

Abstract
We previously reported a unique peptide-peptoid hybrid, PPS1 that specifically recognizes lipid-phosphatidylserine (PS) and a few other negatively charged phospholipids, but not neutral phospholipids, on the cell membrane. The dimeric version of PPS1, i.e., PPS1D1 triggers strong cancer cell cytotoxicity and has been validated in lung cancer models both in vitro and in vivo. Given that PS and other negatively charged phospholipids are abundant in almost all tumor microenvironments, PPS1D1 is an attractive drug lead that can be developed into a globally applicable anti-cancer agent. Therefore, it is extremely important to identify the minimum pharmacophore of PPS1D1. In this study, we have synthesized alanine/sarcosine derivatives as well as truncated derivatives of PPS1D1. We performed ELISA-like competitive binding assay to evaluate the PS-recognition potential and standard MTS cell viability assay on HCC4017 lung cancer cells to validate the cell cytotoxicity effects of these derivatives. Our studies indicate that positively charged residues at the second and third positions, as well as four hydrophobic residues at the fifth through eighth positions, are imperative for the binding and activity of PPS1D1. Methionine at the first position was not essential, whereas the positively charged Nlys at the fourth position was minimally needed, as two derivatives that were synthesized replacing this residue were almost as active as PPS1D1.
Keywords: minimum pharmacophore, peptoids, phosphatidylserine (PS), sarcosine-scan
Graphical Abstract
1. Introduction
Identification of the minimum pharmacophore of a drug lead is an extremely important step in the drug development process. Various pharmacophore models are used to identify the chemical features and spatial arrangement of an active drug-lead responsible for recognizing the binding site of the targeted biomolecule.1–8 Once the residues or moieties that are essential to the binding and activity of the compound are identified, this knowledge can be used to design new derivatives with enhanced drug activity. In addition, this knowledge can be used to increase the water solubility and bio-distribution of the compound with better pharmacokinetic properties.
Typically, proteins such as receptors, enzymes, and hormones, are the exclusive targeted biomolecules for modern-day drugs. Recently, we have reported a peptide-peptoid hybrid named PPS1 that uniquely recognizes lipid-phosphatidylserine (PS) on the cancer cell surface.9 PPS1 was identified via an unbiased selection approach using our unique on-bead two-color cell screen (OBTC), which is capable of recognizing the difference between two cell surfaces.10 In our previous study, we targeted red stained HCC4017 lung cancer cells in the presence of green stained HBEC30KT normal bronchial epithelial cells derived from the same patient. We mixed these two cell types in a 1:1 ratio and exposed to one-bead one-compound peptide-peptoid hybrid library synthesized on tentagel beads. The aim of that study was to identify compounds that bind only with cancer cells in the presence of normal cells. These compounds were given an equal chance to recognize a protein, lipid, or carbohydrate distinct to cancer cells, whereas those bound to normal cells were discarded. PPS1 displayed strong binding to cancer cells over normal cells both in vitro and in vivo, and later was identified as recognizing lipid-PS. This PPS1-PS binding event was validated through ELISA-like, on-bead, lipid dot-blots and liposome-based FACS binding assays.9 The simple dimeric version of PPS1, i.e., PPS1D1, is selectively cytotoxic toward lung cancer cell lines such as HCC4017 and H460 in vitro over normal HBEC cells and had an IC50 value of 5–10 μM. PPS1D1 displayed a strong tumor burden effect in mouse models as a single agent, as well as in combination with other chemotherapeutics.9
Various phospholipids that exist in different forms are distributed asymmetrically over the plasma membrane. Phosphatidylcholine (PC) and sphingomyelin (SM) are distributed mostly on the outer leaflet, whereas amino phospholipids, such as phosphatidylethanolamine (PE) and phosphatidylserine (PS), are concentrated mostly within the inner leaflet.11–14 However, several pathological and physiological conditions, such as apoptosis, cell activation, injury, and malignant transformation, disturb this lipid asymmetry by exposing PS to the outer leaflet.15 This externalization of PS also has been reported as a tumor marker due to intense stress conditions of the tumor microenvironment.15,16 Thus, PS could be a potential target or biomarker for tumor identification and treatment. A small number of PS-binding ligands have been reported, including proteins,17 antibodies,15,18–21 peptides,22–25 and small molecules.26
While PPS1D1 strongly binding to PS, it also recognizes, to a lesser extent, a few other lipids that have an overall negative charge, such as phosphatidic acid (PA), phosphatidylinositol (PI), and phosphatidylglycerol (PG). These are typically found in the outer layer of cancer cells.9 PPS1D1 did not bind to neutral lipids such as PE, which is found in cancer cells, and PC and SM, which are highly abandon in normal cells. This observation strongly suggests that PPS1D1 has a very specific recognition capability for negatively charged phospholipids over neutrally charged lipids. Therefore, it is extremely interesting, as well as important, to identify the individual residues or groups of moieties that are directly responsible for this binding event. The most conventional approach to identify minimum pharmacophore in peptides is the replacement of amino acids one at a time with glycine or alanine (glycine or alanine scan).2,3 For peptoids, the similar process is the replacement of each side chain one at a time with a methyl group (N-methyl), which is called sarcosine scan (Figure 1A). In our previous report, we effectively implemented this approach to identify the pharmacophore of a peptoid that antagonizes vascular endothelial growth factor receptor-2 (VEGFR2).6 Herein, we report a minimum pharmacophore identification study on PPS1D1 to recognize key components responsible for its interaction with PS and to gain insight into developing a modified PPS1D1 with better activity as a drug candidate.
