasive/migratory phenotypes of MDA-MB-231 cells. However, the EGF-induced invasion of MDA-MB-231 cells was decreased by FTI-277, implicating that FTI-277 inhibits breast cell invasion and migration by blocking H-Ras activation. Taken together, the results of the present study suggest that FTase inhibition by FTI-277 may be an effective strategy for targeting H-Ras-mediated proliferation, migration and invasion of breast cells.
Keywords: breast cancer, FTI-277, H-Ras, invasion, proliferation
Introduction
The mutation of RAS is the most frequent event during the development of human cancer (1,2). Four closely related isoforms of the Ras protein exist and consist of H-Ras, K(A)-Ras, K(B)-Ras and N-Ras (3,4). Mutations of distinct Ras isoforms are detected in an organ-specific manner and have different cellular functions (3,4). To be functionally active, the Ras protein must be associated with cellular membranes through several post-translational modifications (5). Farnesyltransferase (FTase) is an essential enzyme for farnesylation on the cysteine of the CAAX motif of cytosolic Ras, resulting in the irreversible step of Ras membrane association (6). Membrane-anchored Ras protein becomes activated by cycling between the GDP-bound inactive state and the GTP-bound active state (5).
Farnesylation by FTase is the first irreversible, rate-limiting step for Ras membrane association and an obligate modification for oncogenic Ras biological activity (7). Initial attempts to inhibit FTase have been made in the development of anti-cancer agents (2,8,9). In vitro and in vivo tumor models have demonstrated that FTase inhibitors (FTIs) are potent against various forms of cancer (2,10). FTI treatment has also been reported to be effective against H-Ras-transformed cells and H-Ras-driven murine tumors (10,11). Following treatment with the FTI lonafarnib, transgenic mice with mammary tumors containing a H-Ras mutation underwent complete tumor regression (10). However, these drugs failed to increase survival in clinical trials of patients with K-Ras mutated pancreatic cancer (12). N-Ras and K-Ras become substrates for geranylgeranyltransferase I, resulting in alternative prenylation and membrane targeting (9,13). By contrast, H-Ras has no alternative prenylation for membrane targeting. Therefore, FTIs are emerging as an effective therapeutic approach for targeting H-Ras in cancer (9,13). FTI-277, an FTI, successfully inhibited Ras membrane association, resulting in prevention of Ras oncogenic signaling from the cell membrane (14).
Breast cancer has been estimated as the most commonly diagnosed form of cancer and the second highest cause of cancer-associated mortality among women in the United States (15). The metastatic spread of breast cancer is a major cause of mortality (16). Despite the low frequency (<5%) of mutated forms of RAS in breast cancer (13), elevated levels of the Ras protein have been identified in 60–70% of human primary breast cancer cases (17). It was previously demonstrated that H-Ras and N-Ras induced a transformed phenotype in human breast epithelial MCF10A cells (18). However, H-Ras, but not N-Ras, led to an invasive phenotype in MCF10A cells (18). The H-Ras-specific Rac1-MKK3/6-p38 mitogen-activated protein kinase pathway is required for breast cell motility and invasion (19–21).
The present study investigated the effect of FTI-277 on H-Ras activation and the migratory/invasive phenotypes of breast cancer cells. The results demonstrated that FTI-277 inhibited activation of H-Ras, but not that of N-Ras. In addition, FTI-277 effectively inhibited the migratory/invasive abilities of breast cancer cells through inhibition of H-Ras activation.
Materials and methods
Cell culture conditions and reagents
H-Ras-MCF10A cells were established and cultured as previously described (18). Hs578T and MDA-MB-231 breast cancer cells were purchased from the Korean Cell Line Bank (Seoul, Korea). Hs578T and MDA-MB-231 cells were cultured in Dulbecco's modified Eagles's medium (GE Healthcare Life Sciences, Salt Lake City, UT, USA) supplemented with 10% fetal bovine serum (Corning Life Sciences, Cambridge, MA, USA) and 100 µg/ml penicillin-streptomycin as previously described (22). FTI-277 (F9803) was purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in distilled water.
