

Abstract
Nonketotic hyperglycinemia, also known as glycine encephalopathy (OMIM #605899), is an autosomal recessive disorder of glycine metabolism resulting from a defect in the glycine cleavage system. We report two novel mutations of the glycine decarboxylase (GLDC) gene observed in a compound heterozygous state in a neonate of mixed Maori and Caucasian parentage: c.395C>T p.(Ser132Leu) in exon 3, and c.256-?_334+?del p.(Ser86Valfs*119), resulting in an out-of-frame deletion of exon 2. Additionally, we describe our experience of implementing the ketogenic diet, alongside standard pharmacological therapy, and highlight its potential therapeutic benefit in severe nonketotic hyperglycinemia, particularly in seizure management.
Keywords: GLDC gene, glycine decarboxylase, glycine metabolism, nonketotic hyperglycinemia, ketogenic diet
Introduction
Nonketotic hyperglycinemia (NKH) is caused by absent or decreased activity of the glycine cleavage system (GCS), and is characterized by the accumulation of a large amount of glycine in body fluids.1 The GCS is a mitochondrial multienzyme system comprising three protein components: glycine decarboxylase (also known as P-protein), aminomethyltransferase (T-protein), and hydrogen carrier protein (H-protein).2 P-protein, T-protein, and H-protein are encoded by the genes GLDC, AMT and GCSH, respectively.3 4 5 Mutations in the GLDC gene account for 75 to 80% of cases of NKH.6 7 The AMT gene is mutated in up to one quarter of cases, whereas mutations in the GCSH gene are responsible for <1% of nonketotic hyperglycemia cases.6 7
Defective cleavage of the amino acid glycine results in its accumulation in different body tissues, most notably in the brain. The neurotransmitter glycine exerts inhibitory effects at the level of the spinal cord and brainstem, and excitatory effects in the cerebral cortex; hence, excess levels can cause hypotonia, apnoea, seizures, and coma.8 NKH typically manifests as an acute encephalopathy in the neonatal period, with 85% displaying a severe form, and 15% presenting with milder forms.9 10 Rare later-onset and atypical cases have also been reported, with onset from childhood to adulthood.11 12 Onset in the neonatal period is associated with a poorer prognosis than onset in infancy; however, there are many exceptions.7 9 Twenty percent of all children presenting as either neonates or infants have a less severe outcome, defined as developmental quotient greater than 20.8 9
The diagnostic characteristic of NKH is an elevated cerebrospinal fluid (CSF) glycine concentration together with an increased CSF:plasma glycine ratio. The prognosis for classical, severe neonatal NKH remains very poor.13 Current therapy is predominantly aimed at decreasing glycine concentrations and blocking its effect at N-methyl-D-aspartate (NMDA) receptors.9 Early treatment appears to be beneficial in certain patients with attenuated forms of NHK.7 14 15 Molecular genetic testing provides a confirmatory diagnosis and enables carrier testing and prenatal testing in affected families.
Case Report
The proband is a male infant born to a nonconsanguineous couple. The mother is of Maori ancestry and the father is Caucasian. The parents have previously had three first-trimester miscarriages and two healthy daughters.
The proband was born at full term following an uneventful antenatal period. He was born in good condition and discharged the following day on full breast feeds. On day three of life, he presented to the regional hospital with lethargy and poor feeding. He developed apnoeas and bradycardias, requiring intubation and ventilation. He subsequently developed seizures in the form of protracted hiccups, lasting approximately ten minutes per episode, necessitating treatment with a loading dose of phenobarbitone at 20 mg/kg. He was very hypotonic with depressed reflexes and extensor plantars.
