

Supplemental Digital Content is available in the text
Abstract
The molecular etiology of nonsyndromic sensorineural hearing loss (SNHL) in subjects with only one detectable autosomal recessive GJB2 mutation is unclear. Here, we report GJB2 single heterozygotes with various final genetic diagnoses and suggest appropriate diagnostic strategies. A total of 160 subjects with SNHL without phenotypic markers were screened for GJB2 mutations. Single-nucleotide variants or structural variations within the DFNB1 locus or in other deafness genes were examined by Sanger sequencing, breakpoint PCR, and targeted exome sequencing (TES) of 129 deafness genes. We identified 27 subjects with two mutations and 10 subjects with only one detectable mutation in GJB2. The detection rate of the single GJB2 mutation among the 160 SNHL subjects in the present study (6.25%) was higher than 2.58% in normal hearing controls in Korean. The DFNB1 was clearly excluded as a molecular etiology in four (40%) subjects: other recessive deafness genes (N = 3) accounted for SNHL and the causative gene for the other non-DFNB1 subject (N = 1) was not identified. The etiology of additional two subjects was potentially explained by digenic etiology (N = 2) of GJB2 with MITF and GJB3, respectively. The contribution of the single GJB2 mutation in the four remaining subjects is unclear. Comprehensive diagnostic testing including TES is prerequisite for understanding GJB2 single heterozygotes.
INTRODUCTION
Extreme etiologic heterogeneity exists in sensorineural hearing loss (SNHL) (http://hereditaryhearingloss.org/). Therefore, molecular genetic testing methods for efficiently screening the various deafness genes are necessary for accurately diagnosing SNHL. Various techniques, such as whole exome sequencing and targeted exome sequencing (TES), have made it possible to screen candidate genes in an extremely high-throughput manner.1 However, making a conclusive molecular diagnosis still requires time and rigorous effort in many cases, particularly in nonsyndromic cases with rare variants. Thus, we recently suggested the importance of phenotype-driven genetic testing focusing on candidate genes according to the clinical phenotypes of the affected subjects.2
The predominant mutations in gap junction protein beta 2 (GJB2) (MIM ID: 121011) account for up to 50% of autosomal recessive nonsyndromic SNHL in some ethnicities. This significantly reduces the molecular genetic testing loads needed to reach a final genetic diagnosis through GJB2 testing alone.3 Although the proportion of nonsyndromic hearing loss and deafness 1 (DFNB1) among nonsyndromic sporadic or autosomal recessive severe to profound SNHL significantly varies, it still accounts for ∼15% of such subjects in Koreans, making GJB2 and solute carrier family 26, member 4 (SLC26A4) the most frequent causative genes of SNHL.3–7 However, it is not always straightforward to diagnose DFNB1 in subjects carrying GJB2 mutations. We occasionally encounter uncertain cases with a monoallelic GJB2 mutation. It is estimated that ∼6% to 20% of identified GJB2 mutations in hearing-impaired subjects were monoallelic mutations,8–12 although the value was reported to be up to 50% in some European populations.13 It has been speculated that a substantial portion of DFNB1 subjects had only one detectable mutation in GJB2 and that an occult large deletion within the DFNB1 locus in trans with the detected GJB2 mutation may cause deafness in these subjects.13–15 However, the large genomic deletion within the DFNB1 locus has not been detected in Koreans to date, suggesting that interpretation of a monoallelic GJB2 mutation should differ depending on ethnicity. Additionally, high carrier frequencies of GJB2 make it difficult to regard these subjects simply as DFNB1 in Korea.4 Moreover, some researchers have reported the digenic pathogenesis of deafness involving the detected single GJB2 mutation in combination with mutations in other deafness genes, further complicating interpretation.16–18
Although numerous studies have examined the clinical implications of single heterozygous mutations in GJB2 on SNHL, there is no consensus regarding the interpretation of the results when one mutation is identified in the GJB2 gene of the subject. Thus, we propose the use of stepwise molecular genetic approaches to clarify the contribution of the detected monoallelic GJB2 mutation to SNHL in Koreans based on our experiences with GJB2 single heterozygotes.
MATERIALS AND METHODS
Ethical considerations
This study was approved by the institutional review boards (IRBs) at Seoul National University Hospital (IRBY-H-0905-041-281) and Seoul National University Bundang Hospital (IRB-B-1007-105-402). Written informed consent was obtained from all of the participating subjects. For children, written informed consent was obtained from their parents or guardians on their behalf.
Clinical Evaluation
A total of 470 subjects with SNHL were subjected to genetic testing for SNHL at the otolaryngology clinics of Seoul National University Hospital and Seoul National University Bundang Hospital from September 2010 to March 2015. All of the recruited subjects were of Korean ethnicity.
Clinical characteristics of the cohort included gender, date of birth, medical history, physical examination, pure tone audiometry, and imaging studies. All of the enrolled subjects underwent audiologic evaluation using pure-tone audiometry, speech audiometry, auditory brainstem response, and auditory state response. The pure-tone thresholds were recorded at 0.25, 0.5, 1, 2, 4, and 8 kHz. However, some infants could be recorded at only limited frequencies because of poor cooperation. The hearing threshold was calculated by averaging the thresholds of 0.5, 1, 2, and 4 kHz, and was classified as subtle (16–25 dB), mild (26–40 dB), moderate (41–70 dB), severe (71–95 dB), or profound (>95 dB).
The presence of any phenotypic markers indicating syndromic SNHL was thoroughly investigated in subjects and their family members. The phenotypic markers included ophthalmologic abnormality, such as dystopia canthorum (lateral displacement of inner canthi) in Waardernburg syndrome,19 depigmented skin lesion, freckled face, or hypopigmentation with respect to hair color. Imaging evaluations were conducted using temporal bone computed tomography or magnetic resonance imaging to identify inner ear anomalies such as an enlarged vestibular aqueduct in subjects. All family members were also investigated for the presence of any type of hearing loss.
Based on the results of the retrieved clinical and audiologic evaluations, 160 bilateral SNHL subjects without audiologic or radiological phenotypic markers and segregating either into a sporadic or possibly autosomal recessive fashion were screened for the presence of GJB2 mutations.
DNA Samples and Sanger Sequencing of GJB2 Coding Region
Genomic DNA was extracted from peripheral blood using standard protocols (Gentra Puregene Blood Kit, Qiagen, Venlo, Limburg, the Netherlands). Sanger sequencing of GJB2 was performed for subjects without noticeable phenotypic markers as described previously.4,6 Further genetic tests were conducted to determine the definitive genetic etiology of SNHL in GJB2 single heterozygotes and their family members as described below (Figure 1).