Immunoblot analysis
Immunoblot analysis was performed as described previously (23). Protein extracts in lysis buffer [50 mM Tris, 2% sodium dodecyl sulfate (SDS), 1 mM ethylenediaminetetraacetic acid (EDTA) and 0.1 M dithiothreitol) containing protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany) were subjected to 12% SDS-polyacrylamide gel electrophoresis. The PVDF membrane (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was probed with rabbit polyclonal anti-human H-Ras [dilution, 1:1,000 in phosphate-buffered saline with Tween (PBST); #sc-520] and anti-N-Ras (dilution, 1:1,000 in PBST; sc-519) antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX, USA). WesternBright ECL kit was used for detection (#K-12045-D50; Advansta Inc., Menlo Park, CA, USA). Relative band intensities were determined by quantification of each band using the Gel Doc™ XR+ system (Bio-Rad Laboratories, Inc.).
Membrane fractions
Membrane fractions were collected as previously described (24). Cells were treated with FTI-277 (50 µM) for 24 h and epidermal growth factor (EGF) (10 ng/ml) for 30 min prior to lysis. Cells were rinsed with PBS buffer, suspended in homogenization buffer (0.25 M sucrose, 25 mM Tris-HCl and 10 µg/ml leupeptin, pH 7.4), and homogenized in a ground-glass dounce homogenizer (Heidolph Instruments, Schwabach, Germany). The lysates were centrifuged for 10 min at 3,000 × g, resulting in the pellet and supernatant. The supernatant was centrifuged again at 12,000 × g for 10 min. The resulting pellet was suspended in chelating buffer (25 mM Tris-HCl, 5 mM EDTA and 10 µg/ml leupeptin, pH 7.4), followed by centrifugation at 12,000 × g for 10 min. The resulting pellet (membrane fraction) was washed and resuspended in incubation buffer (25 mM Tris-HCl and 10 µg/ml leupeptin, pH 7.4).
Ras activity assay
The level of Ras-GTP was measured by Ras assay reagent (Merck Millipore, Darmstadt, Germany) following the manufacturer's protocol, as previously described (22). The cells were treated with 10 ng/ml EGF (Sigma-Aldrich) for 30 min and lysed in Mg2+ lysis/wash buffer. Raf-1 RBD-agarose was added to the lysates and incubated for 45 min at 4°C. The bead pellet was washed 3 times with MLB buffer, boiled in 2X Laemmli sample buffer, and subjected to immunoblot analysis with anti-H-Ras or anti-N-Ras antibodies.
MTT assay
MTT assay was performed as previously described (25). Cells (5×103) were seeded in a 96-well plate and incubated for 24 h at 37°C. Cells were then treated with various concentrations of FTI-277 (0, 10, 20 and 50 µM) for 24 or 48 h. Following incubation, 25 µl 5 mg/ml MTT (Sigma-Aldrich) was added and incubated for 3 h, and formazan was dissolved in 100 µl dimethyl sulfoxide. The optical density was measured at 540 nm using a Synergy 2 Multi-Mode Reader (BioTek Instruments, Inc., Winooski, VT, USA). Each sample was assayed in triplicate.
In vitro invasion/migration assays
In vitro invasion/migration assays were performed using a 24-well Transwell unit with polycarbonate filters (Corning Life Sciences) as previously described (26). The lower side of the filter was coated with type I collagen (Sigma-Aldrich) and the upper side of the filter was coated with Matrigel (BD Biosciences, Bedford, MA, USA). Cells (5×104 cells) suspended in DMEM supplemented with 100 µg/ml penicillin-streptomycin were placed in the upper chamber. The lower chamber was filled with DMEM supplemented with 10% fetal bovine serum and 100 µg/ml penicillin-streptomycin. Following 24 h of incubation, cells that had invaded the lower side of the filter were solubilized with 0.5% crystal violet solution and measured at 595 nm using a micro-ELISA reader. Each sample was assayed in triplicate.