The initial investigations revealed a normal blood pH and ammonia. Amino acid analysis showed highly elevated concentrations of glycine in the plasma at 2,358 µmol/L (reference range: 150–560 µmol/L), cerebrospinal fluid (CSF) at 535.3 µmol/L (reference range: 3.8–8 µmol/L), and urine at 6,370 µmol/mmol creatinine (reference range: <1,097). The CSF:plasma glycine ratio was markedly elevated at 0.23 (reference range: <0.02). The diagnosis of classical NKH was made. An electroencephalogram (EEG) recorded on day four of life was markedly abnormal and showed a discontinuous background with a burst suppression pattern. His cranial ultrasound scan was normal. The initial brain magnetic resonance imaging (MRI) at five days showed a thin corpus callosum and changes suggestive of prior posterior aspect sagittal sinus thrombosis with partial recanalization. A repeat MRI at two weeks of age showed bilateral, symmetrical, abnormal diffusion restriction involving the posterior corona radiata, the posterior limbs of the internal capsule, the cerebral peduncles, and the dorsal brainstem, consistent with areas of vacuolating myelinopathy. The magnetic resonance spectroscopy demonstrated a high glycine peak within the spectra at 3.56 ppm bilaterally within the cerebral white matter.
The poor long-term outcome in survivors of classical neonatal-onset NKH was discussed with the parents, and the critical decision on withdrawing ventilator support was offered to them. Particular emphasis was placed on the need for a decision to be reached within a short timeframe due to the transient nature of respiratory depression.16 The parents chose to defer the decision to withdraw ventilator support. The neonate was treated with oral dextromethorphan at 5 mg/kg/day and sodium benzoate at 300 mg/kg/day,17 18 the latter initially normalising plasma glycine levels. He continued to have hiccups and myoclonic jerks, which responded well to levetiracetam and clonazepam. He gradually gained regular, spontaneous respiratory effort and was extubated at 21 days. Evolving spasticity, brisk reflexes and clonus were evident throughout his stay in neonatal intensive care. He was discharged home on day 33 of life.
A ketogenic diet was considered during the neonatal period as it has been used to reduce seizures in a small cohort of patients with NKH.19 20 However, due to social constraints, it was deferred initially and commenced at 6 months of age in view of refractory seizures. Difficulties in maintaining glycaemic control and weight gain led to a temporary disruption of the ketogenic diet, which was recommenced five weeks later in view of intractable seizures. Plasma glycine levels had also risen despite increased doses of sodium benzoate of up to 510 mg/kg/day and mild dietary protein restriction. Further increments of sodium benzoate were, however, not made due to associated reflux oesophagitis and possible gastritis. The implementation of ketogenic diet led to significant reduction in spasticity, seizure frequency, improvements in EEG, normalisation of plasma glycine levels, and enhanced quality of life in the patient.21 22 23
Currently, at 13 months of age, he remains globally delayed; however, his seizure frequency has significantly reduced. He is able to fix and follow and responds to his parents' voices. He smiles and has some vocalisations.
Molecular Studies
The UCSC genome browser (http://genome.ucsc.edu) was used to obtain the reference transcript of the GLDC gene (NM_000170.2) and its protein product (NP 000161.2; UniProtKB P23378). Primers were designed, checked for underlying single nucleotide polymorphisms, and synthesized as described previously.24
DNA was extracted from peripheral blood on a QIAsymphony SP using the QIAsymphony DSP DNA Midi Kit (Qiagen Pty Ltd, Maryland, United States). DNA quantity and quality were assessed using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific, Wilmington, Delaware, United States). Genomic DNA (50 ng) was subjected to conventional polymerase chain reaction (PCR) and bidirectional DNA sequencing as described previously.24
Variant ReporterSoftware v1.0 (Applied Biosystems) was used to analyze the sequences of the coding regions and flanking upstream and downstream 20 bases of the adjacent introns. The reference sequence was GenBank NM_000170.2, with cDNA number +1 corresponding to the adenosine of the translation initiation codon (codon 1). Each sequence trace had a minimum trace score of 35, which corresponds to an average false base call frequency of 0.031%.
An Agilent 8 × 60K custom comparative genomic hybridization (CGH) array was used for dosage analysis. This bespoke CGH array has been designed against UCSC build HG19 to interrogate the coding regions of 30 genes of interest to our laboratory. The exons, introns, and 1,000 bp of the upstream and downstream untranslated regions of these genes are covered by unique probes at a high density. Genome-wide coverage is achieved with low density ‘backbone’ probes, which are essential to the normalisation of probe intensity during data analysis.
