

Abstract
Cancer metastasis is a multi-step, secondary tumor formation that is responsible for the vast majority of deaths in cancer patients. Animal models have served as one of the major tools for studying metastatic diseases. However, these metastasis models inherently lack the ability to decouple many of the key parameters that might contribute to cancer progression, and therefore ultimately limit detailed, mechanistic investigation of metastasis. Recently, organ-on-a-chip model systems have been developed for various tissue types with the potential to recapitulate major components of metastasis. Here, we discuss recent advances in in vitro biomimetic on-a-chip models for cancer metastasis.
Introduction
Cancer metastasis is the dissemination of a primary tumor to distant sites in the body, often resulting in the formation of many secondary tumor masses. The dissemination process often involves multiple steps, including tumor angiogenesis (the ingrowth of new capillary vessels that feed the growing tumor), intravasation (the migration of cancer cells into the blood stream), extravasation (attachment and escape of cancer cells out of the blood vessels at a distant site), and colonization at a distal tissue [1]. Even after primary tumor resections or a series of chemotherapeutic treatments, cancer metastasis can be lethal and is responsible for 90% of deaths in cancer patients [2].
Animal models have served as one of the primary tools for studying cancer metastasis [3]. Through technologies such as green fluorescent protein (GFP) transgene expression [4] and the development of spontaneous [5], experimental [6], and transgenic mouse metastasis models [7], we have been able to not only track the cancer cells in vivo but also identify key genes for metastatic progression. Although these in vivo studies provide physiologically relevant perspectives on tumor pathology, the inability to isolate the many interacting parameters that probably participate in metastasis in animal models presents disadvantages in identifying clear mechanisms. Metastasis is a complex and dynamic process, influenced by the multiple local tissue microenvironments the cancer cells experience as they transit through the body and involving cross-communication amongst several cell types [8]. These multiple parameters are intimately coordinated, which make it hard to study how each parameter contributes to metastasis using traditional in vivo models. Therefore, there has been a growing interest in developing biomimetic in vitro systems that can (1) recapitulate the key parameters that affect tumor progression (e.g., oxygen tension, nutrient gradients, and tissue stiffness) and (2) provide the flexibility to decouple these parameters in experimental settings.
For tuning certain physical conditions (e.g., oxygen tension, nutrient gradients, and tissue stiffness), in vitro models can be advantageous over in vivo models to study effects of these parameters. For example, oxygen tension and nutrient gradients are determined by the tissue location in the body, the metabolic needs of its parenchyma, and the vascular features, such as vessel density, network, and permeability. Manipulating and monitoring such parameters are therefore challenging in the in vivo setting. By contrast, tools such as hypoxia chambers and chemical inducers of hypoxia inducible factor (HIF) allow easier modulation of oxygen tension in the system and/or hypoxia-related gene expression. Composition of the cell culture media can also be optimized to create desired nutrient gradients. Tissue stiffness of tumor extracellular matrix (ECM) modulates tumor cell migration, aggressiveness, proliferation, chemotherapeutic response, and dormancy [2]. Artificially manipulating the stiffness and the composition of the tumor ECM in vivo is challenging. By contrast, we can easily tune the matrix stiffness and composition in vitro by manipulating crosslinking density and available cell attachment moieties in in vitro scaffolds.
Such models have begun to focus on capturing various aspects of cancer cell migration, adhesion, and proliferation; and cancer cell interaction with other cells in two-dimensional (2D) and three-dimensional (3D) culture. Two-dimensional wound healing models provided gradient-independent migration of cancer cells [9]. Co-culture system in 2D/3D allowed study of cell-to-cell interaction and paracrine signaling [10]. Three-dimensional spheroid models showed tumor proliferation and survival [11]. While these simple in vitro models have helped identifying the basic machinery, recent advances in biomaterials and fabrication techniques [12] have led to the development of various organ-on-a-chip models [13] that provide more physiologically relevant platforms for modeling the tissue microenvironment, including proper gradients of cell types and their paracrine signals, matrices, vessel network, and flow [14]. For example, lung-on-a-chip and bone marrow-on-a-chip recapitulated many aspects of these organs’ properties in vitro [15,16]. Given that lungs and bones are vulnerable target organs for metastasis, these organ-on-a-chip models may serve as in vitro platforms to study organ invasion. In addition, our group and others have developed in vitro blood vessel models to mechanistically study angiogenesis [17,18••,19,20-22]. With advancements and appropriate combination of these systems, we believe biomimetic on-a-chip models can become an imperative platform for studying tissue-specific cancer metastasis that can ultimately provide a better guidance for future clinical studies. In this review, we discuss advances in biomimetic on-a-chip models that have allowed us to begin to investigate key mechanisms of cancer metastasis (Figure 1).
Figure 1.