FIGURE 1.
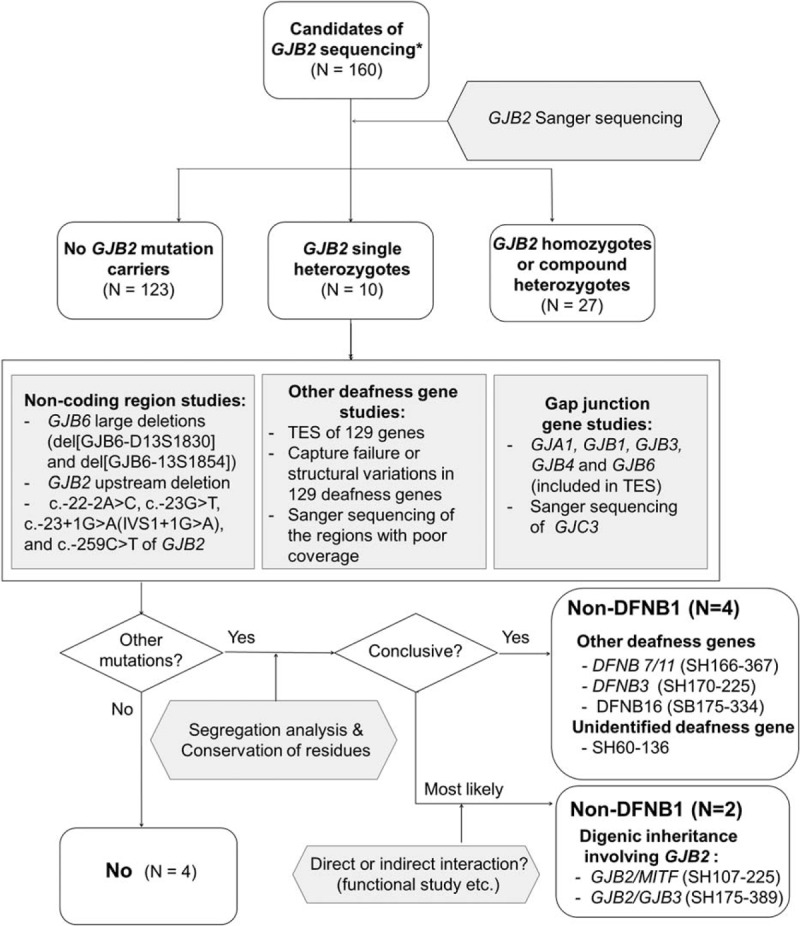
Molecular diagnostic flow of the present study: GJB2 single heterozygotes were analyzed using a stepwise and comprehensive molecular diagnostic protocol including massive parallel sequencing of 129 known deafness genes and Sanger sequencing of additional gap junction genes as well as screening of known GJB6 large deletions. Approximately half of GJB2 single heterozygotes were not DFNB1. Completely different causative genes or even digenic etiology involving the GJB2 mutation accounted for these non-DFNB1 subjects. DFNB1 = nonsyndromic hearing loss and deafness 1, GJB2 = gap junction protein beta 2, GJB6 = gap junction protein beta 6.
Examination of GJB2 Noncoding Region
To exclude the presence of pathogenic GJB2 mutations outside of the coding regions, Sanger sequencing for the four known pathogenic mutations, c.-22-2A>C,20 c.-23G>T,21 c.-23+1G>A (formerly referred to as IVS1+1G>A),22 and c.-259C>T23 was performed using two primer sets: GJB2-AF (5′-GGCGGGAGACAGGTGTTG-3′), GJB2-AR (5′-CCAAGGACGTGTGTTGGTC-3′) and G259F (5′-AGCGCTCATAAATGCCAAGT-3′), G259R (5′-GCCGCAACACCTGTCTCC-3′).
Next, we conducted a multiplex breakpoint PCR assay for two previously reported large genomic deletions (del[GJB6-D13S1830] and del[GJB6-D13S1854]).13 To detect other structural variations involving 5-kb regions upstream of GJB2 and the GJB6 region within the DFNB1 locus, we also verified the raw data of TES using Integrative Genomic Viewer (http://www.broadinstitute.org/igv/home (Figure 2).
FIGURE 2.
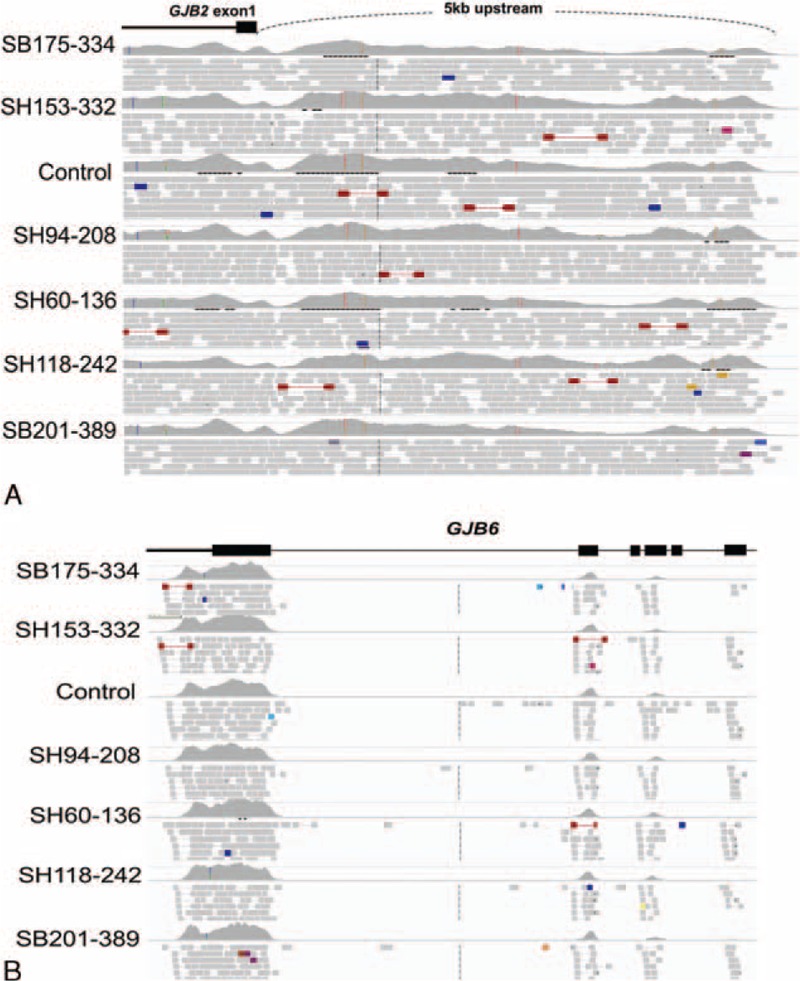
Integrative Genomic Viewer findings of noncoding upstream regions of GJB2 (A) and GJB6 regions (B) from six GJB2 single heterozygotes (five group III subjects and SH60-136) with prelingual SNHL and a control with normal hearing: six GJB2 single heterozygotes with SNHL showed no noticeable difference in terms of coverage in these regions compared with the control, indicating that no structural variations were in trans with the detected single heterozygous GJB2 mutation in these regions. GJB2 = gap junction protein beta 2, GJB6 = gap junction protein beta 6, SNHL = sensorineural hearing loss.
