

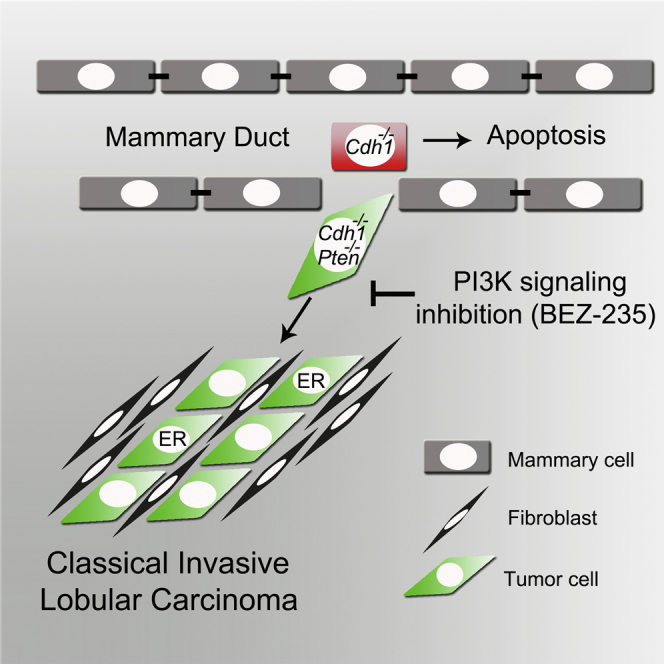
Summary
Invasive lobular carcinoma (ILC) is an aggressive breast cancer subtype with poor response to chemotherapy. Besides loss of E-cadherin, a hallmark of ILC, genetic inactivation of PTEN is frequently observed in patients. Through concomitant Cre-mediated inactivation of E-cadherin and PTEN in mammary epithelium, we generated a mouse model of classical ILC (CLC), the main histological ILC subtype. While loss of E-cadherin induced cell dissemination and apoptosis, additional PTEN inactivation promoted cell survival and rapid formation of invasive mammary tumors that recapitulate the histological and molecular features, estrogen receptor (ER) status, growth kinetics, metastatic behavior, and tumor microenvironment of human CLC. Combined inactivation of E-cadherin and PTEN is sufficient to cause CLC development. These CLCs showed significant tumor regression upon BEZ235-mediated inhibition of PI3K signaling. In summary, this mouse model provides important insights into CLC development and suggests inhibition of phosphatidylinositol 3-kinase (PI3K) signaling as a potential therapeutic strategy for targeting CLC.
Keywords: breast cancer, classical invasive lobular carcinoma, genetically engineered mouse model, Cre-lox, E-cadherin, PTEN, metastasis, tumor microenvironment, BEZ235
Graphical Abstract
Highlights
-
•
PTEN loss rescues apoptosis induced by E-cadherin loss in mouse mammary epithelium
-
•
Combined loss of E-cadherin and PTEN is sufficient to cause mouse mammary tumors
-
•
These mouse mammary tumors closely resemble human classical lobular carcinoma (CLC)
-
•
Mouse CLCs induced by loss of E-cadherin and PTEN regress upon PI3K inhibition
Boelens et al. demonstrate that combined inactivation of E-cadherin and PTEN in mouse mammary epithelium results in rapid formation of classical invasive lobular carcinoma (CLC). This mouse model provides important insights into CLC development and suggests that inhibition of PI3K signaling is a potential strategy for targeting CLC.
Introduction
Invasive lobular carcinoma (ILC) is the second most common subtype of breast cancer, comprising 8%–14% of all breast cancer cases (Li et al., 2003, Pestalozzi et al., 2008). ILC is histologically characterized by single-cell files of tumor cells in a fibrous stroma. Due to its invasive growth behavior, this tumor type does not immediately form a defined tumor mass and, therefore, is difficult to detect by physical examination or mammography (Houssami et al., 2003). Despite being classified as a histologic low-grade tumor type with a low-to-intermediate mitotic index, the infiltrating nature of ILC complicates surgical removal. Moreover, ILCs typically respond poorly to classical regimes of chemotherapy. To improve treatment options, more insight into ILC development is required.