Five hundred nanograms of gDNA were processed according to the manufacturer's instructions (Agilent oligonucleotide array-based CGH for genomic DNA analysis: enzymatic labeling for blood, cells, or tissues v7.2; http://www.agilent.com). Briefly, extracted gDNA from samples and gender matched controls was initially prepared by restriction digestion. Samples were subsequently denatured in the presence of Cyanine 3- (test-) or Cyanine 5- (control-) labeled random primers (product number: 5190–0441, Agilent) and incubated with the Klenow fragment of DNA polymerase, together with dNTPs and reaction Buffer, at 37°C for 2 hours. The enzyme was inactivated by a subsequent incubation at 65°C for 10 minutes. The labeled gDNA was purified using the purification column (Aligent) and quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). Equimolar amounts of the test and control samples were combined with Cot-1 DNA (Agilent), 10x aCGH blocking agent and HI-RPM hybridization buffer (Agilent) to form the hybridization master mix. This mix was subsequently incubated at 95°C for 3 minutes followed by 30 minutes at 37°C. Hybridization was performed in an Agilent SureHyb chamber at 65°C for a period of 24 hours.
Following hybridization, the slides were washed and scanned using a Roche NimbleGen MS 200 microarray scanner. Analysis was performed using Agilent CytoGenomics edition 2.7.22.0 (Default Analysis Method – CGH v2). Regions exhibiting a copy number change within the GLDC gene were examined using the UCSC genome browser (http://genome.ucsc.edu) to determine the location and significance of the change. The inclusion threshold for analysis of a dosage change required a log2 ratio of less than -0.25 over at least three contiguous probes for a deletion, and a log2 ratio of greater than 0.25 over at least three contiguous probes for a duplication (as recommended by the manufacturer).
Carrier testing was subsequently performed in the parents. PCR and DNA sequencing of the relevant exon were performed to test for the presence of the mutation found in the proband. Testing was performed in duplicate alongside the proband as well as a negative control. The Agilent 8 × 60K custom CGH array was used to screen for the exonic deletion event found in the proband.
Results
The sequence traces of the proband's DNA indicated three variants (Table 1). Two of these variants are listed in the Database of Single Nucleotide Polymorphisms (dbSNP; http://www.ncbi.nlm.nih.gov/projects/SNP/) as known polymorphisms and are unlikely to be of pathogenic significance. The other variant, c.395C>T p.(Ser132Leu), has, to our knowledge, not been reported in the literature or mutation databases (Human Gene Mutation Database Professional; HGMD Pro; https://portal.biobase-international.com/cgi-bin/portal/login.cgi?redirect_url=/hgmd/pro/start.php). It was subjected to bioinformatic analysis, which predicted that this nucleotide substitution is pathologically significant (Table 2). Splice site analysis by BDGP (www.fruitfly.org/seq_tools/splice.html) and ASSP (http://wangcomputing.com/assp/index.html) programs offered uninformative predictions in relation to this variant.
Table 1. Variants detected in the DNA of the proband upon sequencing (A) and dosage analysis (B).
| Nucleotide change | Amino acid change | Exon/intron | rs IDa accession number | Variant state | |
|---|---|---|---|---|---|
| A | c.395C>T | p.(Ser132Leu) | Exon 3 | – | Heterozygous |
| c.498T>C | p.(=) | Exon 4 | 150193069 | Heterozygous | |
| c.3063+7G>C | Intron 25 | 2228098 | Heterozygous | ||
| B | c.256-?_334+?del | p.(Ser86Valfs*119) | Deletion of exon 2 | – | Heterozygous |
rs ID: reference single nucleotide polymorphism identity.