Studies of Other Deafness Genes: TES and gap Junction Gene Studies
The presence of causative mutations in other deafness genes was investigated by targeted capture of exons and flanking sequences of 129 deafness genes, followed by massively parallel sequencing of DNA libraries as previously described.24 Variant detection was accomplished as described.24
To exclude the possibility of capture failure or structural variations involving the exons of any of the 129 target genes, we also analyzed the coverage status of TES during the second phase of TES data analysis. Sanger sequencing of exons showing poor coverage (<10 × ) was performed with primer sets of Otoferlin (OTOF), Stereocilin (STRC), and Otoancorin (OTOA) (see Table S1, Supplemental Content, which illustrates primer sequences for Sanger sequencing to screen exons of OTOF, STRC, and OTOA showing poor coverages by TES) so that variants that may reside in these regions could be detected (see Table S2, Supplemental Content, which illustrates regions showing significantly low depth of coverage in TES: OTOF, STRC, and OTOA).
Several gap junction genes, including gap junction protein alpha 1 (GJA1), gap junction protein beta 1 (GJB1), GJB2, gap junction protein beta 3 (GJB3), gap junction protein beta 4 (GJB4), and gap junction protein beta 6 (GJB6), were screened either by TES or Sanger sequencing. Particularly, Sanger sequencing of Gap junction protein gamma 3 (GJC3), which was not included in our TES deafness panel, was separately conducted to explore other mutations in gap junction genes.25 The GJC3 primer sequences were as follows: GJC3-1F (5′-CCTTGGATTAGGAGTGACAAGG-3′), GJC3-1R (5′-CCCTGGGACATCTGTGTTG-3′), GJC3-2F (5′-AAGGCTGCCTGCTTCGAT-3′), GJC3-2R (5′-TCTTTAGGAAAATGGTCTTCTCA-3′), GJC3-3F (5′-CCTGGGGTTGCAGTACCAC-3′), GJC3-3R (5′-TTGTACTTCCCAGAAAGGTGA-3′), GJC3-4F (5′-ATGGGTGGCACCTAAAGTGT-3′), and GJC3-4R (5′-GTCCCAGTTGTCGGTTATGC-3′).
Investigation of Genetic Contributions of Detected Variants and/or Mutations
Segregation analysis, evaluating variants in the Korean normal hearing control group, and evaluating the conservation of residues among species were performed for variations and/or mutations obtained in the genetic studies described earlier. The possibility of digenic inheritance of SNHL resulting from the detected monoallelic GJB2 mutation with other deafness genes was also considered based on previous studies.16–18
RESULTS
Recruitment of GJB2 Single Heterozygotes and Clinical Evaluations
Among the 160 subjects with moderate to profound SNHL without any phenotypic markers, 37 subjects (23.1%, 37/160) harbored at least one mutant allele in GJB2. Twenty-seven subjects were confirmed to be DFNB1 with two definitely pathogenic mutant alleles in GJB2 either as homozygotes or compound heterozygotes. The remaining 10 subjects (27.0%, 10/37) presented monoallelic GJB2 mutations or suspicious variants (p.T123N from SH94-208) (Tables 1 and 2). Five kinds of GJB2 mutations including one possibly pathogenic variant (p.T123N) were detected in these subjects: p.V37I (SB 175-334), p.R143W (SH60-136 and SB201-389), p.V193E (SH118-242, SH184-416, SH170-377, and SH175-389), c.235delC (SH153-332, SH166-387, and SH107-225), and p.T123N (SH94-208).
TABLE 1.
GJB2 Single Heterozygotes where DFNB1 was Excluded as a Final Molecular Diagnosis (Groups I and II)
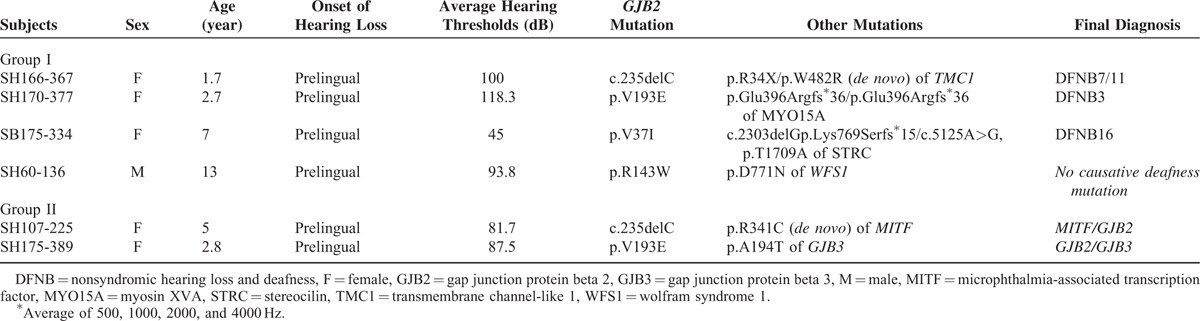
TABLE 2.
GJB2 Single Heterozygotes Where DFNB1 Could Not be Excluded (Group III)

None of the 10 single heterozygotes of GJB2 carried pathogenic structural variations of GJB6/upstream regions of GJB2 (Figure 2) or previously reported single-nucleotide variations in the noncoding regions of GJB2 (data not shown). Next, we analyzed the TES data to identify causative point mutations of other deafness genes, if any, in these subjects.