On the molecular level, ILC is characterized by loss of the cell-adhesion protein E-cadherin (Moll et al., 1993), an event that has been shown to drive tumor invasion. Nevertheless, somatic inactivation of E-cadherin in mouse mammary epithelium does not induce spontaneous mammary tumor development (Boussadia et al., 2002, Derksen et al., 2006), indicating that additional (epi)genetic lesions are required for ILC formation. Activation of the phosphatidylinositol 3-kinase (PI3K) pathway, as displayed by either activating PIK3CA mutations or homozygous deletions or inactivating mutations in PTEN, is frequently observed in ILC (Buttitta et al., 2006, Ciriello et al., 2015, Michaut et al., 2016, Michelucci et al., 2009). PI3K signaling controls multiple cellular processes, such as cell proliferation, survival, and migration, by the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3), driving activation of downstream targets such as AKT. To test whether constitutive activation of PI3K signaling can, indeed, drive the formation of ILC, we generated mice with mammary-specific Cre-conditional inactivation of E-cadherin and PTEN. We found that combined loss of both E-cadherin and PTEN in mouse mammary epithelial cells induces mouse mammary tumors that closely resemble classical invasive lobular carcinoma (CLC), the main subtype of human ILC.
Results
Inactivation of E-Cadherin and PTEN In Vitro Alters Growth and Invasion of Mammary Epithelium
To study the direct consequence of inactivation of E-cadherin (encoded by the Cdh1 gene) and PTEN in primary mammary epithelial cells, we generated mammary organoids from Cdh1F/F;PtenF/F mice (Derksen et al., 2006, Ma et al., 2005). In vitro transduction of organoids using a Cre-recombinase-encoding adenovirus (AdCre) allowed us to test the direct effects of E-cadherin and PTEN loss in mammary epithelium. To confirm efficient AdCre-mediated recombination in vitro, we used organoids from mTmG reporter mice that express tandem dimer Tomato (tdT) prior to recombination and GFP after Cre-mediated excision of tdT (Muzumdar et al., 2007). AdCre transduction of mTmG organoids induced loss of tdT and expression of GFP in nearly all cells (Figure S1A). Importantly, the phenotype of AdCre-transduced mTmG organoids appeared unaltered as compared to non-transduced organoids from the same mouse, indicating that organoid growth rate and phenotype are not affected by AdCre transduction (Figure S1A). This strategy was, therefore, applied to inactivate E-cadherin and/or PTEN in Cdh1F/F, PtenF/F, and Cdh1F/F;PtenF/F organoids, respectively. All non-transduced organoids displayed similar-sized cyst-like structures after 7 days of culture (Figure S1B). The inactivation of E-cadherin altered mammary organoid phenotype by inducing discohesive cellular structures surrounding the organoid cores, while the inactivation of PTEN resulted in enlarged smooth-edged organoids (Figure S1B). Concurrent inactivation of E-cadherin and PTEN revealed a combined phenotype of Cdh1Δ/Δ and PtenΔ/Δ organoids, as Cdh1Δ/Δ;PtenΔ/Δ organoids were both enlarged and showed discohesive cellular structures (Figure S1B). Efficient recombination was confirmed by western blotting and PCR, showing strong reduction of E-cadherin and/or PTEN protein expression and deletion of the Cdh1F and PtenF alleles, respectively (Figures 1A and S1C). In addition, AdCre-transduced PtenF/F and Cdh1F/F;PtenF/F organoids showed a strong induction of AKT phosphorylation, indicating activation of PI3K signaling due to PTEN loss (Figure 1A).
Figure 1.

Inactivation of E-Cadherin and PTEN Alters Mammary Epithelial Morphology and Growth In Vitro
(A) Western blot analyses of E-cadherin, PTEN, and p-AKT(Ser473)/AKT protein expression of untransduced and AdCre-transduced Cdh1F/F, PtenF/F, and Cdh1F/F;PtenF/F mammary organoids.
(B) Representative bright-field images of untransduced and AdCre-transduced Cdh1F/F, PtenF/F, and Cdh1F/F;PtenF/F mammary organoids at day 21 of organoid culture. The dashed circle depicts the surface size of AdCre-transduced Cdh1F/F organoids. Scale bar, 50 μm.