Figure 1.

Alanine (for peptides) and sarcosine (for peptoids) scan. (A) Outline of the sarcosine scan for peptoid residues, (B) Chemical structures of the alanine/sarcosine derivatives of PPS1D1. Residues are numbered starting from the C-terminus of each monomeric unit. Each residue (1–8) on both monomeric units was replaced one at a time with a methyl group (-CH3), which resulted in eight different derivatives of PPS1D1.
2. Results
2.1 Synthesis of PPS1D1 alanine/sarcosine derivatives
As described in the Introduction, the most suitable way to identify the minimum pharmacophore of our peptide-peptoid hybrid is to conduct an alanine/sarcosine scan. The PPS1 monomeric sequence consists of three peptide residues and five peptoid residues. Hence, we first synthesized eight different derivatives by replacing amino acids with alanine (PPS1D1-1 to PPS1D1-3) and peptoids with sarcosine (PPS1D1-4 to PPS1D1-8) one at a time, as shown in Figure 1B. All the dimers were synthesized using the complete on-bead synthesis protocol described in our previous reports.10,27 Briefly, peptide portions were synthesized using the standard Fmoc synthesis approach, and peptoid residues were coupled using the standard microwave-assisted peptoid synthesis protocol28, 29 All compounds were purified using semi-preparative HPLC. Purity was confirmed by analytical HPLC, and synthesis was confirmed by matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (Supplementary Figure S2–S9).
2.2 PS-binding validation of PPS1D1 alanine/sarcosine derivatives
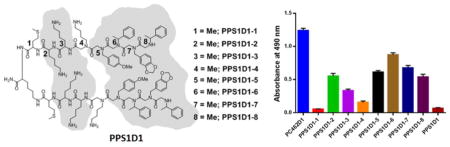
All eight derivatives were first investigated for PS binding capacity using an ELISA-like binding assay, as previously described, with modifications.6,10,30 We performed a competitive ELISA-like binding assay on PS-coated 96-well plates. We first saturated PS-coated wells with fluorescence-labeled parent PPS1D1 (FITC-PPS1D1) at 200 nM concentration, which was then competed out using the eight different alanine/sarcosine derivatives (1 μM concentration of each derivative), as well as our standard non-PS binding control compound PC462D1.9,10 As shown in Figure 2 and Supplementary Figure S19, unmodified PPS1D1 was able to almost completely eradicate bound FITC-PPS1D1, indicating full competition at 1 μM concentration, whereas the control PC462D1 was unable to display any competition. The derivatives PPS1D1-1 and PPS1D1-4 also displayed a certain level of competition, but all other derivatives were unable to compete out FITC-PPS1D1 from the PS-coated plates. These results indicate that Met at the first position (from the C-terminus) and Nlys at the fourth position are not critical for binding to PS. In contrast, two positively charged residues at the second position (amino acid Lys) and third position (peptoid Nlys) are critical for binding. Furthermore, these data indicate that the four hydrophobic residues at the N-terminus are also extremely important for binding, as removal of even one of these residues almost completely reduce the binding capacity of PPS1D1.
Figure 2.

ELISA-like competitive binding assay of PPS1D1 for all eight alanine/sarcosine derivatives and the control compound PC462D1 with FITC-PPS1D1.