GJB2 Single Heterozygotes where DFNB1 was Excluded as a Final Molecular Diagnosis: A Fortuitously Detected GJB2 Mutation (Group I)
There were three subjects (SH166-367, SH170-377, and SB175-334) with two recessive mutations, presumed to be pathogenic, in completely different deafness genes. One of the children with a heterozygous c.235delC mutation (SH 166-367) was identified to carry a predominant founder mutation, p.R34X (c.100C>T) (rs121908073), and a novel variant, p.W482R of Transmembrane channel-like 1 (TMC1) (NM_138691), in a trans configuration (Table 1). The recessive mutation p.R34X was previously reported as a single founder mutation of TMC1.27,28 The other novel missense variant, c.1444T>C (p.W482R), was also strongly considered pathogenic because the residue was highly conserved among various species including zebrafish and Caenorhabditis elegans as indicated by the high GERP score (6.02). This variant was predicted to be “probably damaging” by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/) based on in silico analyses. Furthermore, this variant was not detected among the 544 control chromosomes from normal hearing Korean subjects. Similarly, SH170-377 carrying the p.V193E mutation in GJB2 also contained a previously reported homozygous p.Glu396Argfs∗36 mutant allele in Myosin XVA (MYO15A) (NM_016239) (Table 1).29
Although no other causative deafness mutation was detected in the initial analysis of TES data, Sanger sequencing for the low coverage area (<10×) in TES (see Table S2, Supplemental Content, which illustrates regions showing significantly low depth of coverage in TES: OTOF, STRC, and OTOA) revealed the two known pathogenic STRC mutations as a compound heterozygous configuration in SB175-334 (Table 1).30 To sum up, SH166-367, SH170-377, and SB175-334 which would have been considered DFNB1 without TES were found to be DFNB7/11, DFNB3, and DFNB16, respectively.
Finally, a subject with the heterozygous p.R143W mutation in GJB2 (SH60-136) carried a p.D771N variant in Wolfram syndrome 1 (WFS1) (NM_001145853) according to TES. However, neither p.R143W in GJB2 nor p.D771N in WFS1 was predicted to contribute to SNHL of SH60-136 based on rigorous segregation analysis of the phenotype and the variants (Figure 3). As a result, DFNB1 was excluded for SH60-136.
FIGURE 3.
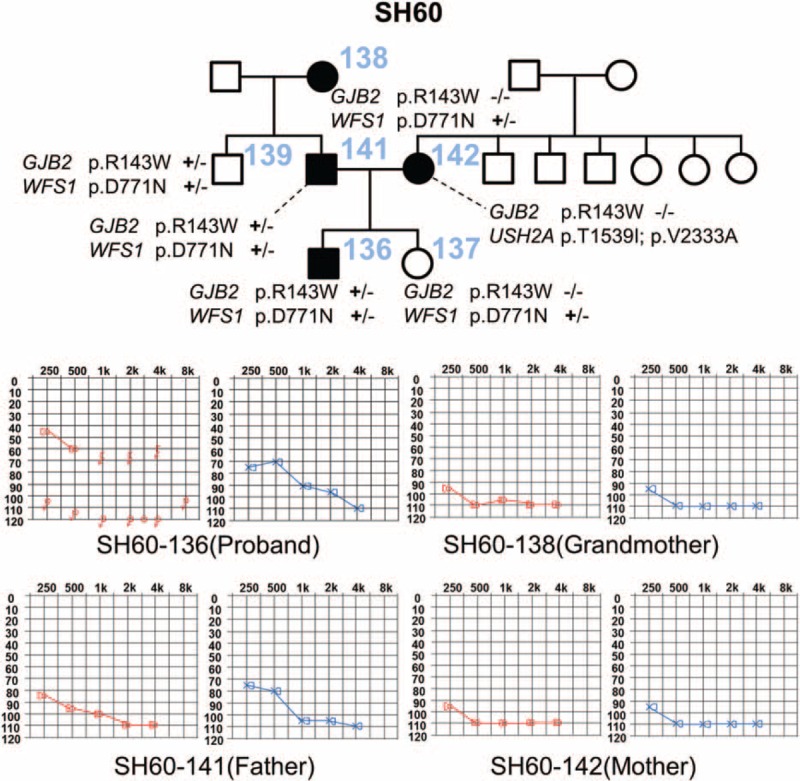
Pedigree and audiograms of SH60 and segregation of variations of GJB2 and WFS1 in this family: two subjects with SNHL, SH60-138 and SH60-142, showed a discrepancy in the GJB2 genotype. Two unaffected subjects, SH60-137 and SH60-139, also carried p.D771N in WFS1. This indicates that neither p.R143W in GJB2 nor p.D771N in WFS1 contributed to SNHL in SH60-136 and that p.R143W in GJB2 was an incidentally detected variant in this subject. GJB2 = gap junction protein beta 2, SNHL = sensorineural hearing loss, WFS1 = wolfram syndrome 1.
Single Heterozygous GJB2 Mutant Allele Possibly Contributing to Deafness via Digenic Inheritance: Double Heterozygosity with Additional Mutation in Other Deafness Genes (Group II)
Interestingly, two subjects (SH107-225 and SH175-389) showed double heterozygosity for a GJB2 mutation and another likely pathogenic mutation in another deafness gene. We detected a de novo Microphthalmia-associated transcription factor (MITF) (NM_000248) variant, p.R341C, in one of the c.235delC carriers (SH107-225) (Figure 4A). She inherited c.235delC of GJB2 from her father and did not have any known large genomic deletions within the DFNB1 locus (Figure 4B). The p.R341 residue of MITF is a well-conserved sequence among species, including zebrafish and tunicates (Figure 4C). Moreover, this MITF variant was not detected in the 666 control chromosomes from normal hearing Korean subjects, supporting the pathogenic potential of p.R341C in MITF in SH107-225. However, symptoms and signs suggesting Waardenburg syndrome type2 (WS2) including retinal abnormalities and pigmentation abnormalities could not be determined due of the patients’ young ages.
FIGURE 4.