(C) Average measurements of surface size (n = 10 per genotype) of untransduced and AdCre-transduced Cdh1F/F, PtenF/F, and Cdh1F/F;PtenF/F organoids at days 7, 14, and 21 of organoid culture.
(D) Expression of Ki67 and CC3 measured by whole-mount immunostaining of AdCre-transduced Cdh1F/F, PtenF/F, and Cdh1F/F;PtenF/F organoids. Scale bars, 50 μm.
(E) Quantification of the fraction of Ki67- and CC3-positive cells over the total cell number (determined by Hoechst) in AdCre-transduced Cdh1F/F, PtenF/F, and Cdh1F/F;PtenF/F (n = 10 per genotype).
(F) Representative bright-field images of a Cdh1F/F;PtenF/F organoid displaying invasive protrusions of single rows of cells (arrows) 1 day after medium change. Scale bars, 25 μm.
To study the long-term consequences of E-cadherin and/or PTEN loss in mammary epithelial cells, we monitored organoid development over a period of 21 days. Non-transduced organoids from each genotype decreased similarly in size and were hardly detectable after 21 days of culture (Figure 1B). The size of Cdh1Δ/Δ organoids remained stable, while the size of PtenΔ/Δ and Cdh1Δ/Δ;PtenΔ/Δ organoids continued to increase (Figures 1B and 1C). Again, the margins of both Cdh1Δ/Δ and Cdh1Δ/Δ;PtenΔ/Δ organoids showed discohesive cellular structures (Figure 1B).
To examine why growth kinetics differed between Cdh1Δ/Δ and Cdh1Δ/Δ;PtenΔ/Δ organoids, we analyzed cell proliferation and apoptosis by immunofluorescence (IF) with antibodies against Ki-67 and cleaved caspase-3 (CC3), respectively. Cdh1Δ/Δ organoids showed a balanced number of cells in proliferation or apoptosis, which might explain their sustained size over time (Figures 1D and 1E). Consistent with a survival role for the PI3K signaling pathway, both PtenΔ/Δ and Cdh1Δ/Δ;PtenΔ/Δ organoids showed almost no apoptotic cells (Figure 1E), resulting in an increase in organoid size over time (Figures 1B and 1C).
In contrast to PtenΔ/Δ organoids, Cdh1Δ/Δ;PtenΔ/Δ organoids displayed an invasive response upon renewal of the cell culture medium (Figures 1F and S1D). These invasive cellular strands displayed single files of cells, indicating an increased invasive potential of Cdh1Δ/Δ;PtenΔ/Δ mammary epithelium upon stimulation by growth factors and/or chemoattractants in the culture medium.
Together, these observations suggest that PI3K pathway activation through the disruption of PTEN can rescue apoptosis induced by E-cadherin loss in mammary epithelial organoids, resulting in invasive and proliferative behavior in vitro.
Inactivation of E-Cadherin and PTEN in Mammary Epithelium In Vivo Induces Rapid Formation of Hyperplastic and Infiltrative Lesions
To study the consequences of E-cadherin and PTEN loss in vivo, we generated Wcre;Cdh1F/F;PtenF/F female mice, in which mammary-epithelium-specific inactivation of E-cadherin and PTEN is induced by whey acidic protein (WAP) gene promoter-driven Cre recombinase (Wcre) (Derksen et al., 2006). We also generated Wcre;Cdh1F/F and Wcre;PtenF/F female mice to study the effects of E-cadherin or PTEN loss alone. Since Wcre activity is first detectable in the fourth week of mammary gland development (data not shown), we analyzed the consequences of somatic inactivation of E-cadherin and/or PTEN during mammary gland development. Histological analysis and immunohistochemistry (IHC) for cytokeratin 8 (CK8; marking luminal mammary epithelial cells) indicated inactivation of E-cadherin to cause hyperplastic and dysplastic ducts, as well as a marked accumulation of dissociated mammary epithelial cells in the lumen of mammary