

Abstract
Objective:
We investigated a series of patients with LGI1 antibody (Ab)–related cognitive deterioration to determine the clinical presentation, long-term outcome, and LGI1 Ab evolution.
Methods:
We retrospectively analyzed the clinical information of 76 patients with LGI1 Ab–related cognitive deterioration. Presenting syndromes were classified as limbic encephalitis (LE), non-LE, or encephalopathy (normal MRI and no CSF pleocytosis). Frequency of relapses and clinical outcome were assessed in 48 patients with prolonged follow-up (median 39 months, range 18–200).
Results:
Sixty-three patients (83%) developed LE, 3 (4%) non-LE, and 10 (13%) encephalopathy. All patients received steroids, IV immunoglobulins (Ig), or both. At 2 years, 17 (35%; 95% CI 21%–49%) fully recovered, 17 (35%) became functionally independent but not at baseline or were unable to return to work, 11 (23%) required assistance because of moderate or severe cognitive deficits, and 3 (6%) died. Predictors of bad outcome included no response to initial immunotherapy (odds ratio 23.0, 95% CI 2.4–215.6, p = 0.006) and clinical relapses (odds ratio 10.2, 95% CI 1.0–100.1, p = 0.047) that occurred in 13 patients (27%). In all patients, the LGI1 Abs were IgG4 and usually detectable in both serum and CSF (only CSF, 8%). Abs remained positive in serum of 4 of 16 patients with long-term follow-up; 3 of these 4 patients fully recovered and none showed class switch to IgG1.
Conclusions:
Up to 13% of patients with LGI1 Abs develop cognitive impairment without criteria of encephalitis. After immunotherapy, only 35% of patients return to their baseline cognitive function. Serum LGI1 Abs may remain detectable after full clinical recovery.
The current knowledge on neurologic manifestations associated with antibodies against leucine rich glioma inactivated 1 protein (LGI1 Abs) has been strongly influenced by the discovery of these Abs in patients with limbic encephalitis (LE), but the landscape of manifestations is suspected to be wider.1–3 For example, it is unknown how many patients with LGI1 Abs develop cognitive deterioration without imaging or CSF evidence of inflammation (encephalopathy).4,5 Moreover, although patients with LGI1 Abs often show a rapid response to immunotherapy, the long-term outcome and frequency of relapses have not been addressed in large studies. Isolated case reports and small series indicate that the clinical outcome is variable and not always benign.6,7
In patients with anti-NMDA receptor (NMDAR) encephalitis, the Ab–immunoglobulin G (IgG) class is predominantly IgG1, Ab detection is more reliable in CSF than in serum, and Ab titers decrease over time regardless of outcome.8 It is unclear whether LGI1 Abs share these features, although preliminary data suggest they are mainly IgG4.9 In the current study, we clarify these clinical and immunologic features in a series of patients with LGI1 Ab–associated cognitive deterioration.
METHODS
Patients.
We reviewed all patients with LGI1 Abs whose serum and CSF were sent for Ab investigations to the laboratories of the Hospital Clinic (Barcelona, Spain) or University of Pennsylvania (Philadelphia) between December 1, 1998, and June 28, 2014. Patients were included if they fulfilled the following criteria: (1) age 18 years or older, (2) adequate clinical information, (3) evidence of cognitive deterioration at diagnosis, and (4) sufficient serum and/or CSF available for Ab studies. Clinical information was obtained from the authors (13 patients) or provided by the referring physicians through a structured written questionnaire. We initially evaluated 83 patients and 7 were finally excluded because the clinical features were isolated seizures (6) or Morvan syndrome (1). The 6 patients with isolated seizures have not developed cognitive deterioration after a median follow-up of 10 months (range 6–21 months).
