

Abstract
Iron is required for most forms of organisms, and it is the most essential element for the functions of many iron-containing proteins involved in oxygen transport, cellular respiration, DNA replication, and so on. Disorders of iron metabolism are associated with diverse diseases, including anemias (e.g., iron-deficiency anemia and anemia of chronic diseases) and iron overload diseases, such as hereditary hemochromatosis and β-thalassemia. Hepcidin (encoded by Hamp gene) is a peptide hormone synthesized by hepatocytes, and it plays an important role in regulating the systematic iron homeostasis. As the systemic iron regulator, hepcidin, not only controls dietary iron absorption and iron egress out of iron storage cells, but also induces iron redistribution in various organs. Deregulated hepcidin is often seen in a variety of iron-related diseases including anemias and iron overload disorders. In the case of iron overload disorders (e.g., hereditary hemochromatosis and β-thalassemia), hepatic hepcidin concentration is significantly reduced.
Since hepcidin deregulation is responsible for iron disorder-associated diseases, the purpose of this review is to summarize the recent findings on therapeutics targeting hepcidin.
Continuous efforts have been made to search for hepcidin mimics and chemical compounds that could be used to increase hepcidin level. Here, a literature search was conducted in PubMed, and research papers relevant to hepcidin regulation or hepcidin-centered therapeutic work were reviewed. On the basis of literature search, we recapitulated recent findings on therapeutic studies targeting hepcidin, including agonists and antagonists to modulate hepcidin expression or its downstream signaling. We also discussed the molecular mechanisms by which hepcidin level and iron metabolism are modulated.
Elevating hepcidin concentration is an optimal strategy to ameliorate iron overload diseases, and also to relieve β-thalassemia phenotypes by improving ineffective erythropoiesis. Relative to the current conventional therapies, such as phlebotomy and blood transfusion, therapeutics targeting hepcidin would open a new avenue for treatment of iron-related diseases.
INTRODUCTION
Iron, as a necessary element, plays an important role in several physiological processes including oxygen carrier, electron transfer in mitochondrial, DNA replication, DNA repair, cell signaling, and free radical production.1 Iron balance is necessary for normal physiology; however, iron disorder is associated with many types of diseases including hereditary hemochromatosis (HH), β-thalassemia, anemia of inflammation, and iron-refractory iron deficiency anemia (IRIDA). In the real world, more than 1 billion people are suffering from iron deficiency.2 Thalassemia major, a representative iron overload disease, is still very popular in the world. There are estimated 56,000 thalassemia major cases annually, and 30,000 of them require regular transfusion to survive.3 These huge numbers of patients present an urgent need to improve their survival and life quality. Nowadays, iron chelation, phlebotomy, splenectomy, bone marrow transplantation, and iron administration are widely accepted therapies; however, serious toxic and side effects (such as secondary iron overload and anemia) are associated with these therapies, which are not satisfactory to all patients.4,5
Previous pathology studies revealed that iron disorder is due to the dysregulation on hepcidin–ferroportin (FPN) axis. Thus, correcting hepcidin–FPN axis would be potential therapeutic strategy for iron disorders. Hepcidin (encoded by Hamp gene) is a 25-amino acid peptide hormone and synthesized in hepatocytes (Figure 1).6 It binds to FPN to promote the latter's degradation, and thus controls iron release from spleen and hepatocytes, and also dietary iron uptake from enterocytes.7,8 Since hepcidin deregulation is closely associated with iron overload or deficiency, fine-tuning Hamp expression would be an efficient strategy to ameliorate iron disorder diseases. In this review, we summarized the iron disorders due to deregulated hepcidin and the development of hepcidin agonists and antagonists for hepcidin regulation.
FIGURE 1.
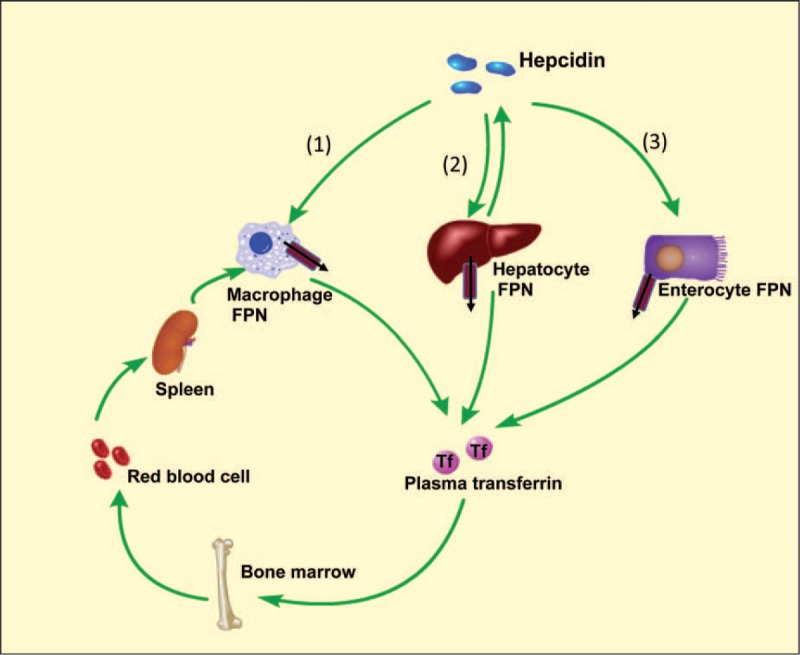
Hepcidin modulates the systemic iron levels. Hepcidin–FPN axis is the key regulator of systemic iron. FPN, the only known iron exporter, is fine-tuned by hepcidin. Hepcidin is synthesized by hepatocytes that promote the degradation of FPN. The regulation of hepcidin is via three causes. (1), Blocking iron release from macrophages. Spleen is the main iron-recycling organ where aged red blood cells are engulfed by macrophages. Fpn1 deficiency induces iron accumulation in spleen. (2), Reducing iron release from hepatocytes. Liver is the main iron storage organ, and FPN degradation would decrease iron transfer to plasma, leading to iron overload. (3), Inhibiting iron absorption by enterocytes. Enterocyte is the main dietary iron uptake site. The degradation of FPN in enterocytes prevents the iron compensation for its loss, including shedding of epithelial cells, hair, sweat, and menstrual blood. FPN = ferroportin.
METHOD
In this systemic review, we performed literature search in Pubmed (http://www.ncbi.nlm.nih.gov/pubmed/). The key words used in searching are as follows: hepcidin, iron overload, hereditary hemochromatosis, anemia of inflammation, and hepcidin regulation. The criterion for exclusions is that the studies are irrelevant to hepcidin regulation or hepcidin-centered therapeutic work. Since no animals or humans were used in the current review paper, ethics statement does not apply here.
MOLECULAR BASIS OF SYSTEMIC IRON HOMEOSTASIS
Hepcidin is a hormone secreted by hepatocytes which plays a crucial role in regulating iron homeostasis.6Hamp deficiency mice (Hamp−/− mice) exhibit severe iron overload; in contrast, Hamp overexpression reversely results in iron deficiency.9 Studies demonstrated that the treatment with hepcidin or its analogs caused dose-dependent hypoferremia.10 In fact, hepcidin regulates iron by binding to its receptor, FPN, and inducing FPN internalization and degradation.11,12 FPN is a multimembrane protein, and it is highly expressed in duodenal enterocytes, liver Kupffer cells, periportal hepatocytes, splenic macrophages, and placental syncytiotrophoblasts.8 FPN is the only known iron exporter in mammals, and its deficiency causes loss of iron absorption from duodenum and reduction of iron egress from liver and spleen macrophages, as evidenced in FPN null mice (Fpn1−/− mice).8,13,14 Thus far, the hepcidin–FPN axis has been considered as the central player in regulating iron homeostasis. We here reviewed the main upstream-signaling pathways responsible for hepcidin regulation, including iron concentration, inflammation, erythropoiesis, and so on (Figure 2).
FIGURE 2.
