

Abstract
High-throughput screening of a subset of the CD3 chemical library (Centre for Drug Design and Discovery; KU Leuven) provided us with a lead compound 1, displaying low micromolar potency against dengue virus and yellow fever virus. Within a project aimed at discovering new inhibitors of flaviviruses, substitution of its central imidazole ring led to synthesis of variably substituted pyrazine dicarboxylamides and phthalic diamides, which were evaluated in cell-based assays for cytotoxicity and antiviral activity against the dengue virus (DENV) and yellow fever virus (YFV). Fourteen compounds inhibited DENV replication (EC50 ranging between 0.5 and 3.4 μM), with compounds 6b and 6d being the most potent inhibitors (EC50 0.5 μM) with selectivity indices (SI) > 235. Compound 7a likewise exhibited anti-DENV activity with an EC50 of 0.5 μM and an SI of >235. In addition, good antiviral activity of seven compounds in the series was also noted against the YFV with EC50 values ranging between 0.4 and 3.3 μM, with compound 6n being the most potent for this series with an EC50 0.4 μM and a selectivity index of >34. Finally, reversal of one of the central amide bonds as in series 13 proved deleterious to the inhibitory activity.
Keywords: Flavivirus inhibitors, Dengue virus, Diamide, Ortho-phtalic acid, Pyrazine dicarboxylic acid
Graphical abstract

Highlights
-
•
Two heterocyclic scaffold series were evaluated for in vitro Flavivirus inhibition.
-
•
Activities at micromolar levels were noted for inhibition of dengue virus.
-
•
Some remarkable inhibitory properties for yellow fever virus were recorded.
-
•
Several pyrazine-2,3-dicarboxylic amides are endowed with anti-flavivirus activity.
-
•
Phthalic acid diamides likewise provide an interesting scaffold for antivirals.
1. Introduction
The dengue virus (DENV), a mosquito-borne disease, is the most common arthropod-borne viral infection in the world [2]. Dengue incidence and prevalence are rising in endemic areas of the tropical and subtropical regions. On the basis of mathematical model estimates, approximately 390 million infections occur each year [2]. Dengue infection occurs in more than 100 countries in the Asia-Pacific, the Americas, the Middle East, and Africa, and cases continue to rise worldwide [3]. Aedes aegypti is the primary mosquito vector; however, other species from the genus Aedes, such as Aedes albopictus, can also be vectors of dengue virus transmission. The viruses have been grouped into four serotypes (DENV-1 to DENV-4) belonging to the genus Flavivirus (family Flaviviridae) with the existence of a fifth subtype being claimed more recently [4]. Each of these serotypes can cause disease symptoms ranging from self-limited febrile illness called dengue fever (DF) to dengue hemorrhagic fever (DHF), or dengue shock syndrome (DSS). Currently, there is no antiviral therapy available for DENV [5], [6].
Many different viral targets to inhibit or control dengue infection already have been envisaged over the last 10–15 years, among which the entry phase where the envelope glycoprotein is targeted by various strategies, or the NS3 helicase, the dengue protease, the RNA dependent RNA polymerase, the NS4b and the methyltransferase. Recently, a new series of 2-aroyl-3-arylquinoline was identified with strong inhibition of DENV2 RNA expression. Some compounds within this series inhibited DENV replication in both viral protein and mRNA levels, without significant cell cytotoxicity [7]. In parallel, a recent review discussed the potential of targeting non-structural proteins and more specifically proteases for combating neglected diseases as caused by arboviruses with in particular Dengue virus [8]. In addition, inhibition of host factors or processes indispensable for viral proliferation have been studied at several instances. An excellent review covering all aspects on the medicinal chemistry of dengue virus of Klein et al. recently appeared [9].
In this communication we discuss our progress on the discovery of new flavivirus inhibitors. Hereto, at first a high-throughput screening (HTS) campaign was conducted with a subset of the CD3 chemical library (Centre for Drug Design and Discovery; KU Leuven) in order to identify novel non-nucleoside inhibitors of DENV (work supported by a Wellcome Trust Seeding Drug Discovery Award). This screening delivered an interesting hit compound 1 which turned to be active in low micromolar range against DENV and YFV (Fig. 1). Hence, a systematic modification program was setup examining substitutions at the four aromatic rings of the lead molecule. In a first report [1], the central imidazole ring B was retained resulting in a first series of analogues with modifications at the aniline moieties. The same paper discusses a second series where the central imidazole ring was substituted for a pyrazine 2,3-dicarboxylic acid moiety. Overall, these efforts allowed to uncover several inhibitors with activities in the low micromolar range. In this communication, we further elaborate on the central pyrazine scaffold, while a second series of compounds focuses on substitution of the original imidazole-4,5-dicarboxylic acid moiety by an ortho-phthalic acid central ring on which the different anilines are attached. The remaining three structural domains allowed for optimization of hit 1: the aromatic moiety (ring A), the substituted aromatic moiety (ring C) and the heterocyclic group (ring D). These efforts resulted in several compounds with enhanced anti-DENV and anti-YFV inhibitory properties (see Fig. 2).
Fig. 1.
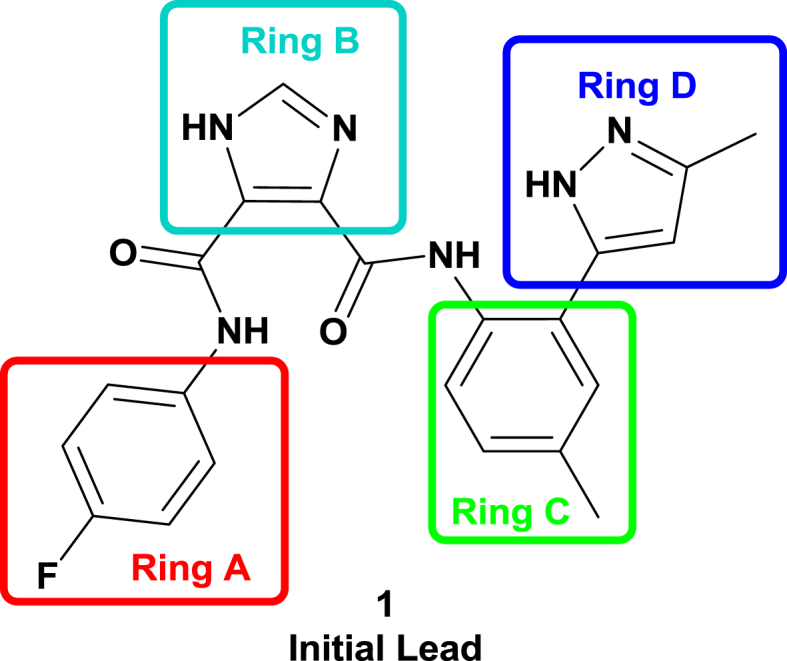
Structure of the initial lead compound endowed with antiviral properties (DENV: EC50 = 2.5 μM; YFV: EC50 = 3.5 μM).
Fig. 2.

Overview of all new compounds most inhibitory to either DENV or YFV as reported in this communication.
2. Results and discussion
2.1. Synthetic aspect
Pyrazines are important pharmacophores present in a number of biologically active compounds such as antimycobacterial, antibacterial, antidiabetic, and hypnotic agents [10], [11], [12]. Functionalization of the pyrazine started from commercial pyrazine-2,3-dicarboxylic acid and a four-step synthesis of the target compound is described in Scheme 1.
Scheme 1.

Reaction Conditions: a) acetic anhydride; b) acetonitrile: water (1:1), aniline, dodecyl hydrogen sulfate sodium salt, rt; c) trifluoroacetic anhydride, TEA, THF; d) aniline, THF, rt.