2.3 Cell cytotoxicity effect of PPS1D1 alanine/sarcosine derivatives
Next, we evaluated the cell cytotoxicity effects of these derivatives using the standard MTS cell viability assay along with unmodified PPS1D1 and the control compound PC462D. The results are shown in Figure 3. Cytotoxicity was investigated at eight concentrations ranging from 0.1 μM to 100 μm (Figure 3A). As expected, the parent PPS1D1 displayed the known cell cytotoxicity effect around 10 μM, whereas all the derivatives exhibited less cytotoxicity. Anyway, PPS1D1-1 and PPS1D1-4 exhibited the smallest reduction in cytotoxicity activity. We analyzed data at 30 μM concentration to better understand the differences between derivatives (Figure 3B). As expected, PPS1D1 exhibited the strongest cytotoxicity, and the control compound PC462D1 did not exhibit any cell-killing effects. However, all the alanine/sarcosine derivatives displayed loss of cytotoxicity compared with PPS1D1, except PPS1D1-1 and PPS1D1-4 (Figure 3B). PPS1D1-1 exhibited similar cytotoxicity to PPS1D1, whereas PPS1D1-4 exhibited a slight loss of cytotoxicity compared with PPS1D1, but still caused considerable cell death. This observation again suggests that Met at the first position (from the C-terminus) and Nlys at the fourth position do not have significant roles in PS recognition, which validates the results obtained from the ELISA-like assay. The cytotoxicity of derivatives PPS1D1-5, PPS1D1-6, PPS1D1-7, and PPS1D1-8, in which hydrophobic residues were replaced with sarcosine, was found to be most affected by the modifications (Figure 3B).
Figure 3.

Standard MTS cell viability assay of HCC4017 lung cancer cells treated with PPS1D1, eight alanine/sarcosine derivatives (PPS1D1-1 to PPS1D1-8), and the control compound PC462D1. (A) Observation of the activity through concentration gradient of 0.1 μM to 100 μM. (B) Absorbance (activity) differences observed at 30μM treatment of all of the compounds mentioned above.
2.4 Cell cytotoxicity effect of fourth-position-modified derivatives of PPS1D1
We further explored the observation that the positively charged Nlys residue at the fourth position is not essential for the overall activity of PPS1D1. We synthesized two derivatives that replace this positively charged residue at the fourth position with relatively neutral residues. PPS1D1-4A was synthesized with Nmea (Figure 4A) and PPS1D1-4B was synthesized with Nall (Figure 4B). We then performed the same MTS assay as used above to validate the cell cytotoxicity effects of PPS1D1-4A and PPS1D1-4B compared to unmodified PPS1D1 and the control compound PC462D1. As shown in Figure 4C, both PPS1D1-4A and PPS1D1-4B displayed cell cytotoxicity similar to that of PPS1D1 (with a slight reduction of activity), whereas the control PC462D1 exhibited no activity.
Figure 4.

Standard MTS cell viability assay of HCC4017 lung cancer cells treated with PPS1D1, PPS1D1-4A, PPS1D1-4B, and the control compound PC462D1.
2.5 Synthesis of truncated derivatives of PPS1D1
As shown in Figures 2 and 3, four hydrophobic residue region at the N-terminus of PPS1D1 is essential for binding activity. We next evaluated how many of these four residues are needed for binding activity. As shown in Figure 5, we synthesized five derivatives of PPS1D1 (3-mer to 7-mer) by truncating one to five residues from the N-terminus. A complete on-bead synthesis protocol, as described in our previous reports, was used, with peptide portions synthesized using the standard Fmoc synthesis approach and peptoid residues coupled using the standard microwave-assisted peptoid synthesis protocol. All compounds were purified using semi-preparative HPLC. Purity was confirmed by analytical HPLC, and synthesis was confirmed by MALDI mass spectrometry (Supplementary Figures S13–S17).
Figure 5.

Truncation array of PPS1D1 from the N-terminals of both monomeric sequences. Dotted lines indicate the residue(s) removed along with the name of the corresponding version of the derivative (3-mer to 7-mer).
2.6 Cell cytotoxicity effect of truncated derivatives of PPS1D1
We evaluated the binding and cell cytotoxicity of the truncated derivatives of PPS1D1 (shown in Figure 5), as well as unmodified PPS1D1, and the control compound PC462D1, using the same ELISA-like competitive assay (for binding) and standard MTS cell viability assay (for cell cytotoxicity) (Figure 6). PPS1D1 exhibited a strong competitive binding (against FITC-PPS1D1), whereas the control PC462D1 had no binding competition (Figure 6A). The 7-mer derivative, which lacked the hydrophobic residue at the eighth position, had a weaker binding than PPS1D1. The binding was completely lost when two or more residues from the N-terminus were eliminated (Figure 6A). This observation confirms the importance of all 4 N-terminus residues for the binding of PPS1D1 to PS. The same pattern was exactly observed when the cytotoxicity assay was performed. PPS1D1 exhibited a strong cell cytotoxic effect, whereas the control PC462D1 had no cell cytotoxic effect (Figure 6B & C). The 7-mer derivative had a weaker cytotoxic effect than PPS1D1. Deleting two or more residues from the N-terminus completely deactivated the molecule. This observation further confirms the importance of all 4 N-terminus residues for the activity of PPS1D1.