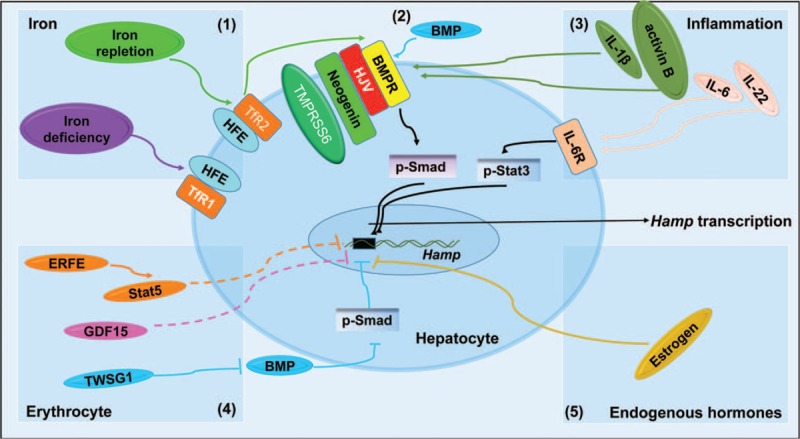
The mechanisms responsible for Hamp transcription. Hamp transcription is regulated by diverse signaling pathways, including iron concentration, inflammation, erythropoiesis, and sex hormone. (1), Iron levels contribute to Hamp transcription through the Smad signaling. In an iron-replete condition, iron binds to TfR2, and then forms a complex with HFE, HJV, and BMPRs to promote Smad1/5/8 activation. In contrast, iron binds to TfR1 and fails to stimulate Hamp transcription in iron deficiency. (2), BMPs are involved in hepcidin regulation. BMPs bind to BMPRs, and thus form BMPs, BMPRs, and HJV complex to activate Smad phosphorylation and promote Hamp transcription. (3), The preinflammatory factors (including IL-6, IL-22, and IL-1β) and activin B incur Hamp up-regulation. IL-6 and IL-22 can bind to its receptor to increase hepcidin expression via Stat3, but IL-1β and activin B (a member of the TGF-β protein superfamily) increase Hamp transcription through the BMP-Smad signaling. (4), Cytokine factors produced by erythroblasts are also implicated in hepcidin regulation. GDF15, TWSG1, and ERFE are recognized to be the hepcidin regulators. GDF15 could modulate hepcidin expression through BMP inhibition, whereas the detailed mechanisms underlying the action of TWSG1 and ERFE in Hamp repression remain unclear. (5) Endogenous hormones (such as estrogen) negatively regulate Hamp transcription by binding to the ERE of Hamp promoter. BMP = bone morphogenetic protein, BMPR = bone morphogenetic protein receptor, ERFE = erythroferrone, GDF = growth differentiation factor, HFE = hemochromatosis protein, HJV = hemojuvelin, IL = interleukin.
Hepcidin is Regulated by Iron Concentration
Iron concentration can regulate Hamp expression in the liver. Under an iron deficiency condition, holo-transferrin will first bind to transferrin receptor 1 (TfR1), and then interact with hemochromatosis protein (HFE). However, the molecular events downstream of this complex are not thoroughly understood. Holo-transferrin will shuttle to transferrin receptor 2 (TfR2) under iron repletion condition, and the holo-transferrin, TfR2, and HFE complex interacts with hemojuvelin (HJV). HJV is a glycosylphosphatidylinositol-linked membrane protein; it promotes Hamp transcription through Smad1/5/8 phosphorylation. As demonstrated in previous studies, Tfr2, Hfe, or Hjv deficiency mice (Tfr2−/− mice, Hfe−/− mice, or Hjv−/− mice) displayed iron overload.15,16 Wu et al's study further demonstrated that Hjv−/− mice exhibited more severe iron overload, compared with Hfe−/− mice, but similar to Hfe−/−Hjv−/− mice.17,18 Wu et al's study also uncovered that mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase does not play a prominent role in this process.17 Overall, these results indicate that HFE is involved in hepcidin regulation in an HJV-dependent manner.
Hepcidin is Regulated by Bone Morphogenetic Protein Signaling
There are nearly 20 bone morphogenetic proteins (BMPs) expressed in mammals, and among these, BMP2, BMP4, BMP5, BMP6, BMP7, and BMP9 can induce Hamp expression.19,20 Studies demonstrated that BMP6 plays a key role in Hamp induction.19,21 It binds to BMP receptor (BMPR) to activate Smad1/5/8 phosphorylation, and the latter together with Smad4 translocates to nucleus and binds to Hamp promoter to induce Hamp transcription.22 This signaling pathway requires HJV, a BMPs coreceptor. Since Hjv−/− mice have severe low hepcidin concentration, iron overload and a low level of p-Smad1/5/8 demonstrated that HJV participates in BMP-stimulated Hamp expression.23,24 To verify the role of HJV in BMP-Smad signaling pathway, HJV expression in hepatocytes of Hjv−/− mice could restore the level of hepcidin, and this finding confirmed the essential role of HJV in Hamp regulation through the Smad signaling pathway.25
In fact, HJV is also modulated by the upper regulators including neogenin and transmembrane protease serine 6 (TMPRSS6, also known as matriptase-2). Neogenin is ubiquitously expressed on cell membrane, and its mutation caused a low level of p-Smad1/5/8 in mice.26 HJV can bind onto the membrane proximal region of neogenin, and this interaction is necessary for BMP4-promoted Hamp transcription.27,28 As the coreceptor of HJV, it is also involved in HJV cleavage; however, this process is more predominantly regulated by TMPRSS6.29 TMPRSS6 is a type of plasma membrane serine protease and it has been recently recognized to have a novel role in regulating Hamp expression and iron homeostasis in both human and mice.30–32 TMPRSS6-mediated hepcidin down-regulation is closely dependent on interaction with HJV that conducts HJV cleavage, and this process needs neogenin to form a trimer complex with HJV and TMPRSS6.33 HJV is also necessary to keep the stability of TMPRSS6, as being demonstrated on liver Tmprss6 mutant mice.34 TMPRSS6 is regulated by hypoxia, BMP6, iron, and inflammation.35–38 Meynard et al35 demonstrated that BMP6 and chronic iron treatment induced the induction of not only Hamp but also Tmprss6. Further research by Zhao et al37 revealed that iron participated in the regulation of TMPRSS6 through protein degradation rather than mRNA regulation.
Meanwhile, activin B, as a member of the TGF-β protein superfamily, induces hepatic hepcidin synthesis through the BMP-Smad signaling.39,40 Additionally, under inflammation conditions, interleukin (IL)-1β was also involved in hepcidin regulation via Smad signaling.41 Recently, homocysteine was identified to play an important role in regulating Hamp expression.42 Homocysteine could up-regulate HAMP via BMP-Smad signaling pathway, and the HAMP regulation was compromised upon siRNA-mediated gene knockdown treatment against BMP6 and its receptors in Hep G2 cells.42
Stat3 Signaling Pathway
Stat3 signaling pathway is another hepcidin regulator, mainly through inflammation. Early study found that treatment of hepatocytes with lipopolysaccharide (LPS) resulted in high hepcidin levels.9 It is contributed to IL-6 and IL-22 productions, but not IL-1β and tumor necrosis factor-α (TNF-α).43,44 Further research confirmed that Hamp promoter contains Stat3-binding site, and siRNA-mediated Stat3 knockdown significantly decreased hepcidin transcription.44 Stat3 signaling pathway is necessary not only under inflammation conditions, but also baseline hepcidin expression. IL-6-mediated hepcidin induction is dependent on IL-6-Stat3 signaling pathway. IL-6 binds to IL-6 receptor (IL-6R), thereafter to glycoprotein 130 (gp130), to activate Stat3 phosphorylation, which promotes Stat3 translocation to nucleus and provokes Hamp transcription.45
Erythropoiesis-related Regulators Involved in Hepcidin Regulation
Recent studies demonstrated that the erythroid-derived proteins including growth differentiation factor 15 (GDF15), twisted gastrulation BMP signaling modulator (TWSG1), and erythroferrone (ERFE) (produced by erythroblasts) are involved in Hamp suppression.46 GDF15 is a member of transforming growth factor-beta superfamily, and it is secreted by late-stage erythroblasts, revealing a role in Hamp suppression in β-thalassemia patients.47 GDF15 is correlated with soluble transferrin receptor, erythropoietin, and ferritin.47 Sera from these patients can also inhibit hepatic Hamp expression ex vivo,47 and another research group confirmed this result in humans as well.48 However, Fertrin et al49 demonstrated that the high concentration of GDF15 is not necessary for Hamp repression. TWSG1 is a bone marrow-binding protein, secreted by early erythroblasts, and it was considered to be the potential erythroid regulator of hepcidin.50 In thalassemic mice, its expression was up-regulated significantly in bone marrow, spleen, and liver.50 Following studies revealed that TWSG1-mediated Hamp reduction is through the Smad1/5/8 signaling.50 ERFE is another erythroid regulator produced by erythroid precursors that participate in hepcidin regulation.51 It belongs to the necrosis factor related family. ERFE was highly expressed to suppress Hamp in β-thalassemia intermedia mice in Epo-ERFE-Stat5 dependent manner.51 Further studies demonstrated that Erfe ablation in thalassemia mice fully restored hepcidin level, suggesting that ERFE contributes to iron overload in β-thalassemic mice.52 Nonetheless, the signaling downstream of Stat5 underlying Hamp regulation is still unclear.