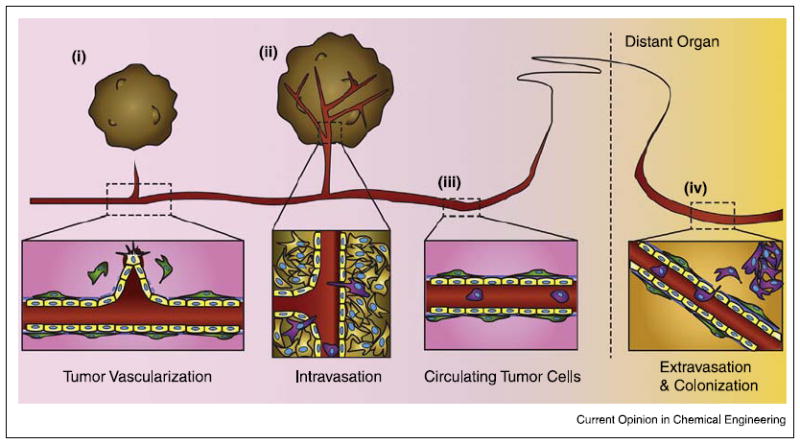
Schematic of the metastic process. (i) Vascularization of growing tumor, Soluble growth factors and cytokines released from cancer cells activate endothelial cells (yellow), resulting in pericyte (green) detachment, basement membrane degradation, and endothelial sprouting. (ii) Intravasation of invasive cancer cells. Cancer cells expressing invasive phenotypes (purple) squeeze through the endothelial cells and enter the circulation. (iii) Circulating tumor cells (CTCs) traveling to a distant tissue. (iv) Extravasation of CTCs and colonization. Various factors induce CTCs to adhere to the vascular wall of a distant organ and enter its stroma. If the microenvironment is suitable for the cancer cells, they proliferate and colonize the organ, Figure not drawn to scale.
Models for tumor angiogenesis
Tumor angiogenesis is formation of new blood vessels in tumor stroma, originating from pre-existing blood vessels in the host [8]. Tumor blood vessels provide cancer cells with oxygen and nutrients, and more importantly, they serve as disseminating routes for metastasis [23]. Physiological angiogenesis includes angiogenic growth factor-induced receptor activation in endothelial cells (ECs), followed by basement membrane degradation by proteases secreted from the activated ECs. Proliferating ECs in the surrounding matrix form new sprouts, and these sprouts make loops to become a vascular lumen [23]. There have been on-a-chip models that recapitulate hallmarks of the angiogenesis, such as vascular sprouting [18••,24], network formation [20,21], perfused lumen formation [18••,19,21], and maturation [25,26]. For example, Nguyen et al. developed endothelial sprouting model system, which induced in vivo-like directed invasion of tip cells with filopodia-guided protrusions, apical–basal polarization of stalk cells, and lumen formation (Figure 2i) [18••]. Three dimensional vascular network formation was demonstrated in an in vitro vasculogenesis model by Alonzo et al. (Figure 2ii) [21]. They reported perfused human capillary networks, and claimed that soluble factors derived from normal human lung fibroblasts (NHLFs) are necessary to form a vascular network derived from endothelial colony forming cell-derived endothelial cells (ECFC-ECs) [21]. Functionally perfused neovessels are finally matured by coverage of pericytes and smooth muscle cells. Jeon et al. mixed mesenchymal stem cells (MSCs) and ECs in fibrin matrix, observed network formation under growth factor stimulation [25]. They tested vascular endothelial growth factor (VEGF), angiopoietin-1 (Ang-1), and transforming growth factor beta 1 (TGF-β1) in the platform (Figure 2iii). Compared to VEGF alone, VEGF and TGF-βl generated a non-interconnected microvasculature; VEGF and Ang-1 promoted functional networks, showing α-SMA (alpha smooth muscle actin) positive cells [25]. The nature and function of the MSCs and α-SMA positive cells, and their mechanistic roles in vascular network formation and barrier function remain to be defined. Another recent paper by Kim et al. employed human placental pericytes, and showed pericyte coverage on their 3D microvascular network [26].
Figure 2.
