

Abstract
Developing therapeutics for neurodegenerative diseases (NDs) prevalent in the aging population remains a daunting challenge. With the growing understanding that many NDs progress by conformational self-templating of specific proteins, the prototypical prion diseases offer a platform for ND drug discovery. We evaluated high-throughput screening hits with the aryl amide scaffold and explored the structure–activity relationships around three series differing in their N-aryl core: benzoxazole, benzothiazole, and cyano. Potent anti-prion compounds were advanced to pharmacokinetic studies, and the resulting brain-penetrant leads from each series, together with a related N-aryl piperazine lead, were escalated to long-term dosing and efficacy studies. Compounds from each of the four series doubled the survival of mice infected with a mouse-passaged prion strain. Treatment with aryl amides altered prion strain properties, as evidenced by the distinct patterns of neuropathological deposition of prion protein and associated astrocytic gliosis in the brain; however, none of the aryl amide compounds resulted in drug-resistant prion strains, in contrast to previous studies on compounds with the 2-aminothiazole (2-AMT) scaffold. As seen with 2-AMTs and other effective anti-prion compounds reported to date, the novel aryl amides reported here were ineffective in prolonging the survival of transgenic mice infected with human prions. Most encouraging is our discovery that aryl amides show that the development of drug resistance is not an inevitable consequence of efficacious anti-prion therapeutics.
Introduction
The single greatest risk factor for most neurodegenerative diseases (NDs), including Alzheimer (AD) and Parkinson (PD) diseases, is age. With increasing life expectancy, the lack of drugs to treat NDs represents an expanding socioeconomic burden. Most NDs appear to be caused by alternatively folded proteins, which undergo self-propagation. This mechanism of conformational propagation was first described for the prototypical prion protein (PrP), which causes uncommon disorders, including Creutzfeldt-Jakob disease (CJD). The conformational conversion of normal cellular, predominantly α-helical PrP (PrPC) into a pathogenic, β-sheet–rich isoform (PrPSc) is accompanied by changes in physical properties—most notably resistance to proteases (Prusiner, 1998).
The PrP prions cause disease not only in humans but also in a number of commercially important animal species, including cattle, sheep, and deer. Moreover, prions can be transmitted to small animals, including mice, hamsters, and rats, to generate experimentally tractable models of disease (Watts and Prusiner, 2014). Since prions replicate by self-templating endogenous protein, they have the sequence of the last host in which they were passaged. Remarkably, despite having the same primary sequence, prions can exist in multiple strains that are phenotypically distinct and can differ in their time to disease onset after inoculation and their resulting neuropathological lesions and biochemical characteristics. Strains are understood to arise from alternative conformations of PrPSc (Bessen and Marsh, 1994; Telling et al., 1996; Collinge and Clarke, 2007).
A limited number of cell lines have been identified that can stably propagate mouse-passaged prions, including N2a, a mouse neuroblastoma line (Butler et al., 1988), fibroblastic LD9 cells, and central nervous system–derived CAD5 cells (Mahal et al., 2007); however, each cell line is susceptible to only a limited number of prion strains, the most permissive being the CAD5 line. In addition to the propagation of mouse-passaged prions in wild-type (WT) mice, transgenic (Tg) mice have been developed in which the endogenous mouse PrP gene is ablated and PrP from a different species is expressed. These Tg lines provide models to test efficacy against natural scrapie in sheep, chronic wasting disease (CWD) in deer and elk, and CJD in humans (Berry et al., 2013; Berry et al., 2015).
Using prion-infected cells for screening assays, multiple compounds have been identified that double the survival of mice inoculated with mouse-passaged prions, including Compound B (Kawasaki et al., 2007), Anle138b (Wagner et al., 2013), and a series with the 2-aminothiazole (2-AMT) chemical scaffold (Berry et al., 2013; Giles et al., 2015). When prions from the brains of mice treated with 2-AMT IND24 (Berry et al., 2013) were used to infect CAD5 cells, the resulting strain was resistant to IND24, raising the specter that small molecule intervention could lead to drug resistance, a concept that had been suggested by other studies (Ghaemmaghami et al., 2009; Oelschlegel and Weissmann, 2013).
In addition to mouse-passaged prion strains, IND24 was effective against CWD prions from deer and elk but was ineffective against two isolates of sheep scrapie (Berry et al., 2013; Berry et al., 2015); however, IND24 and all other compounds tested to date failed to extend the survival of Tg mice expressing either human PrP or a chimeric human/mouse PrP that were each infected with human CJD prions (Berry et al., 2013; Lu et al., 2013; Giles et al., 2015).
The 2-AMT scaffold was identified early in our high-throughput screening (HTS) campaign (Ghaemmaghami et al., 2010a) and was the focus of medicinal chemistry and pharmacokinetic optimization efforts (Gallardo-Godoy et al., 2011; Li et al., 2013b; Silber et al., 2013b). Because of drug resistance and limited prion strain efficacy with this scaffold, however, we sought to identify further lead series.
From an extended HTS of more than 50,000 compounds in a stably transfected cell line overexpressing PrP infected with RML prions, termed ScN2a-cl3 (Ghaemmaghami et al., 2010b), we identified 13 additional chemical scaffolds. The most highly populous of these were the aryl amides (Silber et al., 2013a). Initial medicinal chemistry efforts on this scaffold focused on N-linked aryl piperazines (Li et al., 2013a) and N-linked aryl oxazoles (Lu et al., 2013) and produced compounds with double-digit nanomolar EC50 values and good brain permeability (compounds 1 and 2; Fig. 1).
Fig. 1.

Chemical structures and EC50 values in ScN2a-cl3 cells of aryl amides differing in their N-linked aryl groups, including previously optimized piperazine (1) and oxazole (2) leads, with additional hits identified by HTS, including thiazole (3), benzothiazole (4), imidazothiazoles (5 and 6), benzoxazole (7), and cyano (8).
Here we report further optimization of the aryl amide scaffold and the progress of selected compounds through pharmacokinetic and in vivo efficacy studies, validating a second chemical scaffold of anti-prion therapeutics. Aryl amides from four different series, differing in their N-linked aryl groups, each doubled the survival of prion-infected mice. Moreover, when cells were infected with brain homogenates from treated mice, the resulting strains remained sensitive to the compounds used for the initial treatments, contending that drug resistance is not an inevitable consequence of efficacious anti-prion therapy.
Materials and Methods
Materials.
Phosphate-buffered saline (PBS), fetal bovine serum (FBS), Opti-MEM, bicinchoninic acid (BCA) protein assay kit, polyacrylamide gels, immunofluorescence secondary antibodies, and PermaFluor aqueous mounting medium were purchased from Thermo Fisher Scientific (Waltham, MA); polyethylene glycol (PEG400) was purchased from Hampton Research (Aliso Viejo, CA); and carboxymethylcellulose (CMC) was purchased from Calbiochem (San Diego, CA). All other compounds and reagents were purchased from Sigma (St. Louis, MO) unless otherwise specified.
The synthesis and characterization of compounds 1 and 2 were previously reported (Li et al., 2013a; Lu et al., 2013); other anti-prion compounds reported here were synthesized by similar methods at Sundia MediTech Ltd. (Shanghai, China) or purchased from commercial vendors, including ChemBridge (San Diego, CA), Life Chemicals (Burlington, Ontario, Canada), Specs (Hopkinton, RI), Enamine (Monmouth Junction, NJ), and ChemDiv (San Diego, CA).
Female FVB and CD-1 mice were purchased from Charles River Laboratories (Hollister, CA), and all other mice were bred inhouse. Mice were fed Harlan 2018 rodent chow except for those in long-term dosing studies, which were given a complete liquid diet (Bio-Serv, Frenchtown, NJ). Experiments were initiated in group-housed mice maintained on a 12-hour light/dark cycle (6 AM–6 PM) with free access to food and water. All procedures using mice were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco, and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (Institute for Laboratory Animal Research, Washington, DC).