Other Signaling Pathways That Regulate Hamp Expression
As a sex hormone, estrogen was found to down-regulate Hamp expression through an estrogen receptor element (ERE) in Hamp promoter.53,54 As the negative regulator of Hamp, 17β-estradiol (E2) treatment decreased Hamp expression in HuH7 and Hep G2 cell lines, which could be blocked by ICI 182780, an antagonist of estrogen receptor.53 This negative regulation of E2 was confirmed in both wild-type and Hfe−/− mice.53 In support of this finding, our study revealed that the ERE-binding site of estrogen on Hamp promoter is responsible for the repression of Hamp transcription upon E2 treatment.54 Moreover, estrogen was also delineated to regulate hepcidin in GPR30-BMP6-dependent manner.55 A recent study suggested that progesterone receptor membrane component-1 (PGRMC1), a membrane-bound progesterone receptor, also contributed to hepcidin regulation through Src-family tyrosine kinases (SFKs).56 However, the signaling downstream of SFKs responsible for Hamp expression modulation is still elusive,56 and warrants further detailed investigation.
IRON DISORDERS DUE TO DEREGULATION OF HEPCIDIN
Several diseases exhibited abnormal HAMP and iron metabolism, such as smoking-caused low hepcidin level in pregnant women, non-alcoholic fatty liver disease, sideroblastic anemia and chronic renal failure.57–59 Meanwhile, β-thalassemia and HH are the most typical low HAMP diseases, and anemia of inflammation and IRIDA are the most representative high HAMP diseases.30,60–62
β-thalassemia
β-thalassemia is a disease with genetic mutation resulting in low β-globin production. It is characterized by ineffective erythropoiesis, iron overload, and low hepcidin, caused by a few pathologies. Firstly, insufficient globin causes anemia and hypoxia. Low hepcidin under anemia is associated with elevated erythropoietin in serum, whereas hypoxia can directly reduce hepatic Hamp expression.63–65 Secondly, low β-globin will promote excess α-globin aggregation that can bind to heme to form haemichromes, resulting in damages of cell membrane. Thirdly, α-globin degrades to globin polypeptides, free heme, porhhyrons, and iron, through which excess iron would provoke formation of reactive oxidative stress (ROS), thereafter leading to lipid peroxidation, impairments of membrane integrity, and activation of growth differentiation factor 11 (GDF11), a cytokine implicated in inhibition of erythroid differentiation through Smad2/3.66,67 Lastly, heat shock protein 70 (HSP70) is another cytosolic protein involved in ineffective erythropoiesis.68,69 It can translocate to nucleus, and then bind to GATA1 to protect the latter from cleavage by caspase 3.68 More association of α-globin with HSP70 would sequestrate HSP70. As a consequence, this process will arrest the endpoint maturation and enhance apoptosis of erythrocytes. Recent evidences suggest that 3 proteins (namely ERFE, TWSG1, and GDF15) are dictated to Hamp suppression in β-thalassemia mice.47,50,52 These deregulated signaling pathways together lead to ineffective erythropoiesis and iron overload in a vicious cycle. β-thalassemia patients with stroke showed higher HAMP levels and iron concentrations. These results indicate that β-thalassemia patients with higher HAMP levels and other complications should be paid attention to.70 Blood transfusion and iron chelating are the main therapeutic strategies for β-thalassemia. However, iron chelators have severe side effects, and blood transfusion would cause secondary iron overload.
Hereditary Hemochromatosis
HH carries genetic mutations in Hfe or other genes (including Hamp, Hjv Tfr2, Fpn1), characterized by normal erythropoiesis and iron accumulation in liver, heart, and endocrine glands.15 There are mainly 4 types of HH. Type 1 harbors genetic mutations in Hfe. Type 2A carries Hjv mutations, whereas type 2B has Hamp mutations. Type 3 is caused by Tfr2 mutations, and type 4 is due to Fpn1 mutation, known as FPN disease. HFE, TfR2, and HJV intertwinedly control Hamp transcription, and mutations of these genes are associated with low hepcidin and iron overload in organs. Excessive iron results in organ damage and dysfunction.15 In clinical practice, phlebotomy is the main therapeutic strategy; however, it may cause secondary low hepcidin and excess iron absorption and storage. In addition, this method is not suitable for patients with poor vascular access.
Anemia of Inflammation
Anemia of inflammation is a common disease that exhibits normocytic and normochromic anemia, sometimes with microcytic and hypochromic anemia. Anemia of inflammation is caused by inflammatory diseases, such as infections, rheumatology disorders, inflammatory bowel diseases, chronic kidney disease, and malignance.71,72 Anemia of inflammation manifests a canonical phenotype of hypoferremia, which is due to high hepcidin levels caused by chronic inflammation.73 Inflammation promotes hepcidin induction through the IL-6–Stat3 signaling and activin B-Smad1/5/8 signaling.39,40,74 In addition to IL-6, IL-1β and IL-22 could also stimulate Hamp expression in cultured cells and mice.41,75 However, the mechanisms underlying Hamp induction by IL-6 and IL-1β are different. IL-6 binds to IL-6R and thereafter to gp130, which activates phosphorylation of Stat3 that docks onto Hamp promoter to increase hepcidin expression.23 In contrast, the induction of hepcidin by IL-1β is due to the increase of BMP2 expression and activin B from hepatocytes.41 IL-22 is another positive regulator of hepcidin via the Stat3 signaling pathway, and a significant increase of hepatic hepcidin was observed after the administration of IL-22 receptor agonist.75,76 Based on these previous data, the contribution of ILs to hepcidin modulation under anemia of inflammation still warrants further investigation. Considering the characteristic of anemia of inflammation, blood transfusion, erythropoietin, and iron supplementation are the main strategies to ameliorate the diseases in clinical practice; however, these interventions may be accompanied by the risk of secondary iron overload.77
Iron-refractory Iron Deficiency Anemia
Iron-refractory iron deficiency anemia is an autosomal recessive disorder caused by mutations of Tmprss6, and a few mutation sites have been identified recently.31,78,79 TMPRSS6 is a negative regulator of hepcidin by cleaving HJV.80Tmprss6 deficiency is incapable to promote HJV shedding, leading to excessive Hamp transcription. The canonical phenotypes of IRIDA are hypochromic, microcytic anemia, low transferrin saturation, and serum ferritin; however, serum iron is overmuch relative to the degree of anemia. This disease is refractory to oral iron administration.
To better depict the role of hepcidin in directing systemic iron homeostasis, we summarized the iron-related diseases due to hepcidin deregulation in Table 1.72,81–83
TABLE 1.
Diseases Related to Disordered Hepcidin Levels

THERAPEUTICS TARGETING HEPCIDIN AND RELEVANT MOLECULES
Based on the molecular basis of iron overload diseases, different strategies are being developed to modulate hepcidin expression.
Hepcidin Agonist
Mimi-hepcidin and Derivatives
Hamp mutation would cause severe iron overload, and Hamp overexpression and hepcidin administration both diminished serum iron accumulation in mice, suggesting that increasing hepcidin level should be able to ameliorate iron overload in HH and β-thalassemia.84,85 However, due to a short half-life of natural hepcidin, hepcidin mimics or other drugs that could stimulate hepcidin level are in urgent need.10 On the basis of structural and functional analysis of hepcidin–FPN axis, the aromatic and hydrophobic residues were identified as the binding site between hepcidin and FPN.86 Thus, a series of mini-hepcidin were designed through computer modeling. Detailed molecular mechanisms are delineated in Figure 3. Serum iron was reduced after chronic administration of retro-inverso mini-hepcidin in Hamp−/− mice.86 Furthermore, an optimized mini-hepcidin named PR65 was chosen to examine the benefits and side effects in Hamp−/− mice. PR65 could effectively redistribute tissue iron after the administration for 2 weeks.87 The mini-hepcidin has free sulfhydryl group in position 7, which may cause dermatological side effects. Thus, a new type of mini-hepcidin analog, PR73, is synthesized through S-protected strategy.88 The analog PR73SH reveals a significant ability in FPN degradation both in vitro and in vivo.88 To limit unusual and expensive amino acid in mini-hepcidin, new cyclic mimics of hepcidin are designed, for example, mHS17. Unfortunately, this new cyclic compound is found to be less active in inducing FPN degradation in vitro, and it even increases serum iron in mice.89
FIGURE 3.