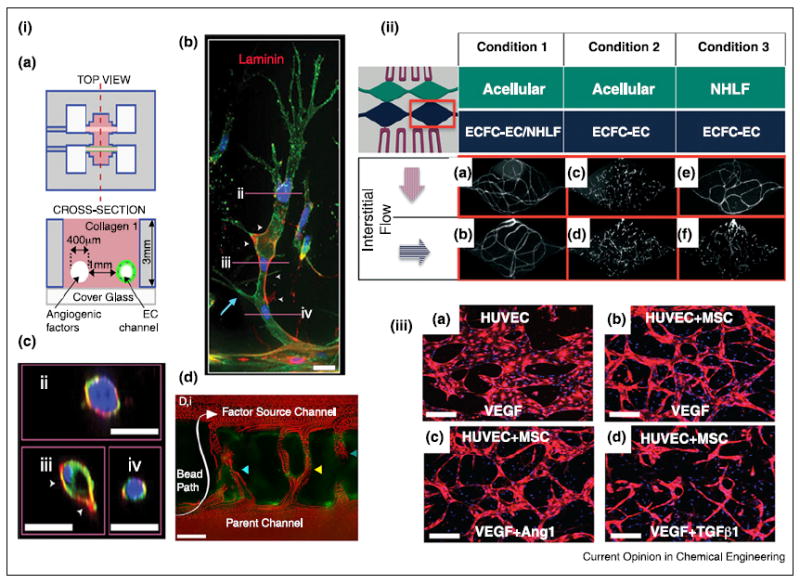
3D models for angiogenesis. (i) Formation of endothelial sprouts and perfusable neovessels in a 3D microfluidic device. (a) Device schematic. Two parallel channels are in a 3D collagen matrix, One channel is coated with ECs and the other channel is perfused with angiogenic factors. (b) Mature sprouts stained for laminin (red), and (c) cross-section of sprouts. Scale bars are 25 μm. (d) Neovessels perfused with 3-μm red fluoescent beads. Scale bar is 100 μm. (ii) Human capillary network formation, confirmed by CD31 staining, depends on interstitial flow-driven communication between endothelial colony forming cell-derived endothelial cells (ECFC-ECs) and normal human lung fibrolast (NHLF). (a,b) Vessel networks developed when ECFC-Ecs and NHLFs were co-cultured regardless of the direction of the interstitial flow. (c,d) In the absence of NHLFs, ECFC-ECs failed to form vessel networks in any directional flow, (e) Vessel network is formed when the interstitial flow direction allows for ECFC-ECs to be exposed to NHLF soluble factors. (f) No significant vessel network formation occurred when the interstitial flow direction was arranged to restrict ECFC-ECs exposure to NHLF soluble factors. (iii) Generation of microvascular network in the presence of bone marrow derived MSCs and HUVEC under growth factor stimulation. Scale bars are 200 μm.
(i) Reproduced from [18••] with permission from the National Academy of Sciences; (ii) reproduced from [21] with permission from the Royal Society of Chemistry; (iii) reproduced from [25] with permission from the Royal Society of Chemistry.
The examples above exhibit phenotypes of physiological angiogenesis. However, tumor angiogenesis is very different from normal angiogenesis: tumor vasculature is characterized by irregular sprouting, tortuous networks of capillaries, and leaky barrier properties [27]. Only in vivo intravital imaging, MRI, and in vivo image-based computer simulation have thus far reported these tumor-specific vasculatures [28-30]. Though underlying pathology could partially be explained by the overproduction of angiogenic growth factors, hypoxia, and abnormal ECM composition in the tumor microenvironment (TME) [27], detailed mechanisms have not been sufficiently well understood. Indeed, the process of generating angiogenesis-on-a-chip models provides the opportunity to understand the relative contributions and interactions of these factors required to recapitulate the unique characteristics of tumor angiogenesis. In this regard, providing relevant tumor angiogenesis models in vitro is a pivotal approach for understanding of the abnormality of tumor endothelium, tumor drug/particle delivery, tumor vascular mimicry, and hematogenous metastasis. However, modeling tumor angiogenesis is not a simple task. The in vitro tumor angiogenesis models described here have not yet been reported to exhibit abnormalities for tumor vessels. One reason may be due to the differences in how these structures form in vitro versus in vivo: in vivo tumor angiogenesis usually occurs when the tumor develops to a certain size and stage, whereas in vitro cancer cells and HUVECs are directly mixed within devices does not recapitulate the time sequence of tumor angiogenesis. While these differences may limit the ability of current approaches to model certain complex steps of tumor development, their ability to nominally mimic the presence of vasculature and organization of tumor cells within a 3D environment may allow the system to recapitulate some aspects of the in vivo processes better than traditional culture approaches. As such, additional investigation is needed.
Models for tumor intravasation
Tumor intravasation is characterized by the migration of cancer cells through the basal membrane into a blood or lymphatic vessels near the tumor stroma [31,32]. Tumor intravasation can be triggered by oxygen tension, chemotactic gradient, ECM condition, impaired endothelial barrier function, and epithelial to mesenchymal transition (EMT) in cancer cells, etc. [8,33,34]. Despite this general notion, it is unclear which element is predominant for intravasation in certain types of cancer and which mechanisms govern the process under a specific treatment such as anti-angiogenic therapy. Developing in vitro models for intravasation will enhance our understanding of tumor intravasation in many different contexts.