Table 2. In silico predictions regarding the effect of the GLDC gene variant p.(Ser132Leu).
| Bioinformatic program | URL | Prediction |
|---|---|---|
| PolyPhen2 | http://genetics.bwh.harvard.edu/pph2/ 25 | Probably damaging |
| PROVEAN | http://provean.jcvi.org/protein_batch_submit.php?species=human 26 | Deleterious effect |
| SIFT | http://sift.bii.a-star.edu.sg/www/SIFT_BLink_submit.html 27 | Not tolerated |
| SNPs&GO | http://snps-and-go.biocomp.unibo.it/snps-and-go/ 28 | Disease causing |
| Mutation Taster | http://www.mutationtaster.org/ 29 | Disease causing |
| Grantham Score | 30 | Moderately radical |
Dosage analysis of the proband revealed an exonic deletion event: c.256-?_334+?del (Fig. 1). The event results in an out-of-frame exonic deletion within the GLDC gene and is predicted to produce a truncated protein: p.(Ser86Valfs*119) (Fig. 2). This mutation has, to our knowledge, not been reported in the online HGMD professional database but it is of the type which is expected to be disease causing.
Fig. 1.
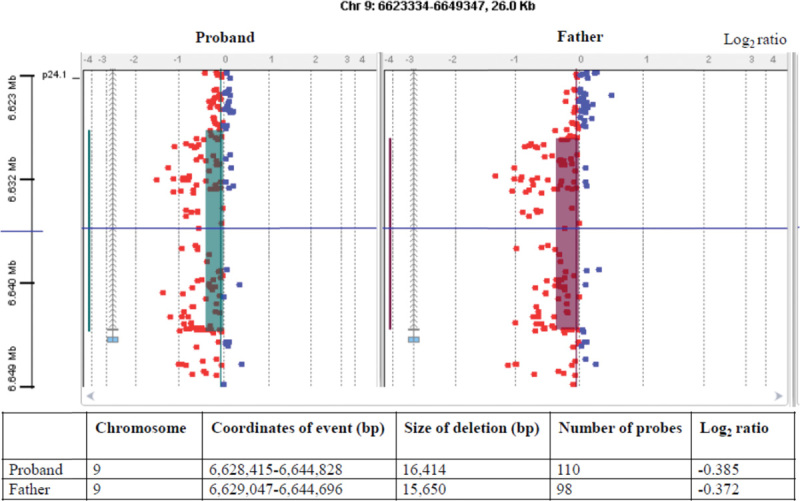
Agilent CytoGenomics Edition 2.7.22.0 software output for the proband and father. Each small square represents an oligo probe. The heterozygous deletion is depicted by probes colored red across the shaded regions, corresponding to a minimum average absolute log2 ratio of less than -0.25 over at least 3 contiguous probes. The UCSC assembly GRCh37/hg19 (http://genome.ucsc.edu) was used to obtain the coordinates of the event.
Fig. 2.
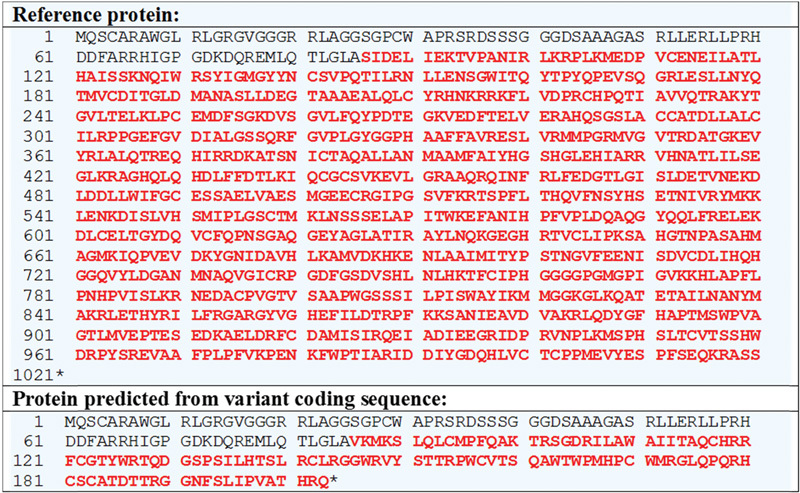
The predicted amino acid sequences of the proteins expressed from the wild type (reference) and mutant (variant) GLDC gene transcripts (Refseq accession number: NM_000170.2). The single letter amino acid code has been used. Amino acids downstream of the mutation are depicted in red, and the asterisk indicates translation termination. This figure was generated through the Mutalyzer 2.0.5 Web site (https://mutalyzer.nl/name-checker).