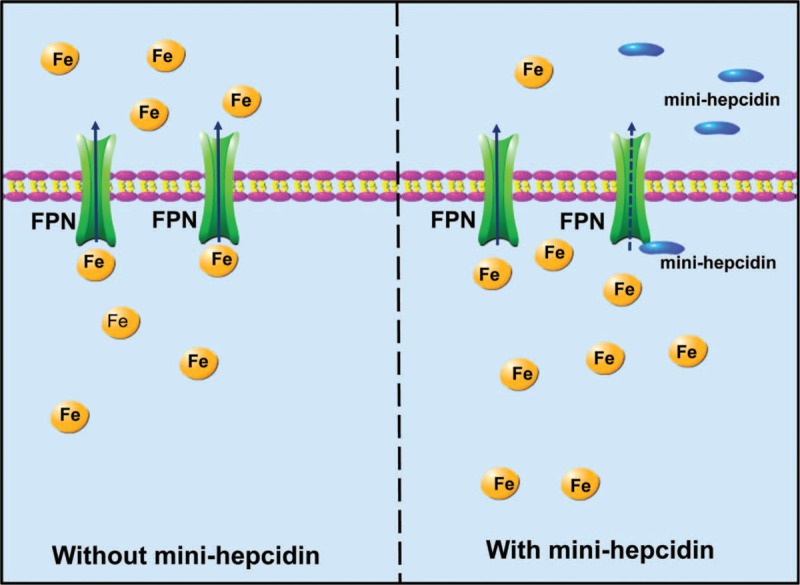
The mechanism underlying mini-hepcidin action. Mini-hepcidin is a synthesized polypeptide, analogs to natural hepcidin. It exhibits a high binding affinity to FPN, which could facilitate the latter's internalization and degradation, and thus diminish iron egress from macrophages and hepatocytes and iron uptake through enterocytes. FPN = ferroportin.
Small Chemical Compounds
Compared with peptide or RNA-based technology, chemical compounds are more cost-effective. Through high-throughput screening, several compounds including genistein and progesterone are identified because they show excellent efficacy in hepcidin induction.56,90
Genistein is an isoflavone compound isolated from plants. The capability of hepcidin induction is screened through zebrafish embryos.90 Its potential to induce hepcidin expression is confirmed in HepG2 cells through Hamp promoter luciferase activity and endogenous mRNA analyses.90 Further studies demonstrated that its hepcidin induction activity is mediated through Smad and Stat3 signaling pathways.90 Unfortunately, whether this compound can ameliorate iron overload in mouse models is still unknown. Nonetheless, these results suggest possible direction for the follow-up studies, in which the natural compounds would be an optional solution to search for more active groups for hepcidin induction.
With the application of high-throughput screening, epitiostanol was identified to degrade FPN from 3120 small chemicals.56 Afterwards, several steroid hormones (e.g., progesterone and mifepristone) were found to have similar capability to diminish FPN concentration through PGRMC1–SFKs signaling pathway.56 Ferristatin II, identified as an iron transport inhibitor, induces the internalization and degradation of TfR1, and reduces intestinal iron uptake and serum iron level.91 A recent study further demonstrated that ferristatin II increased hepcidin through phosphorylation of Stat3 without affecting Smad 1/5 phosphorylation.92
Gaun et al93 constructed a screening study using a firefly reporter plasmid containing the human Hamp promoter for a library of 10,169 chemicals. Only 16 of them were found to induce Hamp expression in endogenous HepG2 cells. These chemicals can induce the BMP-Smad and/or Stat3-dependent gene expression; however, none of them enhanced phosphorylation of Smad1/5/8 or Stat3.93 In addition, some of these chemicals are inhibitors of histone deacetylase and serotonin receptors, highlighting a new strategy for chemical screening or synthesis. However, the detailed molecular mechanisms warrant further investigation.
Additionally, our recent results demonstrated that icariin and its analogs had a robust ability to elevate hepatic hepcidin level and then regulate systemic iron homeostasis. This study signifies the potential application of certain natural compounds in treating iron disorders through regulating hepcidin expression.94
TMPRSS6 Antagonist
As previously described, TMPRSS6 takes part in hepcidin regulation through HJV cleavage.33,95 To induce Hamp expression, TMPRSS6 inhibition would be an alternative strategy. Nai et al96 demonstrated that homozygous loss of Tmprss6 in thalassemia mice could improve ineffective erythropoiesis and reduce splenomegaly and iron load. This study suggested that TMPRSS6 would be a potential target to reduce iron overload and improve ineffective erythropoiesis. Based on this study, oligonucleotides and siRNA are designed.97,98 The molecular mechanism for TMPRSS6 and the potential therapeutic significance are described in Figure 4. antisense oligonucleotides (ASO), an antisense oligonucleotide, is synthesized to against Tmprss6 mRNA.97 Sera and liver iron levels are significantly reduced after ASO administration in Hfe−/− mice.97 In addition, it also improved ineffective erythropoiesis and decreased splenomegaly, with resultant increase of total hemoglobin levels in β-thalassemia mice after ASO treatment for 4 weeks. Another RNA-based technology, namely double-stranded nucleic acid, also showed an excellent effect.98 siRNA molecules are capsuled in a lipid nanoparticle (LNP-RNAi), and the assembled complex displayed a dose-dependent TMPRSS6 inhibition after 24 hours administration.98 It greatly induced Hamp expression and ameliorated HH phenotypes in Hfe−/− mice after the administration for 2 or 6 weeks.98 Furthermore, LNP-RNAi treatment diminished secondary iron overload, and even reduced α-globin expression.98 To achieve a better result, this group treated β-thalassemia mice with LNP-RNAi in combination with oral deferiprone. This combination therapy reached better results than any of the individual ones.99 Recently, a new type of matriptase-2 inhibitors was designed. Kunitz-type inhibitor hepatocyte growth factor activator inhibitor (HAI-1) and HAI-2 were demonstrated to inhibit the function of matriptase-2, and HAI-2 was shown to have better capability in matriptase-2 inhibition.100 These kunitz-type inhibitors indicated a new strategy in negative hepcidin regulation.
FIGURE 4.
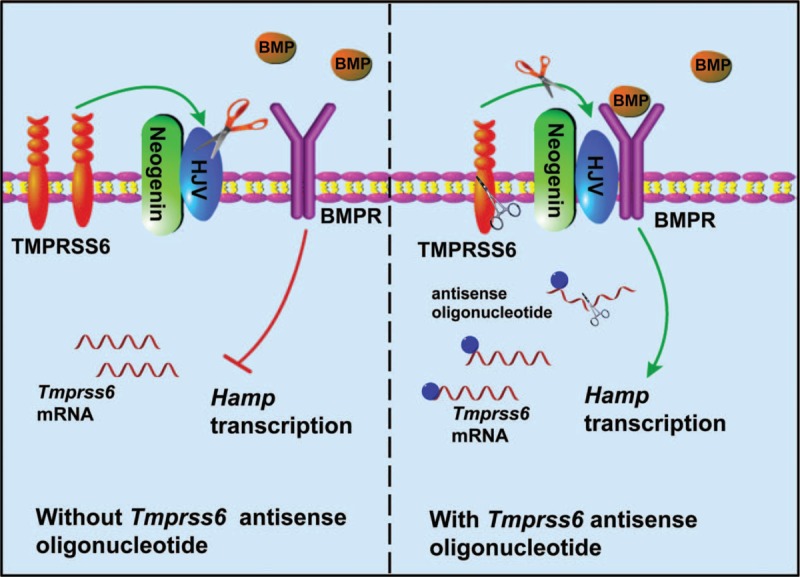
The mechanism of antisense oligonucleotide for hepcidin induction. TMPRSS6 is a transmembrane protease serine 6, and it cleavages HJV to inactivate the Smad signaling. Antisense oligonucleotide molecules could silence the mRNA of Tmprss6, and then down-regulate the expression of Tmprss6 that functions to increases the stability of HJV. HJV is a part of the complex formed by BMPRs to induce Hamp transcription. BMPR = bone morphogenetic protein receptor, HJV = hemojuvelin.