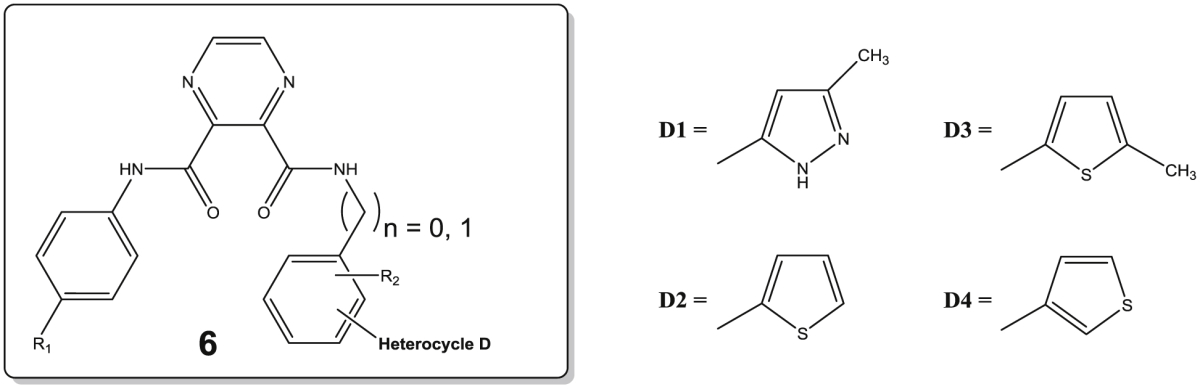
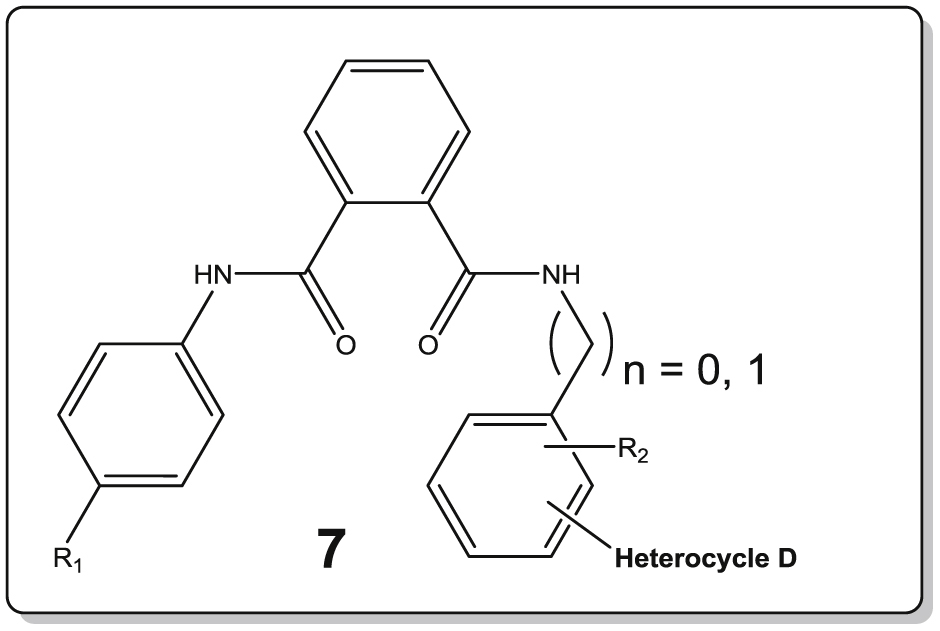
The commercially available pyrazine-2,3-dicarboxilic acid 2a was allowed to reflux with acetic anhydride for 15 min. On cooling to 0 °C, pure anhydride 3a was conveniently found to crystallize out which was collected by filtration, washed with ether and dried to obtain 85% yield. While benzoic anhydride can be directly added to an aqueous solution of the amine, the reaction times are longer and some benzoic anhydride remains unreacted, resulting into lower yields and an impure product. However, better results are obtained by adding acetonitrile to an equimolar amount of the respective anhydride and the appropriate aromatic amine dissolved in water containing sodium dodecyl sulfate (SDS) affording the N-arylphthalamic acids 4a,b. The reaction was usually completed within 15 min. Removal of acetonitrile under reduced pressure led to precipitation of the benzoylated product along with benzoic acid. The desired product was further purified by column chromatography giving excellent yields (75–80%). Further, trifluoroacetic anhydride was added drop wise to a solution of the respective N-arylphthalamic acid and TEA in dioxane at 0 °C. The pale yellow intermediates 5a,b resulting from simple dehydration of N-arylphthalamic acids were obtained by simply pouring the reaction mass into cold water. The precipitate formed was filtered and washed with water and the spectroscopic data were as could be expected. The analogous procedure starting from phthalic acid 2b afforded the intermediates 5c,d. Each of the intermediates 5a–d were subsequently reacted with preformed heterocycle substituted anilines to give the target compounds 6a–n and 7a–n in 60–80% overall yields. All final products were purified by silica gel chromatography and were fully characterized.
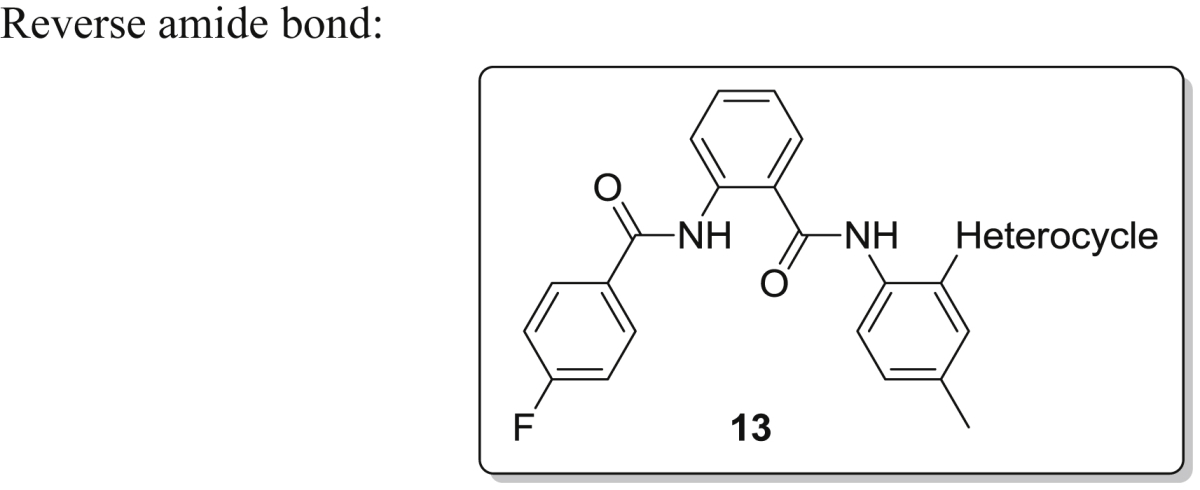
To evaluate whether the orientation of the amide bonds for the lead compound were critical for biological activity, four compounds were synthesized having one of the amide bonds reversed. The 5-step synthesis of the target compound series 13a–d is described in Scheme 2.
Scheme 2.

Reaction Conditions: a) phthalic anhydride, AcOH, reflux, 1 h, 83%; b) SOCl2, DMF(cat.), reflux, 1 h; aniline, DIPEA, CH2Cl2, rt, 1 h, 80%; c) hydrazine hydrate, THF, 60 C, 16 h; d) SOCl2, DMF(cat.), reflux, 2 h; e) DIPEA, CH2Cl2, rt, 2 h; f) K2CO3, water: dioxane (1:1), Pd(TPP)2Cl2, boronic acid.
Hereto, anthranilic acid 8 was converted to its corresponding phthalimide derivative 9 simply by refluxing in presence of phthalic anhydride with acetic acid for 1 h. The obtained product was allowed to reflux with SOCl2 and catalytic DMF to afford the corresponding acid chloride. Anilines/amines subsequently provided straightforward coupling at reduced temperature resulting in 10. Hydrazinolysis at 60 °C resulted in deprotection of the amine which was further coupled to another acid chloride to give target compound 12. Coupling reactions of the latter with four different commercially available heterocyclic boronic acids afforded the desired novel molecules with general structure 13a–13d in 60–70% yield. All final products were purified by silica gel chromatography and fully characterized. The obtained products were evaluated by the laboratory of virology for their inhibitory properties against dengue and yellow fever virus.
2.2. Assessment of antiviral activity
Based on the high throughput lead compound 1, a total of 32 new analogues were synthesized and evaluated for their inhibitory properties against DENV (serotype 2) and YFV using Green monkey kidney cells [Vero-B cells (ECACC for DENV assays and ATCCCCL-81 for YFV assays)]. The 50% effective concentration (EC50), which is defined as the compound concentration that is required to inhibit the virus-induced CPE by 50%, and the 50% cytotoxic concentration (CC50), which is defined as the compound concentration that is required to inhibit the cell growth by 50%, was determined. Original data were obtained as μg/mL concentrations and were recalculated to μM concentrations as shown here. Compound 1 was included as a reference compound.
The initial lead compound 1 comprised the central imidazole ring B. In our efforts to understand SAR properties we replaced this imidazole ring (Fig. 1) with either a six-membered pyrazine (series 6) or a phenyl ring (series 7). Apart from a slightly different orientation of the attached substituents, we also expected this to provide us some information on the requirement for an imidazole ring B to interact with its target, or whether this 5-membered scaffold only serves to perfectly orient the pending substituents. While pyrazine preserves the polarity of the imidazole ring, the former only has H-bond accepting capacity. In contrast, the phenyl ring leads to increased lipophilicity. In addition, both six-membered rings give a slightly different 3D orientation for the attached aromatic moieties compared to the lead structure 1. In another SAR study, we preserved the imidazole core and studied the effect of different aromatic substituents within this series of compounds [1]. As in the present study not all results could be obtained for inhibition of YFV, mainly the inhibitory properties for dengue infections are discussed more thoroughly below.
As to potential interactions of the original central imidazole ring, the pyrazine analogue 6a of the lead structure 1 showed a 10-fold reduction in inhibitory properties against dengue virus (EC50 = 26.5 μM). In contrast however, its phenyl counterpart 7a was 5-fold more inhibitory (EC50 = 0.5 μM) compared to lead compound 1, and is endowed with a selectivity index (SI) of more than 235. Substituting the electron accepting fluorine for a hydrophobic methyl moiety increased the DENV inhibitory properties for 6b 50-fold in comparison with 6a, attaining likewise an EC50 of 0.5 μM with a SI above 235. Comparison of 6c and 6d or 6k with 6l confirmed this finding with a 10-fold increase in activity for the methyl congener, where for other analogous pairs of compounds this activity increase was less clear-cut (e.g. the couples 6g,6h, 6i,6j). In contrast, within series 7 with a central phenyl ring, the analogue 7c carrying the p-tolyl moiety lost 60-fold in DENV inhibitory activity (EC50 = 29 μM) compared to 7a, while for other compound pairs the para-methyl substituent at ring A again slightly outperformed the para-fluorine congener (e.g. 7e,7f and 7g,7h). The YFV inhibitory activity could not be determined for all compounds in view of the more demanding assay, and no analogous conclusions regarding the advantage of a p-tolyl moiety for ring A could be drawn (see Table 1, Table 2).
Table 1.