We classified the patients in one of the following groups: limbic encephalitis (LE), non-LE, or encephalopathy. Patients with LE presented short-term memory loss, seizures, or psychiatric symptoms, involvement of medial aspects of the temporal lobes by MRI, and CSF pleocytosis or EEG with epileptic or slow activity involving the temporal lobes. Non-LE patients showed MRI abnormalities suggesting inflammation outside the limbic system or CSF pleocytosis without criteria of LE. We applied the term encephalopathy to patients with normal MRI and no evidence of CSF pleocytosis.
We assessed the long-term functional status by an in-house scale score designed when the study was planned to detect disability caused by cognitive deficits. This cognitive performance score (CPS) ranged from 0 to 4, where 0 = asymptomatic; 0.5 = minor cognitive complaints that do not prevent the return to previous activities and work; 1 = unable to perform some previous activities or return to work but independent for activities of daily living; 2 = unable to perform most previous activities or return to work and dependent for activities of daily living; 3 = severe cognitive deficits requiring constant assistance at all levels; and 4 = severe cognitive deficits followed by death. This CPS was obtained at 2 time points during the disease: (1) 18–24 months after symptom onset, and (2) at the last follow-up. When we designed the study, we considered a CPS ≤1 a good outcome and a CPS >1 a bad outcome. We defined relapse as clinical worsening noted during treatment after initial improvement but requiring a change of the medication, or clinical worsening noted after recovery and being off medication.
Immunologic studies.
LGI1 Abs were screened by immunohistochemistry on frozen sections of rat brain and confirmed by cell-based assay using HEK293 cells cotransfected with LGI1 and ADAM23.1 Determination of LGI1 IgG class was performed with specific fluorescein isothiocyanate–labeled secondary Abs against the 4 IgG subclasses (IgG1–IgG4; The Binding Site, Birmingham, UK). We assessed LGI1 Ab titers by cell-based assay using serial dilutions of serum and CSF being the titer the last dilution that showed visible reactivity.
Standard protocol approvals, registrations, and patient consents.
The ethics committees of the Hospital Clinic and University of Pennsylvania approved the study. All patients or proxies gave written informed consent for the storage and use of serum, CSF, and clinical information for research purposes. Serum and CSF samples were deposited in the collection of biological samples named “Neuroinmunología” registered in the biobank of Institut d' Investigació Biomèdica August Pi i Sunyer (IDIBAPS), Barcelona, Spain.
Statistics.
We used nonparametric tests (Fisher exact test and Mann–Whitney U test) to compare clinical and immunologic data between 2 independent groups. Independent variables associated with bad outcome (CPS >1) at 2 years were assessed using binary logistic regression. A stepwise regression was performed to explore significant variables (inclusion p value ≤0.05 and exclusion p value >0.1). We used Stata version 13.1 (StataCorp, College Station, TX) for the statistical analysis.
RESULTS
Initial symptoms.
Seventy-six patients, mean age 61 years (range 32–80), 50 (66%) men, were included in the study. Twenty-nine patients (38%, 95% confidence interval [CI] 27%–49%) developed over several weeks or months (median of 2 months, range 0.4–29 months) isolated or predominantly isolated symptoms: 12 had seizures, 9 had mood/behavioral manifestations (4 apathy, 3 depression, and 2 anxiety [see 2 examples in appendix e-1 on the Neurology® Web site at Neurology.org]), 7 had an amnestic syndrome, and one developed a gait disorder. The remaining 47 patients (62%) developed a combination of the above symptoms (figure e-1). The type of initial symptoms did not vary between patients initially characterized as developing encephalitis and those characterized as encephalopathy (see below). In 47 patients (62%), the neurologic symptoms reached the nadir in ≤3 months.
Neurologic symptoms and paraclinical studies at diagnosis.