BMP Protein Administration
The study by Meynard et al21 unearthed that lack of BMP6 caused massive iron overload and undetectable hepcidin. Corradini et al101 concluded that exogenous BMP6 treatment could relieve the HH phenotypes caused by Hfe deficiency. BMP6 administration enhanced hepcidin expression, and thus reduced iron overload in Hfe−/− mice, suggesting that BMP6-like agonists may represent a promising route to ameliorate iron overload disease.101 However, there are still some concerns on the use of BMP6. For example, BMP6 administration could lead to peritoneal calcifications.101 Not only BMP6, but also BMP2, 4, 5, 7, and 9, can induce Hamp transcription as well. Nonetheless, BMP6 elicits the most robust induction of Hamp expression than others.19,20
Hepcidin Antagonists
Hepcidin Blockade by Antibody, Antisense Oligonucleotides, or siRNA
Considering the pathologies of anemia of inflammation and IRIDA, a high level of hepcidin and inflammatory cytokines are the main causes of iron deficiency. Thus, targeting hepcidin mRNA, protein, and its upstream regulators such as IL-6 and IL-6R would be optional strategies.
Targeting hepcidin mRNA or protein would be the direct manner to reduce hepcidin concentration. Thus, Hamp siRNA molecules and antibody were designed.102,103 NOX-H94 (also called Lexaptepid) is an L-oligoribonucleotide with a strong affinity to hepcidin mRNA. The pharmacological study on cynomolgus monkeys showed a reduction of serum hepcidin concentration, and increase of hemoglobin level after NOX-H94 administration under IL-6-induced anemia.104 NOX-H94 also exhibited a great capability to increase serum iron and transferrin saturation in a dose-dependent manner during the phase I clinical trial in humans.105 It also showed a significant effect on blocking the inflammation-associated low iron in volunteers with systemic inflammation.106 NOX-H94 also reveals an excellent efficacy with satisfactory tolerance in a phase II human trial study.107 Anticalin PRS-080 is an efficient peptide that can specifically bind hepcidin.103 Cynomolgus monkeys showed significant mobilization of iron and hyperferremia after PRS-080 administration.103 Additionally, hepcidin antibodies were also developed in animal studies on the model of inflammation of anemia, with promising results for modulating iron mobilization.108
Hepcidin Repression Through Acting on BMP and BMPR Complex
As described above, the BMP–Smad signaling pathway fundamentally regulates hepcidin. Thus, inhibition on BMPs or BMPRs would be able to diminish Hamp expression. sHJV is a soluble fragment of HJV that binds to BMPs to prevent the latter's association with BMPRs (as illustrated in Figure 5). sHJV shows a significant inhibition on BMP6 and BMP2, but to a less extent on other types of BMPs in Hep3B cells.109 Afterwards, a soluble HJV–Fc fusion protein (sHJV.Fc) was synthesized, and it manifested a greater ability to ameliorate anemia of inflammation in rats after peptidoglycan-polyasccharide (PG-APS) injection.110 As a result, the rats exhibited elevated serum iron and hemoglobin concentrations.110
FIGURE 5.
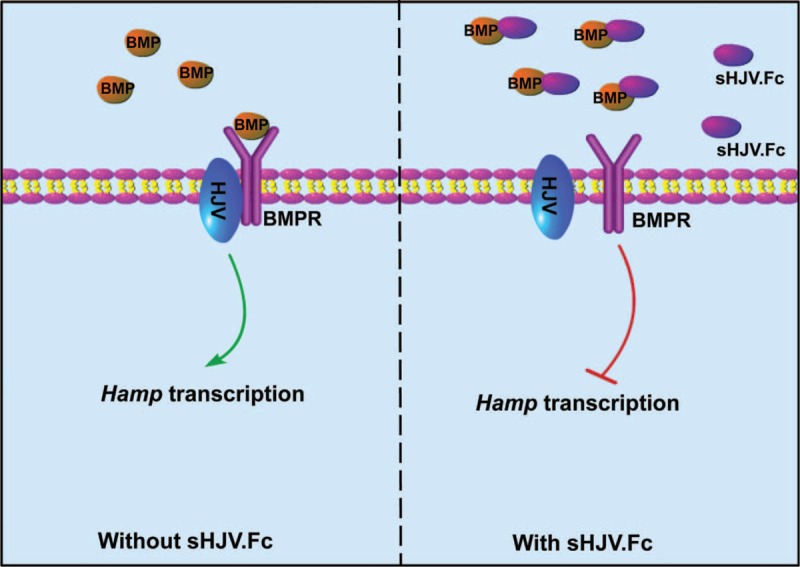
The mechanism by which sHJV.Fc conducts hepcidin suppression. sHJV.Fc is a soluble HJV–Fc fusion protein which can bind to BMPs to prevent the latter's interaction with HJV. HJV is a core composition within the BMP-Smad signaling. The association of sHJV.Fc with BMPs decreases the phosphorylation of Smad1/5/8 and thus reduces Hamp transcription. BMP = bone morphogenetic protein, HJV = hemojuvelin.
Not limited to BMPs, BMPRs are also the target to reduce hepcidin level. A small molecule inhibitor, LDN-193189, selectively antagonizes activin-like kinase type I receptors (ALK2 and ALK3), and increases hemoglobin level in mice.111 However, it can not decrease Hamp expression and increase hemoglobin concentration in a rat model.112,113 HJV, as a cofactor of BMPRs, is a potential target to down-regulate Hamp expression as well. Two monoclonal antibodies, namely ABT-207 and h5F9-AM8, were developed to target HJV/repulsive guidance molecule C.114 Hepatic and serum hepcidin were reduced in rats after a single administration of ABT-207 or h5F9-AM8, and the serum iron concentration was consequentially increased several weeks later.114 Furthermore, TNF-α elicits the down-regulation of hepcidin through suppressing HJV transcription in human hepatoma cells.115 But anti-TNF-α antibody greatly suppressed Hamp expression in patients with rheumatoid arthritis.116
Repression of Hepcidin through IL-6 Signaling
Inflammation caused by IL-6 and other cytokines stimulates Hamp expression via the IL-6–Stat3 or other possible signaling pathways.39,117 Thus far, a few strategies have been developed to target IL-6 or IL-6R, or to inhibit the phosphorylation of Stat3. Tocilizumab is a humanized anti-IL-6R antibody, and the working model is delineated in Figure 6. Tocilizumab can significantly improve anemia in cynomolgus monkeys with arthritis, and reduce Hamp expression after the administration once a week for a total of 4 weeks.118 Additional studies on patients with multicentric Castleman disease suggested that tocilizumab administration resulted in a rapid hepcidin reduction and the long-term administration even normalized the iron status.119 Moreover, anti-TNF-α antibody greatly repressed Hamp expression in patients with rheumatoid arthritis.116 Although anticytokine-based hepcidin repression seems effective; the detrimental and side effects are still under investigation.120,121 Additionally, a previous study demonstrated that AG490 could inhibit the activation of Stat3 signaling to diminish hepcidin expression,122 representing the rationale of hepcidin suppression by blocking the IL-6–Stat3 signaling through chemicals.
FIGURE 6.
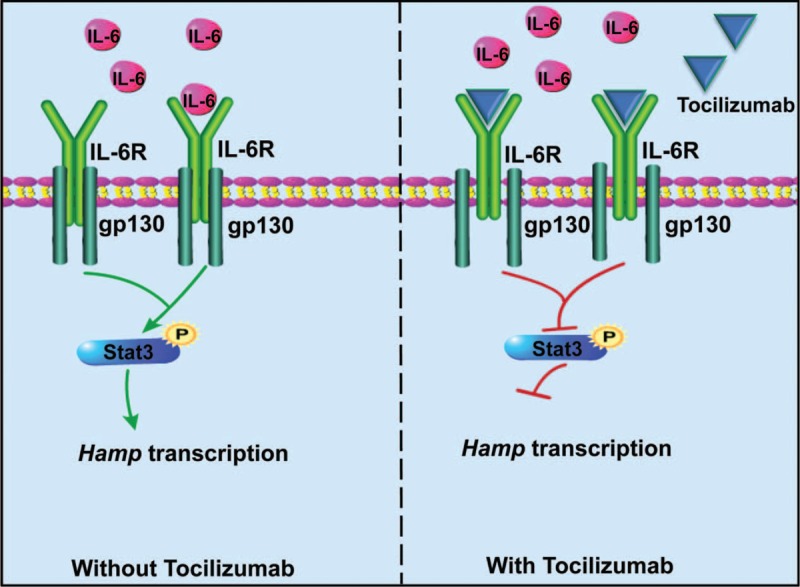
The mechanism of IL-6 Ab in hepcidin suppression. IL-6-provoked proinflammation effects activate Hamp expression through the Stat3 signaling. Blocking IL-6-activated Stat3 signaling would be an optimal strategy to reduce hepcidin level under inflammation. Tocilizumab is a humanized anti-IL-6R antibody, and it works to inactivate the phosphorylation of Stat3. IL = interleukin.