The model systems for tumor intravasation contain tumor and vascular compartments, examining tumor cell migration into the vasculature. Such platforms could be based on microfluidic systems [35], transwells [36•], and pre-vascularized tumor spheroids [37••]. For example, Zervantonakis et al. demonstrated macrophage-mediated tumor intravasation in their microfluidic system that allowed them to study tumor and endothelium interface in 3D. They unveiled that tumor necrosis factor alpha (TNF-α) secreted by macrophages directly impairs endothelial barrier function and enhanced breast tumor intravasation (Figure 3ii) [35]. Similarly, Roh-Johnson et al. reported that direct contact between a macrophage, an EC, and a tumor cell plays a role in tumor transendothelial migration. In their transwell system, they showed that a direct contact of macrophages to the tumor cell activates RhoA in the tumor cell and induces invadopodium formation (Figure 3iii) [36•]. Their extending study on human tumor samples validated that this presence of the three cell types could serve as a marker for breast cancer metastasis [38]. This is a good example of synergy gained by using a biomimetic on-a-chip model and traditional models to understand metastatic diseases.
Figure 3.
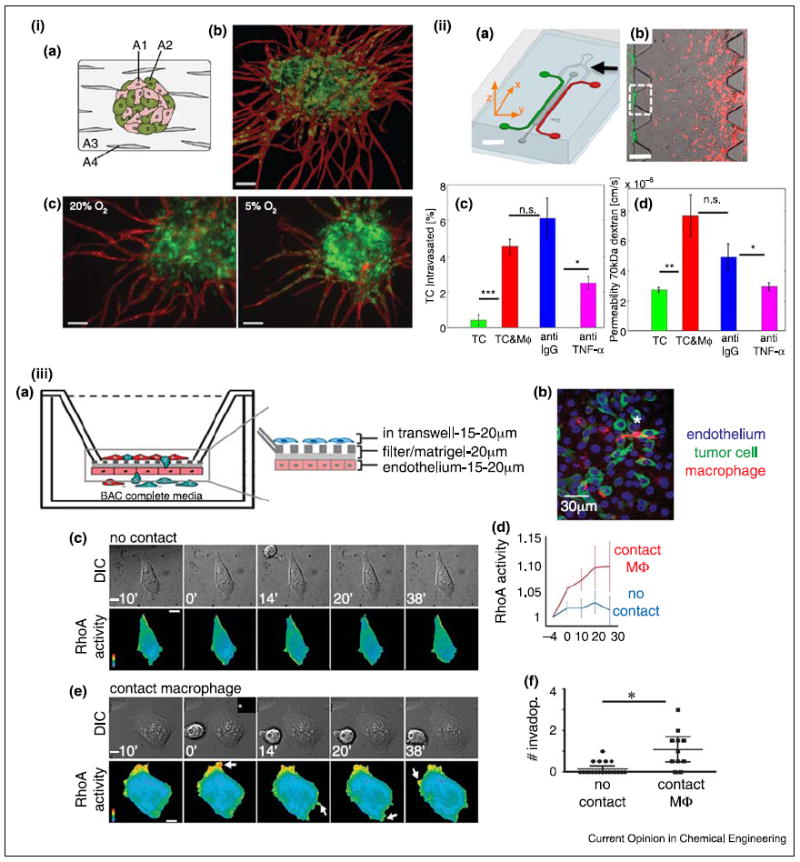
Models for tumor intravasation. (i) Prevascularized tumor (PVT) spheroid model, (a) Schematic of the PVT spheroid model. EC (A1) and tumor cell (A2) spheroids were co-cultured with fibroblasts (A4) in 3D fibrin matrix (A3), (b) PVT spheroid shows radial EC sprouting (CD31, red) from the prevascularized tumor (EGFP-transfected SW620, colon) spheroid. Scale bar is 100 μm. (c) Decreased oxygen tension increases intravasation of the SW620 cells. Scale bar is 100 μm. (ii) Microfluidic tumor-vascular interface model, (a) Schematic of the device. EC channel (green), tumor channel (red). Scale bar is 2 mm. (b) Fibrosarcoma cells (HT1080, red) invade through the ECM toward EC (green). Scale bar is 300 μm. (c) Macrophages enable tumor intravasation though TNF-α signaling, (d) Enhanced tumor intravasation is endothelial permeability dependent. (iii) Transwell in vitro model for tumor intravasation. (a) Schematic of the transendothelial migration of cancer cells in the presence of macrophages. (b) A representative image of apical section of the transwell, (c) RhoA activity in cancer cells in the absence of direct contact of macrophages, (d) RhoA activity in cancer cells with or without direct contact of macrophage, (e) RhoA activity in cancer calls in direct contact of macrophages, (f) Tumor invasion with or without direct contact with macrophage. Scale bars (a,b) are 10 μm. (i) Reproduced from [37••] with permission from the Royal Society of Chemistry; (ii) reproduced from [34] with permission from the National Academy of Sciences; (iii) reproduced from [36•] with permission from the Nature Publishing Group.