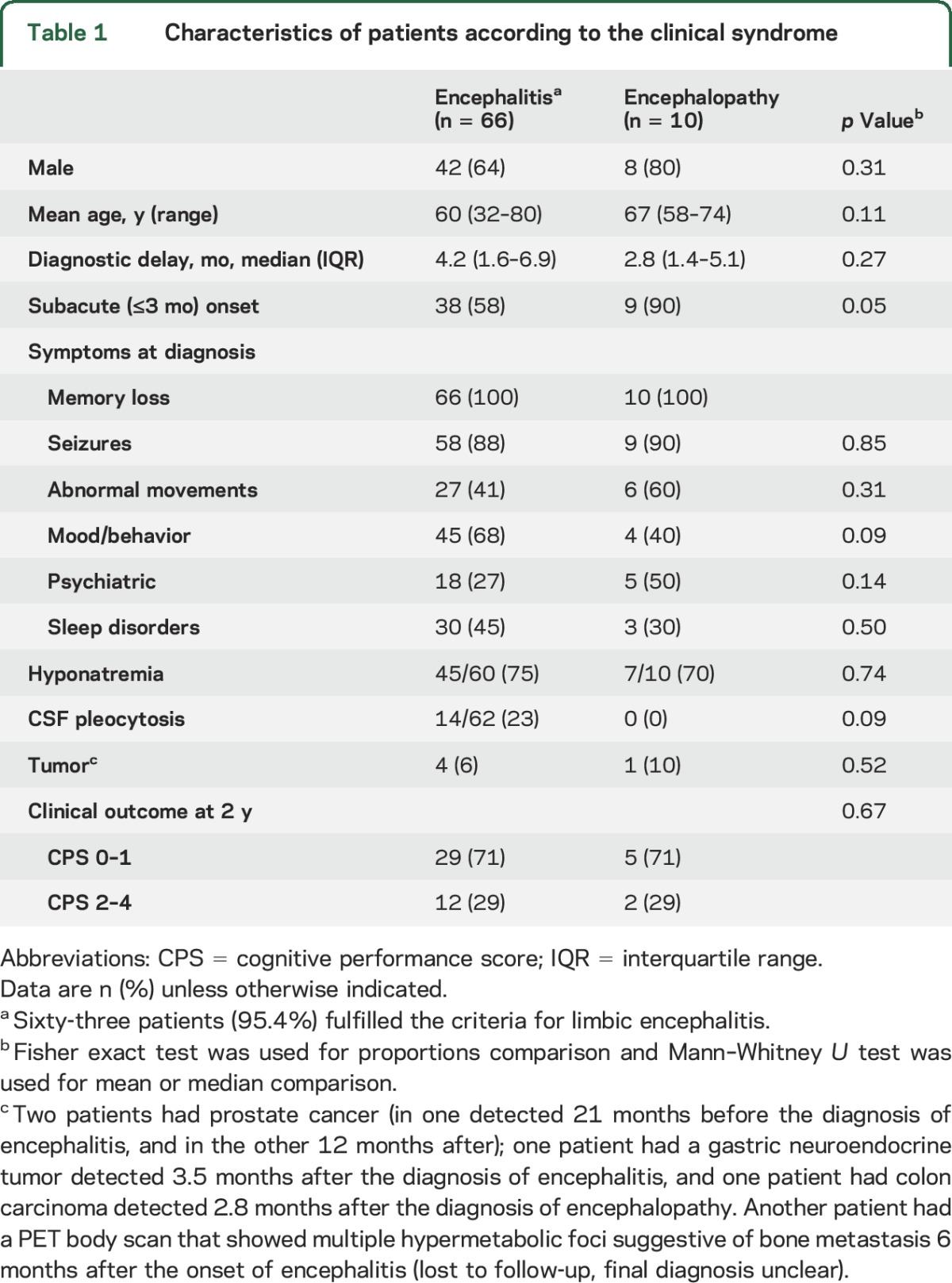
By inclusion criteria, all patients had memory loss and cognitive deterioration. Symptoms at diagnosis are summarized in table 1. Overall, 63 patients (83%) fulfilled the criteria of LE. The MRI findings were unilateral in 15 (24%) and bilateral in 48 (76%). In 8 patients, the initial MRI was considered normal (even with evidence of cognitive deficits) with subsequent MRI studies revealing features of LE. Three patients (4%) were considered to have non-LE: 2 had MRI abnormalities outside the temporal lobes, and the third had a normal MRI with CSF pleocytosis. CSF pleocytosis (median 10 white blood cells/mL, range 6–41) was identified in 14 of 62 patients (23%) with encephalitis. Ten patients (13%, 95% CI 5%–21%) did not have MRI or CSF evidence of inflammation (encephalopathy). Compared to patients with encephalitis, they were more likely to have a subacute onset (58% vs 90%, p = 0.05; table 1). The 10 patients had memory loss and cognitive deterioration, 5 with prominent psychiatric symptoms and 7 with seizures.
