

Abstract
Propylene glycol (PG) is used as a solvent in numerous medications, including trimethoprim/sulfamethoxazole (TMP/SMX) and lorazepam, and is metabolized in the liver to lactic acid. Cases of lactic acidosis related to PG toxicity have been described and always involved large doses of benzodiazepines and PG. We present the first case of severe lactic acidosis after a 3-day course of TMP/SMX alone, involving allegedly safe amounts of PG.
A 31-year-old female with neurofibromatosis and pilocytic astrocytoma, receiving temozolomide and steroids, was admitted to the intensive care unit for pneumonia and acute respiratory failure requiring intubation. Her initial hemodynamic and acid–base statuses were normal. She was treated with intravenous TMP/SMX for possible Pneumocystis jirovecii pneumonia and was successfully extubated on day 2. On day 3, she developed tachypnea and arterial blood gas analysis revealed a severe metabolic acidosis (pH 7.2, PCO2 19 mm Hg, bicarbonates 8 mEq/L) with anion gap of 25 mEq/L and lactate of 12.1 mmol/L. TMP/SMX was discontinued and the lactate decreased to 2.9 mmol/L within 24 hours while her plasma bicarbonates normalized, without additional intervention. The patient never developed hypotension or severe hypoxia, and her renal and liver functions were normal. No other cause for lactic acidosis was identified and it resolved after TMP/SMX cessation alone, suggesting PG toxicity.
Although PG-related lactic acidosis is well recognized after large doses of lorazepam, clinicians should bear in mind that TMP/SMX contains PG as well and should suspect PG toxicity in patients developing unexplained metabolic acidosis while receiving TMP/SMX.
INTRODUCTION
Propylene glycol (PG) is a colorless, odorless organic chemical approved by the US Food and Drug Administration for use as a solvent in food, drugs, and cosmetics. It is included in numerous intravenous and oral medications, some of them being broadly used in critically ill patients, like lorazepam or diazepam, barbiturates, etomidate, phenytoin, or trimethoprim/sulfamethoxazole (TMP/SMX).1 Although PG is generally considered safe, there is an increasing body of evidence suggesting that large doses or prolonged treatments with intravenous medications containing PG can result in severe toxicity.1,2 The reported toxic effects of PG include hyperosmolality and anion gap metabolic acidosis,3 lactic acidosis,4–6 acute kidney injury,3,7 CNS toxicity with seizures,8 and cardio-respiratory arrest.9 The precise mechanism of PG toxicity is presently unknown, however, the accumulation of PG and/or its byproducts may result in hyperosmolality and anion gap metabolic acidosis. Furthermore, the findings of swelling and vacuolization of proximal tubular cells in a PG-exposed patient kidney biopsy10 as well as observed toxicity on cultured human proximal tubular cells11 suggest a mechanism by which PG-related cellular injury results in acute kidney injury.
Although the World Health Organization recommends a maximum consumption of 25 mg/kg/day of PG when used as a food additive,12 this does not address its use as a drug solvent. Among the PG-containing drugs used in ICU settings, intravenous lorazepam involves the largest amounts of PG with respect to the administered dosage 1; as expected, lorazepam has been associated with most cases of PG toxicity3–5,7,13–17 which always involved large doses or prolonged treatments with large amounts of daily or total PG intakes. Some authors have suggested risks factors for PG toxicity, including chronic renal or liver disease, alcohol abuse,2 and have recommended to limit daily intravenous PG intakes to 1 g/kg/d or ∼69 g/day.1,18 Although TMP/SMX contains PG as well, doses usually administered to treat Pneumocystis jirovecii pneumonia (15–20 mg/kg/d) involve <0.5 g/kg/d of PG, well below the reported levels of toxicity. Even though 2 case reports of PG toxicity involved TMP/SMX as a co-administered drug, both patients were receiving large doses of lorazepam which accounted for most of the PG intakes (respectively 552 g over 12 days7 and 540 g over 5 days5). We present a unique case of severe lactic acidosis after only a 3-day course of intravenous TMP/SMX, involving amounts of PG well below the reported toxic levels, in a patient without known risk factor for PG toxicity.