Antiviral activity of compounds 6a–6n.
| Sr. No. | n | R1 | R2 | Heterocycle (positiona) | Dengue |
Yellow fever |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | SI | EC50 (μM) | CC50 (μM) | SI | |||||
| 6a | 0 | F | p-methyl | D1 (ortho) | 26.5 | >116 | >4 | ND | ND | NA |
| 6b | 0 | CH3 | p-methyl | D1 (ortho) | 0.5 | >117 | >235 | ND | ND | NA |
| 6c | 0 | F | p-methyl | D2 (ortho) | 4.6 | >116 | >25 | 23 | >116 | 5 |
| 6d | 0 | CH3 | p-methyl | D2 (ortho) | 0.5 | >117 | >235 | ND | ND | NA |
| 6e | 0 | F | p-methyl | D3 (ortho) | >112 | >112 | 1 | ND | ND | NA |
| 6f | 0 | CH3 | p-methyl | D3 (ortho) | >113 | >113 | 1 | ND | ND | NA |
| 6g | 0 | F | p-methyl | D4 (ortho) | 2.1 | >116 | >55 | 10.6 | >116 | >11 |
| 6h | 0 | CH3 | p-methyl | D4 (ortho) | <0.9 | 26 | >29 | >116 | >116 | 1 |
| 6i | 1 | F | H | D4 (ortho) | 24 | 52 | 2 | 3.9 | 76 | 19 |
| 6j | 1 | CH3 | H | D4 (ortho) | >48 | 48 | 1 | 5.4 | 75 | 14 |
| 6k | 1 | F | H | D4 (para) | >116 | >116 | 1 | 18.5 | >116 | >6 |
| 6l | 1 | CH3 | H | D4 (para) | 14 | >117 | >8 | 21.5 | >117 | >5 |
| 6m | 0 | F | H | D1 (ortho) | 2.2 | >120 | >55 | 7.8 | 77 | 10 |
| 6n | 0 | CH3 | H | D1 (ortho) | 3.4 | 24 | 7 | 0.4 | >13.6 | >34 |
Indicates the attachment point of the heterocycle on the phenyl ring C.
Table 2.
Antiviral activity of compounds 7a–7n.
| Sr. No. | n | R1 | R2 | Heterocycle (positiona) | Dengue |
Yellow fever |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | SI | EC50 (μM) | CC50 (μM) | SI | |||||
| 7a | 0 | F | p-methyl | D1 (ortho) | 0.5 | >117 | >235 | ND | ND | NA |
| 7b | 0 | F | p-methyl | D2 (ortho) | 4.0 | >117 | >29 | 23.2 | >37 | >1.6 |
| 7c | 0 | CH3 | p-methyl | D1 (ortho) | 29 | >117 | >4 | ND | ND | NA |
| 7d | 0 | CH3 | p-methyl | D2 (ortho) | 14 | >117 | >8 | 23.2 | >37 | >1.6 |
| 7e | 0 | CH3 | p-methyl | D3 (ortho) | 2.0 | >114 | >57 | 6.8 | 23 | 3.4 |
| 7f | 0 | F | p-methyl | D3 (ortho) | 2.9 | >113 | >39 | 6.8 | >113 | >17 |
| 7g | 0 | CH3 | p-methyl | D4 (ortho) | 0.7 | 12.9 | 41 | 1.9 | >117 | >60 |
| 7h | 0 | F | p-methyl | D4 (ortho) | 3.0 | 47 | 16 | 6.5 | 67 | 10 |
| 7i | 1 | F | H | D4 (ortho) | 3.0 | 50 | 17 | 1.4 | 75 | 53 |
| 7j | 1 | CH3 | H | D4 (ortho) | 3.0 | 50 | 17 | 2.6 | 61 | 23 |
| 7k | 1 | F | H | D4 (para) | 16 | >116 | >7 | 7.4 | >117 | >16 |
| 7l | 1 | CH3 | H | D4 (para) | 13.8 | >117 | >8 | 3.3 | >117 | >35 |
| 7m | 0 | F | H | D1 (ortho) | >120 | >120 | 1 | 12.0 | >120 | >10 |
| 7n | 0 | CH3 | H | D1 (ortho) | 14.1 | >122 | >9 | 5.8 | >122 | >21 |
indicates the attachment point of the heterocycle on the phenyl ring.
Next, taking a look at the effect of ring C and its various substitutions, we notice that within the pyrazine series 6, using a benzyl moiety (n = 1, compounds 6i–6l) instead of a (substituted) phenyl ring lead to strongly reduced inhibitory activity when comparing 6g versus 6i and 6h versus 6j. Removal of the para methyl substituent of ring C leads to conflicting conclusions within series 6, with a 12-fold improvement for the p-fluorophenyl (ring A) compound 6m over 6a, but a 7-fold reduced inhibitory activity for the p-tolyl containing compound 6n versus 6b. For series 7 carrying a benzene central ring B, removal of the para-methyl moiety on ring C leading to 7m was not well tolerated and led to strongly reduced inhibitory activity for DENV as compared to the initial lead compound 7a. The negative effect of a benzyl substituent for ring C was less outspoken within series 7, with DENV inhibitory activities varying from 3 to 16 μM for all compounds 7i–7l. Likewise, strong inhibitory properties were noted against YFV for this series ranging from 1.4 to 7.4 μM.
The general trend observed for replacement of the five-membered pyrazole moiety (ring D) in the initial lead 1 with other heterocycles led to progressively inferior inhibition [1]. For evaluation of the present series with either a pyrazine (6) or a phenyl (7) moiety as the central core B, mainly the ortho position on ring C has been targeted to attach heterocyclic ring D as was found in the initial lead compound 1. As well the original 3-methylpyrazol-5-yl, thien-2-yl and thien-3-yl ring displayed inhibitory activity with slight preference for the latter within both series 6 and 7. This resulted in strong DENV inhibitory properties for compounds 6d (EC50 = 0.5 μM) and 6h (EC50 = <0.9 μM), resulting in selectivity indices (SI) of >235 and >29, respectively. Introduction of the 5-methylthien-2-yl moiety in contrast induced complete loss of DENV inhibitory activity only within series 6 (6e and 6f in contrast to 7e and 7f). Within the latter series the 5-methylthien-2-yl moiety seems to be accommodated well affording DENV inhibitory activity at 2.0 and 2.9 μM, respectively, as well as strong YFV inhibition. Finally, a 3-thienyl at the para position of ring C proved less rewarding.
In addition, some analogues were synthesized comprising one reversed amide bond starting from anthranilic acid (Scheme 2) as displayed in Table 3. Unfortunately this modification mostly annihilated the inhibitory activity. Only compound 13b displayed nice DENV inhibitory properties (EC50 = 3.2 μM), comparable in strength to its congener 7f where the latter was based on the ortho-phthalic acid central scaffold of series 7.
Table 3.
Antiviral activity of compounds 13a–13d having a reversed amide bond.
| Sr. No. | Heterocycle (positiona) | Dengue |
Yellow fever |
||||
|---|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | SI | EC50 (μM) | CC50 (μM) | SI | ||
| 13a | D4 | >116 | >116 | NA | >116 | >116 | NA |
| 13b | D3 | 3.2 | >113 | >35 | >113 | >113 (12.5 visual) | 1 |
| 13c | Pyridin-4-yl | 17 | >117 | >7 | >117 | >50 (30 visual) | 1 |
| 13d | Pyridin-3-yl | 23 | >117 | >5 | >117 | >50 (39 visual) | 1 |
Indicates the attachment point of the heterocycle on the phenyl ring.
Unfortunately, several attempts have been made to generate resistant viruses without success, and no clue so far has been obtained regarding the target of these polyaromatic compounds. Hence, all “rationalized” activities for the different modifications remain hypothetical, as no clear-cut structure-activity relationship can be worked out in absence of an interacting target.
Summarizing the antiviral results for all synthesized compounds, fourteen of them (6b, 6d, 6g, 6h, 6m, 6n, 7a, 7e–j and 13b) exhibited selective activity against DENV in the low micromolar range (EC50 = 0.5–3.4 μM). In particular, derivatives 6b, 6d and 7g turned out to be most potent against DENV (EC50 = 0.5 μM) and they were found to be non-cytotoxic at concentrations up to >116 μM resulting in selectivity indices above 235. As far as the activity against YFV is concerned, seven of the tested compounds (6i, 6j, 6n, 7g, 7i, 7j and 7l) showed inhibition in the low micromolar range (EC50 = 0.4–3.3 μM). Among these derivatives, 6n and 7i were found to be most potent against YFV with an EC50 = 0.4 μg/mL and 1.4 μM, respectively.
3. Conclusion
We performed the synthesis and SAR of a series of phthalic diamide and pyrazine-2, 3-dicarboxamide derivatives for the inhibition of DENV and YFV. In total, 17 hits have been identified to inhibit either DENV or YFV during in vitro evaluation, five of which inhibit both viruses. Two of these molecules 6b and 6d displayed most interesting activities against DENV with EC50 = 0.5 μM and SI above 235. Compound 7a likewise exhibited anti-DENV activity with an EC50 = 0.5 μM and a SI of >235. In addition, compound 6n demonstrated strong inhibition of YFV growth with an EC50 = 0.4 μM and a selectivity index of >34. Reversal of one of the central amide bonds proved deleterious to the inhibitory activity.
4. Materials and methods
4.1. General procedure for synthesis of 4a–d
[13] Sodium dodecyl sulfate (SDS, 6 mmol) was added to a stirred heterogeneous suspension of amine (5 mmol) in water (20 mL) until a homogeneous solution was formed, (in case of turbidity, the mixture was warmed to obtain a clear solution). Phthalic anhydride (5 mmol) dissolved in acetonitrile (5 mL) was added to this solution in one lot. After stirring for 1 h at room temperature, the acetonitrile was evaporated and the product precipitated from the aqueous layer. To the aqueous solution containing precipitate, solid sodium hydrogen carbonate was added pinch-wise until the effervescence ceased and the pH reached 6.0. The remaining precipitated product was filtered, washed with water (20 mL), and dried in a vacuum desiccator. In cases where the product did not precipitate, the reaction mixture was extracted with ethyl acetate (2 × 25 mL). The combined organic extracts were dried with anhydrous Na2SO4 and the solvent was removed in a rotary evaporator under reduced pressure to yield the pure product, as solids.