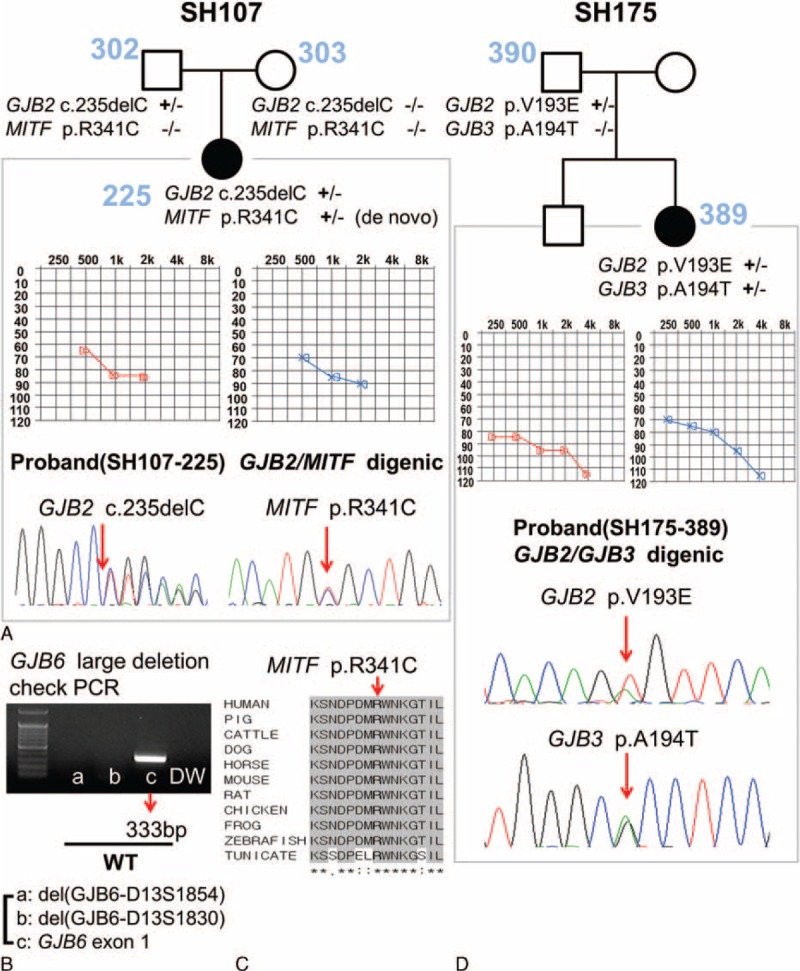
Digenic inheritances of GJB2/MITF and GJB2/GJB3 (group II). (A) In addition to c.235delC in GJB2, the de novo variant of MITF, p.R341C was identified in SH107-225. (B) There was no GJB6 large deletion within the DFNB1 locus. (C) The sequence of the p.R341C variant is well-conserved from humans to tunicates. (D) SH175-389 harbored a monoallelic p.V193E variant of GJB2 and a monoallelic p.A194T variant of GJB3. DFNB1 = nonsyndromic hearing loss and deafness 1, GJB2 = gap junction protein beta 2, GJB3 = gap junction protein beta 3, GJB6 = gap junction protein beta 6, MITF = microphthalmia-associated transcription factor.
By screening other gap junction genes, another subject (SH175-389) carrying a single heterozygous p.V193E in GJB2 allele harbored a single heterozygous p.A194T mutant allele of GJB3 (NM_001005752) (SH175-389) with known pathogenicity (Figure 4D).31 This 2-year-old female showed severe autosomal recessive SNHL with a mean hearing threshold of 87.5 dB HL.
Single Heterozygous GJB2 Mutant Allele with Unknown Contribution to SNHL in Our Cohort (Group III)
A 39-year-old female subject (SH94-208) showed the p.T123N variant of GJB2. The pathogenic potential of the p.T123N variant is controversial. Three variants of USH2A (NM_007123), R5143C, C4870F, and G805A with unknown pathogenic potential were identified using TES (see Table S3, Supplemental Content, which illustrates variants or mutations of Usher syndrome type 2A (USH2A) and Ankyrin 1 (ANK1) identified in SH 94-208). However, this subject showed no retinal abnormalities and only manifested severe SNHL with a mean hearing threshold of 75 dB HL, which was not compatible with type II Usher syndrome. Therefore, these variants of USH2A were excluded as causative deafness mutations. SH94-208 also showed the G1748S variant of ANK1 (NM_000037). Structural variations such as large genomic deletions involving ANK1 at chromosome 8p11.2p12 can lead to contiguous syndrome, with SNHL as one of the symptoms.32 However, the G1748S variant of ANK1 was a point mutation, not a structural variation (see Table S4, Supplemental Content, which illustrates depth of coverage of TES). Moreover, SH94-208 showed no phenotypic markers for syndromic SNHL, such as craniometaphyseal dysplasia. Thus, this ANK1 variant was not likely to be a causative mutation. The remaining three subjects showed no other convincingly pathogenic mutations other than the detected GJB2 mutation (Table 2).
Detection Rates of Single Heterozygous GJB2 Mutation in our SNHL Cohort and a Normal Hearing Control Cohort From the Literature
Based on previously published Korean GJB2 normal carrier rates, we compared the rates of GJB2 single heterozygotes in the hearing-impaired cohort with that of normal hearing controls.4,26 Total rates (5.63% [9/160]) of GJB2 single heterozygotes (N = 9 from group I + II + III excluding a GJB2 variant with controversial pathogenicity [SH94-208]) among all GJB2-sequenced hearing-impaired subjects (N = 160) was significantly higher than that (2.58%) of normal controls (P = 0.03). When we excluded two cases (Group II) with the GJB2 mutation contributing to SNHL possibly through digenic inheritance, we still observed a slightly higher rate of GJB2 single heterozygotes (Group I + III) than in the normal controls (4.38% vs. 2.58%, P = 0.20), although the difference was not statistically significant.
Analysis of Missing Regions of TES Data From GJB2 Single Heterozygotes with Unknown Molecular Etiology
The average depth of coverage for these six subjects, including group III and SH60-136, were 225.73 (see Table S4, Supplemental Content, which illustrates depth of coverage of TES), with more than 97.09% of the target bases covered >20×. Among the 1737 regions that were targeted in this study, 2.17% were covered <20×, including several regions of OTOF (NM_001287489), STRC (NM_153700), and OTOA (NM_144672) (see Table S2, Supplemental Content, which illustrates regions showing significantly low depth of coverage in TES: OTOF, STRC, and OTOA). We confirmed that the 48th exon of OTOF (NM_001287489) showed very high GC content and that STRC and OTOA had pseudogenes (STRCP1 and LOC6537686, respectively), which generated missing regions of the TES data. Sanger sequencing of these exons with significant poor coverage by TES revealed two compound heterozygous STRC mutations in SB175-334 (Table 1). However, no pathogenic variants was detected in the remaining five subjects (SH 153-332, SB201-389, SH94-208, SH60-136, and SH118-242) (data not shown).