CASE REPORT
A 31-year-old Caucasian female (weight 106 kg) with a past medical history of hypothyroidism, neurofibromatosis type 1, and pilocytic astrocytoma (Figure 1) recently treated with metronomic temozolomide and steroids, presented with dyspnea at rest, a 2 week duration of nonproductive cough, and a 6 week history of progressive weakness, ataxia, anorexia, weight loss, and throat pain. Her vital signs on admission to the Emergency Department were as follows: temperature 36.5°C, heart rate 110/minute, respiratory rate 30/minute, blood pressure 114/83 mm Hg, SpO2 79% on room air, and then 88% on oxygen 6 L/min nasal cannula. Initial assessment, including a chest radiograph and lung computed tomography (Figure 2), revealed patchy bilateral parenchymal opacities with consolidation concerning for multifocal pneumonia, in addition to a small left pleural effusion. Laboratory test revealed a white blood cell count of 8.75 × 103/mm3 (normal range 4.0–10.4 × 103/mm3) with 92% neutrophils, 5% lymphocytes, 3% monocytes, 0.3% eosinophils, and no basophils or bands. Serum sodium level was 142 mmol/L (137–145 mmol/L), potassium 2.8 mmol/L (3.5–5.1 mmol/L), chloride 100 mmol/L (96–107 mmol/L), bicarbonate 31 mmol/L (22–30 mmol/L), blood glucose 96 mg/dL (74–106 mg/dL), and baseline blood urea nitrogen (BUN) and serum creatinine were 7 mg/dL (7–20 mg/dL) and 1.02 mg/dL (0.6–1.0 mg/dL), respectively. The patient's initial acid base status was normal, with a venous pH of 7.4. Blood, urine, and sputum cultures were obtained, and the patient was treated empirically with intravenous cefepime, vancomycin, and azithromycin. Given worsening hypoxia requiring oxygen via high flow nasal cannula (FiO2 65%), the patient was admitted to the intensive care unit (ICU) and intubated shortly after for acute hypoxic respiratory failure requiring mechanical ventilation.
FIGURE 1.
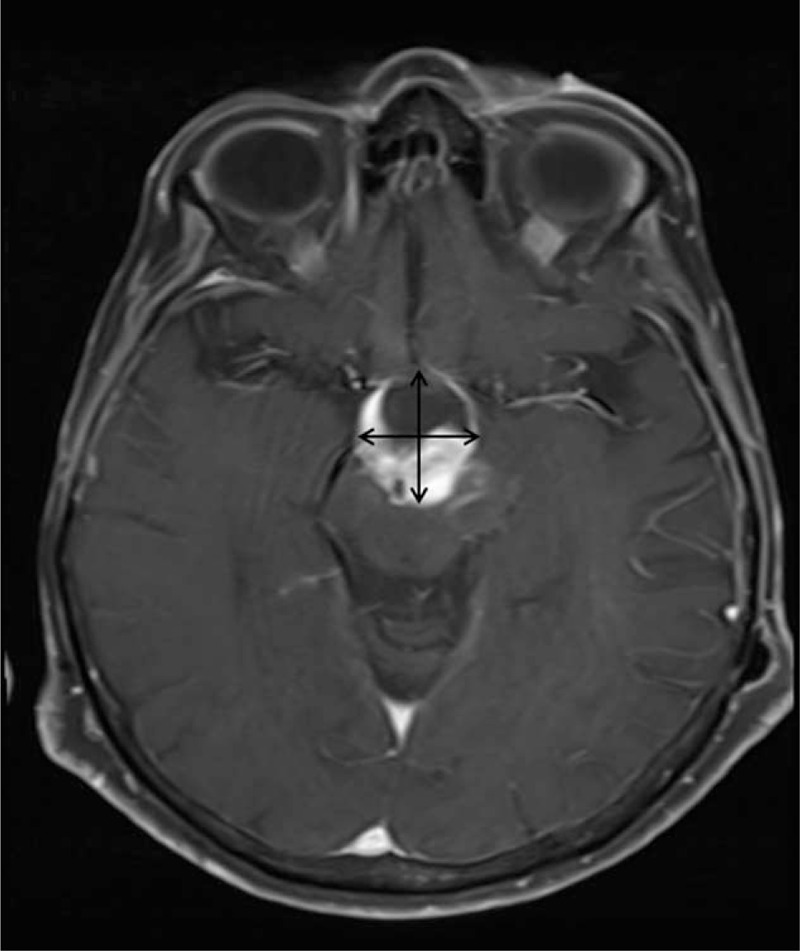
Brain magnetic resonance imaging with intravenous contrast showing the pilocytic astrocytoma as an enhancing cystic lesion (2.5 × 2.9 cm) in the region of the hypothalamus and optic chiasm extending into the brain stem.