Carrier testing in the parents revealed that the mother was heterozygous for the c.395C>T p.(Ser132Leu) mutation (Fig. 3) and the father was heterozygous for the c.256-?_334+?del p.(Ser86Valfs*119) mutation (Fig. 1).
Fig. 3.
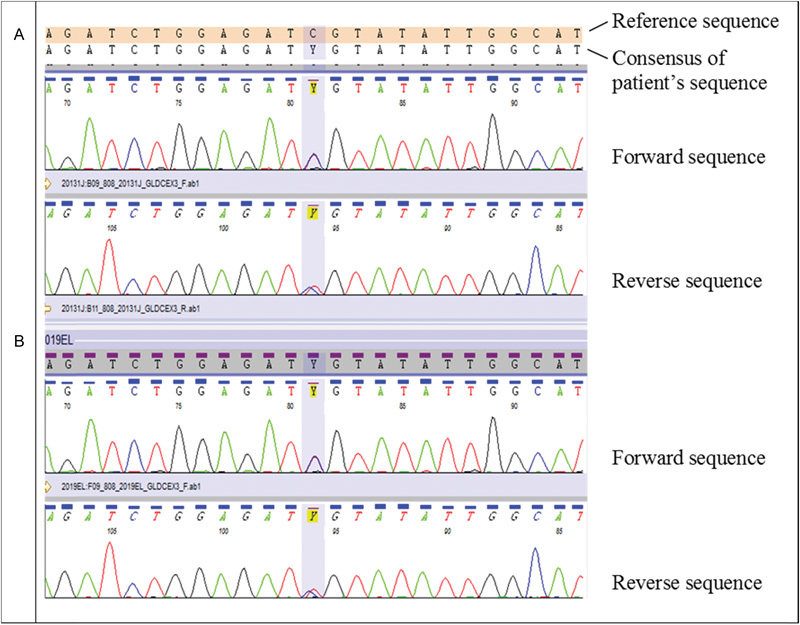
Sequence electropherograms showing the bidirectional traces of a region of exon 3 of the GLDC gene. The proband (A) and mother (B) are heterozygous for the c.395C>T p.(Ser132Leu) mutation.
Discussion
In its classical form, NKH is a rare and devastating neurometabolic disorder with a yet unknown precise worldwide incidence. Its prevalence is, however, relatively high in several geographical areas, including Northern Finland (1 in 12,000 newborns) and British Columbia (1 in 63,000 newborns).31 32 Common mutations are found in these countries and many patients are homozygous for one of these mutations.31 In contrast, mutations among NKH patients outside of these geographical areas are highly heterogeneous, with over 80% of patients found to be compound heterozygotes.7
There are currently 116 mutations in the GLDC gene that are reported to cause NKH in the HGMD pro database. Approximately two-thirds of the reported variants are missense or nonsense mutations; the remainder comprise small insertions/deletions, larger deletions, and mutations that affect splicing.33
The c.395C>T missense mutation in exon 3 reported here leads to the substitution of a polar amino acid serine with a nonpolar amino acid leucine at position 132 of the P-protein. Unfortunately, we do not have resources available to determine whether this mutation abolishes enzyme activity or whether residual enzyme activity remains, but this could be assessed using the P-protein assay.34 This amino acid is, however, conserved across a broad range of species (data available at BLAST, www.ncbi.nlm.nih.gov) and bioinformatic programs predict that the mutation is of pathogenic significance when observed in a homozygous or compound heterozygous state. A nucleotide substitution has previously been reported at this position: c.395C>G p.(Ser132Trp).35 This mutation was found in a Turkish neonate with classical NKH. It also results in a change from a polar to a nonpolar amino acid and is predicted to be damaging by in silico analysis. The deletion of exon 2 of the GLDC gene is a frameshift mutation resulting in the premature termination of the P-protein, and is of the type to be clinically significant. Nineteen gross deletions causative of the NKH phenotype are reported in HGMD pro, the majority of which are multi-exon deletions. The deletion of exon 2 has previously been reported in conjunction with the deletion of exon 1 or in conjunction with the deletion of exons 1 and 3.36 37