Chemical Compounds
Heparin, a type of glycosaminoglycan, is a well-recognized hepcidin inhibitor.123 Heparin functions to sequester BMP6 and to repress Smad activation, leading to supression of Hamp expression.123 In addition to reducing Hamp expression, heparin is also known for its anticoagulant activity. Thus, it would be optimal to maintain the hepcidin-inhibitory activity with low anticoagulant activity. For this purpose, a new generation of heparin, named glycol-split heparins (gs-heparins), are designed.124 Four gs-heparins, termed as RO-82, RO-68, NAc-91, and NAcRO-00, have been synthesized, and all of them repressed Hamp exprssion in HepG2 cells and primary hepatocytes.124 RO-82 and RO-68 were chosen for animal studies, and both of them were proved to reduce hepatic Hamp expresison and serum hepcidin concentration in mice.124
Another research group found that vitamin D could decrease Hamp expression.125 Prohormone 25-hydroxyvitamin D or active 1,25-dihydroxyvitamine D repressed Hamp expression by 50% in hepatocytes and monocytes.125 Further experiments in healthy humans confirmed this reduciton of hepcidin by vitamin D.125 Vitamin D is thus a potential drug to ameilorate diseases with hepcidin overprodction. K-7174 is a synthesized compound that was identified to improve anemia induced by inflammatory cytokines in mice.126 K-7174 was verified to reduce hepcidin expression in human hematoma cells and in mice as well. Further mechanistic studies indicated that the signaling of hepcidin inhibition by K-7174 was through GDF15.126 Ganz et al developed a high-throughput platform to screen hepcidin atangonists, and they identified 14 chemicals out of 7000 that effeciently atangonized hepcidin funtion.127 Fursultiamine, a FDA-approved thiamine can bind to C-326 thiol residue of FPN, which blocks hepcidin binding to FPN.127 However, it fails to interfere with the action of hepcidin in vivo due to its quick covertion to inactive metabolities,127 and thus more efforts are warranted.
Other Options for Hepcidin Repression
Not limited to synthesized chemical compounds, Chinese medicinal plant extracts are another promising source for hepcidin modulation. Guan et al's128 study found that Caulis spatholobi (CS, also named Jixueteng) exerted a potent inhibitory effect on Hamp epxression through suppression of Smad 1/5/8 phosphorylation.128 It incurred significant hepcidin suppression after CS supplemetion for 5 days in animals.128 In addition to chemical compounds, hormones are also able to modulate Hamp expression. For example, testosterone can down-regulate hepatic Hamp expression through disrupting the signaling of Samd1/4-mediated hepcidin induciton.129 Meanwhile, it can increase hemoglobin and tissue iron in mice subjected to the inhibition of hepcidin after testosterone administration. 17-estradiol represents another sex hormone that could suppress hepcidin,53 as discussed above. Therefore, hormones would be additional considerations for developing therapeutics for iron disorders.
CONCLUSIONS
Thus far, strategies targeting hepcidin–FPN axis are being developed to correct iron disorders for diverse diseases. We reviewed the pathogenesis of iron disorders and according molecular mechanisms with relevance to hepcidin. We also recapitulated the current progress on hepcidin modulation including siRNAs, antibodies, chemical compounds, and plant extracts. Compared with conventional therapies (namely phlebotomy and blood transfusion), strategies targeting the hepcidin–FPN axis may open a new avenue for hepcidin regulation through an endogenous physiological way by avoiding secondary iron overload and other implications.
Footnotes
Abbreviations: ALK2/3 = activin-like kinase type I receptor, ASO = antisense oligonucleotides, BMPR = BMP receptor, BMPs = bone morphogenetic proteins, E2 = 17β-estradiol, ERE = estrogen receptor element, ERFE = erythroferrone, Fpn−/− mice = ferroportin null mice, FPN = ferroportin, GDF11 = growth differentiation factor 11, GDF15 = growth differentiation factor 15, Gp130 = glycoprotein 130, HAI = hepatocyte growth factor activator inhibitor, Hamp−/− mice = hepcidin deficiency mice, Hfe−/− mice = hereditary hemochromatosis protein deficiency mice, HFE = hemochromatosis protein, HH = hereditary hemochromatosis, Hjv−/− mice = hemojuvelin deficiency mice, HJV = hemojuvelin, HSP70 = heat shock protein 70, IL-1β = interleukin-1β, IL-22 = interleukin-22, IL-6 = interleukin-6, IL-6R = interleukin-6 receptor, IRIDA = iron-refractory iron deficiency anemia, LPS = lipopolysaccharide, MAPK = mitogen-activated protein kinase, PG-APS = peptidoglycan-polyasccharide, PGRMC1 = progesterone receptor membrane component-1, ROS = reactive oxidative stress, SFKs = Src-family tyrosine kinases, sHJV.Fc = soluble HJV-Fc fusion protein, TfR1 = transferrin receptor 1, Tfr2−/− = micetransferrin receptor 2 deficiency mice, TfR2 = transferrin receptor 2, TMPRSS6 = transmembrane protease serine 6, TNF-α = tumor necrosis factor-α, TWSG1 = twisted gastrulation BMP signaling modulator.
This study was supported by a grant under the national “973” program (grant number: 2014CB932000), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB14000000), and grants from the National Natural Science Foundation of China (grant numbers: 21425731, 21377159, 91543124).
The authors declare no competing financial interests. The authors have nothing to disclose.