FIGURE 2.
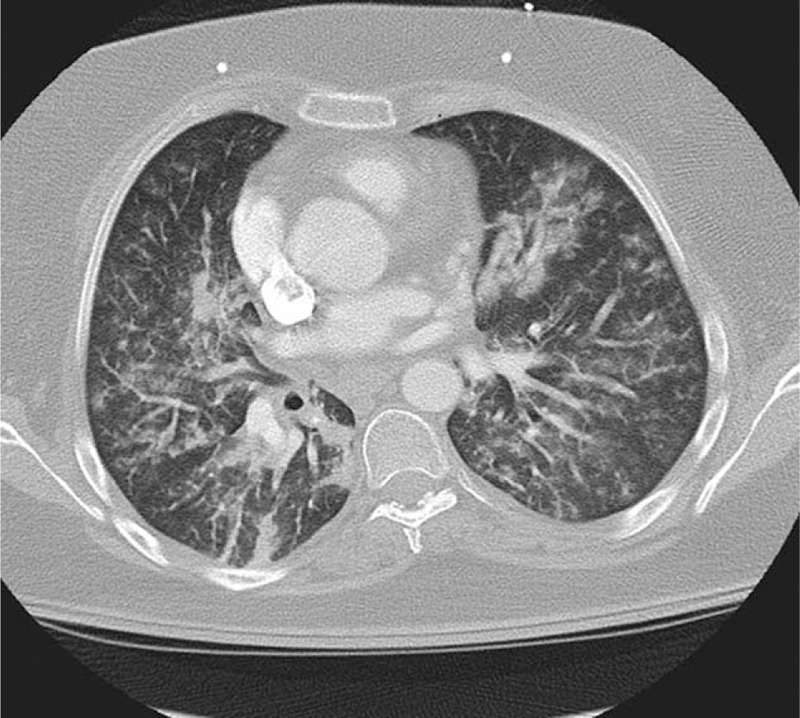
Chest computed tomography showing patchy bilateral parenchymal opacities with trace left pleural effusion, consistent with multifocal pneumonia.