Brain tissue was homogenized with a Precellys 24 bead beater (Bertin Technologies, Île-de-France, France), and liquid chromatography-tandem microscopy (LC/MS/MS) analysis was performed on an API4000 triple-quadrapole mass spectrometer (AB Sciex, Foster City, CA) with a BDS Hypersil C8 column (Thermo Fisher Scientific) using a 75%–95% acetonitrile gradient.
Pharmacokinetic Experiments.
Single-dose pharmacokinetic studies were performed by oral gavage in 9- to 20-week-old female FVB mice, with compounds dosed at 10 mg/kg dissolved in 20% propylene glycol, 5% labrosol, 5% ethanol, and 70% PEG400 and delivered in a 200-µl bolus through a 22-gauge gavage needle. Tissues were collected from two mice following euthanasia with CO2 at 0.5, 2, 4, and 6 hours after dosing (for compounds 3–8 and 14, samples were also collected at 0.25, 1, and 24 hours after dosing) and stored at −80°C until analysis.
Samples were processed as previously described (Giles et al., 2015). Briefly, 20% brain homogenates were prepared by bead beater and extracted with equal parts of a 50/50 (vol/vol) solution of water and methanol and then diluted 4-fold in acetonitrile with 0.1% formic acid. Analysis was performed by LC/MS/MS, and specific methods were developed for each compound to enable absolute concentrations to be determined. Pharmacokinetic parameters, including Cmax, tmax, and area under the curve (AUC)last, were calculated using Prism 6 software (GraphPad Software, Inc., La Jolla, CA).
Measurements of brain-tissue binding for compounds 1, 14, and 41 were conducted by contract research organizations using equilibrium dialysis (AMRI Global, Albany, NY) or ultracentrifugation (WuXi AppTec, Shanghai, China).
For 3-day feeding studies, compounds were dissolved either in PEG400 and diluted 800-fold or in 5% CMC in water and diluted 10-fold into complete liquid diet, as previously described (Lu et al., 2013). Studies were performed in 8- to 9-week-old female CD-1 mice; however, studies with dosing between 5 and 50 mg/kg per day with PEG400 as the excipient were performed in 6- to 11-week-old female FVB mice. Brains were collected at 9 AM on the third day and frozen at −80°C. Compound concentrations were determined as described above.
Efficacy Studies.
Seven-week-old female WT FVB and 8- to 10-week-old female FVB-Tg(MoPrP)4053 mice overexpressing mouse PrP (Carlson et al., 1994) were intracerebrally inoculated with the RML prion strain. Seven- to ten-week old female Tg(MHu2M,M111V,M165V,E167Q)1014 Prnp0/0 mice expressing chimeric human/mouse PrP (Giles et al., 2010) were inoculated with a previously characterized sporadic CJD isolate from a patient with the MM1 disease subtype and no mutations in the PrP open reading frame (Telling et al., 1994). Inoculation and monitoring of mice were performed as described previously (Giles et al., 2015).
Cell Infectivity.
CAD5-RML[IND24] cells propagating the IND24-resistant strain were previously described (Berry et al., 2013). CAD5 cells were also infected with prions from RML-inoculated mice that had been treated with compounds 1, 14, 37, and 41 by the same method (Butler et al., 1988). Infected cells were maintained in Opti-MEM, supplemented with 90 units of penicillin-streptomycin and 9% FBS at 37°C in 100-mm plates, and fed with fresh media every 2 to 3 days. Cells were plated at 10% confluency and then split 1:10 when they became confluent.
To test for resistance, cells were incubated with the respective compound at 0.0032 to 10 µM in half-log steps over 5 days with refeeding. Compounds were diluted in dimethyl sulfoxide with a 50-µl final volume in 10 ml of media. At the end of the 5-day treatment period, cells were lysed with lysis buffer (100 mM Tris⋅HCl, pH 8.0; 150 mM NaCl; 0.5% Nonidet P-40 (NP40); 0.5% sodium deoxycholate), and protein concentrations were measured using the BCA assay kit. Protein extracts were normalized to 1 mg/ml total protein with lysis buffer before proteinase K (PK) digestion. Lysates were digested with 20 µg/ml PK at 37°C for 1 hour. The reaction was stopped with phenylmethylsulfonyl fluoride (2 mM final concentration), and samples were ultracentrifuged at 100,000g for 1 hour, resuspended in loading buffer, and subjected to immunoblot analysis as previously described (Giles et al., 2015).
Biochemical Analysis.
Frozen brains were thawed, and 10% (wt/vol) homogenates were prepared in PBS by bead beater. Detergent extraction was performed by adding NP40 and cholic acid to a final concentration of 0.5% (vol/vol), and samples were mixed and incubated on ice for 30 minutes followed by a 5-minute centrifugation at 1000g. Samples were prepared for electrophoresis by digestion with PK as described above and immunoblotted as previously described (Giles et al., 2015). Control and treated samples were run on the same gel, and isoform band intensities were quantified using ImageJ (Rasband, 1997–2014).
Neuropathological Analysis.
Formalin-fixed brains were coarse-cut into four pieces using a slicing matrix, embedded into a single paraffin block, and processed as previously described (Giles et al., 2015) or stained with H&E. For immunofluorescence, sections were subjected to hydrolytic autoclaving (121°C for 10 minutes in citrate buffer) before blocking with 10% (vol/vol) normal goat serum for 1 hour. Slides were incubated with primary antibodies overnight: anti-glial fibrillary acid protein (GFAP) rabbit polyclonal antibody Z0334 (Dako, Carpiteria, CA) at 1:500 dilution and anti-PrP HuM Fab R2 (Williamson et al., 1998) at a concentration of 4 µg/ml and then washed twice with PBS containing 0.2% Tween-20. Secondary antibodies of goat anti-rabbit Alexa Fluor 647 and goat anti-human DyLight 488 were used at a 1:500 dilution in 10% goat serum and incubated at room temperature for 2 hours, followed by two washes with PBS containing 0.2% Tween-20. Slides were coverslipped with PermaFluor aqueous mounting medium.
Fluorescent 16-bit images of PrP and GFAP dual immunohistochemistry on brain slices were obtained with the AxioScan.Z1 slide scanning system (Carl Zeiss, Oberkochen, Germany) using a 20× objective lens and standardized acquisition settings. All images were processed and analyzed using Zeiss ZEN software with the analysis module (Carl Zeiss). In each dual-color image, PrP and GFAP channels were isolated, and a standardized fluorescent threshold for each signal was applied to all control and experimental treatment groups. Brain regions of interest (ROI) were drawn individually for each section using the spline tool to trace the boundaries of a given ROI using intrinsic neuroanatomical features. The total area occupied by PrP or GFAP fluorescent staining in a given brain ROI above the threshold was expressed as a percentage of the total area of the ROI.
Semiquantitative scoring of spongiform degeneration was performed on H&E slides. A skilled observer (B.N.D.) blinded to group status assigned scores in increments of 0.5 to different brain regions as follows: 0 (no vacuolation), 1 (mild vacuolation), 2 (moderate vacuolation), or 3 (severe vacuolation).
Results
Optimizing Aryl Amide Hits.
HTS in ScN2a-cl3 cells (Silber et al., 2013a) identified multiple N-linked aryl heterocycles, including thiazole (3), benzothiazole (4), imidazothiazoles (5 and 6), and benzoxazole (7) (Fig. 1).
To explore structure–activity relationships (SARs), we purchased additional aryl amides with N-linked benzothiazoles and determined their EC50 values in the prion-infected cell assay (Table 1). The initial benzothiazole hit (4) (Fig. 1), with a 3-methyl-2-furyl amide, had an EC50 of 58 nM. The unsubstituted 2-furalyl amide (9) had similar potency to the parent compound, but analogs lacking the 6-methyl substituent on the benzothiazole ring (10 and 11) had 2- to 3-fold reduced activity. Introduction of a chlorine substituent on the aryl ring (12) nearly abolished activity in the cell assay, whereas modification of the benzothiazole by introduction of a nitrogen into the 7-position to generate a thiazolinopyridine (13) resulted in an EC50 of ∼1 µM. The 5-methyl-2-furyl (14) and 3-furyl (16) analogs of the parent compound had EC50 values of 80 and 92 nM, respectively; however, removal of the 6-methyl substituent on the benzothiazole (15 and 17) reduced activity 3- to 5-fold. Replacement of the furyl amide with a benzofuryl amide resulted in a significant decrease in activity in the presence or absence of the 6-methyl group on the N-linked aryl benzothiazole (18–20). Notably, activity was restored with a furopyridine (21) but not furopyrimidinone (22). Replacing the furan of the thiazolinopyridyl-containing analog 13 with 4-dimethylaminobenzene (23) or 2-fluorobenzene (24) provided improved potency, but these compounds were still less active than the original lead (4).