Several dosage analysis methods are available to the molecular diagnostic laboratory, including multiplex ligation-dependent probe amplification (MLPA), quantitative real-time PCR, and customized fluorescence in situ hybridization.38 These methods are relatively expensive, largely owing to the cost of the probes.38 For small diagnostic laboratories, low sample throughput prevents batching of samples if turnaround times are to be maintained, thus decreasing the cost-effectiveness of gene-specific assays. Furthermore, staff proficiency in a large range of dosage techniques can present difficulty when sample numbers are modest. A custom CGH array provides a robust and cost-effective solution by enabling consolidation of previously separate gene-targeted dosage assays to a single validated technique, allowing analysis of a greater number of genes to be offered in-house and thereby bringing increased revenue into the laboratory. Additionally, as multiple probes cover each exon, array CGH eliminates the risk of false positives occurring due to polymorphisms under primer binding sites, an inherent risk in PCR-based techniques.38 Finally, in contrast to MLPA, array CGH permits the interrogation of intronic as well as exonic regions, enabling more accurate mapping of breakpoints.38
In summary, we describe two novel mutations in the GLDC gene in a patient with classical NKH. The identification of the causative mutations in an affected individual assists in confirming a clinical diagnosis, supports genetic counselling, and enables carrier and prenatal testing to be offered. This case demonstrates the value of a two-tiered approach to mutation screening. DNA sequencing and dosage analysis, using a custom-designed CGH array, offer a robust and cost-effective means of providing comprehensive mutation analysis of the GLDC gene. In our patient, ketogenic diet, in association with standard pharmacological therapy, resulted in dramatic reduction of seizures and improved quality of life. This preliminary data may suggest that ketogenic diet, in combination with standard treatment, could be considered as a potential therapeutic option, particularly in seizure management in classical neonatal NKH. Larger studies are, however, required to further substantiate this.
Acknowledgments
The authors thank PathWest Laboratory Medicine (Western Australia) for the extraction of DNA from the peripheral EDTA blood samples.
Footnotes
Author Contributions S. L. N. wrote the manuscript with input from D. O. P. and D. R. L. D. O. P. completed the initial evaluation of the exon primers and optimized the PCRs. D. O. P. additionally undertook the amplification and sequencing of exons from the proband's DNA sample, analyzed the sequence traces and performed bioinformatic analysis of the variants. P. A. D. undertook dosage testing and analysis, including bioinformatic workup of the variants. J. M. L. performed targeted PCR and DNA sequencing in the parents. D. R. L. supervised the molecular studies. S. B. and M. P. K. were the lead clinicians, counselled the parents and assisted in writing the case presentation. Ethical Approval The work presented here was undertaken with informed consent.