In the ICU, 18 mg/kg/d of intravenous TPM/SMX, corresponding to 0.47 g/kg/d of PG, was added for empiric coverage of Pneumocystis Jirovecii pneumonia, given the patient's immunocompromised state. After intubation and mechanical ventilation, the patient's hypoxia quickly improved and the FiO2 was weaned down to 40% within 24 hours with SpO2 ∼99%. The patient was successfully extubated after 36 hours of mechanical ventilation. Her vital signs after extubation were unremarkable with normal heart rate and blood pressure, respiratory rate at 18/minute, and SpO2 97% on oxygen 2 L with nasal cannula. Twenty-four hours after extubation, the patient developed progressively worsening tachypnea (respiratory rate 32/minute) without worsening hypoxemia (SpO2 97% on oxygen 2 L with nasal cannula) or change in her chest x-ray. This prompted an arterial blood gas analysis which revealed a severe metabolic acidosis with pH 7.2, PCO2 19 mm Hg, PO2 107 mm Hg on 2 L oxygen, bicarbonate 8 mmol/L, an anion gap of 25 mEq/L, and a lactate of 12.1 mmol/L. Blood, urine, and sputum cultures remained negative and the patient's vital signs (temperature, heart rate, and blood pressure) remained normal. As TMP/SMX was suspected as the cause of her lactic acidosis and the P jirovecii, direct fluorescent-antibody test returned negative, TMP/SMX was discontinued. The other medications received between admission and the occurrence of lactic acidosis included cefepime, vancomycin, azithromycin, enoxaparin, fentanyl, propofol, dexmedetomidine, hydrocortisone, levothyroxine, pantoprazole, and lorazepam (1 mg twice), among them only lorazepam included PG (0.8 g for 2 mg of lorazepam). Within 24 hours of stopping the TMP/SMX and without any additional intervention, the lactate decreased from 12.1 to 2.9 mmol/L and serum bicarbonate increased to 23 mmol/L. The renal and liver function tests remained within normal limits. Figures 3 and 4 display the course of relevant vital signs (mean arterial pressure and SpO2) and biological data (serum bicarbonates, creatinine, and lactic acid) during the initial 6-day course. In total, the patient received 54 g (0.5 g/kg) of PG daily for 3 days while being treated with TMP/SMX and an additional 0.8 g with lorazepam. No other cause for the patient's lactic acidosis was identified, and her acid–base status remained normal for the rest of her hospital admission.
FIGURE 3.
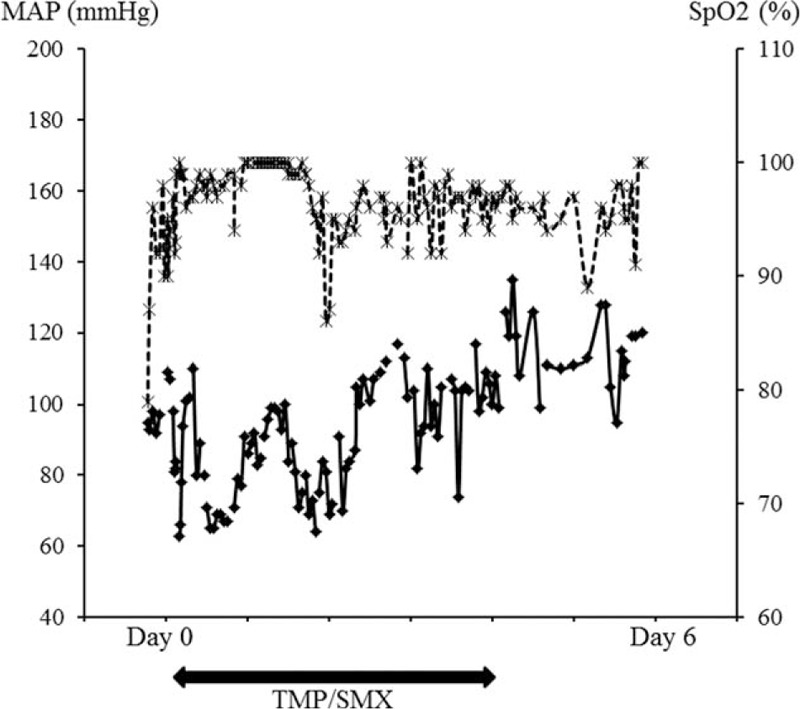
Course of mean arterial pressure (MAP, diamond markers with straight line on the left y-axis) and SpO2 (x markers with dotted line on the right y-axis) over the first 6 days of admission. TMP/SMX was started on admission (day 0) and stopped at day 4. Scales are different for the sake of legibility. Except for initial hypoxemia (SpO2 79% on room air on admission) and a brief (1 hour) episode of desaturation at day 2 (SpO2 86–87% on O2 2 liters), MAP and SpO2 remained above 65 mmHg and 90% respectively.