4.1.1. 3-(p-tolylcarbamoyl)pyrazine-2-carboxylic acid (4a)
Yield: 75%; 1H NMR (300 MHz, DMSO-d6) δ: 10.68 (bs, 1H, COOH), 8.89 (s, 2H), 7.66 (d, 2H, J = 8.4 Hz), 7.18 (d, 2H, J = 8.4 Hz), 2.29 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 166.5, 162.3, 146.5, 145.8, 145.6, 144.5, 136.0, 133.4, 129.3 (2xC), 120.2 (2xC), 20.7; HRMS calcd. for C13H10N3O3 [M-H]−: 256.0728; found: 256.0743.
4.1.2. 3-((4-fluorophenyl)carbamoyl)pyrazine-2-carboxylic acid (4b)
Yield: 70%; 1H NMR (300 MHz, DMSO-d6) δ: 10.86 (bs, 1H, COOH), 8.90 (s, 2H), 7.83–7.79 (m, 2H), 7.25–7.19 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ: 165.9 (br), 164.1, 160.0, 156.8, 144.7 (br), 143.8 (br), 135.5 (JC,F = 6 Hz), 121.6 (2xC, JC,F = 30 Hz), 115.5 (2xC, JC,F = 87 Hz) + missing quaternary signal; HRMS calcd. For C12H7N3O3F1 [M-H]-: 260.0477; found: 260.0480.
4.1.3. 2-(p-tolylcarbamoyl)benzoic acid (4c)
Yield: 72%; 1H NMR (300 MHz, DMSO-d6) δ: 10.69 (bs, 1H, COOH), 9.08 (s, 1H), 8.89 (s, 2H), 7.69 (d, 2H, J = 8.4 Hz), 7.36 (s, 1H), 7.18 (d, 2H, J = 8.4 Hz), 2.29 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 166.5, 162.3, 149.0, 145.7, 144.5, 136.0, 133.4, 130.0, 129.6 (2xC), 129.3, 127.3 (2xC), 120.2, 20.6; HRMS calcd. for C15H14N1O3 [M+H]+: 256.0968; found: 256.0970.
4.1.4. 2-((4-fluorophenyl)carbamoyl)benzoic acid (4d)
Yield: 71%; 1H NMR (300 MHz, DMSO-d6) δ: 13.05 (bs, 1H, COOH), 10.38 (bs, 1H, NH), 7.90–7.21 (m, 6H), 7.18 (t, 2H, J = 9.0 Hz); 13C NMR (75 MHz, DMSO-d6) δ: 167.4, 167.3, 159.7, 156.5, 138.8, 136.0, 131.7, 129.9, 129.5, 127.8, 121.3, 121.2, 115.3, 115.1; HRMS calcd. for C14H9N1O3F1 [M-H]−: 258.0572; found: 258.0571.
4.2. General procedure for synthesis of 5a–d
[14]Trifluoroacetic anhydride (1.5 equiv) was added dropwise to a stirring solution of acid 4a–d (1 equiv) and Et3N (TEA, 3 equiv) in 1,4-dioxane (20 mL) that was kept at 0 °C with an ice bath. After 15 min the yellow solution was allowed to warm to room temperature and was stirred for 30 min, then it was poured in cold H2O (100 mL). A yellowish precipitate formed, which was collected by filtration using a Buchner funnel and washed with H2O. The product was dried overnight under high vacuum.
4.2.1. (Z)-7-(p-tolylimino)furo [3,4-b]pyrazin-5(7H)-one (5a)
Yield: 70%; 1H NMR (300 MHz, CDCl3) δ: 9.08–9.03 (m, 2H), 7.63 (d, 2H, J = 8.1 Hz), 7.29 (d, 2H, J = 6.0 Hz), 2.43 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ: 150.5, 149.0, 129.5, 126.4, 21.0. + missing quaternary C signals; HRMS calcd. for C13H10N3O2 [M+H]+: 240.0766; found: 240.0766.
4.2.2. (Z)-7-((4-fluorophenyl)imino)furo [3,4-b]pyrazin-5(7H)-one (5b)
Yield: 68%; 1H NMR (300 MHz, CDCl3) δ: 9.09–9.05 (m, 2H), 7.76–7.47 (m, 2H), 7.28–7.14 (m, 2H). 13C NMR (75 MHz, CDCl3) δ: 150.9, 149.5, 128.7, 128.6, 116.2, 115.9 + missing quaternary C signals; HRMS calcd. for C12H7N3O2F1 [M+H]+: 244.0517; found: 244.0508.
4.2.3. (Z)-3-(p-tolylimino)isobenzofuran-1(3H)-one (5c)
Yield: 65%; 1H NMR (300 MHz, CDCl3) δ: 8.11–7.98 (m, 2H), 7.86–7.76 (m, 2H), 7.42 (d, 2H, J = 8.4 Hz), 7.23 (d, 2H, J = 8.4 Hz), 2.40 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 164.8, 147.3, 141.6, 136.5, 136.0, 135.5, 133.7, 129.6, 129.1, 127.6, 125.4, 124.1, 123.4, 20.8 + missing C N-Ph signal; HRMS calcd. for C15H12N1O2 [M+H]+: 238.0862; found: 238.0843.
4.2.4. (Z)-3-((4-fluorophenyl)imino)isobenzofuran-1(3H)-one (5d)
Yield: 67%; 1H NMR (300 MHz, DMSO-d6) δ: 8.13–7.88 (m, 4H), 7.42–7.30 (m, 2H), 7.30–7.22 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ: 164.7, 161.8, 158.5, 140.9, 136.3, 136.1, 134.0, 127.9, 125.9, 125.8, 125.5, 123.5, 116.0, 115.7; HRMS calcd. for C14H9N1O2F1 [M+H]+: 242.0612; found: 242.0615.
4.2.5. General procedure for synthesis of 6a–n and 7a–n
[14] Phthalisoimide 5a–d (1 equiv) was added to a stirring solution of the aniline/benzylamine (1.2 equiv) in THF. The mixture was stirred overnight at room temperature. Two alternative work-up procedures were followed depending on whether the product precipitated out of solution or not.
Work-up 1 (for soluble products): The THF was evaporated on a rotavapor. The mixture was diluted with EtOAc (using the triple volume of THF) and washed three times with 1 M HCl. The organic phase was dried with Na2SO4 and evaporated. The product was purified by flash chromatography on silica gel using a MeOH gradient in DCM and affording a greyish precipitate following evaporation.
Work-up 2 (for insoluble products): The precipitate was collected by filtration using a Buchner funnel and washed on the frit with small volumes of 1:1 Et2O/n-hexane until the yellow impurities had disappeared. The solid product obtained from this work-up procedure did not need further chromatographic purification.
4.2.6. N2-(4-fluorophenyl)-N3-(4-methyl-2-(3-methyl-1H-pyrazol-5-yl)phenyl) pyrazine-2,3 dicarboxamide (6a)
Yield: 72%; 1H NMR (300 MHz, DMSO-d6) δ: 13.10 (bs, 1H), 13.00 (bs, 1H), 10.62 (bs, 1H), 8.97–8.90 (bs, 2H), 8.50 (d, 1H, J = 8.1 Hz), 7.70–7.57 (m, 3H), 7.25–7.11 (m, 3H), 6.58 (bs, 1H), 2.33 (s, 6H, 2CH3); 13C NMR (150 MHz, CDCl3) δ: 207.0, 163.1, 161.3, 160.5, 158.9, 149.6, 149.2, 144.7, 143.3, 133.6, 133.4, 129.3, 128.4, 121.8, 121.8, 121.7, 121.6, 121.1, 116.4, 115.8, 103.4, 21.0, 11.0; HRMS calcd. for C23H20F1N6O2 [M+H]+: 431.1626; found: 431.1620.
4.2.7. N2-(4-methyl-2-(3-methyl-1H-pyrazol-5-yl)phenyl)-N3-(p-tolyl)pyrazine-2,3-dicarboxamide (6b)
Yield: 65%; 1H NMR (600 MHz, CDCl3) δ: 11.93 (bs, 0.6H), 9.12 (bs, 0.7H), 8.66–8.59 (bs, 2H), 7.60–7.13 (m, 7H), 6.36 (s, 1H), 2.37 (s, 3H, CH3), 2.32 (s, 3H, CH3), 2.28 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ: 207.0, 163.2, 161.1, 151.3, 149.2, 145.6, 144.6, 143.2, 142.0, 140.3, 134.9, 134.4, 133.4, 129.6, 129.5, 128.3, 121.5, 121.1, 120.0, 119.9, 103.2, 21.0, 20.9, 10.9; HRMS calcd. for C24H22N6O2Na1 [M+Na]+: 449.1697; found: 449.1697.
4.2.8. N2-(4-fluorophenyl)-N3-(4-methyl-2-(thiophen-2-yl)phenyl)pyrazine-2,3-dicarboxamide (6c)
Yield: 73%; 1H NMR (600 MHz, CDCl3) δ: 8.99 (bs, 0.7H, NH), 8.92 (bs, 0.8H, NH), 8.64–8.59 (m, 2H), 8.36 (d, 1H, J = 4.2 Hz), 7.70–7.68 (m, 2H), 7.48–7.42 (m, 2H) + coinciding CHCl3 peak, 7.27–7.15 (m, 3H), 7.09–7.03 (m, 2H), 2.37 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ: 161.7, 149.6, 146.9, 144.2, 144.1, 138.2, 134.7, 133.4, 131.8, 130.5, 129.1, 128.7, 128.3, 128.3, 127.5, 126.6, 123.8121.9, 121.7, 116.4, 115.8, 115.7, 20.9; HRMS calcd. for C23H18F1N4O2S1 [M+H]+: 433.1129; found: 433.1133.