Figure 6.

Binding and activity validation of truncated PPS1D1 derivatives. (A) ELISA-like competitive binding assay of five truncated derivatives of PPS1D1 (3-mer to 7-mer) and the control compound PC462D1 with FITC-PPS1D1. (B) Standard MTS cell viability assay of HCC4017 lung cancer cells treated with PPS1D1, truncated derivatives of PPS1D1 (3-mer to 7-mer), and the control compound PC462D1. Observation of the activity through concentration gradient of 0.1 μM to 100 μM. (C) Absorbance (activity) differences observed at 30μM treatment of all of the compounds mentioned above.
3. Discussion
As mentioned previously, there are only small number of PS-binding ligands have been reported up to date. Annexin V is a coagulation inhibitory protein that is known to have high affinity and specificity toward anionic phospholipids, mainly PS. The coagulation inhibitory effect of annexin V is purely via mechanical binding to reactive surfaces. The key feature is that annexin V requires Ca2+ for binding to PS.27 However, to the best of our knowledge, there are no reports of cytotoxicity effects of annexin V. In contrast, bavituximab is an example of a PS-targeting antibody that has been reported as a potent anti-tumor agent.18,21,31 PS-targeting with bavituximab stimulates innate immune cells (e.g., macrophages) to perform antibody-dependent cellular cytotoxicity, induces the differentiation of myeloid-derived suppressor cells (MDSCs) into M1 macrophages and mature dendritic cells, and facilitates the development of an adaptive immune response.32 The mechanism of action of bavituximab is Fc-dependent, although the involvement of PS receptor(s) is under investigation. Interestingly, bavituximab requires β2-glycoprotein-1 to recognize and bind to PS.15, 28 In other words, bavituximab binds to PS via β2-glycoprotein-1. However, PPS1D1 does not require the support of any ions or proteins to recognize PS. Taken together; this indicates that there are several available mechanisms for recognizing phospholipid binding agents.
Because PPS1D1 is an amphipathic molecule with six positively charged residues (three in each monomer) and a hydrophobic region consisting of eight consecutive aromatic residues (four in each monomer), it was anticipated that PPS1D1 would bind to all phospholipids, as phospholipids have negatively charged phosphate heads and hydrophobic tails that align perfectly with PPS1D1. However, we previously observed that PPS1D1 only recognizes phospholipids that have an ‘overall’ negative charge (i.e., PS, PA, PI, and PG) and does not bind to neutral lipids such as PC, PE, and PG.9 This observation ruled out the possibility that the positively charged residues of PPS1D1 could recognize the negatively charged phosphate head groups, because otherwise PPS1D1 would bind to all lipids indiscriminately. Our current minimum pharmacophore studies indicate that two of the positive charges in the second and third positions of PPS1D1 are critical for binding and activity, whereas the positive charge at the fourth position is the least important. It is noteworthy that although PPS1D1-2 lost binding and cell-killing activity as compared to PPS1D1, PPS1D1-2 still exhibited a moderate effect, which could be attributed to the interaction of the Nlys residue at the third position of PPS1D1-2 with negative charges on PS in the absence of the positive residue at the second position. Replacing the positively charged residue (Nlys) at the fourth position with relatively neutral residues (i.e., Nmea and Nall) did not significantly affect activity, as only a slight reduction was observed (Figure 4). Thus, we believe that positive charges at the second and third positions may interact with the additional negative charge found on PS, PA, PI, and PG. The hydrophobic region of the N-terminus of PPS1D1 appears to interact with the hydrophobic tails of the phospholipids on the outer layer of the cell membrane. Both sarcosine and truncation studies (Figures 3 and 6) indicated that all four residues of this hydrophobic region are critical for PS binding and cytotoxicity. Removal of even a single residue caused a significant reduction in activity.