FIGURE 4.
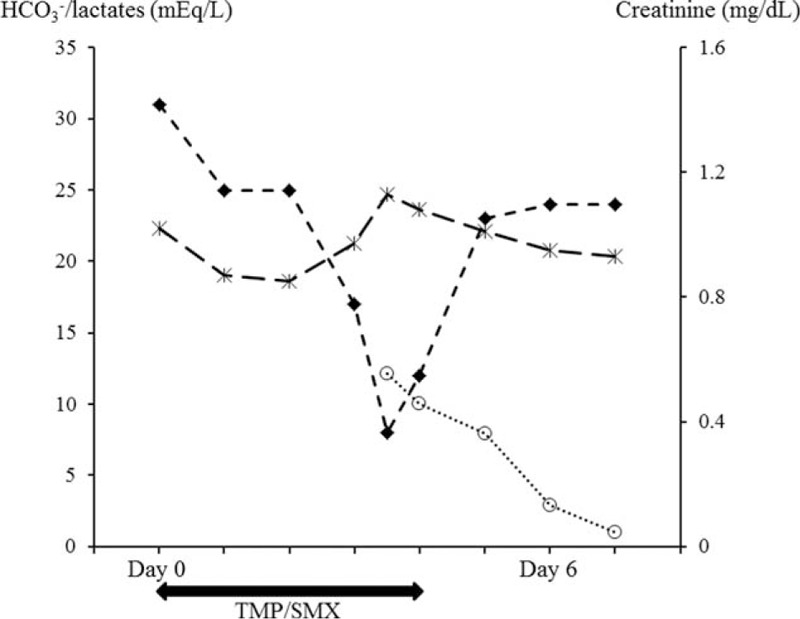
Course of serum bicarbonates (diamonds) and lactates (circles) on the left y-axis and creatinine (x markers) on the right y-axis over the first 6 days of admission. TMP/SMX was started on admission (day 0) and stopped at day 4. Scales are different for the sake of legibility. While serum bicarbonates was normal on admission, a severe metabolic acidosis developed at day 3, concomitant with an increased lactates. After TMP/SMX cessation, serum bicarbonates and lactates levels normalized within 24 hours.
The subsequent patient's hospital course was complicated by persistent encephalopathy ascribed to her brainstem astrocytoma, with recurrent airway mucus plugging requiring re-intubation 10 days after extubation. The patient eventually underwent a tracheostomy for difficult ventilator weaning and was transferred to a rehabilitation facility after 3 weeks.
DISCUSSION
There has been an increasing interest in PG toxicity in recent years,1–3 and multiple adverse reactions have been reported,1,8,18 including hyperosmolality, anion gap metabolic acidosis, lactic acidosis, acute kidney injury, CNS toxicity with seizures, and a sepsis or systemic inflammatory-like syndrome. Although drugs with high PG content, such as benzodiazepines (lorazepam contains as high as 80% v/v of PG), barbiturates, etomidate, phenytoin, and TMP/SMX, are broadly used in the ICU setting, their sometimes life-threatening adverse reactions may be under-recognized. In a prospective observational study, Arroliga and coworkers demonstrated that 6 out of 9 ICU patients being treated with high dose (≥10 mg/h) of intravenous lorazepam developed hyperosmolar high anion gap metabolic acidosis consistent with PG accumulation, the osmolar gap being the strongest predictor of serum PG concentrations.16 Wilson and colleagues also observed that 4 out of 21 patients who had received PG-containing sedative medications (either lorazepam or diazepam) had metabolic abnormalities consistent with PG toxicity.14 These studies suggest that the incidence of PG toxicity is likely much higher than clinically recognized and underscore the need for a better knowledge by clinicians and ICU staff of PG-containing drugs and for a prudent monitoring of patients receiving PG.