DISCUSSION
In the first phase analysis of TES data, we were able to exclude DFNB1 from 4 subjects (SH166-367, SH170-377, SH60-136, and SH175-389) and possibly from 1 subject (SH107-225) among a total of 10 single heterozygotes of GJB2 in a Korean SNHL population. The second phase analysis of TES data which double-checked the exons with poor coverage in OTOA, STRC, and OTOF by Sanger sequencing enable us to reveal DFNB16 from one subject (SB175-334). Detection of the GJB2 mutation in four subjects (SH166-367, SH170-377, SB175-334, and SH60-136) (Group I) was found to be incidental, independently of the phenotype. This is expected based on the similarity of the incidence rates of detection of c.235delC of GJB2 between the one (0.63% (1 [SH166-367]/160 [our total SNHL cohort]) calculated from group I and reported figures from normal hearing Korean subjects (0.44%–1.25%) (Tables 1 and 2).4,27
Our first-line screening of known large genomic deletions involving GJB6 or upstream regions of GJB2 yielded no convincing structural variations in our population, which was in sharp contrast with previous reports of a Spanish population.33 In the Spanish population, 66% of the affected subjects with a monoallelic GJB2 mutation carried the del (GJB6-D13S1830) in GJB6, which was also frequently observed in subjects from France and Israel.33 A completely different molecular etiology was clearly observed or strongly suggested in four of the five subjects in groups I and II, except SH60-136 in the present study, indicating that TES covered the known prevalent deafness genes, including gap junction genes and genes known to cause syndromic deafness such as Usher syndrome and Waardenburg syndrome for the GJB2 single heterozygotes in Korean SNHL subjects.
For the most common GJB2 variant in Koreans, c.235delC, suspicion of other molecular etiologies should be examined, particularly when detected as a single heterozygous state from a child with no residual hearing in Koreans, such as for SH166-367. c.235delC was recently reported to manifest a dynamic range of SNHL and a slightly milder audiologic phenotype compared with other GJB2 variants in Koreans.34 Detection of mutations in MYO15A and TMC1 in group I are relatively common in East Asian populations, including Koreans,2,35–38 indicating that application of panel sequencing covering the genes prioritized based on the ethnicity-specific prevalence would be effective for identifying GJB2 single heterozygotes with severe to profound SNHL in Koreans.
For the family SH60 with a most likely genetic etiology but without a clear result after TES, whole exome sequencing can be used for definitive molecular diagnosis. This family SH60 segregates prelingual or perilingual severe to profound SNHL, likely in an autosomal dominant fashion, although prelingual SNHL of SH60-136 was caused by autosomal recessive mutations in other deafness genes (Figure 3). Further segregation analyses of the two variants (p.R143W and p.D771N) among the six family members of SH60 as well as clinical evaluations including audiograms excluded both p.R143W of GJB2 and p.D771N of WFS1 as a molecular etiology of SH60-136. Specifically, SH60-138 (grandmother of SH60-136) complained of perilingual deafness but did not carry p.R143W of GJB2, eliminating the contribution of this allele to prelingual profound SNHL if it segregated dominantly from SH60-138 to SH60-136 (Figure 3). Alternatively, SNHL of SH60-136 may result from autosomal recessive mutations in the same gene with SH60-142 (mother of SH60-136) if SNHL of SH60-142 was because of autosomal recessive mutations in a certain deafness gene other than GJB2 and if the father (SH60-141) is a carrier of a mutation in the gene. In any case, p.R143W in GJB2 does not contribute to the SNHL of SH60-136 (Figure 3). Therefore, a completely different deafness gene yet to be identified would account for the SNHL of SH60-136, warranting whole exome sequencing in this subject.
The p.V193E variant in GJB2 occurring in complex heterozygosity with a pathogenic GJB3 variant, p.A194T from SH175-389, suggests a possible digenic etiology of SNHL involving two different gap junction proteins, Cx26 and Cx31. Large deletions in GJB6 (del [GJB6-D13S1830] and del [GJB6-D13S1854]) are frequently detected in a trans configuration with a monoallelic GJB2 mutation in certain populations.33,35,39 Based on these findings, it was previously hypothesized that variations in GJB2 and GJB6 in trans can cause SNHL through digenic inheritance.33,40 However, subsequent studies revealed that GJB6 deletions result in an allele-specific lack of GJB2 mRNA expression, contributing to SNHL in a manner not resulting from digenic inheritance.40 Nevertheless, a digenic etiology involving mutations in several gap junction genes has been proposed.18,41 Structurally, the gap junction is composed of two connexons (Cxs), which are formed by the oligomerization of six Cx subunits. Several Cx genes, including Cx26, Cx29, Cx30, Cx31, Cx32, Cx30.3, and Cx43, can induce SNHL.42 These Cx gene families are known to be able to form heteromeric gap junction assemblies.43 In this context, digenic inheritance of SNHL involving GJB2 (Cx26) and GJB3 (Cx31) has been strongly supported by functional studies conducted by Liu et al (2009), which demonstrated a direct physical interaction between Cx26 and Cx31, the presence of heteromeric Cx26/Cx31 connexons, and finally co-assembly of two transfected proteins in the same junction plaques in vitro.31 In their study, two different GJB3 mutations (p.N166S and p.A194T) were identified in three unrelated families among 108 Chinese families with a single pathogenic GJB2 mutation.31 This strongly corroborated a possible digenic etiology of SNHL involving GJB2 and GJB3 in SH175-389. Thus, we excluded DFNB1 as a molecular etiology of SNHL from SH175-389.
Although no direct physical interaction occurs in the heterotypic Cx formation, two different genes can functionally interact with each other, which may result in functional deficit if these interactions are disrupted. MITF is an important gene in the development and regulation of melanocytes and is expressed in melanoblast-derived intermediate cells of the stria vascularis.25 Interestingly, MITF also regulates potassium ion circulation of endolymphatics in the inner ear.44,45 Although the signaling networks between GJB2 and MITF remain unclear, their common final pathway in regulating potassium ion circulation in the inner ear can be significantly disrupted by the digenic effect of MITF and GJB2 mutations. A subject with Waardenburg syndrome type II (WS2) in a large Chinese population had both MITF and GJB2 mutations in a compound heterozygous state.17 The profound SNHL in the subject may have been caused by the digenic effect of GJB2 and MITF mutations, although the WS2 phenotype was caused by the MITF mutation.17 In our studied family, SH107-225 with profound SNHL carried c.235delC in GJB2 and a de novo variant, p.R341C in MITF. DFNB1 as a molecular etiology was excluded from this subject, while digenic inheritance of SNHL can be proposed for this subject because the pathogenic potential of p.R341C was strongly supported by significant conservation of the p.R341 residue among various species and by the absence of this variant among the 666 control chromosomes from normal hearing control subjects. Based on the varying degrees of audiologic phenotypes of MITF-related WS2, including single side deafness,37 bilateral profound SNHL of SH107-225 may have resulted from the additive effect of single heterozygous c.235delC of GJB2, and the MITF mutation through digenic inheritance. Thus, panel sequencing should be conducted to examine syndromic deafness genes as well as gap junction genes for definitive genetic diagnosis of GJB2 single heterozygotes in seemingly nonsyndromic SNHL in Koreans. However, final documentation of digenic inheritance of deafness in our two subjects (SH175-389 and SH107-225) warrants a rigorous functional study.