REFERENCES
- 1.Evstatiev R, Gasche C. Iron sensing and signalling. Gut 2012; 61:933–952. [DOI] [PubMed] [Google Scholar]
- 2.Raiten DJ. Iron: current landscape and efforts to address a complex issue in a complex world. J Pediatr 2015; 167:S3–7. [DOI] [PubMed] [Google Scholar]
- 3.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008; 86:480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter JB. Optimizing iron chelation strategies in beta-thalassaemia major. Blood Rev 2009; 23:3–S7. [DOI] [PubMed] [Google Scholar]
- 5.Pippard MJ, Warner GT, Callender ST, et al. Iron-absorption and loading in beta-thalassemia intermedia. Lancet 1979; 2:819–821. [DOI] [PubMed] [Google Scholar]
- 6.Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001; 276:7806–7810. [DOI] [PubMed] [Google Scholar]
- 7.Ramey G, Deschemin JC, Durel B, et al. Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica 2010; 95:501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200. [DOI] [PubMed] [Google Scholar]
- 9.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001; 276:7811–7819. [DOI] [PubMed] [Google Scholar]
- 10.Rivera S, Liu L, Nemeth E, et al. Hepcidin excess induces the sequestration of iron and exacerbates tumor-associated anemia. Blood 2005; 105:1797–1802. [DOI] [PubMed] [Google Scholar]
- 11.Ganz T, Nemeth E. Hepcidin and disorders of iron metabolism. Annu Rev Med 2011; 62:347–360. [DOI] [PubMed] [Google Scholar]
- 12.Qiao B, Sugianto P, Fung E, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab 2012; 15:918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Zhang F, Guo X, et al. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 2012; 56:961–971. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z, Zhang F, An P, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011; 118:1912–1922. [DOI] [PubMed] [Google Scholar]
- 15.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 2010; 139:393–408.408 e1-2. [DOI] [PubMed] [Google Scholar]
- 16.Wallace DF, Summerville L, Crampton EM, et al. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009; 50:1992–2000. [DOI] [PubMed] [Google Scholar]
- 17.Wu Q, Wang H, An P, et al. HJV and HFE play distinct roles in regulating hepcidin. Antioxid Redox Signal 2015; 22:1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kent P, Wilkinson N, Constante M, et al. Hfe and Hjv exhibit overlapping functions for iron signaling to hepcidin. J Mol Med (Berl) 2015; 93:489–498. [DOI] [PubMed] [Google Scholar]
- 19.Andriopoulos B, Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 2009; 41:482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Truksa J, Peng H, Lee P, et al. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci U S A 2006; 103:10289–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 2009; 41:478–481. [DOI] [PubMed] [Google Scholar]
- 22.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab 2005; 2:399–409. [DOI] [PubMed] [Google Scholar]
- 23.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 2006; 38:531–539. [DOI] [PubMed] [Google Scholar]
- 24.Huang FW, Pinkus JL, Pinkus GS, et al. A mouse model of juvenile hemochromatosis. J Clin Invest 2005; 115:2187–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang AS, Gao J, Koeberl DD, et al. The role of hepatocyte hemojuvelin in the regulation of bone morphogenic protein-6 and hepcidin expression in vivo. J Biol Chem 2010; 285:16416–16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee DH, Zhou LJ, Zhou Z, et al. Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis. Blood 2010; 115:3136–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang F, West AP, Jr, Allendorph GP, et al. Neogenin interacts with hemojuvelin through its two membrane-proximal fibronectin type III domains. Biochemistry 2008; 47:4237–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang AS, Yang F, Wang J, et al. Hemojuvelin-neogenin interaction is required for bone morphogenic protein-4-induced hepcidin expression. J Biol Chem 2009; 284:22580–22589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enns CA, Ahmed R, Zhang AS. Neogenin interacts with matriptase-2 to facilitate hemojuvelin cleavage. J Biol Chem 2012; 287:35104–35117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du X, She E, Gelbart T, et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008; 320:1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat Genet 2008; 40:569–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Folgueras AR, de Lara FM, Pendas AM, et al. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008; 112:2539–2545. [DOI] [PubMed] [Google Scholar]
- 33.Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab 2008; 8:502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frydlova J, Prikryl P, Truksa J, et al. Effect of erythropoietin, iron deficiency and iron overload on liver matriptase-2 (TMPRSS6) protein content in mice and rats. PLoS One 2016; 11:e0148540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meynard D, Vaja V, Sun CC, et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011; 118:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakhal S, Schodel J, Townsend AR, et al. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. J Biol Chem 2011; 286:4090–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao N, Nizzi CP, Anderson SA, et al. Low intracellular iron increases the stability of matriptase-2. J Biol Chem 2015; 290:4432–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meynard D, Sun CC, Wu Q, et al. Inflammation regulates TMPRSS6 expression via STAT5. PLoS One 2013; 8:e82127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Besson-Fournier C, Latour C, Kautz L, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 2012; 120:431–439. [DOI] [PubMed] [Google Scholar]
- 40.Canali S, Core AB, Zumbrennen-Bullough KB, et al. Activin B induces noncanonical SMAD1/5/8 signaling via BMP type I receptors in hepatocytes: evidence for a role in hepcidin induction by inflammation in male mice. Endocrinology 2016; 157:1146–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shanmugam NK, Chen K, Cherayil BJ. Commensal bacteria-induced interleukin 1beta (IL-1beta) secreted by macrophages up-regulates hepcidin expression in hepatocytes by activating the bone morphogenetic protein signaling pathway. J Biol Chem 2015; 290:30637–30647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo X, Luo Z, Zhang Z, et al. Homocysteine upregulates hepcidin expression through BMP6/SMAD signaling pathway in hepatocytes. Biochem Biophys Res Commun 2016; 471:303–308. [DOI] [PubMed] [Google Scholar]
- 43.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113:1271–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verga Falzacappa MV, Vujic Spasic M, Kessler R, et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood 2007; 109:353–358. [DOI] [PubMed] [Google Scholar]
- 45.Yu H, Lee H, Herrmann A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 2014; 14:736–746. [DOI] [PubMed] [Google Scholar]
- 46.Kim A, Nemeth E. New insights into iron regulation and erythropoiesis. Curr Opin Hematol 2015; 22:199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med 2007; 13:1096–1101. [DOI] [PubMed] [Google Scholar]
- 48.Tanno T, Noel P, Miller JL. Growth differentiation factor 15 in erythroid health and disease. Curr Opin Hematol 2010; 17:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fertrin KY, Lanaro C, Franco-Penteado CF, et al. Erythropoiesis-driven regulation of hepcidin in human red cell disorders is better reflected through concentrations of soluble transferrin receptor rather than growth differentiation factor 15. Am J Hematol 2014; 89:385–390. [DOI] [PubMed] [Google Scholar]
- 50.Tanno T, Porayette P, Sripichai O, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009; 114:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet 2014; 46:678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015; 126:2031–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Q, Jian J, Katz S, et al. 17beta-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology 2012; 153:3170–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hou YL, Zhang SP, Wang L, et al. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene 2012; 511:398–403. [DOI] [PubMed] [Google Scholar]
- 55.Ikeda Y, Tajima S, Izawa-Ishizawa Y, et al. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PLoS One 2012; 7:e40465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X, Rhee DK, Malhotra R, et al. Progesterone receptor membrane component-1 regulates hepcidin biosynthesis. J Clin Invest 2016; 126:389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu S, Bennett RG, Kharbanda KK, et al. Lack of hepcidin expression attenuates steatosis and causes fibrosis in the liver. World J Hepatol 2016; 8:211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chelchowska M, Ambroszkiewicz J, Gajewska J, et al. Hepcidin and iron metabolism in pregnancy: correlation with smoking and birth weight and length. Biol Trace Elem Res 2016; 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camaschella C. Iron and hepcidin: a story of recycling and balance. Hematology Am Soc Hematol Educ Program 2013; 2013:1–8. [DOI] [PubMed] [Google Scholar]
- 60.Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007; 109:5027–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vujic Spasic M, Kiss J, Herrmann T, et al. Physiologic systemic iron metabolism in mice deficient for duodenal Hfe. Blood 2007; 109:4511–4517. [DOI] [PubMed] [Google Scholar]
- 62.van Eijk LT, Kroot JJ, Tromp M, et al. Inflammation-induced hepcidin-25 is associated with the development of anemia in septic patients: an observational study. Crit Care 2011; 15:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Q, Davidoff O, Niss K, et al. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest 2012; 122:4635–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ashby DR, Gale DP, Busbridge M, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica 2010; 95:505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sonnweber T, Nachbaur D, Schroll A, et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 2014; 63:1951–1959. [DOI] [PubMed] [Google Scholar]
- 66.Blank U, Karlsson S. The role of Smad signaling in hematopoiesis and translational hematology. Leukemia 2011; 25:1379–1388. [DOI] [PubMed] [Google Scholar]
- 67.Dussiot M, Maciel TT, Fricot A, et al. An activin receptor IIA ligand trap corrects ineffective erythropoiesis in beta-thalassemia. Nat Med 2014; 20:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arlet JB, Ribeil JA, Guillem F, et al. HSP70 sequestration by free alpha-globin promotes ineffective erythropoiesis in beta-thalassaemia. Nature 2014; 514:242–246. [DOI] [PubMed] [Google Scholar]