Table 1.
Characteristics of patients according to the clinical syndrome
LGI1 Ab subtypes, serum and CSF levels at diagnosis, and evolution in follow-up samples.
In 51 patients with paired serum/CSF samples available, we detected LGI1 Abs in both serum and CSF in 47 (92%) and only in the CSF in 4 (8%). All sera available for subclass analysis (57/57) showed IgG4 Abs. LGI1 Abs were exclusively of the IgG4 subclass in 33 patients (57.9%), IgG2 and IgG4 in 14 (24.6%), and IgG1, IgG2, and IgG4 in 10 (17.5%). None of the serum samples had IgG3 subclass Abs (figure 1).
Figure 1. Analysis of IgG subclasses of LGI1 antibodies.

Cell-based assay with HEK293 cells showing the 3 different combinations of IgG subclasses among 57 sera examined at disease onset. Each row represents an anti-LGI1–positive serum revealed by a specific secondary antibody against total human IgG (A, E, I), IgG1 (B, F, J), IgG2 (C, G, K), and IgG 4 (D, H, L). The frequency of each IgG subclass combination is shown on the right side of each row. The green signal indicates reactivity against transfected LGI1. Nuclei counterstained with DAPI (4′,6-diamidino-2-phenylindole). Scale bar = 20 μm. IgG = immunoglobulin G.
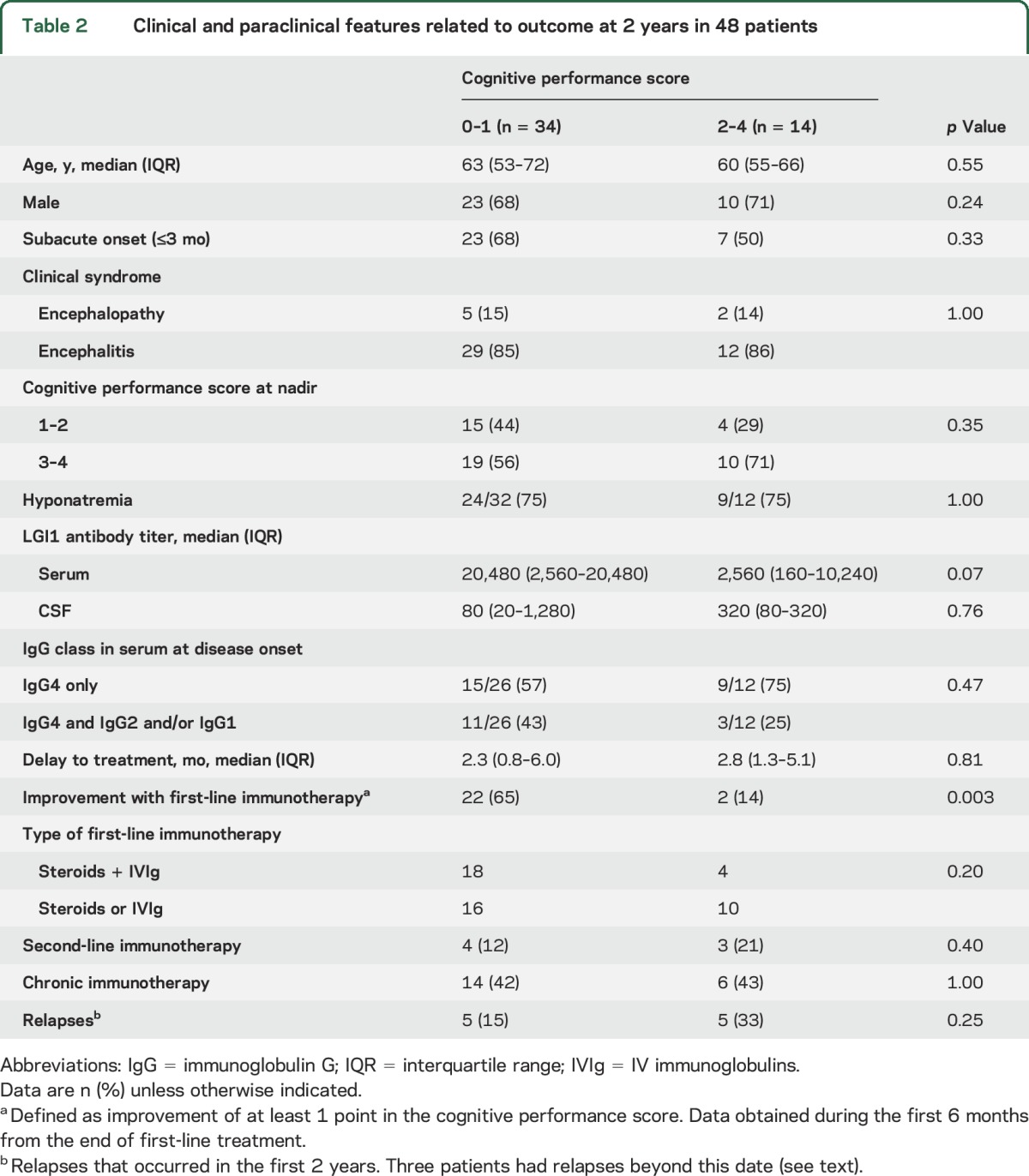
Baseline titers and evolution of LGI1 Abs were assessed in the group of patients with long-term follow-up and sufficient sample volume for evaluation (40 sera and 36 CSF). Serum and CSF titers were not significantly different between patients with good (CPS 0–1) and bad (CPS 2–4) outcome (table 2). Follow-up serum samples were available in 19 patients. In 3 of them, the sample was obtained shortly before or after a