Table 1.
SARs of N-linked aryl benzothiazoles. Mean EC50 values ± S.D. and number of replicates (n) for aryl amides with the N-linked aryl benzothiazole scaffold.
| R1 | R2 | R3 | X | R4 | EC50 (μM) | n | |
|---|---|---|---|---|---|---|---|
| 9 |  |
H | H | CH | Me | 0.063 ± 0.028 | 2 |
| 10 |  |
Me | H | CH | H | 0.13 ± 0.07 | 2 |
| 11 | 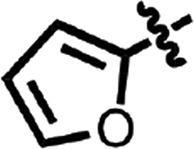 |
H | Me | CH | H | 0.17 ± 0.10 | 2 |
| 12 | 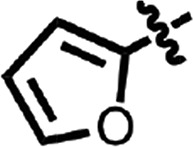 |
H | Cl | CH | H | 7.96 ± 1.71 | 2 |
| 13 | 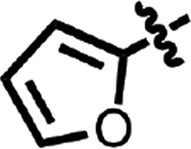 |
H | H | N | H | 0.98 ± 0.12 | 2 |
| 14 | 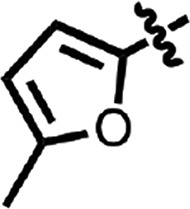 |
H | H | CH | Me | 0.080 ± 0.018 | 3 |
| 15 | 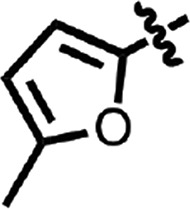 |
H | H | CH | H | 0.27 ± 0.03 | 2 |
| 16 | 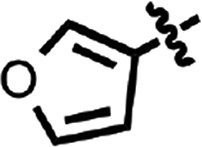 |
H | H | CH | Me | 0.092 ± 0.005 | 2 |
| 17 | 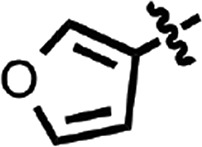 |
H | H | CH | H | 0.47 ± 0.05 | 2 |
| 18 | 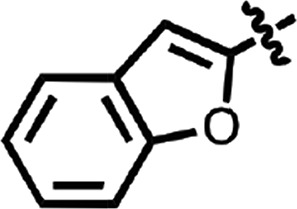 |
Me | H | CH | H | 0.48 | 1 |
| 19 | 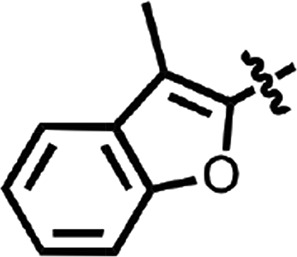 |
H | H | CH | Me | 0.45 ± 0.25 | 2 |
| 20 | 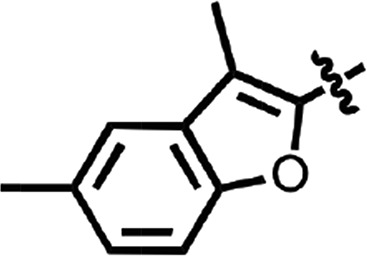 |
H | H | CH | Me | 7.66 ± 2.53 | 2 |
| 21 | 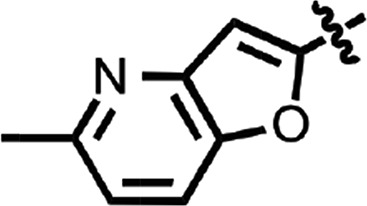 |
H | H | CH | Me | 0.084 ± 0.014 | 2 |
| 22 | 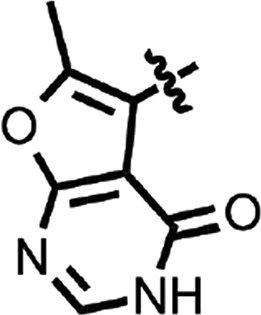 |
H | H | CH | Me | 1.68 ± 0.09 | 2 |
| 23 | 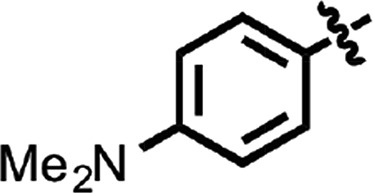 |
H | H | N | H | 0.15 ± 0.01 | 2 |
| 24 | 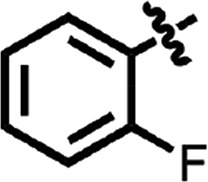 |
H | H | N | H | 0.38 ± 0.04 | 2 |
Despite the ∼100 nM activity of the two N-linked aryl imidazothiazole hits (5 and 6) (Fig. 1), three additional substituted imidazothiazoles containing aliphatic amides (25–27) (Supplemental Table 1) were inactive in the prion cell assay, suggesting that an aryl amide moiety is important for activity.
Benzoxazole (7) (Fig. 1) had an EC50 of 75 nM. Replacement of the anisamide with 3-furyl amide (28) or 2,5-dimethyl-3-furyl amide (29) reduced potency 15- and 4-fold, respectively (Table 2). With a 2-furyl amide, 6-methyl benzoxazole (30) had an EC50 of 73 nM, but a 5-methoxy substitution of the benzoxazole (31) resulted in an EC50 of 320 nM. The benzofuranyl amide (32) was less active. Replacing the amide with nicotimamide analogs had varying effects: 6-isopropoxynicotinamide (33) and 6-(cyanoethoxy)nicotinamide (34) substituents were inactive in the ScN2a-cl3 cell assay, whereas the 6-(2-methoxyethoxy)nicotinamide (35), 6-(dimethylamino)nicotinamide (36), and 6-(4-methylpiperazine-1-yl)nicotinamide (37) had EC50 values of 360, 740, and 170 nM, respectively.
Table 2.