4.2.9. N2-(4-methyl-2-(thiophen-2-yl)phenyl)-N3-(p-tolyl)pyrazine-2,3-dicarboxamide (6d)
Yield: 78%; 1H NMR (600 MHz, CDCl3) δ: 8.95 (bs, 1H, NH), 8.78 (bs, 1H, NH), 8.64–8.59 (m, 2H), 8.38 (d, 1H, J = 4.2 Hz), 7.60 (d, 2H, J = 3.9 Hz), 7.47 (bs, 1H), 7.42–7.38 (m, 1H), 7.28–7.15 (m, 5H), 2.37 (s, 3H, CH3), 2.34 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ: 162.0, 161.3, 147.3, 146.4, 144.2, 143.9, 138.3, 134.8, 134.5, 134.5, 131.9, 130.4, 129.5 (2xC), 129.0, 128.7, 127.4, 126.5, 123.8, 121.9, 120.1, 120.0, 20.9 (2xC); HRMS calcd. for C24H21N4O2S1 [M+H]+: 429.1380; found: 449.1376.
4.2.10. N2-(4-fluorophenyl)-N3-(4-methyl-2-(5-methylthiophen-2-yl)phenyl)pyrazine-2,3-dicarboxamide (6e)
Yield: 69%; 1H NMR (600 MHz, CDCl3) δ: 9.19 (bs, 0.7H, NH), 8.91 (bs, 0.7H, NH), 8.73–8.63 (m, 2H), 8.45–8.38 (m, 1H), 7.71–7.69 (m, 2H), 7.23–7.14 (m, 2H), 7.09–7.03 (m, 3H), 6.75 (bs, 1H), 2.51 (s, 3H, CH3), 2.36 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ: 161.7, 160.3, 158.7, 149.6, 146.5, 144.2, 144.1, 141.2, 136.9, 134.6, 133.2, 131.1, 129.3, 128.0, 127.0, 125.9, 125.0, 122.0, 121.8, 116.4, 115.9, 115.7, 20.9, 15.5; HRMS calcd. for C24H20F1N4O2S1 [M+H]+: 447.1285; found: 447.1283.
4.2.11. N2-(4-methyl-2-(5-methylthiophen-2-yl)phenyl)-N3-(p-tolyl)pyrazine-2,3-dicarboxamide (6f)
Yield: 75%; 1H NMR (600 MHz, CDCl3) δ: 9.07 (bs, 0.7H, NH), 8.89 (bs, 0.7H, NH), 8.67–8.65 (m, 2H), 8.42–8.40 (m, 1H), 7.61 (d, 2H, J = 3.9 Hz), 7.25–7.12 (m, 4H), 7.07 (d, 1H), 6.74 (bs, 1H), 2.50 (s, 3H, CH3), 2.36 (s, 3H, CH3), 2.34 (s, 3H, CH3); 13C NMR (150 MHz, CDCl3) δ: 161.9, 161.4, 147.3, 146.6, 144.2, 143.9, 141.1, 137.0, 134.8, 134.5, 131.8, 131.0, 129.6 (2xC), 129.3, 127.0, 125.9, 125.4, 121.9, 121.8, 120.2, 120.1, 21.0, 20.9, 15.3; HRMS calcd. for C25H21N4O2S1 [M-H]−: 441.1391; found: 441.1367.
4.2.12. N2-(4-fluorophenyl)-N3-(4-methyl-2-(thiophen-3-yl)phenyl)pyrazine-2,3- dicarboxamide (6g)
Yield: 64%; 1H NMR (500 MHz, CDCl3) δ: 8.95 (bs, 1H, NH), 8.89 (bs, 1H, NH), 8.65 (bs, 1H), 8.62 (bs, 1H), 8.38 (d, 1H, J = 7.0 Hz), 7.72–7.69 (m, 2H), 7.46–7.42 (m, 2H), 7.30–7.16 (m, 3H), 7.08–7.05 (t, 2H, J = 7.25 Hz), 2.37 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ: 161.8, 161.6, 160.5, 158.9, 147.1, 146.4, 144.3, 144.0, 138.2, 134.7, 133.4, 131.8, 130.5, 129.1, 127.5, 126.6, 123.8, 122.0, 121.9, 121.8, 115.8, 115.7, 20.9; HRMS calcd. for C23H18F1N4O2S1 [M+H]+: 433.1129; found: 433.1121.
4.2.13. N2-(4-methyl-2-(thiophen-3-yl)phenyl)-N3-(p-tolyl)pyrazine-2,3-dicarboxamide (6h)
Yield: 69%; 1H NMR (500 MHz, CDCl3) δ: 8.94 (bs, 1H, NH), 8.76 (bs, 1H, NH), 8.66 (s, 1H), 8.63 (s, 1H), 8.39 (d, 1H, J = 7.0 Hz), 7.62–7.61 (d, 2H, J = 7.0 Hz), 7.48 (bs, 1H), 7.42–7.41 (m, 1H), 7.28–7.27 (d, 1H, J = 4.0 Hz), 7.22–7.17 (m, 4H), 2.37 (s, 3H, CH3), 2.34 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ: 162.1, 161.3, 149.0, 147.4, 146.3, 144.3, 143.8, 138.3, 134.8, 134.6, 131.9, 130.4, 129.6 (2xC), 129.0, 128.8, 127.4, 126.5, 123.8, 121.8, 120.1 (2xC), 21.0, 20.9; HRMS calcd. for C24H21N4O2S1 [M+H]+: 429.1380; found: 429.1366.
4.2.14. N2-(4-fluorophenyl)-N3-(2-(thiophen-3-yl)benzyl)pyrazine-2,3-dicarboxamide (6i)
Yield: 68%; 1H NMR (300 MHz, DMSO-d6) δ: 10.67 (bs, 1H, NH), 9.44–9.24 (dt, 1H, NH rotamers), 8.91–8.87 (m, 2H), 7.79–7.18 (m, 10H), 4.50 (d, 2H, J = 6.0 Hz, CH2); 13C NMR (75 MHz, DMSO-d6) δ: 164.2, 163.5, 160.0, 156.8, 148.1, 147.6, 146.1, 145.3, 144.9, 144.5, 140.4, 137.4, 136.0, 135.3, 132.4, 129.7, 129.1, 129.0, 128.5, 127.7, 127.4, 127.0, 126.2, 123.8, 122.1, 121.6, 121.6, 121.5, 121.5, 115.6, 115.3, 42.9 (many double peaks as of rotamers); HRMS calcd. for C23H18F1N4O2S1 [M+H]+: 433.1129; found: 433.1121.
4.2.15. N2-(2-(thiophen-3-yl)benzyl)-N3-(p-tolyl)pyrazine-2,3-dicarboxamide (6j)
Yield: 65%; 1H NMR (300 MHz, DMSO-d6) δ: 10.65 (bs, 1H, NH), 9.39 (t, 1H), 9.24 (t, 1H), 8.91–8.86 (m, 2H), 7.67–7.15 (m, 10H), 4.50 (d, 2H, J = 6.0 Hz, CH2), 2.29 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 164.4, 163.4, 148.3, 147.8, 146.2, 145.2, 144.9, 144.6, 140.5, 137.5, 136.5, 135.1, 132.9, 132.4, 129.7, 129.3, 129.1, 128.6, 127.8, 127.5, 127.0, 126.3, 123.9, 122.2, 119.8, 42.9, 20.6; HRMS calcd. for C24H21N4O2S1 [M+H]+: 429.1380; found: 429.1380.
4.2.16. N2-(4-fluorophenyl)-N3-(4-(thiophen-3-yl)benzyl)pyrazine-2,3-dicarboxamide (6k)
Yield: 72%; white solid; 1H NMR (300 MHz, DMSO-d6) δ: 10.65 (bs, 1H, NH), 9.34 (t, 1H, J = 5.3 Hz), 8.89–8.87 (m, 2H), 7.85–7.54 (m, 7H), 7.40 (d, 2H, J = 8.1 Hz), 7.21 (t, 2H, J = 8.5 Hz), 4.50 (d, 2H, J = 6.3 Hz, CH2); 13C NMR (75 MHz, DMSO-d6) δ: 164.0, 163.7, 160.1, 156.9, 148.2, 145.5, 145.2, 144.5, 141.4, 138.1, 135.5, 133.9, 128.0 (2xC), 127.2, 126.3, 126.1 (2xC), 121.7, 121.6, 120.8, 115.7, 115.4, 42.2; HRMS calcd. for C23H18F1N4O2S1 [M+H]+: 433.1129; found: 433.1122.
4.2.17. N2-(4-(thiophen-3-yl)benzyl)-N3-(p-tolyl)pyrazine-2,3-dicarboxamide (6l)
Yield: 75%; white solid; 1H NMR (300 MHz, DMSO-d6) δ: 10.49 (bs, 1H, NH), 9.36 (t, 1H, J = 5.3 Hz), 8.88–8.85 (m, 2H), 7.85–7.84 (m, 1H), 7.68–7.54 (m, 6H), 7.40 (d, 2H, J = 8.1 Hz), 7.17 (d, 2H, J = 8.4 Hz), 4.49 (d, 2H, J = 6.3 Hz), 2.29 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 164.0, 163.6, 148.4, 145.6, 145.1, 144.4, 141.4, 138.1, 136.6, 133.9, 132.8, 129.2 (2xC), 128.0 (2xC), 127.1, 126.3, 126.1 (2xC), 120.8, 119.8, 119.7, 42.2, 20.7; HRMS calcd. for C24H21N4O2S1 [M+H]+: 429.1380; found: 429.1375.
4.2.18. N2-(4-fluorophenyl)-N3-(2-(3-methyl-1H-pyrazol-5-yl)phenyl)pyrazine-2,3-dicarboxamide (6m)
Yield: 65%; white solid; 1H NMR (300 MHz, DMSO-d6) δ: 13.20 (bs, 1H, NH), 13.00 (bs, 1H, NH), 10.64 (bs, 1H, NH), 8.98–8.91 (m, 2H), 8.62–8.60 (m, 1H), 7.76–7.70 (m, 3H), 7.33–7.15 (m, 4H), 6.60 (s, 1H), 2.32 (s, 3H, CH3); 13C NMR (75 MHz,, DMSO-d6) δ: 164.1, 161.7, 159.4, 157.4, 150.2, 149.7, 149.1, 146.1, 144.1, 143.7, 139.9, 135.3, 128.0, 127.9, 124.1, 121.8, 121.4, 120.3, 115.6, 115.5, 102.7, 10.4; HRMS calcd. for C22H16F1N6O2 [M-H]-: 415.1324; found: 415.1308.