clinical relapse and all were LGI1 Ab positive. Of the remaining 16 patients, 12 became Ab negative (median follow-up 20 months, range 5–62 months) and all but one had a good outcome (CPS 0–1). Four patients remained LGI1 Ab positive 20 months after treatment even though 3 of them had substantial clinical recovery (CPS 0–1). None of these 4 patients showed Ab class switch to IgG1.
Table 2.
Clinical and paraclinical features related to outcome at 2 years in 48 patients
Immunotherapy and outcome.
A follow-up of at least 18 months was available for 48 patients (median [interquartile range] time of 39.2 [22.0–58.3] months). First-line immunotherapy included oral (3 patients) or IV (19) steroids in 22 patients (46%), IVIg in 4 (8%), and steroids combined with IVIg in 22 (46%). With this treatment, 24 patients (50%) improved at least 1 point in the CPS. Seven patients were additionally treated with second-line immunotherapy (4 rituximab, 1 cyclophosphamide, 2 both). Twenty patients (42%) underwent chronic oral immunosuppression (13 prednisone, 6 azathioprine, and 1 mycophenolate mofetil) for a median (interquartile range) time of 17.4 (10.9–32.2) months (table e-1).
At 24 months, 34 patients (71%, 95% CI 57%–84%) had a good outcome with a CPS of 0 to 0.5 (17) or 1 (17), and 14 patients had a bad outcome: 8 (17%) CPS of 2, 3 (6%) CPS of 3, and 3 (6%) CPS of 4. Among the 35 patients with a follow-up >2 years, 4 continued to improve and returned to their premorbid baseline status, 2 worsened because of late relapses, and 2 died (one from complications of the disease); the clinical status of the other patients remained unchanged or normal.