For most protein-binding molecules, typically there are only a few residues or moieties that are critical for binding and activity.33,34 In particular, for small organic compounds, binding and activity could depend on a single atom.35,36 Typically, removal or replacement of these residues or atoms completely disrupts the function of the molecule, mainly because proteins have well-defined structures, and ligand-binding events occur through specific charge-charge or hydrophobic (i.e., van der Waals) interactions. For example, there may be a hydrogen bond between the ligand and the protein through specific atoms. However, small phospholipid molecules do not have structures that are as well-defined as proteins. These small phospholipid molecules are arranged in a bilayer that forms the cell membrane and are not structurally rigid. This arrangement of multiple lipid types on the cell membrane largely varies and constantly interchanges. Therefore, defining the parameters of a specific ligand-lipid interaction is extremely difficult and challenging, in particular with respect to the structures of targeted phospholipids. For example, as described above, the residues at the second, third, and fifth through eighth positions are the most important for the binding and activity of PPS1D1 (Figure 7). However, closer inspection of the data in Figure 3 reveals that removal of each of those residues one-by-one affects activity by a maximum of 50%, except for the residue at the sixth positon (PPS1D1-6), the removal of which causes approximately 75% reduction in activity. This observation indicates that some of these residues contribute to the binding activity of PPS1D1 in a cooperative manner, rather than depending on a specific single-residue or atomic-level interactions with a single phospholipid on the cell membrane. For example, two positive charges at the second and third position may collectively interact with the additional negative charge on PS, PA, PI, and PG. Also, the four hydrophobic residues (fifth through eighth position) may form a collective hydrophobic patch that interacts with the hydrophobic tails of phospholipids. Nonetheless, we are planning to conduct very specific studies using various platforms such as liposomes, as well as computer simulations studies, to further understand this very interesting and specific interaction of PPS1D1 with negatively charged phospholipids. In particular, we are carefully evaluating the possibility of interactions through additional negative charges found in regions of phospholipids other than the phosphate head-groups.
Figure 7.

Minimum pharmacophore of PPS1D1. Residues that are predicted to be essential for binding and activity at the second, third and fifth through eighth positions are highlighted in gray.
4. Conclusion
Eight alanine/sarcosine derivatives of PPS1D1 were investigated to identify the minimum pharmacophore of PPS1D1, which has strong affinity for PS and other negatively charged phospholipids. The ELISA-like binding and MTS cell viability assays for all the mutated derivatives revealed that residues at the second, third, and fifth through eighth positions are most important for the binding and activity of PPS1D1. Although it seems like the positive groups at the second and third positions of PPS1D1 interact with additional negative groups displayed on PS and other negatively charged phospholipids, further careful and focused studies are needed to evaluate the exact interactions between those predicted moieties. All four hydrophobic residues at the N-terminus seem to be essential for the interaction with hydrophobic tail regions of phospholipids.
5. Experimental
5.1 General
Novasyn TGR was purchased from EMD Millipore (Billerica, MA, USA). All Fmoc-protected amino acids and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from EMD Millipore. All primary amines, bromoacetic acid, N,N′-diisopropylcarbodiimide (DIC), N,N-diisopropylethylamine (DIPEA), piperidine, NMM, trifluoroacetic acid (TFA), dichloromethane (DCM) and N,N-dimethylformamide (DMF) were obtained from Sigma-Aldrich (St. Louis, MO, USA). All chemical reagents and solvents from commercial sources were used without further purification. Five-mL disposable reaction columns (Intavis AG, Tübingen, Germany) were used as reaction vessels for solid-phase synthesis. Syntheses of peptoids under microwave conditions were performed in a 1000 W microwave oven with 10% power.
All purifications were completed on a Waters HPLC system, Waters Corporation, MA, USA. Mass spectra were recorded on an Applied Biosystems Voyager DE Pro mass spectrometer using alpha-cyano-4-hydroxycinnamic acid as the matrix.
5.2 Synthesis
5.2.1 Synthesis of PPS1D1
PPS1D1 was synthesized on Novasyn TGR (EMD Millipore). The resin was swelled in DMF for 1 h prior to use, and no de-protection was required for terminal amine groups. The resin was first coupled to Fmoc-Lys(Fmoc)-OH using 5.0 eq HBTU and 5.0 eq HOBt as coupling reagents in the presence of 10.0 eq of DIPEA for overnight. Both Fmoc were removed with the help of 20% piperidine solution in DMF [2 × (2 mL × 10 min)]. The Fmoc de-protection of both amine groups produced two N-terminals simultaneously to build two copies of peptide-peptoid hybrid. Three amino acids, Fmoc Met-OH, Fmoc D-Lys(Boc)-OH, and Fmoc-Lys(Boc)-OH, were introduced using the same peptide-coupling protocol (HBTU/HOBt/DIPEA), washing 10 times with DMF and removing the Fmoc group at each step. After removal of the final Fmoc, five peptoid residues were then coupled using a two-step peptoid coupling procedure (acylation and amination) under a microwave (1000 W)-assisted synthesis protocol.29 For the acylation step, beads were treated with 2M bromoacetic acid (1 mL) and 3.2M DIC (1 mL), and microwaved at 10% power (2 × 15 s) with gentle shaking in between for 30 s. After washing with DMF, beads were treated with 2M Boc-diaminobutane (2 mL), and coupling was performed again in the microwave oven as described above. The procedure was repeated again to attach the remaining four residues [4-methoxybenzylamine, (R)-methylbenzylamine, piperonylamine, and (R)-methylbenzylamine] with DMF washing at every step. At the end, beads were washed with DCM and dried under vacuum before cleavage. Beads were then treated with a cleavage cocktail of TFA/H2O/tri-isopropylsilane (95%/2.5%/2.5%) for 2 h. The desired compound was purified using HPLC and analyzed by MALDI-TOF (Voyager DE Pro, AB Systems), USA.