4.2.19. N2-(2-(3-methyl-1H-pyrazol-5-yl)phenyl)-N3-(p-tolyl)pyrazine-2,3-di-carboxamide (6n)
Yield: 62%; white solid; 1H NMR (300 MHz,, DMSO-d6) δ: 13.17 (bs, 1H, NH), 13.00 (bs, 1H, NH), 10.48 (bs, 1H, NH), 8.97–8.90 (m, 2H), 8.62–8.60 (m, 1H), 7.76–7.74 (m, 1H), 7.57 (d, 2H, J = 8.5 Hz), 7.30–7.16 (m, 4H), 6.59 (s, 1H), 2.32 (s, 3H, CH3), 2.29 (s, 3H, CH3); 13C NMR (75 MHz,, DMSO-d6) δ: 164.0, 161.7, 150.2, 149.9, 146.1, 144.0, 143.8, 139.9, 136.6, 135.3, 132.8, 129.3 (2xC), 128.0, 127.9, 124.1, 121.8, 120.3, 119.6 (2xC), 102.7, 20.6, 10.4; HRMS calcd. for C23H21N6O2 [M+H]+: 413.1720; found: 413.1727.
4.2.20. N1-(4-fluorophenyl)-N2-(4-methyl-2-(3-methyl-1H-pyrazol-5-yl)phenyl)phthal-amide (7a)
Yield: 65%; 1H NMR (600 MHz, DMSO-d6) δ: 12.90 (bs, 1H), 12.27 (bs, 1H), 10.50 (bs, 1H, NH), 10.44 (s, 1H), 8.46 (d, 1H, J = 8.4 Hz), 7.83 (m, 1H), 7.75–7.53 (m, 6H), 7.17–7.08 (m, 3H), 6.61 (s, 1H), 2.32 (s, 3H, CH3), 2.28 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 166.7, 166.5, 165.7, 159.0, 157.4, 150.7, 139.7, 137.6, 136.7, 136.4, 135.9, 133.9, 132.3, 130.4, 130.1, 128.4, 128.3, 127.2, 121.5 (2xC), 120.5, 119.8, 115.3 (2xC), 102.6, 20.6, 10.4; HRMS calcd. for C25H20F1N4O2 [M-H]-: 427.1576; found: 427.1577.
4.2.21. N1-(4-fluorophenyl)-N2-(4-methyl-2-(thiophen-2-yl)phenyl)phthalamide (7b)
Yield: 68%; 1H NMR (600 MHz, DMSO-d6) δ: 10.43 (s, 0.4H), 9.78 (s, 1H), 9.12 (bs, 0.4H), 8.12–7.00 (m, 13H), 7.68–7.52 (m, 1H), 2.50 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 167.7, 167.6, 165.0, 155.5, 145.5, 141.0, 139.7, 137.3–115.1 (complex aromatic pattern), 22.5; HRMS calcd. for C25H20F1N2O2S1 [M+H]+: 431.1223; found: 431.1221.
4.2.22. N1-(4-methyl-2-(3-methyl-1H-pyrazol-5-yl)phenyl)-N2-(p-tolyl)phthalamide (7c)
Yield: 65%; 1H NMR (600 MHz, DMSO-d6) δ: 12.89 (bs, 0.7H), 12.24 (bs, 0.7H), 10.31 (s, 1H), 8.46 (bs, 0.7H), 8.03–7.52 (m, 7H), 7.23–7.00 (m, 3H), 6.73–6.68 (m, 1H), 2.33 (s, 3H, CH3), 2.26 (s, 3H, CH3), 2.18 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 170.1, 166.9, 165.9, 139.5, 137.8, 137.0, 136.5, 133.9, 132.3, 130.3, 130.0, 129.7, 129.0 (2xC), 128.4, 128.0, 127.9, 127.1, 119.7 (2xC), 115.7, 102.6, 22.1, 21.2, 14.1; HRMS calcd. for C26H25N4O2 [M+H]+: 425.1972; found: 425.1971.
4.2.23. N1-(4-methyl-2-(thiophen-2-yl)phenyl)-N2-(p-tolyl)phthalamide (7d)
Yield: 69%; 1H NMR (500 MHz, DMSO-d6) δ: 13.05 (bs, 1H), 8.89 (bs, 0.6H), 7.85 (d, 1H), 7.78–6.95 (m, 11H), 6.75–6.55 (m, 2H), 2.23 (s, 3H, CH3), 2.15 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 167.6 (2xC), 154.5, 146.0, 141.1, 137.4, 137.2, 132.1, 131.1, 130.7, 130.6, 130.2, 129.4, 129.1 (2xC), 128.6, 128.3 (2xc), 125.3, 123.8, 122.7, 122.4, 120.1, 119.6, 20.9, 20.8; HRMS calcd. for C26H23N2O2S1 [M+H]+: 427.1475; found: 427.1471.
4.2.24. N1-(4-methyl-2-(5-methylthiophen-2-yl)phenyl)-N2-(p-tolyl)phthalamide (7e)
Yield: 78%; 1H NMR (600 MHz, DMSO-d6) δ: 10.66 (bs, 1H), 10.26 (s, 1H), 9.09 (s, 1H), 8.92 (d, 1H), 8.89 (d, 1H), 7.76–7.73 (m, 3H), 7.32 (d, 1H), 7.25–7.17 (m, 5H), 6.82 (d, 1H), 2.51 (s, coinciding with DMSO-d5, CH3), 2.45 (s, 3H, CH3), 2.33 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 163.8, 163.4, 162.3, 159.2, 157.7, 149.0, 147.8, 145.5, 144.8, 144.6, 140.2, 136.9, 135.4, 131.2, 129.9, 128.6, 128.3, 126.8, 126.3, 125.5, 121.7 (2xC), 115.6 (2xC), 20.6 (2xC), 15.0; HRMS calcd. for C27H24N2O2S1Na1 [M+Na]+: 463.1451; found: 463.1450.
4.2.25. N1-(4-fluorophenyl)-N2-(4-methyl-2-(5-methylthiophen-2-yl)phenyl)phthalamide (7f)
Yield: 65%; 1H NMR (500 MHz, DMSO-d6) δ: 10.40 (s, 1H, NH), 9.87 (s, 1H, NH), 7.74–7.05 (m, 12H), 6.79 (d, 1H), 2.45 (s, 3H, CH3), 2.32 (s, 3H, CH3); 13C NMR (150 MHz, DMSO-d6) δ: 167.2, 167.0, 159.0, 157.3, 139.7, 137.6, 137.0, 136.2, 136.0, 132.8, 131.7, 130.5, 129.9, 129.4, 128.7, 128.4, 128.0, 127.5, 126.3, 125.9, 121.4 (2xC), 115.3 (2xC), 115.2, 20.6, 15.0; HRMS calcd. for C26H21F1N2O2S1Na1 [M+Na]+: 467.1200; found: 467.1198.
4.2.26. N1-(4-methyl-2-(thiophen-3-yl)phenyl)-N2-(p-tolyl)phthalamide (7g)
Yield: 68%; 1H NMR (500 MHz, CDCl3) δ: 8.85 (bs, 1H, NH), 8.08 (d, 1H, J = 7.8 Hz), 7.84 (d, 1H, J = 6 Hz), 7.80 (m, 1H), 7.53–7.15 (m, 12H), 2.37 (s, 3H, CH3), 2.34 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ: 168.1, 165.8, 138.2, 135.4, 135.2, 134.2, 131.8, 130.7, 130.5, 129.7 (multiple), 129.0, 128.7, 128.2, 127.2, 126.8, 123.7 (2xC), 122.7, 120.2 (2xC), 20.9 (2xC); HRMS calcd. for C26H22N2O2S1Na1 [M+Na]+: 449.1294; found: 449.1289.
4.2.27. N1-(4-fluorophenyl)-N2-(4-methyl-2-(thiophen-3-yl)phenyl)phthalamide (7h)
Yield: 64%; 1H NMR (500 MHz, CDCl3) δ: 9.30 (bs, 1H, NH), 8.07 (d, 1H, J = 7.0 Hz), 7.80 (d, 2H, J = 7.0 Hz), 7.60 (m, 2H), 7.50–7.35 (m, 3H), 7.30 (m, 2H), 7.20–7.12 (m, 3H), 7.00–6.94 (m, 2H), 2.36 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ: 168.2, 165.9, 160.2, 158.6, 156.0, 138.2, 135.3, 135.1, 134.2, 131.8, 130.7, 130.5, 129.0, 128.7, 128.2, 127.0 (2xc), 123.7, 122.6, 121.9, 121.8, 115.6, 115.4, 20.9; HRMS calcd. for C25H19F1N2O2S1Na1 [M+Na]+: 453.1044; found: 453.1038.
4.2.28. N1-(4-fluorophenyl)-N2-(2-(thiophen-3-yl)benzyl)phthalamide (7i)
Yield: 74%; 1H NMR (500 MHz, DMSO-d6) δ: 10.41 (bs, 1H, NH), 9.00–8.83 (dt, 1H, NH rotamers), 7.75–7.14 (m, 15H), 4.44 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ: 168.1, 167.1, 159.1, 157.2, 140.6, 137.7, 137.1, 137.0, 136.5, 136.0, 135.8, 135.3, 132.3, 130.0, 129.7, 129.6, 129.1, 128.9, 128.8, 127.9, 127.7, 127.5, 126.9, 126.2, 123.8, 122.1, 121.3 (2xC), 115.5, 115.3, 43.1 (many double peaks as of rotamers); HRMS calcd. for C25H19F1N2O2S1Na1 [M+Na]+: 453.1044; found: 453.1046.