Results
FTI-277 inhibits EGF-induced H-Ras activation in MDA-MB-231 cells
The present study examined the effect of FTI-277 on the localization and activation of H-Ras and N-Ras in breast cancer cells. Membrane fractions were extracted from MDA-MB-231 cells in which both H-Ras and N-Ras may be activated by EGF (22,27,28). H-Ras-GTP was induced by EGF in the membrane fractions (Fig. 1A). Following treatment with 50 µM FTI-277 for 24 h, H-Ras-GTP was almost completely abolished in the membrane fractions. However, in whole cell lysates, FTI-277 did not inhibit H-Ras-GTP induced by EGF. Unfarnesylated H-Ras (upper band) was detected following FTI-277 treatment in whole cell lysates, but not in the membrane fractions (Fig. 1A). These results indicate that FTI-277 inhibited farnesylation, and therefore also inhibited the membrane localization of H-Ras in MDA-MB-231 cells.
Figure 1.
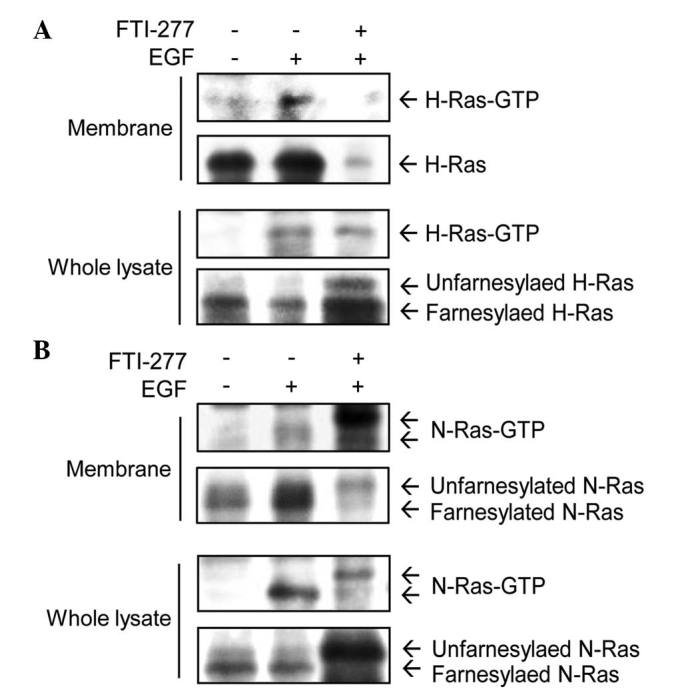
FTI-277 inhibits EGF-induced H-Ras activation in MDA-MB-231 cells. MDA-MB-231 cells treated with 50 µM FTI-277 for 24 h were exposed to 10 ng/ml EGF for 30 min prior to cell lysis. Membrane fractions and whole cell lysates were extracted and then subjected to Ras activity assay and immunoblot analysis with (A) anti-H-Ras or (B) anti-N-Ras antibodies. FTI, farnesyltransferase inhibitor; EGF, epidermal growth factor.
EGF treatment induced activation of N-Ras in membrane fractions and whole cell lysates (Fig. 1B). The level of N-Ras-GTP in membrane fractions was not decreased by treatment with FTI-277. These results suggest that N-Ras has an alternative prenylation for membrane targeting, unlike H-Ras. In combination, these data demonstrate that FTI-277 inhibited the EGF-induced activation of H-Ras, but not that of N-Ras, in membrane fractions of MDA-MB-231 cells.