SARs of N-linked aryl benzoxazoles. Mean EC50 values ± S.D. and number of replicates (n) for aryl amides with the N-linked aryl benzoxazole scaffold.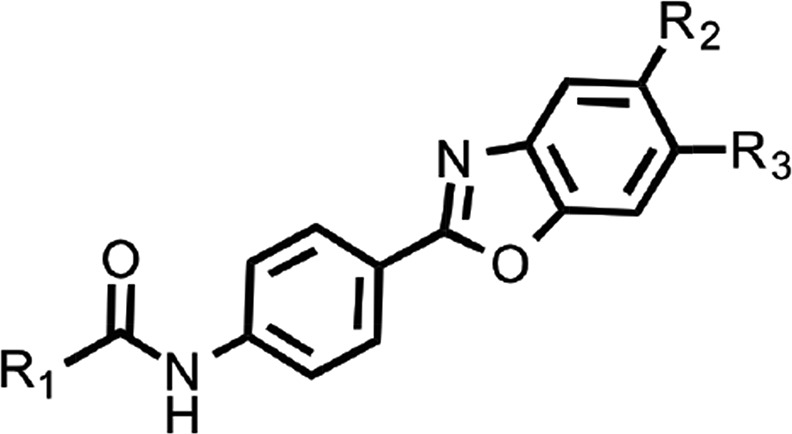
| R1 | R2 | R3 | EC50 (μM) | n | |
|---|---|---|---|---|---|
| 28 | 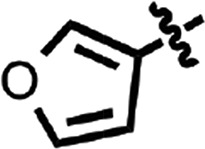 |
H | H | 1.18 ± 0.01 | 2 |
| 29 | 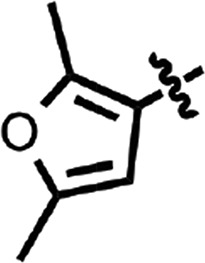 |
H | H | 0.31 ± 0.00 | 2 |
| 30 | 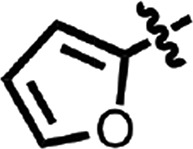 |
H | Me | 0.073 ± 0.019 | 2 |
| 31 | 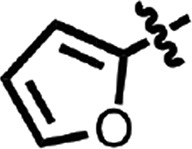 |
OMe | H | 0.32 ± 0.11 | 2 |
| 32 | 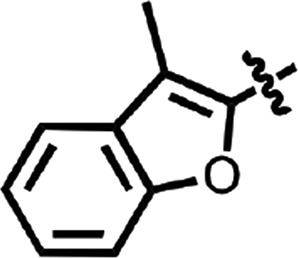 |
Cl | H | 0.64 ± 0.24 | 2 |
| 33 | 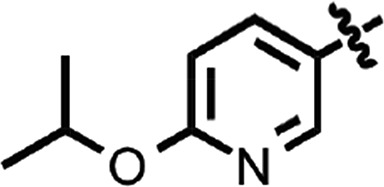 |
H | H | >10 | 2 |
| 34 | 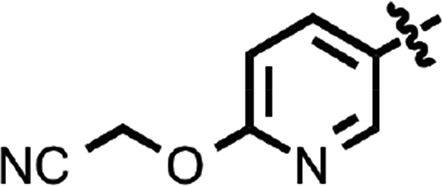 |
H | H | > 10 | 1 |
| 35 | 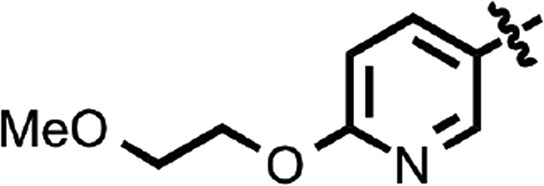 |
H | H | 0.36 | 1 |
| 36 | 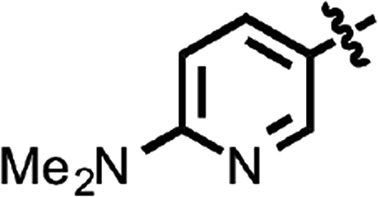 |
H | H | 0.74 ± 0.04 | 2 |
| 37 | 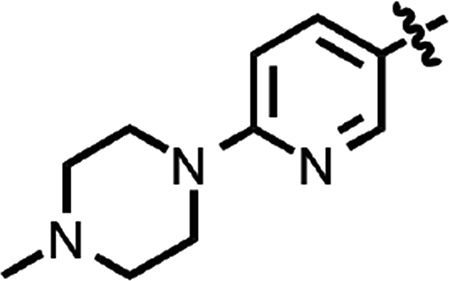 |
H | H | 0.17 ± 0.01 | 2 |
N-(4-cyanophenyl)-4-phenylbenzamide (8) (Fig. 1) was also identified as a hit in the HTS. Through a series of commercially available and synthesized analogs, we explored the SARs around the 4-phenyl substituent of the benzamide (Table 3). Replacement with pyridines (38–40) had little effect on potency; however, further derivatization of the 3-phenylpyridyl (39) led to a dramatic improvement in activity. The 2-methoxyethyl ether (41), dimethylamine (42), morpholine (43), and methylpiperazine (44) each had EC50 values less than 100 nM in the ScN2a-cl3 cell assay. Conversely, the cyanomethyl ether (45) was inactive. Replacing the 4-phenyl substituent with an oxazole resulted in reduced potency. The oxazol-2-yl analog (46) was inactive, and the oxazol-5-yl analog (47) had an EC50 of 0.81 µM. The dimethyl-aminophenyl amide (48) had an EC50 of ∼2 µM.
Table 3.
SARs of N-linked cyanobenzyls. Mean EC50 values ± S.D. and number of replicates (n) for aryl amides with the N-linked cyanobenzyl scaffold.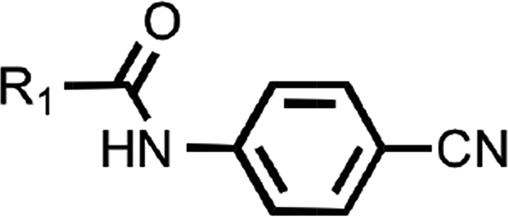
| R1 | EC50 (μM) | n | |
|---|---|---|---|
| 38 | 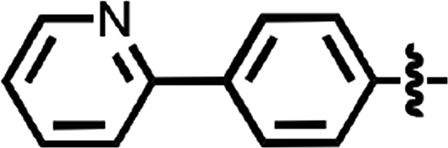 |
0.50 | 1 |
| 39 | 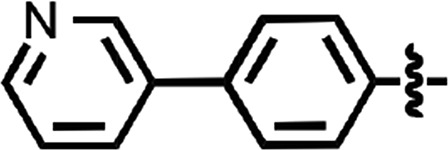 |
1.39 | 1 |
| 40 | 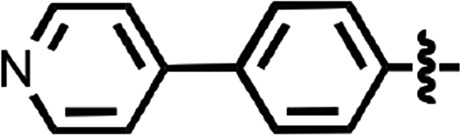 |
1.1 | 1 |
| 41 | 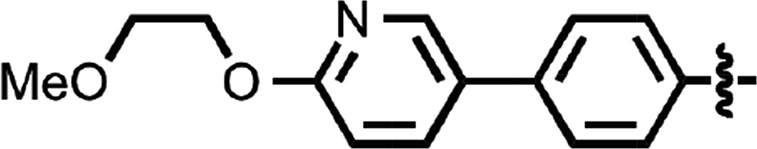 |
0.055 ± 0.001 | 2 |
| 42 | 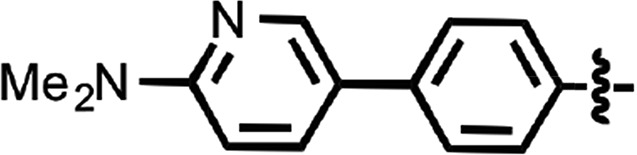 |
0.036 ± 0.004 | 3 |
| 43 | 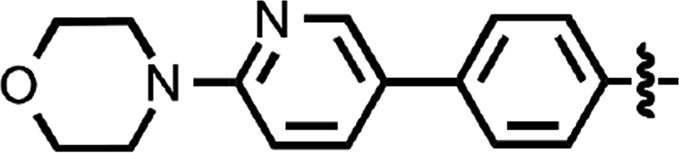 |
0.035 ± 0.001 | 3 |
| 44 | 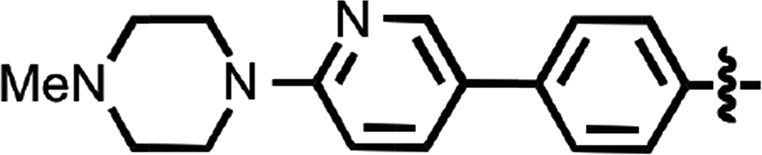 |
0.066 ± 0.013 | 2 |
| 45 | 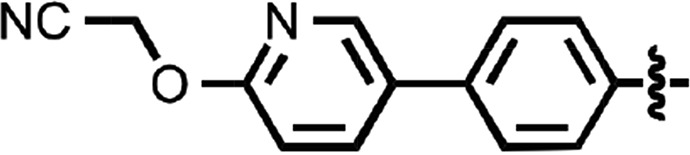 |
>10 | 2 |
| 46 | 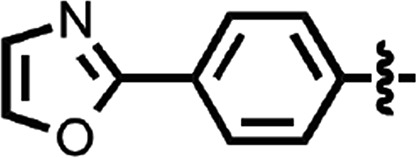 |
>10 | 2 |
| 47 | 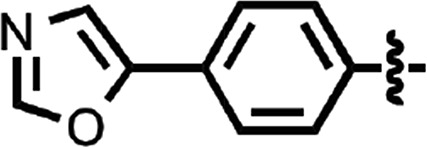 |
0.81 ± 0.03 | 2 |
| 48 | 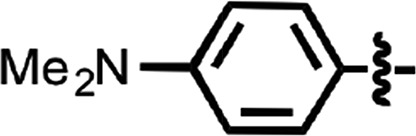 |
2.2 | 1 |
Aryl Amides Exhibit Good Brain Penetration.