4.2.29. N1-(2-(thiophen-3-yl)benzyl)-N2-(p-tolyl)phthalamide (7j)
Yield: 68%; 1H NMR (300 MHz, DMSO-d6) δ: 10.27 (bs, 1H, NH), 8.98–8.81 (m, 1H), 7.69–7.50 (m, 9H), 7.30–7.08 (m, 6H), 4.44 (d, 2H, J = 5.7 Hz), 2.28 (s, 3H, CH3); 13C NMR (75 MHz,, DMSO-d6) δ: 168.3, 167.0, 140.6, 137.8, 137.1, 136.5, 136.2, 135.9, 135.4, 132.3, 129.9, 129.6, 129.1, 128.9, 128.8, 128.0, 127.9, 127.8, 127.5, 126.9, 126.2, 123.8, 122.2, 119.7, 43.2, 20.6; HRMS calcd. for C26H21N2O2S1 [M-H]-: 425.1329; found: 425.1333.
4.2.30. N1-(4-fluorophenyl)-N2-(4-(thiophen-3-yl)benzyl)phthalamide (7k)
Yield: 69%; 1H NMR (500 MHz, DMSO-d6) δ: 10.39 (bs, 1H, NH), 8.94 (t, 1H, J = 5.0 Hz), 7.80 (d, 1H), 7.73–7.68 (m, 2H), 7.65–7.50 (m, 8H), 7.36 (d, 2H, J = 7 Hz), 7.16 (t, 2H, J = 7.5 Hz), 4.42 (d, 2H, J = 5.0 Hz); 13C NMR (125 MHz, DMSO-d6) δ: 168.2, 167.5, 159.3, 157.7, 141.7, 138.7, 137.5, 136.4, 134.0, 130.2 (2xC), 130.0, 128.2 (2xC), 127.5, 126.5 (2xC), 121.7, 121.6 (2xC), 115.7, 115.5, 42.4; HRMS calcd. for C25H19F1N2O2S1Na1 [M+Na]+: 453.1044; found: 453.1044.
4.2.31. N1-(4-(thiophen-3-yl)benzyl)-N2-(p-tolyl)phthalamide (7l)
Yield: 61%; 1H NMR (300 MHz, DMSO-d6) δ: 10.25 (bs, 1H, NH), 8.90 (t, 1H, J = 6.2 Hz), 7.82 (dd, 1H, J = 2.7 Hz and 1.2 Hz), 7.70–7.45 (m, 10H), 7.38 (d, 2H, J = 8 Hz), 7.13 (d, 2H, J = 8.4 Hz), 4.44 (d, 2H, J = 6.0 Hz), 2.27 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 168.0, 167.0, 141.4, 138.4, 137.2, 136.1, 133.7, 132.3, 129.8, 129.6, 129.1 (2xC), 127.9 (2xc),127.1, 126.2, 126.0 (2xC), 120.7 (2xC), 119.7, 42.4, 20.6; HRMS calcd. for C26H22N2O2S1Na1 [M+Na]+: 449.1294; found: 449.1288.
4.2.32. N1-(4-fluorophenyl)-N2-(2-(3-methyl-1H-pyrazol-5-yl)phenyl)phthalamide (7m)
Yield: 72%; 1H NMR (500 MHz, DMSO-d6) δ: 12.94 (bs, 1H, NH), 12.40 (bs, 1H, NH), 10.51 (bs, 1H, NH), 8.58 (d, 1H, J = 6.5 Hz), 7.85–7.60 (m, 8H), 7.29 (t, 1H, J = 6.5 Hz), 7.20–7.10 (m, 3H), 6.64 (s, 1H), 2.29 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 167.1, 166.1, 159.0, 157.4, 150.7, 139.9, 137.6, 136.3, 135.9, 130.5, 130.2, 128.5, 127.9 (2xC), 127.3, 123.5, 121.4 (2xC), 120.6, 119.8, 115.4, 115.2, 102.6, 10.4; HRMS calcd. for C24H19F1N4O2Na1 [M+Na]+: 437.1384; found: 437.1379.
4.2.33. N1-(2-(3-methyl-1H-pyrazol-5-yl)phenyl)-N2-(p-tolyl)phthalamide (7n)
Yield: 61%; 1H NMR (500 MHz, DMSO-d6) δ: 12.94 (bs, 1H, NH), 12.37 (bs, 1H, NH), 10.34 (bs, 1H, NH), 8.60 (d, 1H, J = 5.0 Hz), 7.82 (t, 1H), 7.77 (dd, 1H), 7.68–7.62 (m, 3H), 7.54 (d, 2H, J = 7 Hz), 7.28 (t, 1H, J = 6.5 Hz), 7.15–7.05 (m, 2H), 6.64 (s, 1H), 2.28 (s, 3H, CH3), 2.26 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δ: 166.9, 166.2, 150.7, 139.9, 138.0, 137.1, 136.6, 136.4, 132.4, 130.5, 130.1, 129.1 (2xC), 128.5, 128.0, 127.2, 123.5, 120.5, 119.8, 119.7 (2xC), 102.7, 20.6, 10.4; HRMS calcd. for C25H23N4O2 [M+H]+: 411.1815; found: 411.1811.
4.3. 2-(1,3-dioxoisoindolin-2-yl)benzoic acid (9)
A mixture of anthranilic acid (1.0 mmol), phthalic anhydride (1.1 mmol) and acetic acid was refluxed for 1 h. After cooling, the precipitated product was filtered and washed with water. The obtained product was dried thoroughly in vacuo.
Yield: 83%; white solid; 1H NMR (300 MHz, DMSO-d6) δ: 11.53 (bs, 1H, COOH), 8.62 (d, 1H, J = 8.1 Hz), 8.03 (d, 1H, J = 7.8 Hz), 7.87 (d, 1H, J = 7.5 Hz), 7.70–7.60 (m, 4H), 7.21 (t, 1H, J = 7.2 Hz); 13C NMR (75 MHz, DMSO-d6) δ: 169.8, 167.7, 167.1, 141.2, 138.2, 134.3, 132.0, 131.3, 130.6, 130.3, 129.8, 127.4, 123.1, 120.0, 116.6; HRMS calcd. for C15H8N1O4 [M-H]-: 266.0459; found: 266.0463.
4.4. N-(2-bromo-4-methylphenyl)-2-(1,3-dioxoisoindolin-2-yl)benzamide (10)
To a solution of 9 (1 mmol) in dry DCM (5 mL), SOCl2 (1.5 mmol) and catalytic DMF were added to obtain a clear solution. The reaction mixture was allowed to reflux for 1.5 h. The reaction mixture was evaporated to dryness and dried for 1 h. The reaction mixture was rediluted with DCM and cooled to 0 °C. DIPEA and 2-bromotoludine were added and the reaction was allowed to stir at rt. for 2 h after which the mixture was concentrated to dryness. The oily residue was purified by silica gel column chromatography to give pure compound 10 (80%) as a white solid.
1H NMR (300 MHz, CDCl3) δ: 8.20 (bs, 2H), 7.95–7.35 (m, 9H), 7.08–7.05 (dd, 1H), 2.30 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 167.0 (2xC), 165.1, 137.9, 134.9, 134.8 (2xC), 133.4, 132.8, 131.8, 131.4, 130.3, 129.8, 128.9, 128.7, 128.3, 127.6, 123.6, 123.5 (2xC),120.4, 20.2 (some double peaks as of rotamers); HRMS calcd. for C22H16Br1N2O3 [M+H]+: 435.0339; found: 435.0344.
4.5. 2-amino-N-(2-bromo-4-methylphenyl)benzamide (11)[15]
To a solution of 10 (1.0 mmol) in anhydrous THF (5 mL), was added hydrazine monohydrate (1.1 mmol) at rt. and the mixture was allowed to stir at 60 °C for 16 h. The reaction mixture was evaporated to dryness and the oily residue was purified by silica gel column chromatography to give pure compound 12 as a white solid.
Yield 79%; 1H NMR (300 MHz, DMSO-d6) δ: 9.66 (bs, 1H, NH), 7.72 (d, 1H, J = 8 Hz), 7.53 (d, 1H, J = 1.2 Hz), 7.41 (d, 1H, J = 7.8 Hz), 7.23–7.18 (m, 2H), 6.75 (d, 1H, J = 8.1 Hz), 6.57 (t, 1H, J = 8.1 Hz), 6.43 (bs, 2H, NH2), 2.33 (s, 3H, CH3); HRMS calcd. for C14H14Br1N2O1 [M+H]+: 305.0284; found: 305.0295.
4.6. N-(2-bromo-4-methylphenyl)-2-(4-fluorobenzamido)benzamide (12)
To a solution of 11 (1.0 mmol) and DIPEA (1.5 mmol) in dry DCM (5 mL), was added 4-fluorobenzoyl chloride (obtained from the acid with thionyl chloride) at 0 °C. The reaction mixture was allowed to stir at rt. for 2 h. The reaction mixture was evaporated to dryness and the oily residue was purified by silica gel column chromatography to afford title compound 12 as a white solid.
Yield 72%; 1H NMR (500 MHz, CDCl3) δ: 12.15 (bs, 1H, NH), 8.90 (dd, 2H, J = 8 Hz), 8.60 (dd, 2H, J = 8.1 Hz), 8.38 (d, 1H, J = 7 Hz), 7.75–7.68 (m, 2H), 7.43 (dd, 2H, J = 4 Hz and J = 2.5 Hz), 7.22–7.15 (m, 2H), 7.10–7.03 (m, 2H), 2.35 (s, 3H, CH3); 13C NMR (125 MHz, CDCl3) δ: 161.6 (2xC), 160.5, 158.9, 147.1, 146,4, 144.2, 144.0, 138.2, 134.7, 133.4, 131.8, 130.5, 129.0, 128.7, 127.5, 126.6, 123.8, 122.0, 121.8, 115.9, 115.7, 20.9; HRMS calcd. for C21H17Br1F1N2O2 [M+H]+: 427.0452; found: 427.0445.