Although PG is generally considered safe, the reported cases of mild forms of lactic acidosis following the use of large doses of PG diluted medications led to recommendations to limit PG intakes to 69 g/d or ∼1 g/kg/d,14,18 a dose presumed safe in the absence of risk factors for PG toxicity.1 The precise mechanism by which PG causes toxicity is unclear, as no convincing evidence supports a direct cytotoxicity of either PG or its byproducts. After intravenous administration, PG has a elimination half-life of 2.3 ± 0.7 hours19 and is excreted by two mechanisms: the liver metabolizes approximately half to lactate, acetate, and pyruvate via oxidation by alcohol dehydrogenase and aldehyde dehydrogenase, and the other half is excreted unchanged by the kidneys.3 The renal clearance of PG decreases as the dose administered increases19 (from 390 mL/min/1.73 m2 for a PG dose of 5 g/d to 144 mL/min/1.73 m2 for a dose of 21 g/d); this may explain why patients with chronic kidney diseases or acute renal failure may be predisposed to PG toxicity,1 but our patient's renal function was normal. The present literature suggests a broad spectrum in the severity of clinical manifestations of PG toxicity, ranging from mild metabolic abnormalities14 to life-threatening clinical conditions.7,9,13 Moreover, PG toxicity has been documented over a wide range of serum PG levels (1220 −13,080 17 mg/dL) and significant overlap exists between levels resulting in only mild metabolic abnormalities as compared with critical clinical deterioration. For instance, in the study by Wilson et al, PG plasma levels ranged from 58 to 127 mg/dL for patients presenting only metabolic abnormalities and from 104 to 144 mg/dL in patients with clinical deterioration because of PG toxicity.14 There is no clear correlation between cumulative lorazepam (and therefore PG) dose and serum PG concentration16 and PG pharmacokinetic studies have shown considerable intra- and interpatient variation in serum PG concentrations for a given dosage administered.19,21 A hypothetical critical PG concentration beyond which clinical deterioration may be observed is presently unknown and appears to be patient dependent, suggesting a patient-specific variability in the metabolism, excretion, and thus toxicity of PG.
Most, if not all of the case reports on PG toxicity involved benzodiazepines (mostly lorazepam), which contain the largest amounts of PG among the drugs routinely used in ICU setting. In 2 published cases, patients were receiving TMP/SMX5,7 but the TMP/SMX accounted for only a minor fraction of total PG intake as patients were also receiving large doses of lorazepam. In these cases, the total PG intake was well above the threshold generally considered as safe. For instance, in the case reported by Hayman et al, the patient had received a total of 552 g of PG within 12 days, only 123 g (20%) of which administered with TMP/SMX during the last 6 days.7 The second case involved a PG load of 540 g within 5 days administered with lorazepam infusion in a patient who was also receiving TMP/SMX at unknown dosages.5 Here we report a unique case of severe lactic acidosis developing after only a 3-day course of intravenous TMP/SMX, involving relatively low amounts of PG (162 g within 3 days), well below the levels generally considered as toxic.1 Applying the Naranjo adverse drug reaction probability scale, PG was determined to be the probable cause for her lactic acidosis with a score of 722: we assigned 1 point for previous conclusive reports on this reaction, 2 points for the time between drug administration and the occurrence of lactic acidosis, 1 point for the improvement in lactic acidosis after discontinuation of TMP/SMX, 2 points for the absence of alternative causes, and 1 point for the objective evidence confirming the reaction. Unfortunately our patient's serum osmolality was not measured and we were unable to obtain a serum PG level to add further evidence of PG toxicity.