FTI-277 exerts an anti-proliferative effect in breast cells expressing active H-Ras
The current study subsequently examined the effect of FTI-277 on the proliferation of breast cells. H-Ras-activated breast H-Ras-MCF10A cell lines, in which a constitutively active mutant of H-Ras (G12D) was stably introduced (18), and Hs578T cells, which endogenously express active H-Ras (G12D), were analyzed (29). MDA-MB-231 cells harboring wild-type H-Ras and N-Ras were also used (30). MTT assay demonstrated that FTI-277 inhibited proliferation of the H-Ras-MCF10A, Hs578T and MDA-MB-231 cells in a dose-dependent manner (Fig. 2). FTI-277 exerted a strong anti-proliferative effect on the H-Ras-MCF10A and Hs578T cells with 50% inhibitory concentration (IC50) values of 6.84 and 14.87 µM for 48 h, respectively. FTI-277 treatment inhibited proliferation of the MDA-MB-231 cells with an IC50 value of 29.32 µM for 48 h. The results suggest that breast cells in which H-Ras is activated may be more susceptible to FTI-277 compared with the cells with wild-type H-Ras.
Figure 2.
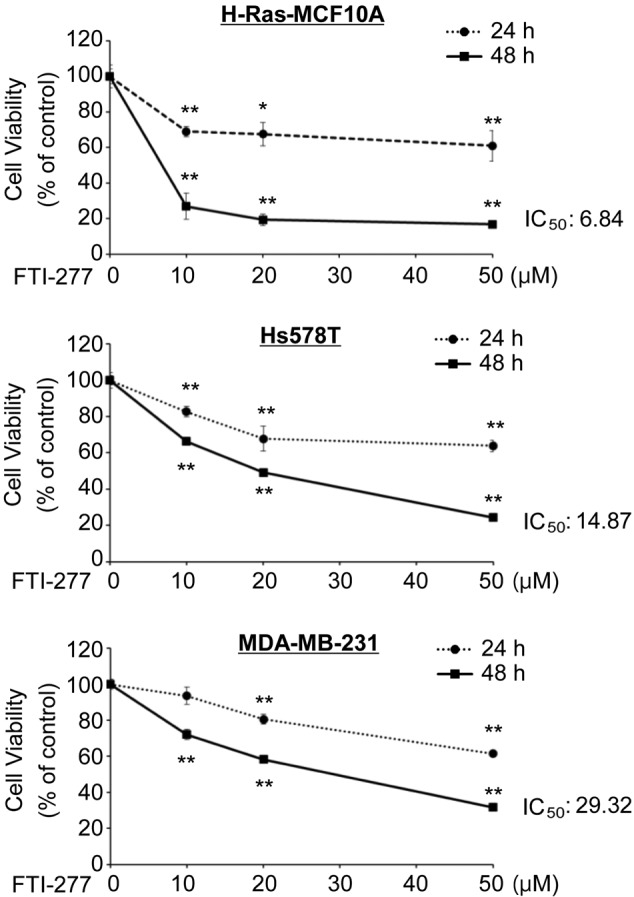
FTI-277 exerts anti-proliferative effects in breast cells. H-Ras-MCF10A, Hs578T and MDA-MB-231 cells were subjected to MTT assay with various concentrations (0, 10, 20 and 50 µM) of FTI-277. The results are presented as the mean ± standard error of triplicates. *P<0.05 and **P<0.01 vs. control. FTI, farnesyltransferase inhibitor; IC50, 50% inhibitory concentration.
FTI-277 inhibits invasive and migratory phenotypes of breast cells expressing active H-Ras
Next, the present study investigated the effect of FTI-277 on the invasive phenotypes of H-Ras-MCF10A, Hs578T and MDA-MB-231 cells, which have been previously demonstrated to be highly invasive (18,19,30). The invasiveness of the H-Ras-MCF10A and Hs578T cells was significantly inhibited by FTI-277 (Fig. 3A). By contrast, FTI-277 did not affect the invasive ability of the MDA-MB-231 cells.
Figure 3.