Several exons in OTOF, STRC, and OTOA were not fully covered (see Table S2, Supplemental Content, which illustrates regions showing significantly low depth of coverage in TES: OTOF, STRC, and OTOA) by our TES data. We tried to minimize the possibility that pathogenic mutations reside in these regions by performing Sanger sequencing of all exons showing poor coverage (<10 × coverage). Considering that STRC-related SNHL (DFNB16) is generally presented as mild to moderate SNHL,30 two moderate SNHL subjects (SB175-334 and SB201-389) in our cohort were initially hypothesized to have either occult structural variations or point mutations in the poorly covered regions of STRC. In accordance with our assumption, one moderate SNHL subject (SB175-334) with a single heterozygous p.V37I turned out to carry two mutant alleles of STRC. However, it is still possible that the remaining moderate SNHL subject (SB201-389) has pathogenic structural variations in the STRC gene. Copy number variations of the STRC gene were reported to be common among deafness genes.46 Designing probes with different or multiple tiling on the probes may improve the capture efficiency; arrayCGH may be another option if a subject is strongly suspected to have STRC-related structural variations.
The contribution of the detected monoallelic GJB2 mutations in four subjects in group III is unclear in the present study. The rate of GJB2 single heterozygotes in our hearing-impaired cohort was significantly higher than that in normal hearing controls, suggesting that a substantial portion of the detected monoallelic GJB2 mutations contributes to SNHL. Moreover, the detected GJB2 mutation from five subjects in group I were not found to contribute to their SNHL, and thus the slightly higher rate of GJB2 single heterozygotes in groups I + III compared with that of controls (4.38% vs 2.58%) suggests that at least some of the monoallelic GJB2 mutations in group III contribute to SNHL.
Genetic approaches such as targeted resequencing of whole noncoding regions of GJB2 in group III may reveal occult mutations in these regions. RNA-seq or microarray mainly focusing on gap junction genes using patient-derived lymphoblastoid cell lines may detect low or absent expression of gap junction genes either because of novel occult large deletions in these genes or disruption of regulatory elements by occult mutations in some GJB2 single heterozygotes in group III.
At least 40% of GJB2 single heterozygotes in Korean SNHL subjects appear to be nonDFNB1. Digenic etiology involving the GJB2 mutation may increase the proportion. TES to cover prevalent autosomal recessive deafness genes should be employed to elucidate these non-DFNB1 GJB2 single heterozygotes in Koreans. Similarly, a recent study also showed that ∼33.3% (4/12) of the 12 Han Chinese subjects with the monoallelic GJB2 mutations were identified as the non-DFNB1; 2 patients with MYO15A mutations, 1 patient with Potassium voltage-gated channel subfamily KQT member 4 (KCNQ4) mutation, and 1 patient with SLC26A4 mutations (under minor revision from “Medicine” by personal communications). Therefore, our results can be probably extrapolated to at least whole East Asian populations and our diagnostic approaches for the GJB2 single heterozygotes can be applied to broader populations beyond Koreans. Because there have been only a small number of study subjects of GJB2 single heterozygotes thus far, further studies with various ethnic groups and more number of subjects are warranted to generalize our diagnostic strategies. Our stepwise molecular genetic approaches presented in the present study serve as an example for understanding single heterozygotes of other autosomal recessive genes as well as of GJB2.
Supplementary Material
Acknowledgments
We thank all the subjects of this study for their participation in this research.
Footnotes
Abbreviations: ANK1 = ankyrin 1, Cx = connexon, DFNB = nonsyndromic hearing loss and deafness, GJA1 = gap junction protein alpha 1, GJB1 = gap junction protein beta 1, GJB2 = gap junction protein beta 2, GJB3 = gap junction protein beta 3, GJB4 = gap junction protein beta 4, GJB6 = gap junction protein beta 6, GJC3 = gap junction protein gamma 3, KCNQ4 = potassium voltage-gated channel subfamily KQT member 4, MITF = microphthalmia-associated transcription factor, MYO15A = myosin XVA, OTOA = otoancorin, OTOF = otoferlin, SLC26A4 = solute carrier family 26, member 4, SNHL = sensorineural hearing loss, STRC = stereocilin, TES = targeted exome sequencing, TMC1 = transmembrane channel-like 1, USH2A = Usher syndrome type 2A, WFS1 = wolfram syndrome 1, WS2 = Waardenburg syndrome type2.
Funding: This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C1867 to B.Y. Choi).
SYK and ARK contributed equally to this work.