- 69.Arlet JB, Ribeil JA, Guillem F, et al. HSP70 regulates ineffective erythropoiesis in beta-thalassaemia. Med Sci (Paris) 2015; 31:9–11. [DOI] [PubMed] [Google Scholar]
- 70.Petrova J, Manolov V, Dimitrov G, et al. Serum hepcidin levels and stroke in thalassemia patients. Int J Stroke 2016; doi: 10.1177/1747493015623557. [DOI] [PubMed] [Google Scholar]
- 71.Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med 2015; 21:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nemeth E, Ganz T. Anemia of inflammation. Hematol Oncol Clin North Am 2014; 28:671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cherayil BJ. Pathophysiology of iron homeostasis during inflammatory states. J Pediatr 2015; 167:15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sakamori R, Takehara T, Tatsumi T, et al. STAT3 signaling within hepatocytes is required for anemia of inflammation in vivo. J Gastroenterol 2010; 45:244–248. [DOI] [PubMed] [Google Scholar]
- 75.Smith CL, Arvedson TL, Cooke KS, et al. IL-22 regulates iron availability in vivo through the induction of hepcidin. J Immunol 2013; 191:1845–1855. [DOI] [PubMed] [Google Scholar]
- 76.Armitage AE, Eddowes LA, Gileadi U, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011; 118:4129–4139. [DOI] [PubMed] [Google Scholar]
- 77.Poggiali E, Migone De Amicis M, Motta I. Anemia of chronic disease: a unique defect of iron recycling for many different chronic diseases. Eur J Intern Med 2014; 25:12–17. [DOI] [PubMed] [Google Scholar]
- 78.Kodama K, Noguchi A, Adachi H, et al. Novel mutation in the TMPRSS6 gene with iron-refractory iron deficiency anemia. Pediatr Int 2014; 56:41–44. [DOI] [PubMed] [Google Scholar]
- 79.Jaspers A, Caers J, Le Gac G, et al. A novel mutation in the CUB sequence of matriptase-2 (TMPRSS6) is implicated in iron-resistant iron deficiency anaemia (IRIDA). Br J Haematol 2013; 160:564–565. [DOI] [PubMed] [Google Scholar]
- 80.Casu C, Aghajan M, Oikonomidou PR, et al. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica 2016; 101:e8–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salgia RJ, Brown K. Diagnosis and management of hereditary hemochromatosis. Clin Liver Dis 2015; 19:187–198. [DOI] [PubMed] [Google Scholar]
- 82.Rund D. Thalassemia 2016: modern medicine battles an ancient disease. Am J Hematol 2015; 91:15–21. [DOI] [PubMed] [Google Scholar]
- 83.De Falco L, Sanchez M, Silvestri L, et al. Iron refractory iron deficiency anemia. Haematologica 2013; 98:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nicolas G, Viatte L, Lou DQ, et al. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet 2003; 34:97–101. [DOI] [PubMed] [Google Scholar]
- 85.Moran-Jimenez MJ, Mendez M, Santiago B, et al. Hepcidin treatment in Hfe−/− mice diminishes plasma iron without affecting erythropoiesis. Eur J Clin Invest 2010; 40:511–517. [DOI] [PubMed] [Google Scholar]
- 86.Preza GC, Ruchala P, Pinon R, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest 2011; 121:4880–4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramos E, Ruchala P, Goodnough JB, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood 2012; 120:3829–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fung E, Chua K, Ganz T, et al. Thiol-derivatized mini-hepcidins retain biological activity. Bioorg Med Chem Lett 2015; 25:763–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chua K, Fung E, Micewicz ED, et al. Small cyclic agonists of iron regulatory hormone hepcidin. Bioorg Med Chem Lett 2015; 25:4961–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhen AW, Nguyen NH, Gibert Y, et al. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology 2013; 58:1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Byrne SL, Buckett PD, Kim J, et al. Ferristatin II promotes degradation of transferrin receptor-1 in vitro and in vivo. PLoS One 2013; 8:e70199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alkhateeb AA, Buckett PD, Gardeck AM, et al. The small molecule ferristatin II induces hepatic hepcidin expression in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol 2015; 308:1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gaun V, Patchen B, Volovetz J, et al. A chemical screen identifies small molecules that regulate hepcidin expression. Blood Cells Mol Dis 2014; 53:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang M, Liu J, Guo W, et al. Icariin regulates systemic iron metabolism by increasing hepatic hepcidin expression through stat3 and smad1/5/8 signalling. Int J Mol Med 2016; (in press). [DOI] [PubMed] [Google Scholar]
- 95.Lee P. Role of matriptase-2 (TMPRSS6) in iron metabolism. Acta Haematol 2009; 122:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nai A, Pagani A, Mandelli G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood 2012; 119:5021–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Guo S, Casu C, Gardenghi S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest 2013; 123:1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmidt PJ, Toudjarska I, Sendamarai AK, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 2013; 121:1200–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schmidt PJ, Racie T, Westerman M, et al. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of beta-thalassemia intermedia. Am J Hematol 2015; 90:310–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beckmann AM, Maurer E, Lulsdorff V, et al. En route to new therapeutic options for iron overload diseases: matriptase-2 as a target for Kunitz-type inhibitors. Chembiochem 2016; 17: [DOI] [PubMed] [Google Scholar]
- 101.Corradini E, Schmidt PJ, Meynard D, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010; 139:1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Poli M, Asperti M, Ruzzenenti P, et al. Hepcidin antagonists for potential treatments of disorders with hepcidin excess. Front Pharmacol 2014; 5:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hohlbaum A, Gille H, Christian J, et al. Iron mobilization and pharmacodynic marker measurements in non-human primates following administration of Prs-080: a novel and highly specific anti-hepcidin therapeutic. Am J Hematol 2013; 88:41–141. [Google Scholar]
- 104.Schwoebel F, van Eijk LT, Zboralski D, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood 2013; 121:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Riecke K, Zollner S, Boyce M, et al. Single and repeated dose first-in-human study with the anti-hepcidin Spiegelmer Nox-H94. Am J Hematol 2013; 88:225–226. [Google Scholar]
- 106.van Eijk LT, John ASE, Schwoebel F, et al. Effect of the antihepcidin Spiegelmer lexaptepid on inflammation-induced decrease in serum iron in humans. Blood 2014; 124:2643–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vater A, Klussmann S. Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer(R) therapeutics. Drug Discov Today 2015; 20:147–155. [DOI] [PubMed] [Google Scholar]
- 108.Sasu BJ, Cooke KS, Arvedson TL, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood 2010; 115:3616–3624. [DOI] [PubMed] [Google Scholar]
- 109.Nili M, Shinde U, Rotwein P. Soluble repulsive guidance molecule c/hemojuvelin is a broad spectrum bone morphogenetic protein (BMP) antagonist and inhibits both BMP2- and BMP6-mediated signaling and gene expression. J Biol Chem 2010; 285:24783–24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood 2011; 118:4977–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cuny GD, Yu PB, Laha JK, et al. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg Med Chem Lett 2008; 18:4388–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Steinbicker AU, Sachidanandan C, Vonner AJ, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood 2011; 117:4915–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun CC, Vaja V, Chen S, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant 2013; 28:1733–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boser P, Seemann D, Liguori MJ, et al. Anti-repulsive guidance molecule C (RGMc) antibodies increases serum iron in rats and cynomolgus monkeys by hepcidin downregulation. AAPS J 2015; 17:930–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Salama MF, Bayele HK, Srai SS. Tumour necrosis factor alpha downregulates human hemojuvelin expression via a novel response element within its promoter. J Biomed Sci 2012; 19:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Doyle MK, Rahman MU, Frederick B, et al. Effects of subcutaneous and intravenous golimumab on inflammatory biomarkers in patients with rheumatoid arthritis: results of a phase 1, randomized, open-label trial. Rheumatology (Oxford) 2013; 52:1214–1219. [DOI] [PubMed] [Google Scholar]
- 117.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006; 108:3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hashizume M, Uchiyama Y, Horai N, et al. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int 2010; 30:917–923. [DOI] [PubMed] [Google Scholar]
- 119.Song SN, Tomosugi N, Kawabata H, et al. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood 2010; 116:3627–3634. [DOI] [PubMed] [Google Scholar]
- 120.Lang VR, Englbrecht M, Rech J, et al. Risk of infections in rheumatoid arthritis patients treated with tocilizumab. Rheumatology (Oxford) 2012; 51:852–857. [DOI] [PubMed] [Google Scholar]
- 121.van de Vosse E, van Agtmael MA. Targets of anticytokine therapy and the risk of infections in humans and mice. Curr Opin Rheumatol 2007; 19:626–635. [DOI] [PubMed] [Google Scholar]
- 122.Zhang SP, Wang Z, Wang LX, et al. AG490: an inhibitor of hepcidin expression in vivo. World J Gastroenterol 2011; 17:5032–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Poli M, Girelli D, Campostrini N, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood 2011; 117:997–1004. [DOI] [PubMed] [Google Scholar]
- 124.Poli M, Asperti M, Naggi A, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood 2014; 123:1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bacchetta J, Zaritsky JJ, Sea JL, et al. Suppression of iron-regulatory hepcidin by vitamin D. J Am Soc Nephrol 2014; 25:564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fujiwara T, Ikeda T, Nagasaka Y, et al. A low-molecular-weight compound K7174 represses hepcidin: possible therapeutic strategy against anemia of chronic disease. PLoS One 2013; 8:75568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fung E, Sugianto P, Hsu J, et al. High-throughput screening of small molecules identifies hepcidin antagonists. Mol Pharmacol 2013; 83:681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guan Y, An P, Zhang ZZ, et al. Screening identifies the Chinese medicinal plant caulis spatholobi as an effective HAMP expression inhibitor. J Nutr 2013; 143:1061–1066. [DOI] [PubMed] [Google Scholar]
- 129.Guo W, Bachman E, Li M, et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell 2013; 12:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]