In parallel with the cellular EC50 determinations, we performed single-dose pharmacokinetic studies on selected compounds. After oral gavage at 10 mg/kg, tissue concentrations were measured. The Cmax achieved in the brain, the time after dosing that Cmax was observed (tmax), and the area under the concentration–time curve to the last point measured (AUClast) were determined (Table 4).
TABLE 4.
Pharmacokinetic parameters of aryl amides after oral gavage at 10 mg/kg. Two female FVB mice (9–20 weeks old) were collected per time point; maximum brain concentration (Cmax) reported as mean ± S.D.
| Tissue | Cmax | tmax | AUClast | |
|---|---|---|---|---|
| μM | h | μM/h | ||
| 1a | Brain | 2.52 ± 0.27 | 2 | 7.48 |
| Plasma | 0.27 ± 0.01 | 2 | 0.97 | |
| 3 | Brain | 0.36 ± 0.24 | 0.25 | 0.79 |
| 4 | Brain | 1.34 ± 0.36 | 0.5 | 0.18 |
| 5 | Brain | 0.19 ± 0.04 | 0.25 | 0.18 |
| 6 | Brain | 0.07 ± 0.01 | 0.25 | 0.03 |
| 7 | Brain | 0.95 ± 0.20 | 0.25 | 2.45 |
| 8 | Brain | 3.59b | 0.25 | 5.60 |
| 14 | Brain | 2.59 ± 0.72 | 2 | 16.35 |
| Plasma | 0.34 ± 0.16 | 0.5 | 1.15 | |
| 35 | Brain | 0.94 ± 0.11 | 0.5 | 2.18 |
| 37 | Brain | 5.75 ± 1.34 | 2 | 23.97 |
| Plasma | 0.50 ± 0.05 | 0.5 | 2.18 | |
| 38 | Brain | 0.45 ± 0.15 | 0.5 | 0.59 |
| 41 | Brain | 2.49 ± 2.03 | 2 | 7.81 |
| Plasma | 0.98 ± 0.01 | 2 | 3.65 | |
| 42 | Brain | 0.23 ± 0.24 | 0.5 | 0.60 |
| 43 | Brain | 0.29 ± 0.03 | 0.5 | 1.14 |
| 44 | Brain | 0.52 ± 0.02 | 2 | 1.67 |
Includes data previously reported (Li et al., 2013a).
Second mouse died before collection.
In pharmacokinetic studies of the six screening hits (3–8), Cmax measurements for the thiazole (3) and one of the imidazothiazoles (6) did not reach their respective EC50 values, and Cmax for the second imidazothiazole (5) was only slightly above its EC50. The benzothiazole (4), benzoxazole (7), and cyanobenzyl (8) all reached their respective Cmax values within 30 minutes. In contrast, the most potent benzothiazole identified from SAR studies (14) showed a brain Cmax more than 30-fold higher than its EC50 and an AUClast value around 20-fold that of the original hit. For two benzoxazoles tested in PK studies, the Cmax/EC50 ratios were 2.6 (35) and 34 (37). Four N-linked aryl cyanobenzyls with double-digit nanomolar EC50 values (41–44) had very different PK concentrations; compound 41 had a Cmax almost 5-fold higher than any of the others. For each scaffold, we selected examples with the highest ratio of AUClast to EC50.
Compounds 1, 14, 37, and 41 were advanced to longer term dosing studies and were administered to mice for 3 days via a complete liquid diet at a range of concentrations using either CMC or PEG400 as the excipient (Fig. 2). For compound 1, exposure was greater than dose proportional in CMC but approximately dose proportional in PEG400. For compounds 14 and 41, exposure was dose proportional, and with compound 37, it was ∼5-fold greater than dose proportional. Brain protein binding was determined for compounds 1, 14, and 41. Rapid equilibrium dialysis (RED) gave values of 99.3% ± 0.1% for compound 1 and 99.9% ± 0.0% for compound 41; however, compound 14 did not reach the RED percent equilibrium criteria. Using an ultracentrifugation method, compound 14 was determined to be >99.0% bound in the brain.
Fig. 2.

Role of excipient and compound concentration on brain and plasma levels in 3-day feeding studies. Six- to 11-week old female mice were dosed for 3 days with compounds at the indicated concentrations, using either 0.5% CMC (closed symbols) or 0.125% PEG400 (open symbols) as the excipient. Levels of compound in the brain (blue) and plasma (red) were determined by extraction of tissue with methanol. Each point represents the mean and S.D. of three mice. The EC50 value in ScN2a-cl3 cells is shown as a dotted line for each compound.
Aryl Amides Double Survival of Prion-Infected Mice.
Long-term dosing of compound 1 at 200 mg/kg per day in WT mice infected with the RML prion strain was lethal after ∼30 days, suggesting compound toxicity; however, dosing at 25 mg/kg per day doubled the survival of RML-infected WT mice (Fig. 3A; Table 5). We subsequently found that long-term dosing with CMC as the excipient in prion-infected mice could limit efficacy (Giles et al., 2015), and so further studies were done with PEG400. Despite their different pharmacokinetic profiles, dosing compounds 14, 37, and 41 at 200 mg/kg per day in RML-infected Tg4053 mice overexpressing mouse PrP led to a similar relative extension in survival. To control for the variation in survival extension between different mouse models and prion strains, we introduced the term survival index, which is normalized to survival of vehicle-treated controls (Berry et al., 2013). Each compound doubled the incubation period compared with vehicle-treated controls, with survival indices of 220, 233, and 227 for compounds 14, 37, and 41, respectively (Fig. 3B; Table 5).
Fig. 3.

Survival of prion-infected mice upon treatment with aryl amides. Seven- to 10-week old female FVB WT (A) and FVB-Tg4053 mice (B) were inoculated with RML prions and treated with aryl amides. (A) Compound 1 dosed at 25 mg/kg per day (red) doubled survival compared with the vehicle-treated control (black). Four mice are represented per arm. (B) Compounds 14 (orange), 37 (blue), and 41 (green) dosed at 200 mg/kg per day all doubled survival compared with the vehicle-treated control (black). Eight mice are represented per arm.
TABLE 5.
Efficacy of aryl amides in prion-infected mice. Four to eight 7- to 10-week old female mice were intracerebrally inoculated with RML or sCJD(MM1) prions, and dosing was started the following day.
| Line | Inoculum | Compound | Dose | Excipient | Mean Incubation Period ± S.E.M. | n/n0 | Survival Index ± S.E.M. |
|---|---|---|---|---|---|---|---|
| mg/kg daily | Per day | ||||||
| FVB | RML | None | CMC | 115 ± 2a | 4/4 | 100 | |
| 1 | 200 | CMC | Toxic | ||||
| 1 | 25 | CMC | 213 ± 3 | 4/4 | 181 ± 3 | ||
| Tg4053 | RML | None | PEG400 | 51 ± 3b | 8/8 | 100 | |
| 14 | 200 | PEG400 | 112 ± 4 | 8/8 | 220 ± 15 | ||
| 37 | 200 | PEG400 | 119 ± 3 | 8/8 | 233 ± 15 | ||
| 41 | 200 | PEG400 | 116 ± 4 | 8/8 | 227 ± 16 | ||
| Tg1014 | sCJD(MM1) | None | PEG400 | 78 ± 1b | 7/7 | 100 | |
| 1 | 25 | CMC | 76 ± 2 | 4/4 | 97 ± 3 | ||
| 14 | 200 | CMC | 74 ± 0 | 4/4 | 95 ± 1 | ||
| 37 | 200 | PEG400 | 76 ± 1 | 8/8 | 97 ± 2 | ||
| 41 | 200 | CMC | 76 ± 2 | 4/4 | 97 ± 3 |
n, number of ill mice; n0, number of inoculated mice.
Data previously reported (Giles et al., 2015).
Data previously reported (Berry et al., 2013).
Aryl Amide Treatment Changes Strain Characteristics.