References
- 1.Tada K, Narisawa K, Yoshida T, Konno T, Yokoyama Y. Hyperglycinemia: a defect in glycine cleavage reaction. Tohoku J Exp Med. 1969;98(3):289–296. doi: 10.1620/tjem.98.289. [DOI] [PubMed] [Google Scholar]
- 2.Kikuchi G. The glycine cleavage system: composition, reaction mechanism, and physiological significance. Mol Cell Biochem. 1973;1(2):169–187. doi: 10.1007/BF01659328. [DOI] [PubMed] [Google Scholar]
- 3.Isobe M, Koyata H, Sakakibara T, Momoi-Isobe K, Hiraga K. Assignment of the true and processed genes for human glycine decarboxylase to 9p23-24 and 4q12. Biochem Biophys Res Commun. 1994;203(3):1483–1487. doi: 10.1006/bbrc.1994.2352. [DOI] [PubMed] [Google Scholar]
- 4.Nanao K, Takada G, Takahashi E. et al. Structure and chromosomal localization of the aminomethyltransferase gene (AMT) Genomics. 1994;19(1):27–30. doi: 10.1006/geno.1994.1007. [DOI] [PubMed] [Google Scholar]
- 5.Kure S, Kojima K, Kudo T. et al. Chromosomal localization, structure, single-nucleotide polymorphisms, and expression of the human H-protein gene of the glycine cleavage system (GCSH), a candidate gene for nonketotic hyperglycinemia. J Hum Genet. 2001;46(7):378–384. doi: 10.1007/s100380170057. [DOI] [PubMed] [Google Scholar]
- 6.Tada K, Hayasaka K. Non-ketotic hyperglycinaemia: clinical and biochemical aspects. Eur J Pediatr. 1987;146(3):221–227. doi: 10.1007/BF00716464. [DOI] [PubMed] [Google Scholar]
- 7.Kure S, Kato K, Dinopoulos A. et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat. 2006;27(4):343–352. doi: 10.1002/humu.20293. [DOI] [PubMed] [Google Scholar]
- 8.Hennermann J B. Clinical variability in glycine encephalopathy. Future Neurology. 2006;1(5):621–630. [Google Scholar]
- 9.Hennermann J B, Berger J-M, Grieben U, Scharer G, Van Hove J LK. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. 2012;35(2):253–261. doi: 10.1007/s10545-011-9398-1. [DOI] [PubMed] [Google Scholar]
- 10.Hoover-Fong J E, Shah S, Van Hove J L, Applegarth D, Toone J, Hamosh A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology. 2004;63(10):1847–1853. doi: 10.1212/01.wnl.0000144270.83080.29. [DOI] [PubMed] [Google Scholar]
- 11.Brunel-Guitton C, Casey B, Coulter-Mackie M. et al. Late-onset nonketotic hyperglycinemia caused by a novel homozygous missense mutation in the GLDC gene. Mol Genet Metab. 2011;103(2):193–196. doi: 10.1016/j.ymgme.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Chiong M A, Procopis P, Carpenter K, Wilcken B. Late-onset nonketotic hyperglycinemia with leukodystrophy and an unusual clinical course. Pediatr Neurol. 2007;37(4):283–286. doi: 10.1016/j.pediatrneurol.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Chien Y H, Hsu C C, Huang A. et al. Poor outcome for neonatal-type nonketotic hyperglycinemia treated with high-dose sodium benzoate and dextromethorphan. J Child Neurol. 2004;19(1):39–42. doi: 10.1177/08830738040190010702. [DOI] [PubMed] [Google Scholar]
- 14.Dinopoulos A, Matsubara Y, Kure S. Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab. 2005;86((1–2):):61–69. doi: 10.1016/j.ymgme.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 15.Korman S H, Boneh A, Ichinohe A. et al. Persistent NKH with transient or absent symptoms and a homozygous GLDC mutation. Ann Neurol. 2004;56(1):139–143. doi: 10.1002/ana.20159. [DOI] [PubMed] [Google Scholar]
- 16.Boneh A, Allan S, Mendelson D, Spriggs M, Gillam L H, Korman S H. Clinical, ethical and legal considerations in the treatment of newborns with non-ketotic hyperglycinaemia. Mol Genet Metab. 2008;94(2):143–147. doi: 10.1016/j.ymgme.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Alfadhel M, Al-Thihli K, Moubayed H, Eyaid W, Al-Jeraisy M. Drug treatment of inborn errors of metabolism: a systematic review. Arch Dis Child. 2013;98(6):454–461. doi: 10.1136/archdischild-2012-303131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamosh A, Maher J F, Bellus G A, Rasmussen S A, Johnston M V. Long-term use of high-dose benzoate and dextromethorphan for the treatment of nonketotic hyperglycinemia. J Pediatr. 1998;132(4):709–713. doi: 10.1016/s0022-3476(98)70365-8. [DOI] [PubMed] [Google Scholar]