5.2.2 Synthesis of PPS1D1-1 to PPS1D1-8
The general peptide synthesis procedure described in section 5.2.1 was used to couple three peptides. Amino acids at the first, second, and third positions were replaced with alanine for PPS1D1-1, PPS1D1-2, and PPS1D1-3, respectively, and methylamine was used to replace the normal amines at the fourth through eighth positions in each of the five derivatives, PPS1D1-4, PPS1D1-5, PPS1D1-6, PPS1D1-7, and PPS1D1-8, respectively.
5.2.3 Synthesis of FITC-PPS1D1
Fmoc-Cys(Trt)-OH was coupled as the first amino acid onto the resin using the same peptide protocol as described above section 5.2.1. The remaining PPS1D1 synthesis was conducted as described previously. After washing with DMF and DCM, beads were treated with a cleavage cocktail of TFA/H2O/tri-isopropylsilane (95%/2.5%/2.5%) for 2 h. TFA was then evaporated, and the resulting solid compound was dissolved in 1:1 water:acetonitrile (ACN) mixture. This solution was subjected to HPLC purification. The purified compound was lyophilized to obtain the dry product. Fluorescein-5–maleimide (Thermo r Scientific, Germany) was then coupled to this compound (1 M:1 M ratio) in buffer solution at a pH of 7. The coupled FITC-PPS1D1 compound was purified using HPLC.
5.2.4 Synthesis of PPS1D1-4A and PPS1D1-4B
Fmoc-Lys(Fmoc)-OH was coupled overnight as the central linker, and both Fmoc groups were removed simultaneously, allowing two copies of the sequence to be built on two amine groups of this central Lys. Then, the first three amino acids, Fmoc-Met-OH, Fmoc–D-Lys(Boc)-OH, and Fmoc-Lys(Boc)-OH, were loaded onto the resin using the previously described peptide-coupling protocol, removing the Fmoc group each time. Allyamine (Nall) for PPS1D1-4A and 2-methoxyethylamine (Nmea) for PPS1D1-4B were introduced as first peptoid residues using the same microwave-assisted peptoid synthesis protocol as described previously followed by remaining peptoid residues. After washing with DMF and DCM, beads were then treated with a cleavage cocktail of TFA/H2O/tri-isopropylsilane (95%/2.5%/2.5%) for 2 h. Compounds were purified using HPLC. Synthesis was confirmed using MALDI-TOF MS (Voyager DE Pro, AB Systems).
5.2.5 Synthesis of truncated derivatives
Fmoc-Lys(Fmoc)-OH was coupled overnight as the central linker, and both Fmoc groups were removed simultaneously, allowing two copies of the sequence to be built on two amine groups of this central Lys. Then, the first three amino acids, Fmoc-Met-OH, Fmoc–D-Lys(Boc)-OH, and Fmoc-Lys-OH, were loaded onto the resin using peptide coupling protocol, removing the Fmoc group each time using the peptide-coupling protocol. For the 3-mer, synthesis was stopped after three peptide couplings, and beads were treated with the cleavage cocktail of TFA/H2O/tri-isopropylsilane (95%/2.5%/2.5%) for 2 h. Compounds were purified using HPLC and confirmed by MALDI-TOF MS. The 4-mer was achieved by coupling one peptoid residue after overnight Fmoc-Lys(Fmoc)-OH coupling and three peptide residues using the same peptide-coupling protocol as described above. Similarly, the 5-mer, 6-mer, and 7-mer derivatives were synthesized by coupling 2, 3, and 4 corresponding peptoid residues using the microwave protocol as described above. After washing with DMF and DCM, beads were then treated with a cleavage cocktail of TFA/H2O/tri-isopropylsilane (95%/2.5%/2.5%) for 2 h. Compounds were purified using HPLC. Synthesis was confirmed using MALDI-TOF MS.