A recent study by Ehsan et al. reported that pre-vascularized tumor spheroids in vitro showed endothelial sprouting mimicking tumor angiogenesis. In the platform, hypoxia enhanced intravasation via Slug-dependent EMT signaling in the cancer cells (Figure 3i) [37••]. Hypoxia may contribute to tumor intravasation in other ways. For example, hypoxia induces ECM remodeling by HIF-1 regulated synthesis of ECM-modifying enzymes, which promote degradation of basement membrane and alignment of collagen fibers to facilitate tumor invasion to blood vasculatures [34], Further in vitro studies, examining hypoxia-mediated changes in ECM composition, alignment, stiffness, and cytokine production by cancer and stromal cells will make a significant progress in our understanding of tumor intravasation.
In addition to blood vessels, intravasation via lymphatic vessels is also an important model to be further developed in in vitro on-a-chip models. Tumor lymphatic vessels are formed by tumor secreted lymphangiogenic growth factors and are central routes for most carcinoma metastasis [8,32]. Tumor lymphatic vessels recruit cancer cells via several chemokine axes [39-41], promote angiogenesis and tumor growth by expressing growth factors [41,42]. Tumor invasion into lymphatic vessels is relatively under-investigated compared to the tumor invasion into blood vessels, and therefore on-a-chip models studying the interaction of tumor-lymphatic system would be of great interest as well.
Models for tumor extravasation
Cancer cells entering blood stream are referred to as circulating tumor cells (CTCs) [43]. CTCs in the blood stream finally lodge at secondary organs, mediated by capillaries in the organs. After the CTCs adhere to the endothelium, they transmigrate through the endothelium, which is referred to as tumor extravasation [44]. Extravasated tumor cells must adapt themselves in the new organ microenvironment to survive and form micrometastases [1]. According to Steven Paget’s ‘seed and soil’ hypothesis, each organ has its unique environment, so that different cancer types may prefer to seeding themselves in a certain type of organ microenvironments [45]. This hypothesis has been supported by clinical observations: for examples, prostate cancer prefers to metastasize to bone; head and neck cancer often results in metastases in cervical lymph nodes and salivary glands, but not the brain despite a spatial proximity [8]. Despite these observations, underlying mechanisms determining these organ-specific preferences are not well understood.
Organ-on-a-chip systems can mimic organ microenvironments in vitro by organizing organ parenchymal or stromal cells, and posing diverse biochemical and physical cues. The systems therefore potentially serve as pre-metastatic organ in vitro to unveil mechanisms of organ-specific metastasis. There have been organ-on-a-chip systems based on transwells [46•], capillary bed platforms [47••], and vascular channel microfluidic devices [48] to study organotypic metastasis.
A transwell approach has been used for modeling brain metastases. Tominaga et al. modeled blood–brain barrier (BBB) in transwells by employing brain ECs, brain pericytes and astrocytes (Figure 4ii) [46•]. This model showed that breast cancer cell derived extracellular vesicles (EVs) break down the BBB through the change in actin dynamic of the ECs in vitro, and the cancer cell derived EVs also promoted brain metastasis in vivo [46•].
Figure 4.
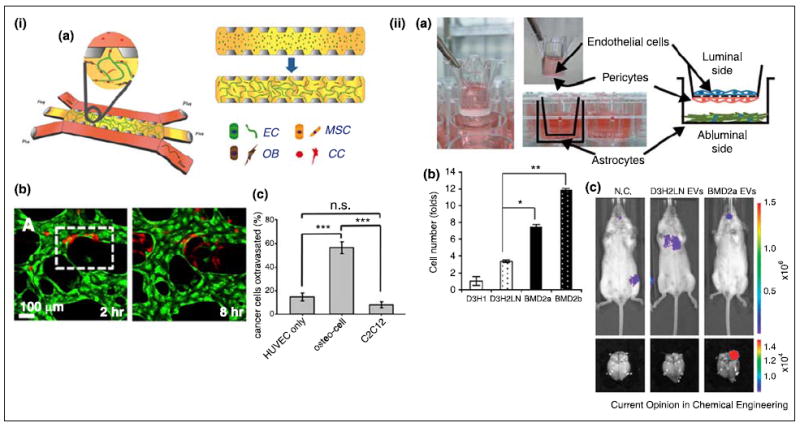
Models for tumor extravasation. (i) Breast tumor extravasation in the bone-mimicking microenvironment (BMi). (a) Schematic of the microfluidic device. ECs, MSCs, and osteoblasts (OBs) were initially seeded in fibrin gel. After formation of vascularized BMi, cancer cells (CC) were added to the side channels. (b) Cancer cell (red) extravasation through HUVEC (green) network in the BMi. (c) Cancer cell extravasation enhanced by osteocells. Myoblasts, C2C12, were used as a control. (ii) Breast tumor extravasation in the brain microenvironment. (a) Schematic of the in vitro model of blood–brain barrier (BBB). (b) Tumor extravasation in the presence of extracellular vesicles (EVs) from different tumor cells (D3H1: primary tumor cells, D3H2LN: lymph node metastases, BMD2a and 2b: brain metastases). Brain metastases derived EVs enhanced breast tumor extravasation. (c) Bioluminescence images of D3H2LN and BMD2a derived EVs injected mice (negative control, N.C.). BMD2a-derived EVs promoted brain metastasis in vivo. (i) Reproduced from [47••] with permission from the National Academy of Sciences and (ii) reproduced from [46•] with permission from the Nature Publishing Group.