Although mice treated with aryl amides survived twice as long as controls, they ultimately developed clinical signs of prion disease and showed the characteristic neuropathological lesions of PrP immunoreactivity after hydrolytic autoclaving, astrocytic gliosis (as demonstrated by GFAP staining), and neuropil vacuolation (Fig. 4, A and B). One of the more noticeable differences between aryl amide–treated mice and controls was the appearance of dense PrPSc aggregates along the corpus callosum after treatment with compound 14, which were apparent in six of eight treated mice compared with none of six controls (Fig. 4C).
Fig. 4.

Neuropathological characterization of RML-infected mice treated with aryl amides. Representative sections shown from the sensory cortex and hippocampus of WT (A) and Tg4053 (B) mice treated with aryl amides. Double immunolabeling (upper panels) against PrP (green) and GFAP (red) and H&E staining (lower panels) show that the characteristic neuropathology of prion diseases remains upon treatment. (C) PrP immunoreactivity in the hippocampus and surrounding cortex from a mouse treated with compound 14 shows intensity in the corpus callosum (white arrows), which is not observed in the vehicle-treated control. Scale bars for the sensory cortex and hippocampus represent 25 and 50 µM, respectively, and apply to all panels for those regions.
To quantify immunohistochemical staining after aryl amide treatment, we determined the percentage area of immuno-staining over multiple brain regions and compared these areas to those for vehicle-treated controls (Fig. 5). In vehicle-treated WT mice, PrPSc deposition was moderate but observed in many brain regions (most robustly in the sensory and motor cortex), whereas GFAP immunoreactivity was strong throughout the brain (Fig. 5A). Treatment with compound 1 showed increased PrPSc staining but with a similar neuroanatomical distribution to vehicle-treated controls. GFAP immunohistochemistry after treatment with compound 1 showed increased staining in the cortex (Fig. 5A). In RML-infected Tg4053 mice treated with vehicle, PrPSc staining appeared predominantly in the sensory and motor cortices, with lower levels in the hippocampus, thalamus, and brainstem, whereas GFAP upregulation was robust throughout much of the brain (Fig. 5B). Tg4053 mice treated with aryl amides demonstrated more limited PrPSc staining than control mice. Compound 14 resulted in reduced PrPSc throughout the cortex, and mice treated with compound 37 had lower levels of PrPSc staining throughout the brain. Treatment with compound 41 resulted in reduced PrPSc levels in the sensory cortex and hippocampus and increased levels in the striatum (Fig. 5B, left panel). GFAP immunoreactivity after treatment with compound 14 was similar to controls in the hippocampus, striatum, cerebellum, and pons and reduced in other areas. Treatment with compounds 37 and 41 resulted in similar GFAP distributions with reduced immunoreactivity in the sensory cortex and hippocampus compared with controls (Fig. 5B, right panel).
Fig. 5.

Quantitation of neuropathological lesions in RML-infected mice treated with aryl amides. Radar plots of lesion profiles representing the average percentage area stained for PrP (left panels) and GFAP (right panels) in multiple brain regions of WT (A) and Tg4053 (B) mice (n = 4–8 for each treatment). (A) Treatment with compound 1 (red) showed greatly increased PrP staining but similar neuroanatomical distribution compared with vehicle-treated control (black). GFAP staining was more intense in the cortex of mice treated with compound 1. (B) Treatment with compounds 14 (orange), 37 (blue), and 41 (green) all changed the intensity and distribution of neuropathological lesions compared with vehicle-treated control (black). Sc, sensory cortex; Hp, hippocampus; Th, thalamus; Mc, motor cortex; St, striatum; Cb, cerebellum; Po, pons.
The degree and spatial distribution of vacuolation were quantified in the brains of vehicle- and compound-treated mice (Supplemental Table 2), and scores for each region were compared using the nonparametric Kruskal-Wallis test with Dunn’s post-test, using a significance threshold of P < 0.05. WT mice treated with vehicle or compound 1 exhibited mild spongiform changes in cortical gray matter, the hippocampus, and the thalamus; however, because of interanimal variability and small sample size (n = 4 for each group), statistically significant difference could not be determined. In Tg4053 mice, treatment with compound 14 significantly reduced vacuolation in the thalamus (P < 0.05), and treatment with compounds 14, 37, and 41 significantly decreased vacuolation in the cerebellar white matter compared with vehicle-treated controls (P < 0.05 for each).
Brain homogenates from vehicle- and compound-treated mice were analyzed by immunoblotting. Differences in total PK-resistant PrP and glycoform ratios were compared by unpaired two-tailed t tests without correction for multiple comparisons using a significance threshold of P < 0.05. No significant differences were observed in glycoform ratios for any of the compounds, and only marginally significant differences were observed in total PrP for treatments with compounds 14 (P = 0.013) and 37 (P = 0.017) compared with controls.
Aryl Amides Do Not Lead to the Development of Drug Resistance.
When the brains of IND24-treated mice were used to infect CAD5 cells, the resulting strain was found to be resistant to IND24 (Berry et al., 2013). To determine whether the development of drug resistance is a general phenomenon limiting prion efficacy, we infected CAD5 cells with brain homogenates from RML-infected mice treated with compounds 1, 14, 37, and 41. Five independent infections were attempted for each brain homogenate, and in all cases, infected cells were obtained, as determined by the presence of PK-resistant PrP for each compound. Three of the resulting infected cultures for each condition were then incubated with the same compound used in the original treatment. The resulting EC50 values did not differ substantially from the original EC50 values for each of these compounds, demonstrating that the treatments did not lead to drug resistance (Table 6). We next determined whether aryl amides were effective against the IND24-resistant strain. CAD5-RML[IND24] cells were treated with compounds 1, 14, and 41 at 10 µM and with compound 37 at 3.2 µM for 5 days and then analyzed by immunoblot. In all cases, there was no reduction in the level of RML[IND24] PK-resistant PrP, suggesting that these compounds are not effective against IND24-resistant prions (Supplemental Table 3).
TABLE 6.
Resistance of CAD5 cells infected with brain homogenates from mice treated with aryl amides to the compounds used in the original treatments. To determine whether the strain had changed upon treatment with aryl amides, CAD5 cells were incubated with brain homogenates from terminal aryl amide –treated RML-infected mice. The resulting cells were then incubated with the same aryl amide at a range of concentrations to measure an EC50 value.
| Mouse Line | Compound | EC50 in N2a Cells by ELISA | No. of Attempts/Successful Cell Infections | No. of Independent Treatments | EC50 in CAD5 Cells by Immunoblot |
|---|---|---|---|---|---|
| μM | μM | ||||
| FVB | IND24 | 1.27a | 6/6b | 2 | >20b |
| FVB | 1 | 0.022 | 5/5 | 3 | 0.026 |
| Tg4053 | 14 | 0.077 | 5/5 | 3 | 0.024 |
| Tg4053 | 37 | 0.17 | 5/5 | 3 | <0.32 |
| Tg4053 | 41 | 0.055 | 5/5 | 3 | 0.097 |
ELISA, enzyme-linked immunosorbent assay.
Data previously reported (Silber et al., 2013b).
Data previously reported (Berry et al., 2013).
Aryl Amides Are Not Efficacious against CJD Prions.
None of the compounds reported to date that extend survival of mice infected with mouse-passaged prion strains shows efficacy against mice inoculated with CJD prions (Berry et al., 2013; Lu et al., 2013; Giles et al., 2015). We therefore inoculated Tg1014 mice expressing a chimeric human/mouse PrP (Giles et al., 2010) with sCJD(MM1) prions and treated them with each of the aryl amides at the same dose shown to be effective against RML prions. None of the treatments extended the survival of CJD-infected mice (Table 5).
Discussion
Prions in Neurodegenerative Diseases.