5.2.6 Synthesis of control PC462D1
Fmoc-Lys(Fmoc)-OH was coupled overnight as the first peptide residue, and both Fmoc groups were removed simultaneously, allowing two copies of the sequence to be built on two amine groups of this central Lys. Then, the first three amino acids Fmoc-Met-OH, Fmoc–D-Lys(Boc)-OH, and Fmoc-Gly-OH were loaded onto the resin using peptide coupling protocol, removing Fmoc group each time. Then, the 5-mer peptoid region containing Nall and Nmea was synthesized using the microwave-assisted peptoid synthesis protocol as described previously. After washing with DMF and DCM, beads were then treated with the cleavage cocktail of TFA/H2O/tri-isopropylsilane (95%/2.5%/2.5%) for 2 h. The compound was purified using HPLC. Synthesis was confirmed using MALDI-TOF MS (Voyager DE Pro, AB Systems).
5.3 Biological assays
5.3.1 ELISA-like binding competition assay
Lipid-PS (Avanti Polar Lipids) was dissolved in Hexane at 10 μg/ml concentration and coated onto 96 well-plates (7005, U Microfluor Microtiter Microplates). Hexane was allowed to evaporate at room temperature, and each well was blocked with 200 μl of 1% BSA in PBS for 1 h. Wells were washed with 3 × PBS, and 100 μL of FITC-PPS1D1 dissolved in blocking buffer (200 nM) was added to each well and incubated for 1 h at room temperature. Wells were then washed again with 5 × PBS. Then, 100 μL of each 1 μM PPS1D1, PC462D1 and all alanine/sarcosine derivatives were added to separate wells in triplicate and incubated for 1 h. Similarly, in truncated derivative studies, the FITC-PPS1D1 coated plate was treated with 100 μL of each 1 μM PPS1D1, PC462D1 & all truncated derivatives and incubated for 1 h. Each well was washed with 3 × PBS and subjected to fluorescence reading. Remaining fluorescence was detected at 520 nm using a spectrophotometer (Spectramax i3, Molecular Devices, Sunnyvale, CA, USA).
5.3.2 Cell cytotoxicity assay
HCC4017 cells were grown in clear-bottom 96-well plates. On the 2nd day, lung cancer cells were treated with PPS1D1, PPS1D1-1, PPS1D1-2, PPS1D1-3, PPS1D1-4, PPS1D1-5, PPS1D1-6, PPS1D1-7, PPS1D1-8, 3-mer, 4-mer, 5-mer, 6-mer, 7-mer, and control PC462D1 in RPMI medium with 10% FBS containing 3% BSA. Eight graded concentrations ranging from 0.1 μM to 100 μM were used for all compounds, and each concentration was tested in triplicate. On day 3, 20 μl of CellTiter 96® AQueous One Solution (Promega, Fitchburg, WI, USA) was added to each well, and absorbance was measured at 490 nm.
Supplementary Material
Acknowledgments
This work was supported by National Cancer Institute (NCI) at National Institute of Health (NIH) grant 1R01CA175779 and Cancer Prevention and Research Institute of Texas Grant RP130258 and funding from University of Houston.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/xxx.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang SY. Drug discovery today. 2010;15:444–450. doi: 10.1016/j.drudis.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 2.Hruby VJ. Nature reviews. Drug discovery. 2002;1:847–858. doi: 10.1038/nrd939. [DOI] [PubMed] [Google Scholar]
- 3.Morrison KL, Weiss GA. Curr Opin Chem Biol. 2001;5:302–307. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- 4.Park M, Wetzler M, Jardetzky TS, Barron AE. PloS one. 2013;8:e58874. doi: 10.1371/journal.pone.0058874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jamieson AG, Boutard N, Sabatino D, Lubell WD. Chem Biol Drug Des. 2013;81:148–165. doi: 10.1111/cbdd.12042. [DOI] [PubMed] [Google Scholar]
- 6.Udugamasooriya DG, Dunham G, Ritchie C, Brekken RA, Kodadek T. Bioorg Med Chem Lett. 2008;18:5892–5894. doi: 10.1016/j.bmcl.2008.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim HS, Archer CT, Kim YC, Hutchens T, Kodadek T. Chem Commun (Camb) 2008:1064–1066. doi: 10.1039/b717861a. [DOI] [PubMed] [Google Scholar]