Bone microenvironment was mimicked in a capillary bed platform to study breast cancer seeding to the bone (Figure 4i) [47••]. Bone is one of the most vulnerable organs for metastasis in breast or prostate cancer. The device developed by Jeon et al., featured a fibrin gel compartment next to microfluidic channels, where ECs, osteoblasts, and bone-marrow derived MSCs formed perfusable capillary beds in a bone mimicking microenvironment. As a control group, they also prepared muscle mimicking microenvironment by using myoblast cell line, C2C12. Breast tumor cells exhibited better extravasation in the bone mimicking environment than in a muscle mimicking one; and the anti-metastatic mechanism in the muscle microenvironment involved secretion of adenosine by the muscle cells [47••].
Bersini et al. recapitulated bone metastasis using vascular channel microfluidic devices [48]. Their 3D device included an endothelial channel and surrounding collagen 1 matrix embedded with osteoblasts and MSCs. Tumor cells were introduced inside the endothelial channel, and the extravasation of tumor cells was observed. The system revealed the extravasation was mediated by the CXCL5–CXCR2 axis. These bone mimicking models need to be explored further in prostate cancer or other bone metastatic cancer to investigate whether there are general mechanisms or molecular targets for bone metastasis.
Beyond the examples above, recently developed organ-on- a-chip models need to be further explored for their utility in metastasis research. For example, on-a-chip models for bone marrow [16], lung [15], liver [49], and brain [50] could be combined with capillary bed or vascular channel models to serve as novel platforms for organ metastasis. These models could serve as in vitro pre-metastatic organ platforms, for example to screen anti-metastatic drug agents as well as to study disease mechanisms.
Conclusions
In vitro organ-on-a-chip models for metastasis have just started gaining attention, and there are numerous topics still to be explored using these models. Several challenges still need to be addressed for on-chip platforms before they can be widely adopted. Many of the current models utilize purified collagen and/or fibrin from non-human sources as their ECM, which often contains residual growth factors and undefined components. Not only do these ECM sources make it challenging to decouple and manipulate biochemical parameters of the cellular microenvironment (cell adhesion peptide density, for example), they also lack physiological fibrillar structure and stiffness that are observed in in vivo TME. Development of highly controllable biomimetic synthetic materials or better sources of in vivo matrix would therefore significantly enhance our ability to make the in vitro models physiologically relevant. In addition, as the on-chip platforms are becoming more and more complex, the point at which an organ mimicry is both ‘functional enough’ for clinical relevance and ‘simple enough’ for practical experimentation remains to be defined. Given that human cancer is enormously complex and variable even between patients (e.g., with different outcomes often observed in the same tumor types depending on disease status, location, and the patient’s age or genetic background), it is unreasonable to expect to model every facet of human cancer metastasis in vitro. Therefore, identifying the universal key aspects of pathology and deducing what is ‘enough’ for an in vitro on-chip model would help set feasible engineering goals and measures of success. Despite more than 100 year of history of molecular biology, animal studies, and genetics in cancer metastasis, defeating it has not yet been accomplished. Successful interconnection between the biomimetic organ-on-a-chip models and traditional models will promise better understanding and curing malignant metastatic diseases.