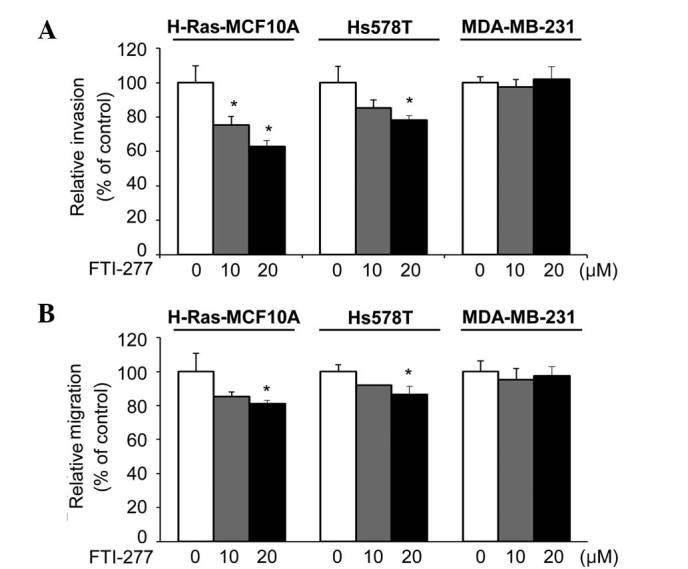
FTI-277 inhibits invasive and migratory phenotypes in breast cells expressing active H-Ras. H-Ras-MCF10A, Hs578T and MDA-MB-231 cells were subjected to in vitro (A) invasion and (B) migration assays with various concentrations (0, 10 and 20 µM) of FTI-277. The results are presented as the mean ± standard error of triplicates. *P<0.05 vs. control. FTI, farnesyltransferase inhibitor.
Similarly, FTI-277 treatment significantly inhibited the migratory phenotypes of the H-Ras-MCF10A and Hs578T cells, but not that of the MDA-MB-231 cells (Fig. 3B). Given that the active state of H-Ras is important in invasive phenotypes of H-Ras-MCF10A and Hs578T cells (18,19,22), the results suggest that FTI-277 may inhibit the invasive and migratory abilities of breast cells through inhibition of H-Ras.
FTI-277 inhibits EGF-induced invasion of MDA-MB-231 cells
To further examine if the inhibitory effect of FTI-277 on breast cell invasion involves inhibition of H-Ras activation, the present study analyzed whether FTI-277 affects the invasive phenotype of MDA-MB-231 cells treated with EGF. As presented in Fig. 4, the invasive phenotype of the MDA-MB-231 cells was enhanced by EGF stimulation. FTI-277 significantly decreased the EGF-enhanced invasion of the MDA-MB-231 cells in a concentration-dependent manner (Fig. 4). Given that FTI-277 inhibited the EGF-induced H-Ras activation in MDA-MB-231 cells (Fig. 1A), the present data indicates that FTI-277 inhibits breast cell invasion, possibly through inhibition of H-Ras activation.
Figure 4.
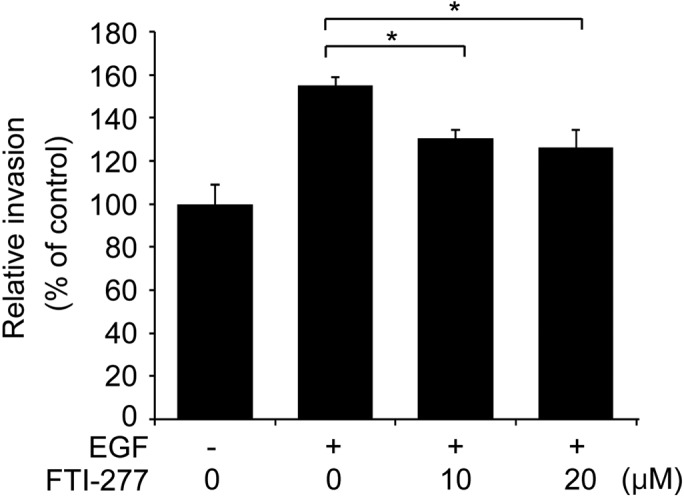
FTI-277 inhibits EGF-induced invasion of MDA-MB-231 cells. MDA-MB-231 cells were subjected to in vitro invasion assay with 10 ng/ml EGF and various concentrations (0, 10 and 20 µM) of FTI-277 in the lower chamber. The results are presented as the mean ± standard error of triplicates. *P<0.05 vs. EGF-treated controls. EGF, epidermal growth factor; FTI, farnesyltransferase inhibitor.
