

PEARLS
Niemann-Pick disease type C (NPC) has heterogeneous clinical presentations.
Although NPC usually affects infants or young adolescents, it should be considered in the differential diagnosis of vertical supranuclear gaze palsy, regardless of age at presentation.
Adult-onset NPC may rarely present with myoclonus.
Disease-specific therapy with miglustat has been shown to stabilize the neurologic progression in patients with NPC.
OY-STERS
NPC can present as late as the 6th–7th decade.
Although more common disorders like progressive supranuclear palsy (PSP) should be suspected, NPC should also be considered in the differential diagnosis of late-onset vertical gaze palsy, thereby minimizing the diagnostic delay.
Rare symptoms of myoclonus and sensorineural hearing loss can mislead the clinician away from the diagnosis of NPC.
CASE REPORT
A 65-year-old, right-handed man of Polish ancestry developed insidious onset, gradually progressive gait ataxia at the age of 55. Three years later, he started dropping things due to myoclonic jerks involving the trunk and upper extremities (video on the Neurology® Web site at Neurology.org). Worsening ataxia and frequent falls resulted in the use of a wheelchair by age 64. His medical history was positive for bilateral hearing loss since his early 50s. There was no history of seizures, dysphagia, mood disorders, psychosis, or any other significant medical illness in the past, besides being a 40-pack-year smoker. He was born of nonconsanguineous parents, and his sister died of an unknown illness at the age of 13. On neurologic examination, cognition was normal and speech was dysarthric. Although visual acuity was normal, there was reduced vertical gaze (more pronounced on downgaze), which responded to oculocephalic maneuver, consistent with a vertical supranuclear gaze palsy (VSGP). Horizontal eye movements were intact, and there was no nystagmus. Bilateral sensorineural hearing loss was detected, right more than left. Generalized myoclonic jerks were present, predominantly involving the upper extremities on posture maintenance. There were no features suggestive of parkinsonism. He had marked gait ataxia with milder appendicular ataxia and dysdiadochokinesia. The remainder of his neurologic examination was normal. Abdominal examination did not reveal any visceromegaly. The NPC suspicion index score1 was 61. Laboratory workup showed normal complete blood count, serum electrolytes, creatinine, glucose, liver function test, serum lactate, and EEG. Spinocerebellar ataxia gene panel was negative for SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, and SCA17. Genetic testing for NPC was positive for 2 pathogenic variants (p.P1007A and p.1077Q) of NPC1 gene. MRI brain showed cortical and midbrain atrophy with subcortical nonspecific white matter changes (figure). His myoclonic jerks responded to valproate 500 mg oral twice daily. He was started on miglustat 100 mg oral twice daily titrated to 200 mg oral 3 times a day.
Figure. MRI brain.

MRI brain shows cortical and midbrain atrophy on T1-weighted sequence (A), and cortical atrophy along with subcortical nonspecific white matter changes on fluid-attenuated inversion recovery sequence (B).