It is important to emphasize that not all prions cause disease. The list of proteins that adopt alternative conformations that become self-propagating but not disease-causing continues to expand (Cai and Chen, 2014; Si, 2015; Wickner et al., 2015; Chakrabortee et al., 2016). In addition, the number of proteins that undergo self-propagation and become disease-causing continues to widen (Walker and Jucker, 2015). Notably, these include the proteins that define the neuropathology of AD and PD. In all cases, the prion forms are β-sheet rich and differ physicochemically from their cellular forms or precursors. Of special importance are the implications of pathologically folded proteins for developing effective AD and PD therapeutics. Unfortunately, however, the development of meaningful drugs for the treatment of these devastating diseases remains disappointing. Identification of anti-prion compounds therefore offers a novel strategy for the development of AD and PD therapeutics.
Although the sequence of the PrP protein is very different from that of Aβ, tau, and α-synuclein, all four proteins are capable of folding into alternative conformations that undergo self-propagation; however, the prototypical PrP prion diseases, for which there are accurate animal and cell models, provide excellent tools for anti-prion drug discovery efforts. Here we report the identification, optimization, and efficacy of a new class of compounds against PrP prion diseases.
Optimizing Aryl Amide Efficacy and Brain Exposure.
The aryl amide scaffold was identified in an HTS campaign for anti-prion compounds (Silber et al., 2013a), and we initially focused our medicinal chemistry efforts on N-linked aryl piperazines (Li et al., 2013a) and N-linked aryl oxazoles (Lu et al., 2013). We have extended these studies into investigations of N-linked aryl benzothiazoles and benzoxazoles and the cyanobenzyl series.
Within the limited chemical space of commercially available compounds, we were able to define SARs for each of the scaffolds, suggesting action on a specific target. For example, a 6-methyl substituent on the benzothiazole ring consistently increased potency. In the analogs where the benzothiazole was converted to a thiazolinopyridine, a 4-dimethylamiobenene substituent on the amide (23) was favored over the 2-furyl, with an ∼6-fold difference in potency. On the amide, 2-furyl, 3-methyl-2-furyl, and 3-furyl substituents were similarly tolerated, but larger groups tended to reduce potency. In the benzoxazole series, a similar flexibility was observed in furyl amide substituents, as was lower potency with larger substituents. The 2-(methylpiperayl)nicotinamide (37) was less potent than the original hit (7); however, it had 10-fold higher exposure after oral gavage (Table 4). Interestingly, a pair of benzothiazole and benzoxazole analogs (9 and 30) had similar potency, and a second pair (17 and 28) was within ∼2-fold of one another, suggesting these heterocyclic substituents have similar SARs. In the cyanobenzyl series, the smaller N-linked substituent needed to be compensated by a larger group on the carbonyl side. Derivatization of the 3-phenylpyridyl group, particularly the para positioning of electron-rich substituents, improved potency. Importantly, this series produced some of the most potent compounds observed (41–44); however, brain exposure after gavage varied over a more than 10-fold range for these compounds (Table 4). Optimized compounds from each series, together with the N-linked aryl piperazine lead (1) (Li et al., 2013a), were advanced to 3-day dosing studies.
All compounds for which the free fraction was measured were discovered to be highly protein bound in the brain. Since the cell assay contains serum proteins (10% FBS), the intrinsic potency of the compounds may be able to overcome their low free fraction. Assuming similar nonspecific binding in FBS and mouse brain, we roughly estimate that an exposure 10 times the EC50 is required to achieve efficacy. For compounds 1 and 37, 3-day dosing predicted values above this, but estimates for compounds 14 and 41 fell below this cutoff. When dosing was initiated the day after inoculation, each of the four aryl amides doubled the survival of mice infected with the mouse-passaged RML prion strain (Table 5), suggesting that brain levels may have been higher overnight when animals were feeding more. We cannot discount the possibility that the mechanism in cell culture is not precisely predictive of that in mouse brain, however, or that our assumptions with respect to free fraction measurements were overly conservative.
Aryl Amides Do Not Lead to Drug Resistance.
Previously, we showed that treatment with 2-AMTs can change the physicochemical and neuropathological phenotypes, suggesting a change in prion strain (Berry et al., 2013). Biochemical determination of glycoform ratios showed no significant differences with any of the treatments; however, neuropathological analysis suggested strains may have been altered (Figs. 4 and 5 and Supplemental Table 2). Quantitation of PrP immunohistochemistry and astrocytic gliosis, as determined by GFAP upregulation, from multiple brain regions of multiple animals enabled direct comparisons. Brains of mice treated with compound 1 had higher levels of PrP staining than control FVB mice, whereas mice treated with compounds 14 and 37 had lower levels than their respective Tg4053 controls (Fig. 5). After treatment with compound 41, overall PrP levels were similar to those of the control, but the profile of neuropathological lesions appeared to differ (Fig. 5). The identification of neuropathological differences but similar efficacy in terms of survival may indicate the formation of different prion strains by the compounds.
Treatment with IND24 led to the generation of a strain that was resistant to further treatment with the compound (Berry et al., 2013). Resistance of RML[IND24] in CAD5 cells was predictive of resistance in vivo; conversely, ME7[IND24] and CWD[IND24] did not show resistance in CAD5 cells or in mice (Berry et al., 2013; Berry et al., 2015). Infecting CAD5 cells with brain homogenates from RML-infected mice treated with aryl amides propagated strains that were sensitive to the respective compounds, contending that drug resistance is not an inevitable feature of efficacious therapy; however, all cell lines reported to date have only been shown to propagate a limited subset of prion strains. If CAD5 cells were refractive to a novel drug-resistant strain, or it propagated more slowly than the drug-sensitive strain, it may not be observed in cells. Whereas none of the aryl amides tested was effective against the RML[IND24] strain in cell culture, whether combinations or sequential dosing of 2-AMT and an aryl amide could extend survival of prion-infected mice longer than either compound alone remains to be determined.
Here we report only aryl amide dosing in RML-infected mice starting the day after inoculation. Studies with IND24 showed unexpected effects on efficacy when dosing was started later and when the mouse models and prion strains were varied (Berry et al., 2013; Berry et al., 2015; Giles et al., 2015). In addition, prophylactic dosing with IND24 was more effective at extending survival than continuous dosing starting after inoculation (Giles et al., 2015). Uncovering the full utility of aryl amides in prion disease will require many additional bioassay experiments.
Although the conformational transition of PrPC to PrPSc is central in prion diseases, the cellular mechanisms by which this leads to neurodegeneration are poorly understood. The compounds reported here that reduced PrPSc in cells also led to extension in survival in prion-infected mice, suggesting that they function upstream of these neurodegenerative processes. PrPSc reduction can arise from decreasing precursor (i.e., lowering PrP expression or increasing its degradation), by interfering with PrPC-to-PrPSc conversion, or by increasing clearance of PrPSc. Because of the prion strain specificity shown by these compounds, interfering with conversion seems most plausible. This could arise by direct interaction with PrP in some transition state or with a putative chaperone involved in the refolding.
Validating the Drug-Discovery Paradigm.
The compound quinacrine reduced PrPSc levels in RML-infected cells (Doh-ura et al., 2000; Korth et al., 2001), but this finding failed to translate to efficacy in mice infected with RML prions or other mouse-passaged prion strains (Collins et al., 2002; Barret et al., 2003; Doh-ura et al., 2004; Ghaemmaghami et al., 2009) or to humans with CJD (Haïk et al., 2004; Collinge et al., 2009; Geschwind et al., 2013), demonstrating that some cell models are not necessarily predictive of in vivo efficacy.
Using a stably transfected cell line overexpressing PrP and infected with RML prions, we performed an HTS campaign and screened more than 50,000 compounds (Silber et al., 2013a). Structural optimization of these hits coupled with pharmacokinetic studies (Gallardo-Godoy et al., 2011; Li et al., 2013a,b; Silber et al., 2013b) led to compounds that were highly potent in cells and reached high concentrations in the brains of mice when dosed orally. Such compounds were extended to long-term efficacy studies. We have now reported efficacy for 10 compounds in RML-infected mice: six 2-AMTs (Berry et al., 2013; Giles et al., 2015) and the four aryl amides described here. Remarkably, all 10 extend the survival of prion-infected mice, many by 2-fold or more.