The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Kim NK, Kim AR, Park KT, et al. Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet Med 2015; 17:901–911. [DOI] [PubMed] [Google Scholar]
- 2.Park JH, Kim NK, Kim AR, et al. Exploration of molecular genetic etiology for Korean cochlear implantees with severe to profound hearing loss and its implication. Orphanet J Rare Dis 2014; 9:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen MB, Willems PJ. Non-syndromic, autosomal-recessive deafness. Clin Genet 2006; 69:371–392. [DOI] [PubMed] [Google Scholar]
- 4.Han SH, Park HJ, Kang EJ, et al. Carrier frequency of GJB2 (connexin-26) mutations causing inherited deafness in the Korean population. Human Genet 2008; 53:1022–1028. [DOI] [PubMed] [Google Scholar]
- 5.Shin JW, Lee SC, Lee HK, et al. Genetic Screening of GJB2 and SLC26A4 in Korean cochlear implantees: experience of soree ear clinic. Clin Exp Otorhinol 2012; 5 Suppl 1:S10–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SY, Park G, Han KH, et al. Prevalence of p.V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PloS One 2013; 8:e61592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagong B, Baek JI, Oh SK, et al. A rapid method for simultaneous screening of multi-gene mutations associated with hearing loss in the Korean population. PloS One 2013; 8:e57237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hutchin T, Coy NN, Conlon H, et al. Assessment of the genetic causes of recessive childhood non-syndromic deafness in the UK—implications for genetic testing. Clin Genet 2005; 68:506–512. [DOI] [PubMed] [Google Scholar]
- 9.Dai P, Yu F, Han B, et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med 2009; 7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan Y, Yu F, Wang G, et al. Prevalence of the GJB2 IVS1+1G >A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. J Transl Med 2010; 8:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Danilenko N, Merkulava E, Siniauskaya M, et al. Spectrum of genetic changes in patients with non-syndromic hearing impairment and extremely high carrier frequency of 35delG GJB2 mutation in Belarus. PloS One 2012; 7:e36354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao J, Lu Y, Wei Q, et al. A systematic review and meta-analysis of 235delC mutation of GJB2 gene. J Transl Med 2012; 10:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.del Castillo FJ, Rodriguez-Ballesteros M, Alvarez A, et al. A novel deletion involving the connexin-30 gene, del (GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet 2005; 42:588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerer I, Sagi M, Ben-Neriah Z, et al. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: a novel founder mutation in Ashkenazi Jews. Hum Mut 2001; 18:460. [DOI] [PubMed] [Google Scholar]
- 15.Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med 2002; 4:258–274. [DOI] [PubMed] [Google Scholar]
- 16.Tang HY, Fang P, Ward PA, et al. DNA sequence analysis of GJB2, encoding connexin 26: observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet Part A 2006; 140:2401–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan X, Zhang T, Wang Z, et al. A novel mutation in the MITF may be digenic with GJB2 mutations in a large Chinese family of Waardenburg syndrome type II. J Genet Genomics 2011; 38:585–591. [DOI] [PubMed] [Google Scholar]
- 18.Kooshavar D, Tabatabaiefar MA, Farrokhi E, et al. Digenic inheritance in autosomal recessive non-syndromic hearing loss cases carrying GJB2 heterozygote mutations: assessment of GJB4, GJA1, and GJC3. Int J Pediatr Otorhinolaryngol 2013; 77:189–193. [DOI] [PubMed] [Google Scholar]
- 19.Hughes AE, Newton VE, Liu XZ, et al. A gene for Waardenburg syndrome type 2 maps close to the human homologue of the microphthalmia gene at chromosome 3p12-p14.1. Nat Genet 1994; 7:509–512. [DOI] [PubMed] [Google Scholar]
- 20.Gandia M, Del Castillo FJ, Rodriguez-Alvarez FJ, et al. A novel splice-site mutation in the GJB2 gene causing mild postlingual hearing impairment. PloS one 2013; 8:e73566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mani RS, Ganapathy A, Jalvi R, et al. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur J Ham Genet 2009; 17:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sirmaci A, Akcayoz-Duman D, Tekin M. The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J Genet 2006; 85:213–216. [DOI] [PubMed] [Google Scholar]
- 23.Matos TD, Caria H, Simoes-Teixeira H, et al. A novel hearing-loss-related mutation occurring in the GJB2 basal promoter. J Med Genet 2007; 44:721–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi BY, Park G, Gim J, et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PloS One 2013; 8:e68692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locher H, de Groot JC, van Iperen L, et al. Development of the stria vascularis and potassium regulation in the human fetal cochlea: insights into hereditary sensorineural hearing loss. Dev Neurobiol 2015; 75:1219–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SY, Lee BY, Lim JH, et al. Determination of the carrier frequencies of selected GJB2 mutations in the Korean population. Int J Audiol 2011; 50:694–698. [DOI] [PubMed] [Google Scholar]
- 27.Ben Said M, Hmani-Aifa M, Amar I, et al. High frequency of the p.R34X mutation in the TMC1 gene associated with nonsyndromic hearing loss is due to founder effects. Genet Test Mol Biomarkers 2010; 14:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Searle C, Mavrogiannis LA, Bennett CP, et al. The common TMC1 mutation c.100C>T (p.Arg34X) is not a significant cause of deafness in British Asians. Genet Test Mol Biomarkers 2012; 16:453–455. [DOI] [PubMed] [Google Scholar]
- 29.Bashir R, Fatima A, Naz S. Prioritized sequencing of the second exon of MYO15A reveals a new mutation segregating in a Pakistani family with moderate to severe hearing loss. Eur J Med Genet 2012; 55:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vona B, Hofrichter MA, Neuner C, et al. DFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics. Clin Genet 2015; 87:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu XZ, Yuan Y, Yan D, et al. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum Genet 2009; 125:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cau M, Congiu R, Origa R, et al. New case of contiguous gene syndrome at chromosome 8p11.2p12. Am J Med Genet: Part A 2005; 136:221–222. [DOI] [PubMed] [Google Scholar]
- 33.del Castillo I, Villamar M, Moreno-Pelayo MA, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 2002; 346:243–249. [DOI] [PubMed] [Google Scholar]
- 34.Kim SY, Kim AR, Han KH, et al. Residual hearing in DFNB1 deafness and its clinical implication in a Korean population. PloS One 2015; 10:e0125416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bolz H, Schade G, Ehmer S, et al. Phenotypic variability of non-syndromic hearing loss in patients heterozygous for both c.35delG of GJB2 and the 342-kb deletion involving GJB6. Hear Res 2004; 188:42–46. [DOI] [PubMed] [Google Scholar]
- 36.Kalay E, Karaguzel A, Caylan R, et al. Four novel TMC1 (DFNB7/DFNB11) mutations in Turkish patients with congenital autosomal recessive nonsyndromic hearing loss. Hum Mut 2005; 26:591. [DOI] [PubMed] [Google Scholar]
- 37.Yang S, Dai P, Liu X, et al. Genetic and phenotypic heterogeneity in Chinese patients with Waardenburg syndrome type II. PloS One 2013; 8:e77149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei Q, Zhu H, Qian X, et al. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J Transl Med 2014; 12:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esteves MC, de Lima Isaac M, Francisco AM, et al. Analysis of the presence of the GJB6 mutations in patients heterozygous for GJB2 mutation in Brazil. Eur Arch Otorhinolaryngol 2014; 271:695–699. [DOI] [PubMed] [Google Scholar]
- 40.Pallares-Ruiz N, Blanchet P, Mondain M, et al. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet 2002; 10:72–76. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Paris J, Tamayo ML, Gelvez N, et al. Allele-specific impairment of GJB2 expression by GJB6 deletion del (GJB6-D13S1854). PloS One 2011; 6:e21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum Mut 2000; 16:190–202. [DOI] [PubMed] [Google Scholar]
- 43.Ahmad S, Chen S, Sun J, et al. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem Biophys Res Commun 2003; 307:362–368. [DOI] [PubMed] [Google Scholar]
- 44.Price ER, Fisher DE. Sensorineural deafness and pigmentation genes: melanocytes and the Mitf transcriptional network. Neuron 2001; 30:15–18. [DOI] [PubMed] [Google Scholar]
- 45.Tachibana M. Cochlear melanocytes and MITF signaling. J Investig Dermatol Symp Pro 2001; 6:95–98. [DOI] [PubMed] [Google Scholar]
- 46.Shearer AE, Kolbe DL, Azaiez H, et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med 2014; 6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.