Discussion
FTase has been recently suggested as a drug target in the development of anti-cancer therapy (7,9). In the present study, it was demonstrated that FTI-277 exerted a more potent anti-proliferative effect on breast cells expressing an active mutant of H-Ras (H-Ras-MCF10A and Hs578T cells) than breast cells expressing wild-type H-Ras (MDA-MB-231 cells). Although H-Ras mutation is infrequent compared with K-Ras or N-Ras (13), the current data suggests that FTIs may serve as an effective strategy for targeting H-Ras-mediated proliferation of breast cancer cells. A promising FTI, tipifarnib, demonstrated anti-cancer activity against breast cancer in a phase II study, which warrants further study (9).
In addition, the present study observed that FTI-277 treatment markedly decreased the level of H-Ras-GTP, but not that of N-Ras-GTP, in membrane fractions of MDA-MB-231 cells stimulated by EGF. As Ras proteins trigger signaling pathways and activate downstream effector molecules when they are located in membrane (31), the results imply that FTI-277 effectively blocked H-Ras activation by inhibiting H-Ras trafficking to the cell membrane of breast cancer cells.
Furthermore, in whole cell lysates, FTI-277 was not able to inhibit H-Ras-GTP or N-Ras-GTP in the current study. It was previously reported that FTI-277 induced the accumulation of Ras-Raf complexes in the cytoplasm where Ras is GTP-bound, but Raf kinase was not activated (14). Hence, it is plausible that the H-Ras-GTP in the cytoplasm of MDA-MB-231 cells may not be active.
There is increasing evidence that FTIs inhibit the migration, invasion and metastasis of various forms of cancer, including breast, head and neck, pancreatic and colon cancer (32–35). The present study demonstrated that FTI-277 effectively inhibited the invasive/migratory phenotypes of H-Ras-MCF10A and Hs578T cells, but not those of MDA-MB-231 cells. Consistent with these results, a previous study observed that FTI-277 did not inhibit the Transwell invasion of MDA-MB-231 cells (36). By contrast, it has been reported that the transendothelial invasion of MDA-MB-231 cells was mildly inhibited by FTI-277 (33). These results suggest that FTI-277 exerts mild or no effect on the invasion of MDA-MB-231 cells without EGF stimulation, depending on the different methods of invasion assays.
Notably, EGF-enhanced invasion of MDA-MB-231 cells was significantly decreased by FTI-277 treatment in the current study. Given that EGF enhances the invasive phenotypes of MDA-MB-231 cells by activation of H-Ras and K-Ras (22), these results indicate that FTI-277 may partially inhibit EGF-increased invasion mediated by H-Ras.
In conclusion, the present study demonstrated that FTI-277 exerted a potent inhibitory effect on the proliferation and invasive/migratory abilities of breast cells expressing active H-Ras. In addition, FTI-277 inhibited EGF-induced H-Ras activation and invasion of breast cancer cells. This suggests that FTase inhibition may be an effective therapeutic approach for targeting H-Ras-mediated proliferation, migration and invasion of breast cells.
Acknowledgements
The present study was supported by the Priority Research Centers Program (grant no. 2016R1A6A1A03007648) and the Bio & Medical Technology Development Program (grant no. 2015M3A9B6074045) of the National Research Foundation of Korea funded by the Korean government.