epithelium from 6-week-old Wcre;Cdh1F/F mice (Figure 2A). Inactivation of PTEN alone did not reveal distinct alterations in mammary epithelial composition in Wcre;PtenF/F (Figure 2A). In contrast, mammary epithelium from 6-week-old Wcre;Cdh1F/F;PtenF/F mice displayed multifocal neoplastic lesions infiltrating the surrounding stroma (Figure 2A). Analysis of over 20 nulliparous Wcre;Cdh1F/F;PtenF/F mice confirmed a robust synergy between loss of E-cadherin and PTEN in driving formation of these mammary epithelial lesions, as all mice displayed neoplastic lesions at 6 weeks of age. The infiltration pattern of these neoplastic lesions showed strong resemblance with the characteristic trabecular and discohesive growth pattern of tumor cells in human CLC. We confirmed Wcre-mediated inactivation of E-cadherin and/or PTEN in luminal epithelial cells by IF (Figures 2B and S2A). Analysis of Wcre;Cdh1F/F glands showed that E-cadherin-deficient cells in the lumen are CK8 positive, indicating that these cells indeed dissociated from the luminal epithelium (Figure S2A). This phenomenon is likely caused by reduced junctional integrity following E-cadherin loss (Shamir et al., 2014). Even though Wcre;PtenF/F glands did not display neoplastic lesions within 6 weeks, we clearly observed stochastic loss of PTEN in luminal epithelial cells (Figure S2A). The neoplastic lesions observed in Wcre;Cdh1F/F;PtenF/F mammary glands displayed discohesive CK8-marked luminal epithelial cells that, indeed, lost E-cadherin and PTEN, whereas surrounding stromal cells retained PTEN expression (Figure 2B). Moreover, E-cadherin-deficient cells still expressed CK8, indicating that these cells did not undergo epithelial-to-mesenchymal transition (EMT), as also previously observed (Derksen et al., 2006).
Figure 2.

Inactivation of E-Cadherin and PTEN in Mammary Epithelium In Vivo Induces Rapid Formation of Neoplastic Lesions
(A) H&E staining and IHC of CK8 in mammary glands from 6-week-old wild-type, Wcre;Cdh1F/F, Wcre;PtenF/F, and Wcre;Cdh1F/F;PtenF/F mice. Scale bars, 150 μm.
(B) IF analysis of CK8, E-cadherin, and PTEN in mammary glands from 6-week-old Wcre;Cdh1F/F;PtenF/F mice. Scale bar, 20 μm. Zooms of the asterisk-marked area reveal CK8-positive neoplastic cells that are negative for of E-cadherin and PTEN (indicated by arrows), flanked by PTEN-positive stromal cells.
(C) IF analysis of CK8 (green) and CC3 (red) in mammary glands from 6-week-old Wcre;Cdh1F/F;PtenF/F mice. Arrows mark CC3-positive apoptotic cells in the mammary duct. Scale bar, 20 μm.
(D) Average numbers of CC3-positive shedded cells per 500-μm2 area in mammary gland sections from Wcre;Cdh1F/F, Wcre;PtenF/Fand Wcre;Cdh1F/F;PtenF/F mice.
Since inactivation of PTEN rescued apoptosis and promoted survival of E-cadherin-deficient mammary epithelial cells in Cdh1Δ/Δ;PtenΔ/Δ organoids, we analyzed apoptosis by CC3 IF staining in mammary gland sections from 6-week-old Wcre;Cdh1F/F, Wcre;PtenF/F, and Wcre;Cdh1F/F;PtenF/F mice. While no apoptosis was observed in Wcre;PtenF/F and Wcre;Cdh1F/F;PtenF/F mammary glands, CK8-positive cells in the lumen of mammary ducts in Wcre;Cdh1F/F mice displayed CC3-marked apoptosis (Figures 2C and 2D). Compared to Cdh1Δ/Δ organoids, Wcre;Cdh1F/F mammary glands showed only limited numbers of CC3-marked cells, most likely due to rapid clearance of apoptotic cells in the lumen of mammary ducts. These data underscore the notion that the loss of PTEN promotes survival of E-cadherin-deficient mammary epithelial cells through inhibition of apoptosis.