Thirteen patients (27%, 95% CI 14%–40%) had relapses that required further treatment. In 9 of these patients, relapses occurred during the first 6 months of the disease (range 1.7–6 months), 6 of them related to steroid tapering. Four patients had late relapses 12, 27, 65, and 66 months after disease onset. Relapses consisted of reappearance of cognitive or behavioral symptoms with or without seizures, or recurrent seizures after a period of months to years without seizures (3 patients). Table 2 summarizes the main features of patients grouped according to good (CPS 0–1) or bad (CPS 2–4) outcome after a follow-up of 2 years. In the multivariate analysis, failure to respond to first-line immunotherapy (odds ratio 23.0, 95% CI 2.4–215.6, p = 0.006) and clinical relapses (odds ratio 10.2, 95% CI 1.0–100.1, p = 0.047) were the only predictors of bad outcome.
DISCUSSION
In this study, we examined the long-term clinical outcome of a large cohort of patients with LGI1 Ab–associated cognitive deterioration, which provided several important findings with practical implications: (1) up to 13% of the patients presented with encephalopathy without MRI or CSF features of encephalitis; (2) 29% had moderate to severe cognitive problems 2 years after disease onset; (3) relapses occurred in 27% of the patients, usually within the first 6 months of the disease, and were an independent predictor of bad outcome; (4) the sensitivity of LGI1 Ab testing was higher in CSF than in serum; and (5) after clinical recovery, serum LGI1 Abs may remain positive.
Our findings show that although most patients with LGI1 Ab–associated symptoms respond to immunotherapy, only 35% are able to return to work or to all premorbid activities. The study was observational and the final outcome was provided by the referring neurologists. We cannot rule out that formal neurocognitive evaluation would detect more patients with subtle cognitive deficits.10 However, the endpoints that we chose when the study was designed were a reasonable objective (patients were able to return to their job or, if retired, to their usual previous daily activities) to confirm that just a third of patients return to normal.
Clinical relapses occurred in 27% of the patients. This frequency is higher than previously reported1 but may reflect the criteria used. We included as relapses cases of clinical worsening noted during treatment (e.g., steroid taper) and requiring a change or redosing of the medication. Even though patients with relapses usually improved after treatment modification, this event and the lack of response to first-line immunotherapy were independent predictors of bad outcome. Taken together, these data suggest that patients who do not respond or worsen during first-line immunotherapy should receive second-line immunotherapy, as shown in patients with anti-NMDAR encephalitis.11 Rituximab has been considered particularly effective in IgG4-mediated disorders9 but there are limited data for LGI1 Ab–associated disorders and no firm recommendations can be made.12–14
A substantial number of patients did not fulfill the recently proposed criteria of autoimmune encephalitis either because the MRI and CSF findings did not suggest an inflammatory disorder or because the presentation was atypical (e.g., >3 months).15 However, most patients with prolonged symptom presentation showed MRI abnormalities in the medial temporal lobes suggesting autoimmune encephalitis. It is of interest that the initial brain MRI was reported normal in 8 patients who subsequently were found to have typical MRI changes of LE in a repeat study.