We believe that this case is clinically relevant and should raise awareness among clinicians, as (1) TMP/SMX is broadly used in critically ill immunocompromised patients and the fact that it contains PG is unrecognized, (2) critically ill patients often have conditions (shock, acute renal failure, and lactic acidosis) that may mimic PG toxicity but also contribute to it, (3) immediate recognition and cessation of TMP/SMX is crucial to prevent the development of life-threatening complications in patients already prone to multiorgan failure. Intravenous TMP/SMX is often used in the setting of immunosuppression, either to treat documented P jirovecii pneumonia or as an empiric treatment in immunocompromised patients presenting with acute respiratory failure and diffuse pulmonary infiltrates concerning for P jirovecii pneumonia. In ICU settings, such patients may be easily exposed to PG toxicity as they are prone to develop acute renal or liver failure, reported as risk factors for PG toxicity.1 On the other hand, patients receiving TMP/SMX often present with signs of sepsis, and any clinical or biological deterioration will easily be attributed to the ongoing sepsis, while potentially severe adverse drug reactions may be overlooked. Despite a severe lactic acidosis, our patient's clinical condition remained stable (except for the acidosis-related tachypnea) as well as her renal function. The normalization of her serum bicarbonates and lactate levels within 24 hours after TMP/SMX cessation is consistent with PG half-life. Without TMP/SMX cessation, the lactic acidosis could have been complicated by progressive multiorgan failure and cardiorespiratory arrest.9 Our patient did not present with acute renal or liver failure, and had no history of chronic kidney, liver disease, or alcohol abuse, described as risks factors for PG toxicity.18 Although TMP/SMX is a known PG-containing drug, PG intake for our patient were below toxic levels and she did not receive other PG-containing drugs except for 2 mg of lorazepam, which only accounted for 0.8 g of PG and likely played a minor role in her lactic acidosis. Our patient had findings on chest imaging concerning for pneumonia; however all of her cultures remained sterile and she never developed hypotension or shock (her mean arterial pressure remained above 65 mm Hg the whole time, as shown in Figure 3), making hypoperfusion and resulting lactic acidosis secondary to sepsis extremely unlikely. Similarly, her severe hypoxemia on presentation (SpO2 79% on room air) is unlikely to account for the delayed lactic acidosis, as it was rapidly corrected with additional oxygen and mechanical ventilation (see Figure 4), and metabolic acidosis developed 48 hours later while her oxygenation had been normal on mechanical ventilation.
CONCLUSION
To our knowledge, severe lactic acidosis after a 3-day course of TMP/SMX, involving allegedly safe doses of PG, has never been described. Although PG-related toxicity following large doses of lorazepam is now well acknowledged, physicians should bear in mind that TMP/SMX also contains PG, and suspect PG toxicity in patients developing metabolic acidosis while receiving TMP/SMX, even at the recommended dosage and without risk factors for toxicity. Immediate cessation of the drug is crucial to prevent the development of worsening multiorgan failure.
Footnotes
Abbreviations: CNS = central nervous system, ICU = intensive care unit, PG = propylene glycol, TMP/SMX = Trimethoprim/sulfamethoxazole
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Zar T, Graeber C, Perazella MA. Recognition, treatment, and prevention of propylene glycol toxicity. Semin Dial 2007; 20:217–219. [DOI] [PubMed] [Google Scholar]
- 2.Liamis G, Milionis HJ, Elisaf M. Pharmacologically-induced metabolic acidosis: a review. Drug Saf 2010; 33:371–391. [DOI] [PubMed] [Google Scholar]