DISCUSSION
NPC is a rare lysosomal lipid storage disorder characterized by heterogeneity in the age at onset, clinical presentations, and disease course.2 It has an incidence of 1 per 120,000 live births.1 While mutation in the NPC1 gene (located on chromosome 18) results in >95% of cases, mutation in the NPC2 gene (located on chromosome 14) occurs in approximately 4% of cases.1 The mutations cause abnormal intracellular transport and accumulation of cholesterol and glycosphingolipids in the brain and other tissues.1,3 Based on the age at onset, patients with NPC are categorized into 5 subgroups: prenatal/perinatal (age between 0 and 3 months), early-infantile (<2 years), late-infantile (2 to <6 years), juvenile (6 to <15 years), and adolescent/adult (≥15 years).1,3 Although previously considered a childhood-onset disorder, growing awareness along with advancements in biochemical and genetic diagnostic methods have increased detection of late-onset cases.4 Adolescent/adult NPC cases comprised 17%–27% of the 309 patients reported in 2 separate studies.2,4 Of the 4 reported patients with NPC with an age >50 years at diagnosis,5,6 3 presented with neurologic or psychiatric manifestations.5 Our patient had onset of neurologic symptoms at age 55 years, second only to a case reported with onset of symptoms at 59 years of age.7
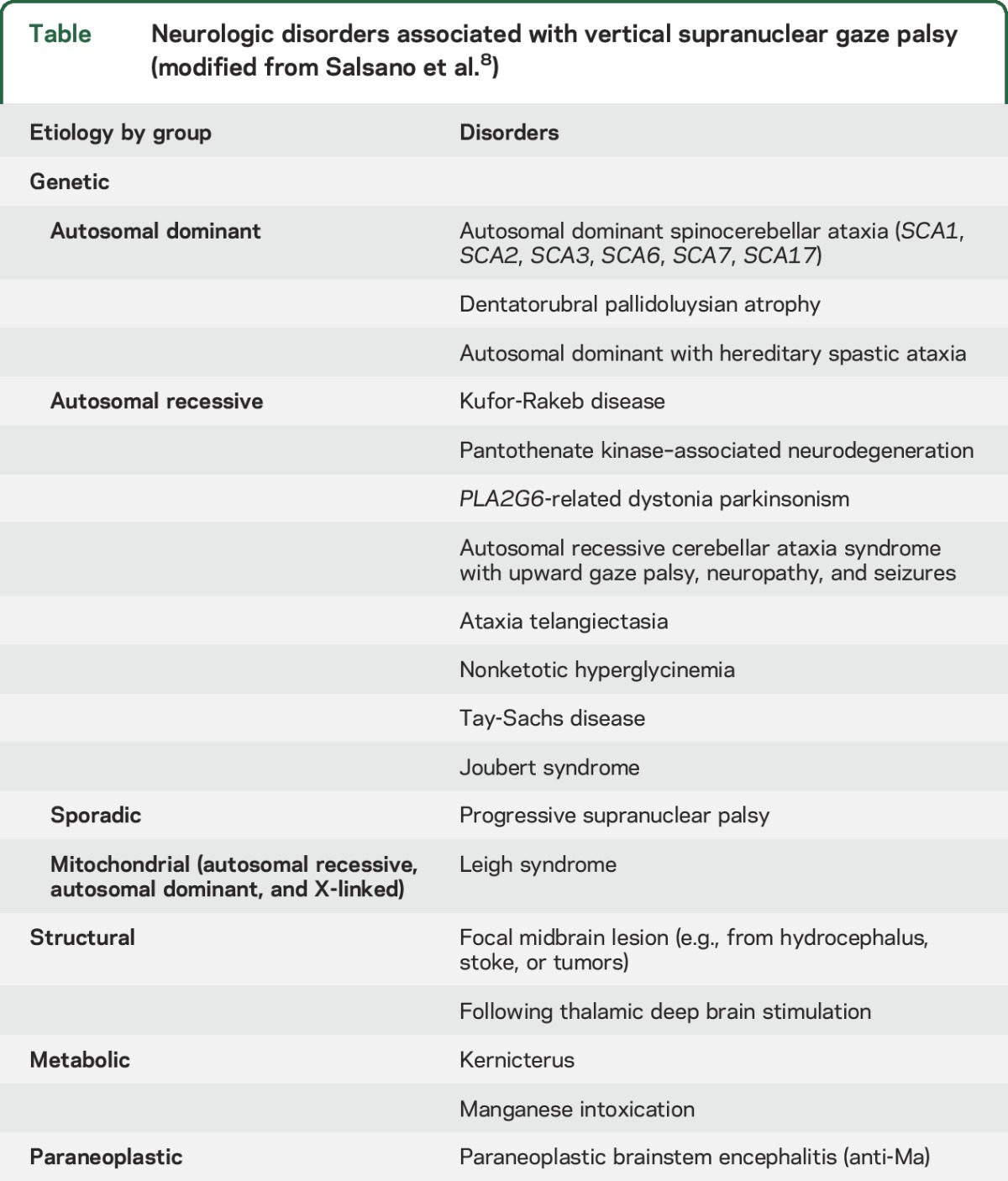
The heterogeneous clinical manifestations of NPC include systemic, neurologic, and psychiatric features.1 Systemic features like neonatal jaundice, hepatosplenomegaly, or isolated splenomegaly are common in early-onset disease, and always precede neurologic manifestations.1,3 Almost 15% of all patients with NPC, and half of those with adult-onset disease, have minimal or no hepatosplenomegaly.3 The presence of isolated splenomegaly without any hepatic derangement in patients with a neurodegenerative or psychiatric illness favors NPC.1 The most common neurologic manifestations include cognitive or motor developmental delay in childhood-onset cases, VSGP, ataxia, dysarthria, dysphagia, dystonia, seizures, and gelastic cataplexy.1,3 Although VSGP may be seen in other neurologic disorders (table),8 it is one of the earliest features in NPC, and is present in 70%–80% of patients across all age at onset categories.1,2,4 With disease progression, horizontal saccadic eye movements are also affected, leading to complete ophthalmoplegia, reflecting progressive brainstem degeneration.1 Myoclonus is rare in adult-onset NPC,1 but it was a major neurologic manifestation in our patient, along with VSGP, ataxia, and bilateral sensorineural hearing loss. Sensorineural hearing loss is commonly seen in clinical practice but remains underreported in patients with NPC.1 Frontal-subcortical cognitive deficits and schizophrenia-like psychosis are usually seen in patients with adolescent/adult-onset NPC.1,2 The combination of phenotypic and genetic heterogeneity precludes formation of genotype–phenotype correlations in patients with NPC.4
Table.