In some previously reported patients with LGI1 Abs, the subacute onset of cognitive deterioration with myoclonus and absent CSF inflammatory changes led to the misdiagnosis of Creutzfeldt-Jakob disease (CJD).5 This confusion was fueled by the description of VGKC Abs (without further antigen characterization) in a few true cases of CJD.16,17 In contrast, LGI1 Abs have not been reported in patients with CJD.18 We found that 13% of our patients with LGI1 Abs developed an encephalopathy that could potentially mimic CJD. However, most of these patients developed seizures in addition to cognitive deterioration, which is unusual in CJD,19 and the MRI findings were different from the cortical and basal ganglia abnormalities characteristic of CJD.20,21 Taken together, our findings suggest that the confusion between CJD and LGI1 Ab encephalopathy should rarely occur. In a recent survey of 384 autopsies of patients with suspected CJD, 6 had definite autoimmune encephalitis associated with neuronal Abs, none of them against LGI1.22
In a previous study of patients with Morvan syndrome, it was suggested that LGI1 Abs in a few of the patients were of the IgG4 subclass.23 We confirmed this finding in all of our patients suggesting that, as in other IgG4-mediated syndromes, the IgG4 LGI1 Abs may alter neuronal function through a disruption of the interaction between LGI1 and its ligand (ADAMs) leading to a secondary dysregulation of synaptic receptors, as recently proposed.24
Similar to anti-NMDAR encephalitis,8 we found that determination of LGI1 Abs in CSF was more sensitive (100%) than in serum (92%). A possible reason why LGI1 Abs are only detectable in CSF in some cases is that the serum levels may be below screening detection. This is supported by a preliminary study suggesting predominant B cell affinity maturation in the brain leading to increased levels of LGI1 Abs in the CSF.25 In any case, our findings emphasize the need to test both serum and CSF in patients with suspected CNS autoimmune disorders.
In this study, serum LGI1 Abs disappeared after a median follow-up of 2 years. The persistent detection of serum LGI1 Abs did not associate with a poor outcome. However, we did not find IgG subclass switch in patients with good outcome. This subclass switch was described in a patient with myasthenia and MuSK Abs during remission of the disease.26
Considering the limitations posed by the observational, retrospective nature of this study, our findings indicate that the final outcome of patients with cognitive deterioration and LGI1 Abs is far from optimal despite an initial improvement with steroids alone or IVIg. Early diagnosis before the development of cognitive dysfunction, recognition of the disorder in patients without criteria of encephalitis, and probably more aggressive upfront immunotherapy are important steps toward an improved outcome. The persistence of LGI1 Abs in patients who fully recover challenges the value of routine surveillance of Ab titers to evaluate the clinical outcome.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Mercè Alba and Sheila León for excellent technical assistance. The authors are indebted to all contributors (listed on the Neurology® Web site at Neurology.org) for providing clinical information of their patients.
GLOSSARY
- Ab
antibody
- CI
confidence interval
- CJD
Creutzfeldt-Jakob disease
- CPS
cognitive performance score
- Ig
immunoglobulin
- LE
limbic encephalitis
- NMDAR
NMDA receptor
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Design/conceptualization of the study: H.A., T.A., F.G., J.D. Analysis/interpretation of the data: all authors. Statistical analysis and figure development: H.A. (academic), L.S. Drafting the manuscript: H.A., F.G., J.D. Revising the manuscript: all authors. All authors gave final approval of the version to be published.
STUDY FUNDING
This study was supported in part by grants from Instituto Carlos III (CM14/00081, T.A.; CD14/00155, E.M.-H.) and Fondo de Investigaciones Sanitarias, FEDER (FIS 15/00377, F.G.; FIS 14/00203, J.D.), Madrid, Spain, NIH RO1NS077851 (J.D.), and Fundació Cellex (J.D.).