- 19.Cusmai R, Martinelli D, Moavero R. et al. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur J Paediatr Neurol. 2012;16(5):509–513. doi: 10.1016/j.ejpn.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 20.Bzduch V Behulova D Kolnikova M Payerova J Fabriciova K Ketogenic diet in nonketotic hyperglycinemia J Inherit Metab Dis 201033(S1):S31 [Google Scholar]
- 21.Queit L The use of ketogenic diet in classical neonatal nonketotic hyperglycinemia Oral presentation at: 39th Annual Scientific Meeting of the Human Genetics of Australasia. August 8–11, 2015; Perth, Australia
- 22.Kava M Balasubramaniam S Robertson A Greed L Walsh P Nagarajan L Ketogenic diet in refractory epilepsy in a child with classical neonatal meeting of the hyperglycinemia Poster presented at: 4th Annual Scientific Meeting of the Australian and New Zealand Child Neurology Society. August 26–28, 2015; Melbourne, Australia
- 23.Balasubramaniama S Robertsona A Kava M The use of ketogenic diet in classical neonatal hyperglycinemia (NKH) Poster presented at: Annual Symposium of the Society for the study of Inborn Errors of Metabolism; September 1-4, 2015; Lyon, France
- 24.Love J M, Prosser D, Love D R, Chintakindi K P, Dalal A B, Aggarwal S. A novel glycine decarboxylase gene mutation in an Indian family with nonketotic hyperglycinemia. J Child Neurol. 2014;29(1):122–127. doi: 10.1177/0883073812471432. [DOI] [PubMed] [Google Scholar]
- 25.Adzhubei I A, Schmidt S, Peshkin L. et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi Y, Sims G E, Murphy S, Miller J R, Chan A P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7(10):e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng P C, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calabrese R, Capriotti E, Fariselli P, Martelli P L, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum Mutat. 2009;30(8):1237–1244. doi: 10.1002/humu.21047. [DOI] [PubMed] [Google Scholar]
- 29.Schwarz J M, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 30.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 31.von Wendt L, Hirvasniemi A, Similä S. Nonketotic hyperglycinemia. A genetic study of 13 Finnish families. Clin Genet. 1979;15(5):411–417. doi: 10.1111/j.1399-0004.1979.tb01773.x. [DOI] [PubMed] [Google Scholar]
- 32.Applegarth D A, Toone J R, Lowry R B. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105(1):e10. doi: 10.1542/peds.105.1.e10. [DOI] [PubMed] [Google Scholar]
- 33.Stenson P D, Ball E V, Mort M. et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21(6):577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 34.Motokawa Y, Kikuchi G. Glycine metabolism by rat liver mitochondria. IV. Isolation and characterization of hydrogen carrier protein, an essential factor for glycine metabolism. Arch Biochem Biophys. 1969;135(1):402–409. doi: 10.1016/0003-9861(69)90556-6. [DOI] [PubMed] [Google Scholar]
- 35.Conter C, Rolland M O, Cheillan D, Bonnet V, Maire I, Froissart R. Genetic heterogeneity of the GLDC gene in 28 unrelated patients with glycine encephalopathy. J Inherit Metab Dis. 2006;29(1):135–142. doi: 10.1007/s10545-006-0202-6. [DOI] [PubMed] [Google Scholar]
- 36.Applegarth D A, Toone J R. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab. 2001;74(1–2):139–146. doi: 10.1006/mgme.2001.3224. [DOI] [PubMed] [Google Scholar]
- 37.Takayanagi M, Kure S, Sakata Y. et al. Human glycine decarboxylase gene (GLDC) and its highly conserved processed pseudogene (psiGLDC): their structure and expression, and the identification of a large deletion in a family with nonketotic hyperglycinemia. Hum Genet. 2000;106(3):298–305. doi: 10.1007/s004390051041. [DOI] [PubMed] [Google Scholar]
- 38.Gouas L, Goumy C, Véronèse L, Tchirkov A, Vago P. Gene dosage methods as diagnostic tools for the identification of chromosome abnormalities. Pathol Biol (Paris) 2008;56(6):345–353. doi: 10.1016/j.patbio.2008.03.010. [DOI] [PubMed] [Google Scholar]