Whereas none of these compounds (or those developed by other groups) has shown efficacy against CJD prions, the repeated success of the compounds identified by this drug-discovery paradigm is promising. Developing a cell model for the replication of human CJD prions remains a top priority for the field. Applying the drug-discovery strategy outlined above to such a model will more likely identify compounds that translate to efficacy in humans.
Supplementary Material
Acknowledgments
The authors thank Drs. Joel Gever and Satish Rao for EC50 determinations and pharmacokinetic studies, respectively, on some of the initial compounds, Rigoberto Roman-Albarran for histopathological assistance, and Dr. Julian Castaneda and the staff at the Hunters Point Animal Facility for help with the mouse studies.
Abbreviations
- AD
Alzheimer disease
- AUC
area under curve
- CJD
Creutzfeldt-Jakob disease
- CMC
carboxymethylcellulose
- CWD
chronic wasting disease
- FBS
fetal bovine serum
- GFAP
glial fibrillary acid protein
- HTS
high-throughput screening
- ND
neurodegenerative disease
- PBS
phosphate-buffered saline
- PD
Parkinson disease
- PEG400
polyethylene glycol
- PK
proteinase K
- PrP
prion protein
- ROI
region of interest
- SAR
structure–activity relationship
- Tg
transgenic
- 2-AMT
2-aminothiazole
- WT
wild-type
Authorship Contributions
Participated in research design: Giles, Berry, Li, Silber, Prusiner.
Conducted experiments: Berry, Oehler, Bhardwaj, Elepano, Guan.
Contributed new reagents or analytic tools: Li, Silber.
Performed data analysis: Giles, Berry, Condello, Dugger, Elepano, Olson, Prusiner.
Wrote or contributed to the writing of the manuscript: Giles, Berry, Condello, Dugger, Olson, Prusiner.
Footnotes
This work was supported by the National Institutes of Health National Institute on Aging [Grants AI064709, AG002132, AG010770, AG021601, and AG031220] and by gifts from the Sherman Fairchild Foundation, the Rainwater Charitable Foundation, and the G. Harold and Leila Y. Mathers Charitable Foundation.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Barret A, Tagliavini F, Forloni G, Bate C, Salmona M, Colombo L, De Luigi A, Limido L, Suardi S, Rossi G, et al. (2003) Evaluation of quinacrine treatment for prion diseases. J Virol 77:8462–8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D, Giles K, Oehler A, Bhardwaj S, DeArmond SJ, Prusiner SB. (2015) Use of a 2-aminothiazole to treat chronic wasting disease in transgenic mice. J Infect Dis 212 (Suppl 1):S17–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DB, Lu D, Geva M, Watts JC, Bhardwaj S, Oehler A, Renslo AR, DeArmond SJ, Prusiner SB, Giles K. (2013) Drug resistance confounding prion therapeutics. Proc Natl Acad Sci USA 110:E4160–E4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 68:7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DA, Scott MRD, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB. (1988) Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 62:1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chen ZJ. (2014) Prion-like polymerization as a signaling mechanism. Trends Immunol 35:622–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Ebeling C, Yang S-L, Telling G, Torchia M, Groth D, Westaway D, DeArmond SJ, Prusiner SB. (1994) Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci USA 91:5690–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabortee S, Kayatekin C, Newby GA, Mendillo ML, Lancaster A, Lindquist S. (2016) Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc Natl Acad Sci USA 113:6065–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. (2007) A general model of prion strains and their pathogenicity. Science 318:930–936. [DOI] [PubMed] [Google Scholar]
- Collinge J, Gorham M, Hudson F, Kennedy A, Keogh G, Pal S, Rossor M, Rudge P, Siddique D, Spyer M, et al. (2009) Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol 8:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SJ, Lewis V, Brazier M, Hill AF, Fletcher A, Masters CL. (2002) Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann Neurol 52:503–506. [DOI] [PubMed] [Google Scholar]
- Doh-ura K, Ishikawa K, Murakami-Kubo I, Sasaki K, Mohri S, Race R, Iwaki T. (2004) Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol 78:4999–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh-ura K, Iwaki T, Caughey B. (2000) Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol 74:4894–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR. (2011) 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem 54:1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind MD, Kuo AL, Wong KS, Haman A, Devereux G, Raudabaugh BJ, Johnson DY, Torres-Chae CC, Finley R, Garcia P, et al. (2013) Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology 81:2015–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Ahn M, Lessard P, Giles K, Legname G, DeArmond SJ, Prusiner SB. (2009) Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog 5:e1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, May BCH, Renslo AR, Prusiner SB. (2010a) Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol 84:3408–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Ullman J, Ahn M, St Martin S, Prusiner SB. (2010b) Chemical induction of misfolded prion protein conformers in cell culture. J Biol Chem 285:10415–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Berry DB, Condello C, Hawley RC, Gallardo-Godoy A, Bryant C, Oehler A, Elepano M, Bhardwaj S, Patel S, et al. (2015) Different 2-aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. J Pharmacol Exp Ther 355:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K, Glidden DV, Patel S, Korth C, Groth D, Lemus A, DeArmond SJ, Prusiner SB. (2010) Human prion strain selection in transgenic mice. Ann Neurol 68:151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haïk S, Brandel JP, Salomon D, Sazdovitch V, Delasnerie-Lauprêtre N, Laplanche JL, Faucheux BA, Soubrié C, Boher E, Belorgey C, et al. (2004) Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology 63:2413–2415. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. (2007) Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol 81:12889–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korth C, May BCH, Cohen FE, Prusiner SB. (2001) Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci USA 98:9836–9841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Rao S, Gever JR, Widjaja K, Prusiner SB, Silber BM. (2013a) Optimization of arylamides as novel, potent and brain-penetrant antiprion lead compounds. ACS Med Chem Lett 4:647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Silber BM, Rao S, Gever JR, Bryant C, Gallardo-Godoy A, Dolghih E, Widjaja K, Elepano M, Jacobson MP, et al. (2013b) 2-Aminothiazoles with improved pharmacotherapeutic properties for treatment of prion disease. ChemMedChem 8:847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Giles K, Li Z, Rao S, Dolghih E, Gever JR, Geva M, Elepano ML, Oehler A, Bryant C, et al. (2013) Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J Pharmacol Exp Ther 347:325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. (2007) Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA 104:20908–20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlegel AM, Weissmann C. (2013) Acquisition of drug resistance and dependence by prions. PLoS Pathog 9:e1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. (1998) Prions. Proc Natl Acad Sci USA 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS. ( 1997–2014) ImageJ, U.S. National Institutes of Health, Bethesda, MD. [Google Scholar]
- Si K. (2015) Prions: what are they good for? Annu Rev Cell Dev Biol 31:149–169. [DOI] [PubMed] [Google Scholar]
- Silber BM, Gever JR, Li Z, Gallardo-Godoy A, Renslo AR, Widjaja K, Irwin JJ, Rao S, Jacobson MP, Ghaemmaghami S, et al. (2013a) Antiprion compounds that reduce PrPSc levels in dividing and stationary-phase cells. Bioorg Med Chem 21:7999–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber BM, Rao S, Fife KL, Gallardo-Godoy A, Renslo AR, Dalvie DK, Giles K, Freyman Y, Elepano M, Gever JR, et al. (2013b) Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice. Pharm Res 30:932–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. (1996) Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082. [DOI] [PubMed] [Google Scholar]
- Telling GC, Scott M, Hsiao KK, Foster D, Yang S-L, Torchia M, Sidle KCL, Collinge J, DeArmond SJ, Prusiner SB. (1994) Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91:9936–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J, Ryazanov S, Leonov A, Levin J, Shi S, Schmidt F, Prix C, Pan-Montojo F, Bertsch U, Mitteregger-Kretzschmar G, et al. (2013) Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol 125:795–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Jucker M. (2015) Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci 38:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Prusiner SB. (2014) Mouse models for studying the formation and propagation of prions. J Biol Chem 289:19841–19849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, Dayani Y, Bezsonov EE. (2015) Yeast prions: structure, biology, and prion-handling systems. Microbiol Mol Biol Rev 79:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. (1998) Mapping the prion protein using recombinant antibodies. J Virol 72:9413–9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.