- 8.Kodadek T. Curr Opin Chem Biol. 2010;14:713–720. doi: 10.1016/j.cbpa.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai TJ, Toombs JE, Minna JD, Brekken RA, Udugamasooriya DG. Oncotarget. 2016;7:30678–30690. doi: 10.18632/oncotarget.8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matharage JM, Minna JD, Brekken RA, Udugamasooriya DG. ACS Chem Biol. 2015;10:2891–2899. doi: 10.1021/acschembio.5b00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balasubramanian K, Schroit AJ. Annu Rev Physiol. 2003;65:701–734. doi: 10.1146/annurev.physiol.65.092101.142459. [DOI] [PubMed] [Google Scholar]
- 12.Devaux PF. Biochemistry. 1991;30:1163–1173. doi: 10.1021/bi00219a001. [DOI] [PubMed] [Google Scholar]
- 13.Rothman JE, Lenard J. Science. 1977;195:743–753. doi: 10.1126/science.402030. [DOI] [PubMed] [Google Scholar]
- 14.Zachowski A. Biochem J. 1993;294(Pt 1):1–14. doi: 10.1042/bj2940001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran S, Downes A, Thorpe PE. Cancer Res. 2002;62:6132–6140. [PubMed] [Google Scholar]
- 16.Ran S, Thorpe PE. Int J Radiat Oncol Biol Phys. 2002;54:1479–1484. doi: 10.1016/s0360-3016(02)03928-7. [DOI] [PubMed] [Google Scholar]
- 17.Stace CL, Ktistakis NT. Biochim Biophys Acta. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Ran S, He J, Huang X, Soares M, Scothorn D, Thorpe PE. Clin Cancer Res. 2005;11:1551–1562. doi: 10.1158/1078-0432.CCR-04-1645. [DOI] [PubMed] [Google Scholar]
- 19.He J, Luster TA, Thorpe PE. Clin Cancer Res. 2007;13:5211–5218. doi: 10.1158/1078-0432.CCR-07-0793. [DOI] [PubMed] [Google Scholar]
- 20.DeRose P, Thorpe PE, Gerber DE. Immunotherapy. 2011;3:933–944. doi: 10.2217/imt.11.87. [DOI] [PubMed] [Google Scholar]
- 21.He J, Yin Y, Luster TA, Watkins L, Thorpe PE. Clin Cancer Res. 2009;15:6871–6880. doi: 10.1158/1078-0432.CCR-09-1499. [DOI] [PubMed] [Google Scholar]
- 22.Burtea C, Laurent S, Lancelot E, Ballet S, Murariu O, Rousseaux O, Port M, Vander Elst L, Corot C, Muller RN. Mol Pharmcol. 2009;6:1903–1919. doi: 10.1021/mp900106m. [DOI] [PubMed] [Google Scholar]
- 23.Igarashi K, Kaneda M, Yamaji A, Saido TC, Kikkawa U, Ono Y, Inoue K, Umeda MJ. Biol Chem. 1995;270:29075–29078. doi: 10.1074/jbc.270.49.29075. [DOI] [PubMed] [Google Scholar]
- 24.Laumonier C, Segers J, Laurent S, Michel A, Coppee F, Belayew A, Elst LV, Muller RN. J Biomol Screen. 2006;11:537–545. doi: 10.1177/1087057106288220. [DOI] [PubMed] [Google Scholar]
- 25.Thapa N, Kim S, So IS, Lee BH, Kwon IC, Choi K, Kim IS. J Cell Mol Med. 2008;12:1649–1660. doi: 10.1111/j.1582-4934.2008.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanshaw RG, Smith BD. Bioorg Med Chem. 2005;13:5035–5042. doi: 10.1016/j.bmc.2005.04.071. [DOI] [PubMed] [Google Scholar]
- 27.Hooks JC, Matharage JP, Udugamasooriya DG. Peptide Science. 2011;96:567. doi: 10.1002/bip.21596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 29.Olivos HJ, Alluri PG, Reddy MM, Salony D, Kodadek T. Org Lett. 2002;4:4057–4059. doi: 10.1021/ol0267578. [DOI] [PubMed] [Google Scholar]
- 30.Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J Am Chem Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 31.Huang X, Bennett M, Thorpe PE. Cancer Res. 2005;65:4408–4416. doi: 10.1158/0008-5472.CAN-05-0031. [DOI] [PubMed] [Google Scholar]
- 32.Yin Y, Huang X, Lynn KD, Thorpe PE. Cancer Immunol Res. 2013;1:256–268. doi: 10.1158/2326-6066.CIR-13-0073. [DOI] [PubMed] [Google Scholar]
- 33.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 34.Hajduk PJ, Greer J. Nat Rev Drug Discov. 2007;6:211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Yang PL, Gray NS. Nature reviews. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 36.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.