References
- 1.Barbacid M. Ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Appels NM, Beijnen JH, Schellens JH. Development of farnesyl transferase inhibitors: A review. Oncologist. 2005;10:565–578. doi: 10.1634/theoncologist.10-8-565. [DOI] [PubMed] [Google Scholar]
- 3.Carbone A, Gusella GL, Radzioch D, Varesio L. Human Harvey-ras is biochemically different from Kirsten- or N-ras. Oncogene. 1991;6:731–737. [PubMed] [Google Scholar]
- 4.Maher J, Baker DA, Manning M, Dibb NJ, Roberts IA. Evidence for cell-specific differences in transformation by N-, H- and K-ras. Oncogene. 1995;11:1639–1647. [PubMed] [Google Scholar]
- 5.Hancock JF, Parton RG. Ras plasma membrane signalling platforms. Biochem J. 2005;389:1–11. doi: 10.1042/BJ20050231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reiss Y, Goldstein JL, Seabra MC, Casey PJ, Brown MS. Inhibition of purified p21ras farnesyl: Protein transferase by Cys-AAX tetrapeptides. Cell. 1990;62:81–88. doi: 10.1016/0092-8674(90)90242-7. [DOI] [PubMed] [Google Scholar]
- 7.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: Back to the future for Anti-RAS drug discovery? Clin Cancer Res. 2015;21:1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunner TB, Hahn SM, Gupta AK, Muschel RJ, McKenna WG, Bernhard EJ. Farnesyltransferase inhibitors: An overview of the results of preclinical and clinical investigations. Cancer Res. 2003;63:5656–5668. [PubMed] [Google Scholar]
- 9.Vasan N, Boyer JL, Herbst RS. A RAS renaissance: Emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res. 2014;20:3921–3930. doi: 10.1158/1078-0432.CCR-13-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu M, Bryant MS, Chen J, Lee S, Yaremko B, Lipari P, Malkowski M, Ferrari E, Nielsen L, Prioli N, et al. Antitumor activity of SCH 66336, an orally bioavailable tricyclic inhibitor of farnesyl protein transferase, in human tumor xenograft models and wap-ras transgenic mice. Cancer Res. 1998;58:4947–4956. [PubMed] [Google Scholar]
- 11.Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, Giuliani EA, Gomez RP, Graham SL, Hamilton K, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med. 1995;1:792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 12.Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, Schoffski P, Post S, Verslype C, Neumann H, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430–1438. doi: 10.1200/JCO.2004.10.112. [DOI] [PubMed] [Google Scholar]
- 13.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, Cox AD, Der CJ, Hamilton AD, Sebti SM. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995;270:26802–26806. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 15.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 16.Hudis CA, Gianni L. Triple-negative breast cancer: An unmet medical need. Oncologist. 2011;16(Suppl 1):S1–S11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 17.Clair T, Miller WR, Cho-Chung YS. Prognostic significance of the expression of a ras protein with a molecular weight of 21,000 by human breast cancer. Cancer Res. 1987;47:5290–5293. [PubMed] [Google Scholar]
- 18.Moon A, Kim MS, Kim TG, Kim SH, Kim HE, Chen YQ, Kim HR. H-ras, but not N-ras, induces an invasive phenotype in human breast epithelial cells: A role for MMP-2 in the H-ras-induced invasive phenotype. Int J Cancer. 2000;85:176–181. doi: 10.1002/(SICI)1097-0215(20000115)85:2%3C176::AID-IJC5%3E3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 19.Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003;63:5454–5461. [PubMed] [Google Scholar]
- 20.Shin I, Kim S, Song H, Kim HR, Moon A. H-Ras-specific activation of Rac-MKK3/6-p38 pathway: Its critical role in invasion and migration of breast epithelial cells. J Biol Chem. 2005;280:14675–14683. doi: 10.1074/jbc.M411625200. [DOI] [PubMed] [Google Scholar]