Neurologic disorders associated with vertical supranuclear gaze palsy (modified from Salsano et al.8)
In order to facilitate diagnosis of NPC in suspected patients, the NPC suspicion index was developed, which incorporates visceral, neurologic, and psychiatric features, along with the family history.1 A score ≥70 suggests immediate testing for NPC, and scores from 40 to 69 indicate a need for further follow-up.1 The probability of NPC is very low with scores below 40.1 Because of the NPC suspicion index score falling in the gray zone (40–69), along with the heterogeneity in clinical presentation, long diagnostic delays occur in patients with adult-onset NPC,1–3 as seen in our patient. In the 3 reported patients with NPC with neurologic illness and age at diagnosis >50 years, the mean delay in diagnosis was 13.5 years for the 2 cases where data was available.5 In our patient, there was a delay of almost 10 years in reaching the diagnosis of NPC. Thus, in adult patients with progressive VSGP, ataxia, dysarthria, dysphagia, cognitive decline, and psychiatric symptoms, one should suspect NPC. An accompanying family history suggestive of NPC is helpful but not necessary. The clinical diagnosis may be supported by brain imaging findings, which alone are nondiagnostic.1,3 MRI brain may show cerebral or cerebellar atrophy, white matter hyperintensities, and midbrain atrophy,1 as was seen in our patient. There is a lesser degree of increase in pontine-to-midbrain ratio in adult patients with NPC than that seen in PSP.9
Patients with NPC with onset of neurologic disease in early childhood develop rapid disease progression, and die at a younger age, as compared to those with late-onset neurologic involvement.1,4 Progressive dysphagia leading to repeated aspirations and bronchopneumonia causes the majority of deaths in patients with NPC.1 Symptomatic treatment for various neurologic and psychiatric manifestations improves quality of life in patients with NPC.1 The myoclonic jerks in our patient improved on valproate. Miglustat is the only disease-specific therapy approved to treat progressive neurologic manifestations in pediatric and adult patients with NPC.10 It competitively inhibits glucosylceramide synthase, and reduces glycosphingolipid accumulation in the brain, thereby stabilizing the neurologic features like ambulation, manipulation, swallowing, and language.1,10 It is advocated for all patients with neurologic, psychiatric, and cognitive manifestations at the time of diagnosis of NPC.1 The usual recommended dose for adolescent/adult patients is 200 mg oral 3 times a day and should be adjusted according to body surface area in children.1,10 The clinical improvement is noticeable by 6 months to 1 year of drug use. The commonly observed side effects are diarrhea, flatulence, abdominal discomfort, weight loss, and tremor.1,10 While its use in hepatic impairment has not been evaluated, it should be avoided in patients with severe renal impairment (creatinine clearance rate of <30 mL/min/1.73 m2).10 Our patient was started on miglustat once genetic diagnosis was made to stabilize his neurologic disease.
We report the second oldest patient at diagnosis with NPC. Myoclonus is not commonly seen in adult-onset NPC, but our patient had myoclonus as one of the major disabling features, along with more commonly reported ataxia and VSGP. Thus, NPC should be suspected in the presence of myoclonus and VSGP. Although increased awareness and improved diagnostic tools have raised the detection rate of NPC, diagnostic delays are still a major concern, especially when substrate reduction therapy using miglustat may stabilize the progression of neurologic disease.
Supplementary Material
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Kumar: conception, design, and writing the first manuscript. Dr. Rizek: conception, design, review, and critique. Dr. Mohammad: conception, review, and critique. Dr. Jog: review and critique.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
N. Kumar, P. Rizek, and Y. Mohammad report no disclosures relevant to the manuscript. M. Jog receives speaker and consultant honoraria from Merz Pharmaceuticals, Allergan, and AbbVie. He also receives research grants from CIHR, AMOSO, Allergan, Merz Pharmaceuticals, and Lawson Health Research Institute and is part of the AGE-WELL Network of Centers of Excellence (NCE) of Canada program. From time to time, he serves on advisory boards of Allergan, Boston Scientific, AbbVie, and Merz Pharmaceuticals. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 2012;106:330–344. [DOI] [PubMed] [Google Scholar]
- 2.Patterson MC, Mengel E, Wijburg FA, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis 2013;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol 2015;15:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sevin M, Lesca G, Baumann N, et al. The adult form of Niemann-Pick disease type C. Brain 2007;130:120–133. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg CR, Barnes JG, Kogan S, Seargeant LE. A rare case of Niemann-Pick disease type C without neurological involvement in a 66-year-old patient. Mol Genet Metab Rep 2015;3:18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO, Pentchev PG. Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification: a collaborative study on 70 patients. Clin Genet 1988;33:331–348. [DOI] [PubMed] [Google Scholar]
- 8.Salsano E, Umeh C, Rufa A, Pareyson D, Zee DS. Vertical supranuclear gaze palsy in Niemann-Pick type C disease. Neurol Sci 2012;33:1225–1232. [DOI] [PubMed] [Google Scholar]
- 9.Walterfang M, Macfarlane MD, Looi JCL, et al. Pontine-to-midbrain ratio indexes ocular-motor function and illness stage in adult Niemann-Pick disease type C. Eur J Neurol 2012;19:462–467. [DOI] [PubMed] [Google Scholar]
- 10.Lyseng-Williamson KA. Miglustat: a review of its use in Niemann-Pick disease type C. Drugs 2014;74:61–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.