DISCLOSURE
H. Ariño, T. Armangué, M. Petit-Pedrol, L. Sabater, E. Martinez-Hernandez, and M. Hara report no disclosures relevant to the manuscript. E. Lancaster taught courses for Grifols, Inc., and consulted for MedImmune, Inc. A. Saiz has received compensation for consulting services and speaker honoraria from Bayer Schering, Merck Serono, Biogen Idec, Sanofi-Aventis, Teva Pharmaceutical Industries Ltd., and Novartis. J. Dalmau receives royalties from Athena Diagnostics for the use of Ma2 as autoantibody tests and from EUROIMMUN for the use of NMDAR as an autoantibody test. F. Graus receives royalties from EUROIMMUN for the use of IgLON5 as a diagnostic test. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004;127:701–712. [DOI] [PubMed] [Google Scholar]
- 4.Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900. [DOI] [PubMed] [Google Scholar]
- 5.Geschwind MD, Tan KM, Lennon VA, et al. Voltage-gated potassium channel autoimmunity mimicking Creutzfeldt-Jakob disease. Arch Neurol 2008;65:1341–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013;265:75–81. [DOI] [PubMed] [Google Scholar]
- 7.Malter MP, Frisch C, Schoene-Bake JC, et al. Outcome of limbic encephalitis with VGKC-complex antibodies: relation to antigenic specificity. J Neurol 2014;261:1695–1705. [DOI] [PubMed] [Google Scholar]
- 8.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2014;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huijbers MG, Querol LA, Niks EH, et al. The expanding field of IgG4-mediated neurological autoimmune disorders. Eur J Neurol 2015;22:1151–1161. [DOI] [PubMed] [Google Scholar]
- 10.Bettcher BM, Gelfand JM, Irani SR, et al. More than memory impairment in voltage-gated potassium channel complex encephalopathy. Eur J Neurol 2014;21:1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irani SR, Gelfand JM, Bettcher BM, Singhal NS, Geschwind MD. Effect of rituximab in patients with leucine-rich, glioma-inactivated 1 antibody-associated encephalopathy. JAMA Neurol 2014;71:896–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown JW, Martin PJ, Thorpe JW, et al. Long-term remission with rituximab in refractory leucine-rich glioma inactivated 1 antibody encephalitis. J Neuroimmunol 2014;271:66–68. [DOI] [PubMed] [Google Scholar]
- 14.Cash SS, Larvie M, Dalmau J. Case records of the Massachusetts General Hospital. Case 34-2011: a 75-year-old man with memory loss and partial seizures. N Engl J Med 2011;365:1825–1833. [DOI] [PubMed] [Google Scholar]
- 15.Graus F, Titulaer MJ, Balu R, et al. Clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016;15:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newey CR, Appleby BS, Shook S, Sarwal A. Patient with voltage-gated potassium-channel (VGKC) limbic encephalitis found to have Creutzfeldt-Jakob disease (CJD) at autopsy. J Neuropsychiatry Clin Neurosci 2013;25:E05–E07. [DOI] [PubMed] [Google Scholar]
- 17.Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013;70:919–922. [DOI] [PubMed] [Google Scholar]
- 18.Grau-Rivera O, Sanchez-Valle R, Saiz A, et al. Determination of neuronal antibodies in suspected and definite Creutzfeldt-Jakob disease. JAMA Neurol 2014;71:74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Valle R, Nos C, Yague J, Graus F, Dominguez A, Saiz A. Clinical and genetic features of human prion diseases in Catalonia: 1993–2002. Eur J Neurol 2004;11:649–655. [DOI] [PubMed] [Google Scholar]
- 20.Tschampa HJ, Kallenberg K, Urbach H, et al. MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease: a study on inter-observer agreement. Brain 2005;128:2026–2033. [DOI] [PubMed] [Google Scholar]
- 21.Heine J, Pruss H, Bartsch T, Ploner CJ, Paul F, Finke C. Imaging of autoimmune encephalitis: relevance for clinical practice and hippocampal function. Neuroscience 2015;309:68–83. [DOI] [PubMed] [Google Scholar]
- 22.Maat P, de Beukelaar JW, Jansen C, et al. Pathologically confirmed autoimmune encephalitis in suspected Creutzfeldt-Jakob disease. Neurol Neuroimmunol Neuroinflamm 2015;2:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012;72:241–255. [DOI] [PubMed] [Google Scholar]
- 24.Ohkawa T, Fukata Y, Yamasaki M, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci 2013;33:18161–18174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irani SR, Lehmann-Horn K, Geschwind M, Wang S, Vincent A, von Budingen HC. The active intrathecal B-cell response in LGI1-antibody encephalitis. Lancet 2015;385(suppl 1):S46. [DOI] [PubMed] [Google Scholar]
- 26.Niks EH, van Leeuwen Y, Leite MI, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol 2008;195:151–156. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.