- 21.Song H, Ki SH, Kim SG, Moon A. Activating transcription factor 2 mediates matrix metalloproteinase-2 transcriptional activation induced by p38 in breast epithelial cells. Cancer Res. 2006;66:10487–10496. doi: 10.1158/0008-5472.CAN-06-1461. [DOI] [PubMed] [Google Scholar]
- 22.Koh MS, Moon A. Activation of H-Ras and Rac1 correlates with epidermal growth factor-induced invasion in Hs578T and MDA-MB-231 breast carcinoma cells. Biochem Biophys Res Commun. 2011;406:25–29. doi: 10.1016/j.bbrc.2011.01.092. [DOI] [PubMed] [Google Scholar]
- 23.Lee HM, Moon A. Amygdalin regulates apoptosis and adhesion in Hs578T triple-negative breast cancer cells. Biomol Ther (Seoul) 2016;24:62–66. doi: 10.4062/biomolther.2015.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang S, Kim ES, Moon A. Simvastatin and lovastatin inhibit breast cell invasion induced by H-Ras. Oncol Rep. 2009;21:1317–1322. doi: 10.3892/or_00000357. [DOI] [PubMed] [Google Scholar]
- 25.Kim YT, Kim SK, Jeon YJ, Park SJ. Seahorse-derived peptide suppresses invasive migration of HT1080 fibrosarcoma cells by competing with intracellular α-enolase for plasminogen binding and inhibiting uPA-mediated activation of plasminogen. BMB Rep. 2014;47:691–696. doi: 10.5483/BMBRep.2014.47.12.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng Y, Zhong Y, Li G. Tubeimoside-1 suppresses breast cancer metastasis through downregulation of CXCR4 chemokine receptor expression. BMB Rep. 2016 May 9; doi: 10.5483/BMBRep.2016.49.9.030. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egan SE, Giddings BW, Brooks MW, Buday L, Sizeland AM, Weinberg RA. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature. 1993;363:45–51. doi: 10.1038/363045a0. [DOI] [PubMed] [Google Scholar]
- 28.Ehrhardt A, David MD, Ehrhardt GR, Schrader JW. Distinct mechanisms determine the patterns of differential activation of H-Ras, N-Ras, K-Ras 4B, and M-Ras by receptors for growth factors or antigen. Mol Cell Biol. 2004;24:6311–6323. doi: 10.1128/MCB.24.14.6311-6323.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraus MH, Yuasa Y, Aaronson SA. A position 12-activated H-ras oncogene in all HS578T mammary carcinosarcoma cells but not normal mammary cells of the same patient. Proc Natl Acad Sci USA. 1984;81:5384–5388. doi: 10.1073/pnas.81.17.5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007;5:195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 31.Prior IA, Hancock JF. Compartmentalization of Ras proteins. J Cell Sci. 2001;114:1603–1608. doi: 10.1242/jcs.114.9.1603. [DOI] [PubMed] [Google Scholar]
- 32.Oh SH, Kang JH, Kyu Woo J, Lee OH, Kim ES, Lee HY. A multiplicity of anti-invasive effects of farnesyl transferase inhibitor SCH66336 in human head and neck cancer. Int J Cancer. 2012;131:537–547. doi: 10.1002/ijc.26373. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Kusama T, Mukai M, Tatsuta M, Nakamura H, Inoue M. Inhibition of transendothelial migration and invasion of human breast cancer cells by preventing geranylgeranylation of Rho. Int J Oncol. 2006;29:217–223. [PubMed] [Google Scholar]
- 34.Kusama T, Mukai M, Tatsuta M, Matsumoto Y, Nakamura H, Inoue M. Selective inhibition of cancer cell invasion by a geranylgeranyltransferase-I inhibitor. Clin Exp Metastasis. 2003;20:561–567. doi: 10.1023/A:1025898316728. [DOI] [PubMed] [Google Scholar]
- 35.Nam JS, Ino Y, Sakamoto M, Hirohashi S. Ras farnesylation inhibitor FTI-277 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Jpn J Cancer Res. 2002;93:1020–1028. doi: 10.1111/j.1349-7006.2002.tb02479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Denoyelle C, Hong L, Vannier JP, Soria J, Soria C. New insights into the actions of bisphosphonate zoledronic acid in breast cancer cells by dual RhoA-dependent and -independent effects. Br J Cancer. 2003;88:1631–1640. doi: 10.1038/sj.bjc.6600925. [DOI] [PMC free article] [PubMed] [Google Scholar]