4.7. General procedure for synthesis of 13a–d
To a solution of 12 (1.0 mmol) in 5 mL dioxane: water (1:1), was added anhydrous K2CO3 (1.5 mmol), the respective aryl boronic acid (1.2 mmol) and Pd(TPP)2Cl2 (0.03 mmol). The mixture was heated at 100 °C for 16 h. All reactions and manipulations were run under argon atmosphere. After completion, the solvent was evaporated under reduced pressure and the residue was purified by column chromatography on silica gel to afford the desired product.
4.7.1. 2-(4-fluorobenzamido)-N-(4-methyl-2-(thiophen-3-yl)phenyl)benzamide (13a)
Yield: 62%; 1H NMR (300 MHz, CDCl3) δ: 12.15 (bs, 1H, NH), 8.83 (d, 1H, J = 8.4 Hz), 8.19–8.05 (m, 4H), 7.58–7.06 (m, 10H), 2.41 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ: 166.9, 166.4, 164.2, 163.1, 140.1, 138.0, 134.9, 132.8, 131.4, 130.4, 129.6, 129.5, 128.8, 128.3, 127.9, 126.9, 125.9, 123.4, 122.8, 122.1, 121.5, 120.1, 115.6, 115.3, 20.6; HRMS calcd. for C25H18F1N2O2S1 [M-H]−: 429.1078; found: 429.1082.
4.7.2. 2-(4-fluorobenzamido)-N-(4-methyl-2-(5-methylthiophen-2-yl)phenyl)benzamide (13b)
Yield: 65%; 1H NMR (300 MHz, CDCl3) δ: 12.09 (bs, 1H, NH), 8.84 (d, 1H, J = 8.7 Hz), 8.32 (bs, 1H, NH), 8.14–8.05 (m, 3H), 7.60–7.46 (m, 2H), 7.28–7.10 (m, 5H), 6.95 (d, 1H, J = 3.6 Hz), 6.75 (m, 1H), 2.52 (s, 3H, CH3), 2.40 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ: 167.1, 166.4, 164.2, 163.0, 141.3, 140.1, 136.3, 134.9, 132.8, 131.3, 130.9, 129.6, 129.5, 129.0, 126.6, 126.4, 126.2, 125.7, 122.8, 122.5, 121.5, 120.3, 115.6, 115.3, 20.6, 15.0; HRMS calcd. for C26H20F1N2O2S1 [M-H]−: 443.1235; found: 443.1229.
4.7.3. 2-(4-fluorobenzamido)-N-(4-methyl-2-(pyridin-4-yl)phenyl)benzamide (13c)
Yield: 59%; 1H NMR (300 MHz, CDCl3) δ: 11.95 (bs, 1H, NH), 8.79 (d, 1H, J = 8.7 Hz), 8.68 (d, 2H, J = 5.1 Hz), 8.06–7.90 (m, 4H), 7.57–7.52 (m, 1H), 7.38–7.03 (m, 8H), 2.45 (s, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ: 167.4, 166.4, 164.2, 163.1, 150.1 (2xC), 146.1, 140.0, 136.1, 133.0, 132.0, 130.6, 130.3, 130.0, 129.6, 125.9, 124.2, 123.5, 122.8, 121.6, 119.7, 115.6, 115.3, 20.7; HRMS calcd. for C26H19F1N3O2 [M-H]−: 424.1467; found: 424.1468.
4.7.4. 2-(4-fluorobenzamido)-N-(4-methyl-2-(pyridin-3-yl)phenyl)benzamide (13d)
Yield: 60%; 1H NMR (300 MHz, CDCl3) δ: 11.99 (bs, 1H, NH), 8.75 (d, 1H, J = 8.4 Hz), 8.51–8.47 (m, 2H), 8.41 (bs, 1H), 8.04–7.71 (m, 4H), 7.51–6.99 (m, 8H), 2.44 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6) δ: 162.4, 155.9, 138.9, 138.3, 137.0, 136.0, 135.1, 133.1, 132.7, 131.5, 130.6, 129.9, 129.6 (2xC), 129.4, 128.4 (2xC), 127.0, 126.0, 124.8, 124.2, 124.0, 120.9, 119.4 (2xC), 20.6; HRMS calcd. for C26H19F1N3O2 [M-H]−: 424.1467; found: 424.1472.
4.8. Antiviral activity determination for DENV and YFV
Antiviral activities were determined as described before by Saudi et al. [1] and reflect the activities as determined versus Dengue virus serotype 2.
Acknowledgements
Milind Saudi was beneficiary of a scholarship of the Erasmus Mundus Cooperation Window. Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225–7). The antiviral work was supported by the Wellcome Trust (grant 089328), EU FP7 project SILVER (contract no HEALTH-F3-2010-260644) and the Marie Curie Initial Training Network “EUVIRNA” EU FP7 [FP7/2007-2013] project EUVIRNA under grant agreement n° [264286], while the chemistry part was supported by the Rega Foundation. We are indebted to Luc Baudemprez for taking numerous NMR spectra on the 500 or 600 MHz platform, and to C. Biernaux for final typesetting.
Footnotes
This is part 4 in a series on Flavivirus inhibitors, part 3 being reference [1].
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejmech.2016.05.043.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Saudi M., Zmurko J., Kaptein S., Rozenski J., Neyts J., Van Aerschot A. Synthesis and evaluation of imidazole-4,5- and pyrazine-2,3-dicarboxamides targeting dengue and yellow fever virus. Eur. J. Med. Chem. 2014;87:529–539. doi: 10.1016/j.ejmech.2014.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., Moyes C.L., Drake J.M., Brownstein J.S., Hoen A.G., Sankoh O., Myers M.F., George D.B., Jaenisch T., Wint G.R., Simmons C.P., Scott T.W., Farrar J.J., Hay S.I. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cuong H.Q., Hien N.T., Duong T.N., Phong T.V., Cam N.N., Farrar J., Nam V.S., Thai K.T., Horby P. Quantifying the emergence of dengue in Hanoi, Vietnam: 1998–2009. PLoS Negl. Trop. Dis. 2011;5:e1322. doi: 10.1371/journal.pntd.0001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasilakis N. 2013. Researchers Identify Fifth Dengue Subtype. [Google Scholar]
- 5.Stevens A.J., Gahan M.E., Mahalingam S., Keller P.A. The medicinal chemistry of dengue fever. J. Med. Chem. 2009;52:7911–7926. doi: 10.1021/jm900652e. [DOI] [PubMed] [Google Scholar]
- 6.Mehlhorn H., Wu Z., Ye B. Springer; 2013. Treatment of Human Parasitosis in Traditional Chinese Medicine. [Google Scholar]
- 7.Tseng C.H., Lin C.K., Chen Y.L., Hsu C.Y., Wu H.N., Tseng C.K., Lee J.C. Synthesis, antiproliferative and anti-dengue virus evaluations of 2-aroyl-3-arylquinoline derivatives. Eur. J. Med. Chem. 2014;79:66–76. doi: 10.1016/j.ejmech.2014.03.074. [DOI] [PubMed] [Google Scholar]
- 8.Bhakat S., Karubiu W., Jayaprakash V., Soliman M.E. A perspective on targeting non-structural proteins to combat neglected tropical diseases: Dengue, West Nile and Chikungunya viruses. Eur. J. Med. Chem. 2014;87:677–702. doi: 10.1016/j.ejmech.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Behnam Mira A.M., Nitsche Christoph, Boldescu Veaceslav, Klein Christian D. The medicinal chemistry of dengue virus. J. Med. Chem. Artic. ASAP. 2016 doi: 10.1021/acs.jmedchem.5b01653. [DOI] [PubMed] [Google Scholar]
- 10.Beck H.C., Hansen A.M., Lauritsen F.R. Novel pyrazine metabolites found in polymyxin biosynthesis by Paenibacillus polymyxa. FEMS Microbiol. Lett. 2003;220:67–73. doi: 10.1016/S0378-1097(03)00054-5. [DOI] [PubMed] [Google Scholar]
- 11.Dickschat J.S., Helmke E., Schulz S. Volatile organic compounds from Arctic bacteria of the cytophaga-flavobacterium-bacteroides group: a retrobiosynthetic approach in chemotaxonomic investigations. Chem. Biodivers. 2005;2:318–353. doi: 10.1002/cbdv.200590014. [DOI] [PubMed] [Google Scholar]
- 12.Niculescu-Duvaz I., Roman E., Whittaker S.R., Friedlos F., Kirk R., Scanlon I.J., Davies L.C., Niculescu-Duvaz D., Marais R., Springer C.J. Novel inhibitors of the v-raf murine sarcoma viral oncogene homologue B1 (BRAF) based on a 2, 6-disubstituted pyrazine scaffold. J. Med. Chem. 2008;51:3261–3274. doi: 10.1021/jm070776b. [DOI] [PubMed] [Google Scholar]
- 13.Naik S., Bhattacharjya G., Talukdar B., Patel B.K. Chemoselective acylation of amines in aqueous media. Eur. J. Org. Chem. 2004;2004:1254–1260. [Google Scholar]
- 14.Pignataro L., Boghi M., Civera M., Carboni S., Piarulli U., Gennari C. Rhodium-catalyzed asymmetric hydrogenation of olefins with phthalaPhos, a new class of chiral supramolecular ligands. Chem. Eur. J. 2012;18:1383–1400. doi: 10.1002/chem.201102018. [DOI] [PubMed] [Google Scholar]
- 15.Majumdar K., Ganai S. CuI/L-proline-catalyzed intramolecular aryl amination: an efficient route for the synthesis of 1, 4-benzodiazepinones. Synlett. 2011;2011:1881–1887. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.