- 3.Barnes BJ, Gerst C, Smith JR, et al. Osmol gap as a surrogate marker for serum propylene glycol concentrations in patients receiving lorazepam for sedation. Pharmacotherapy 2006; 26:23–33. [DOI] [PubMed] [Google Scholar]
- 4.Neale BW, Mesler EL, Young M, et al. Propylene glycol-induced lactic acidosis in a patient with normal renal function: a proposed mechanism and monitoring recommendations. Ann Pharmacother 2005; 39:1732–1736. [DOI] [PubMed] [Google Scholar]
- 5.Arbour R, Esparis B. Osmolar gap metabolic acidosis in a 60-year-old man treated for hypoxemic respiratory failure. Chest 2000; 118:545–546. [DOI] [PubMed] [Google Scholar]
- 6.Kelner MJ, Bailey DN. Propylene glycol as a cause of lactic acidosis. J Anal Toxicol 1985; 9:40–42. [DOI] [PubMed] [Google Scholar]
- 7.Hayman M, Seidl EC, Ali M, et al. Acute tubular necrosis associated with propylene glycol from concomitant administration of intravenous lorazepam and trimethoprim-sulfamethoxazole. Pharmacotherapy 2003; 23:1190–1194. [DOI] [PubMed] [Google Scholar]
- 8.MacDonald MG, Getson PR, Glasgow AM, et al. Propylene glycol: increased incidence of seizures in low birth weight infants. Pediatrics 1987; 79:622–625. [PubMed] [Google Scholar]
- 9.Fligner CL, Jack R, Twiggs GA, et al. Hyperosmolality induced by propylene glycol. A complication of silver sulfadiazine therapy. JAMA 1985; 253:1606–1609. [PubMed] [Google Scholar]
- 10.Yorgin PD, Theodorou AA, Al-Uzri A, et al. Propylene glycol-induced proximal renal tubular cell injury. Am J Kidney Dis 1997; 30:134–139. [DOI] [PubMed] [Google Scholar]
- 11.Morshed KM, Jain SK, McMartin KE. Propylene glycol-mediated cell injury in a primary culture of human proximal tubule cells. Toxicol Sci 1998; 46:410–417. [DOI] [PubMed] [Google Scholar]
- 12.Additives F-WECoF. Toxicological evaluation of certain food additives with a review of general principles and of specifications. Seventeenth report of the joint FAO-WHO Expert Committee on Food Additives. World Health Organ Tech Rep Ser 1974; 539:1–40. [PubMed] [Google Scholar]
- 13.Parker MG, Fraser GL, Watson DM, et al. Removal of propylene glycol and correction of increased osmolar gap by hemodialysis in a patient on high dose lorazepam infusion therapy. Intensive Care Med 2002; 28:81–84. [DOI] [PubMed] [Google Scholar]
- 14.Wilson KC, Reardon C, Theodore AC, et al. Propylene glycol toxicity: a severe iatrogenic illness in ICU patients receiving IV benzodiazepines: a case series and prospective, observational pilot study. Chest 2005; 128:1674–1681. [DOI] [PubMed] [Google Scholar]
- 15.Nelsen JL, Haas CE, Habtemariam B, et al. A prospective evaluation of propylene glycol clearance and accumulation during continuous-infusion lorazepam in critically ill patients. J Intensive Care Med 2008; 23:184–194. [DOI] [PubMed] [Google Scholar]
- 16.Arroliga AC, Shehab N, McCarthy K, et al. Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults. Crit Care Med 2004; 32:1709–1714. [DOI] [PubMed] [Google Scholar]
- 17.Al-Khafaji AH, Dewhirst WE, Manning HL. Propylene glycol toxicity associated with lorazepam infusion in a patient receiving continuous veno-venous hemofiltration with dialysis. Anesth Analg 2002; 94:1583–1585.table of contents. [DOI] [PubMed] [Google Scholar]
- 18.Lim TY, Poole RL, Pageler NM. Propylene glycol toxicity in children. J Pediatr Pharmacol Ther 2014; 19:277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speth PA, Vree TB, Neilen NF, et al. Propylene glycol pharmacokinetics and effects after intravenous infusion in humans. Ther Drug Monit 1987; 9:255–258. [DOI] [PubMed] [Google Scholar]
- 20.Cawley MJ. Short-term lorazepam infusion and concern for propylene glycol toxicity: case report and review. Pharmacotherapy 2001; 21:1140–1144. [DOI] [PubMed] [Google Scholar]
- 21.Yu DK, Elmquist WF, Sawchuk RJ. Pharmacokinetics of propylene glycol in humans during multiple dosing regimens. J Pharm Sci 1985; 74:876–879. [DOI] [PubMed] [Google Scholar]
- 22.Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther 1981; 30:239–245. [DOI] [PubMed] [Google Scholar]