Acknowledgments
We thank Jeroen Eyckmans for his constructive comments on the manuscript. The authors acknowledge support by grants from the National Institutes of Health (EB00262, EB08396, UH3EB017103, UC4DK104196), the National Science Foundation (CMMI-1462710), the RESBIO Technology Resource for Polymeric Biomaterials (P41-EB001046), and the Biological Design Center at Boston University.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
- 1.Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P, Sliva D, et al. Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol. 2015 doi: 10.1016/j.semcancer.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 2.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McClatchey Al. Modeling metastasis in the mouse. Oncogene. 1999;18:5334–5339. doi: 10.1038/sj.onc.1203086. [DOI] [PubMed] [Google Scholar]
- 4.Chishima T, Yang M, Miyagi Y, Li L, Tan Y, Baranov E, Shimada H, Moossa AR, Penman S, Hoffman RM. Governing step of metastasis visualized in vitro. Proc Natl Acad Sci U S A. 1997;94:11573–11576. doi: 10.1073/pnas.94.21.11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee E, Pandey NB, Popel AS. Pre-treatment of mice with tumor- conditioned media accelerates metastasis to lymph nodes and lungs: a new spontaneous breast cancer metastasis model. Clin Exp Metastasis. 2014;31:67–79. doi: 10.1007/s10585-013-9610-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang S, Zhang JJ, Huang XY. Mouse models for tumor metastasis. Methods Mol Biol. 2012;928:221–228. doi: 10.1007/978-1-62703-008-3_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 8.Lee E, Pandey NB, Popel AS. Crosstalk between cancer cells and blood endothelial and lymphatic endothelial cells in tumour and organ microenvironment. Expert Rev Mol Med. 2015;17:e3. doi: 10.1017/erm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riahi R, Yang Y, Zhang DD, Wong PK. Advances in wound-healing assays for probing collective cell migration. J Lab Autom. 2012;17:59–65. doi: 10.1177/2211068211426550. [DOI] [PubMed] [Google Scholar]
- 10.Miki Y, Ono K, Hata S, Suzuki T, Kumamoto H, Sasano H. The advantages of co-culture over mono cell culture in simulating in vivo environment. J Steroid Biochem Mol Biol. 2012;131:68–75. doi: 10.1016/j.jsbmb.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 11.LaBarbera DV, Reid BG, Yoo BH. The multicellular tumor spheroid model for high-throughput cancer drug discovery. Expert Opin Drug Discov. 2012;7:819–830. doi: 10.1517/17460441.2012.708334. [DOI] [PubMed] [Google Scholar]
- 12.Song HH, Park KM, Gerecht S. Hydrogels to model 3D in vitro microenvironment of tumor vascularization. Adv Drug Deliv Rev. 2014;79–80:19–29. doi: 10.1016/j.addr.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benam KH, Dauth S, Hassell B, Herland A, Jain A, Jang KJ, Karalis K, Kim HJ, MacQueen L, Mahmoodian R, Musah S, et al. Engineered in vitro disease models. Annu Rev Pathol Mech Dis. 2015;10:195–262. doi: 10.1146/annurev-pathol-012414-040418. [DOI] [PubMed] [Google Scholar]
- 14.Sackmann EK, Fulton AL, Beebe DJ. The present and future role of microfluidics in biomedical research. Nature. 2014;507:181–189. doi: 10.1038/nature13118. [DOI] [PubMed] [Google Scholar]
- 15.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torisawa YS, Spina CS, Mammoto T, Mammoto A, Weaver JC, Tat T, Collins JJ, Ingber DE. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat Methods. 2014;11:663–669. doi: 10.1038/nmeth.2938. [DOI] [PubMed] [Google Scholar]
- 17.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DH, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, et al. Rapid casting of patterned vascular networks for perfusable engineered three-dimensional tissues. Nat Mater. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18••.Nguyen DH, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, Chen CS. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc Natl Acad Sci U S A. 2013;110:6712–6717. doi: 10.1073/pnas.1221526110.. This work shows in vivo-like angiogenic sprouting and perfusable neovessel formation in 3D collagen matrix. The microfluidic device utilizes acupuncture needles to prepare size-controllable cylindrical channels in the matrix. Two channels respectively serve as an endothelial channel, as an angiogenic factor channel to demonstrate directed angiogenesis for evaluation of angiogenic growth factor cocktails and for screening of the anti-angiogenic agents.
- 19.Kim S, Lee H, Chung M, Jeon NL. Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip. 2013;13:1489–1500. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- 20.Morgan JP, Delnero PF, Zheng Y, Verbridge SS, Chen J, Craven M, Choi NW, Diaz-Santana A, Kermani P, Hempstead B, Lopez JA, et al. Formation of microvascular networks in vitro. Nat Protoc. 2013;8:1820–1836. doi: 10.1038/nprot.2013.110. [DOI] [PubMed] [Google Scholar]
- 21.Alonzo LF, Moya ML, Shirure VS, George SC. Microfluidic device to control interstitial flow-mediated homotypic and heterotypic cellular communication. Lab Chip. 2015;15:3521–3529. doi: 10.1039/c5lc00507h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whisler JA, Chen MB, Kamm RD. Control of perfusable microvascular network morphology using a multiculture microfluidic system. Tissue Eng Part C Methods. 2014;20:543–552. doi: 10.1089/ten.tec.2013.0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song JW, Munn LL. Fluid forces control endothelial sprouting. Proc Natl Acad Sci U S A. 2011;108:15342–15347. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeon JS, Bersini S, Whisler JA, Chen MB, Dubini G, Charest JL, Moretti M, Kamm RD. Generation of 3D functional microvascular networks with human mesenchymal stem cells in microfluidic systems. Integr Biol (Camb) 2014;6:555–563. doi: 10.1039/c3ib40267c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Chung M, Kim S, Jo DH, Kim JH, Jeon NL. Engineering of a biomimetic pericyte-covered 3D microvascular network. PLoS One. 2015;10:e0133880. doi: 10.1371/journal.pone.0133880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fukumura D, Duda DG, Munn LL, Jain RK. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation. 2010;17:206–225. doi: 10.1111/j.1549-8719.2010.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown E, Munn LL, Fukumura D, Jain RK. In vivo imaging of tumors. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5452. pdb prot5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emblem KE, Farrar CT, Gerstner ER, Batchelor TT, Borra RJ, Rosen BR, Sorensen AG, Jain RK. Vessel caliber – a potential MRI biomarker of tumour response in clinical trials. Nat Rev Clin Oncol. 2014;11:566–584. doi: 10.1038/nrclinonc.2014.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim E, Stamatelos S, Cebulla J, Bhujwalla ZM, Popel AS, Pathak AP. Multiscale imaging and computational modeling of blood flow in the tumor vasculature. Ann Biomed Eng. 2012;40:2425–2441. doi: 10.1007/s10439-012-0585-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Labelle M, Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012;2:1091–1099. doi: 10.1158/2159-8290.CD-12-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pereira ER, Jones D, Jung K, Padera TP. The lymph node microenvironment and its role in the progression of metastatic cancer. Semin Cell Dev Biol. 2015;38:98–105. doi: 10.1016/j.semcdb.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blonska M, Agarwal NK, Vega F. Shaping of the tumor microenvironment: stromal cells and vessels. Semin Cancer Biol. 2015 doi: 10.1016/j.semcancer.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wirtz D, Konstantopoulos K, Searson PC. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc Natl Acad Sci USA. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36•.Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H, Hodgson L, Condeelis J. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene. 2014;33:4203–4212. doi: 10.1038/onc.2013.377.. This work demonstrates that a physical contact between a macrophage, an EC, and a tumor cell, plays a role in tumor intravasation. A direct contact of the macrophage to the tumor cell activates RhoA in the tumor cell and induces invadopodium formation, finally promoting tumor intravasation.
- 37••.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CC, George SC. A three-dimensional in vitro model of tumor cell intravasation. Integr Biol (Camb) 2014;6:603–610. doi: 10.1039/c3ib40170g.. This paper shows radial sprouting of endothelium from prevascularized tumor spheroids embedded in fibroblast-containing fibrin gel. In this process tumor cells invade into the blood vessels, which are dependent on oxygen tension. Hypoxic condition triggers Slug-mediated EMT in tumor cells.
- 38.Rohan TE, Xue X, Lin HM, D’Alfonso TM, Ginter PS, Oktay MH, Robinson BD, Ginsberg M, Gertler FB, Glass AG, Sparano JA, et al. Tumor microenvironment of metastasis and risk of distant metastasis of breast cancer. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Issa A, Le TX, Shoushtari AN, Shields JD, Swartz MA. Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res. 2009;69:349–357. doi: 10.1158/0008-5472.CAN-08-1875. [DOI] [PubMed] [Google Scholar]
- 40.Das S, Sarrou E, Podgrabinska S, Cassella M, Mungamuri SK, Feirt N, Gordon R, Nagi CS, Wang YR, Entenberg D, Condeelis J, et al. Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J Exp Med. 2013;210:1509–1528. doi: 10.1084/jem.20111627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB, Popel AS. Breast cancer cells condition lymphatic endothelial cells within premetastatic niches to promote metastasis. Nat Commun. 2014;5:4715. doi: 10.1038/ncomms5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee E, Pandey NB, Popel AS. Lymphatic endothelial cells support tumor growth in breast cancer. Sci Rep. 2014;4:5853. doi: 10.1038/srep05853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joosse SA, Gorges TM, Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015;7:1–11. doi: 10.15252/emmm.201303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–870. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 45.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 46•.Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K, Lotvall J, Nakagama H, Ochiya T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood–brain barrier. Nat Commun. 2015;6:6716. doi: 10.1038/ncomms7716.. This study mimics blood–brain barrier (BBB) in transwells using brain ECs, brain pericytes, and astrocytes. This model shows interaction between breast cancer cell derived extracellular vesicles and the BBB, revealing actin-mediated BBB breakdown and tumor extravasation.
- 47••.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc Natl Acad Sci U S A. 2015;112:214–219. doi: 10.1073/pnas.1417115112.. This paper shows breast cancer extravasation to the bone mimicking microenvironment that comprises ECs, mesenchymal stem cells, and osteoblasts. These cell types form vascularized bone microenvironment in the fibrin gel. Breast cancer cells are perfused through the vascular network and transmigrate toward the outside of the vascular network.
- 48.Bersini S, Jeon JS, Dubini G, Arrigoni C, Chung S, Charest JL, Moretti M, Kamm RD. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials. 2014;35:2454–2461. doi: 10.1016/j.biomaterials.2013.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prodanov L, Jindal R, Bale SS, Hegde M, McCarty WJ, Golberg I, Bhushan A, Yarmush ML, Usta OB. Long term maintenance of a microfluidic 3-D human liver sinusoid. Biotechnol Bioeng. 2015 doi: 10.1002/bit.25700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]