To further investigate the effects of concomitant inactivation of E-cadherin and PTEN on mammary gland development and functionality, we analyzed mammary gland outgrowth during puberty and mammary gland maturation during pregnancy in nine Wcre;Cdh1F/F;PtenF/F mice. Whole-mount analysis of mammary glands from 8-week-old Wcre;Cdh1F/F;PtenF/F mice showed, beside neoplastic lesions and hyperplastic ducts, a delay in mammary gland development, as compared to age-matched wild-type mammary glands (Figure S2B). Impaired mammary gland maturation was also observed during pregnancy, as pups from five 12-week-old Wcre;Cdh1F/F;PtenF/F mice grew poorly and died 6 days after birth, while littermates fostered onto wild-type dams showed normal weight and survival (Figure S2C). This phenomenon is likely caused by the reduced numbers of milk-producing glands in Wcre;Cdh1F/F;PtenF/F mice, as compared to wild-type foster dams (Figure S2D). Thus, both the development and maturation of Cdh1Δ/Δ;PtenΔ/Δ mammary epithelium is strongly impaired, which is likely caused by the loss of mammary epithelial integrity and formation of multi-focal neoplasia.
Inactivation of E-Cadherin and PTEN Drives Formation of Mammary Tumors that Closely Resemble Human CLC
To examine potential synergy between E-cadherin and PTEN loss in mammary tumorigenesis, we monitored tumor formation, as detected by palpation, in Wcre;PtenF/+, Wcre;PtenF/F, Wcre;Cdh1F/F;PtenF/+, and Wcre;Cdh1F/F;PtenF/F mice. In line with previous literature, female Wcre;PtenF/+ and Wcre;PtenF/F mice developed heterozygous or homozygous mammary tumors, with median latencies of 615 and 250 days, respectively (Figure 3A) (Li et al., 2002). Additional inactivation of E-cadherin significantly decreased mammary tumor latency to 109 days of age in Wcre;Cdh1F/F;PtenF/F mice, demonstrating strong synergy between E-cadherin and PTEN loss in mammary tumorigenesis (Figure 3A). Interestingly, Wcre;Cdh1F/F;PtenF/+ and Wcre;PtenF/+ female mice showed a similar tumor-free survival, indicating that complete loss of PTEN is required for malignant transformation of E-cadherin-deficient mammary epithelial cells (Figure 3A).
Figure 3.

Somatic Inactivation of E-Cadherin and PTEN Induces Mammary Tumors that Closely Resemble Human CLC
(A) Mammary tumor-free survival plot of Wcre;PtenF/+ (n = 26), Wcre;Cdh1F/F;PtenF/+ (n = 39), Wcre;PtenF/F (n = 23), and Wcre;Cdh1F/F;PtenF/F (n = 60) mice. Latency is shown in days of age. n.s., not significant.
(B) Pie charts showing the distribution of mammary tumor phenotypes in Wcre;PtenF/+ (n = 7), Wcre;Cdh1F/F; PtenF/+ (n = 23), Wcre;PtenF/F (n = 28), and Wcre;Cdh1F/F;PtenF/F (n = 238) mice.
(C) Representative H&E staining and IHC of CK8, Vimentin, E-cadherin, PTEN, and ER in mammary tumors from 12-week-old Wcre;Cdh1F/F;PtenF/F mice. Scale bars, 25 μm. The zoom of the asterisk-marked area reveals PTEN-negative tumor cells flanked by PTEN-positive stromal cells.
(D) Quantification of ER-positive tumor cells detected by IHC of ER in Brca1Δ/Δ;Trp53Δ/Δ (n = 10) and Cdh1Δ/Δ;PtenΔ/Δ (n = 14) mammary tumors.
(E) Unsupervised hierarchical clustering of 956 human breast cancer samples, 22 Brca1Δ/Δ;Trp53Δ/Δ mammary tumor samples, and 20 Cdh1Δ/Δ;PtenΔ/Δ mCLC samples. Sidebars indicate tumor subtype according to PAM50 for the human breast tumors, genotypes for the mouse mammary tumors, and Cdh1 mutation status for all tumors.
To analyze the mammary tumors for each genotype, mice were sacrificed when tumors reached 225 mm2, except for Wcre;Cdh1F/F;PtenF/F mice, which were sacrificed after a median period of 101 days after tumor detection due to cumulative burden of multifocal tumors, abnormal locomotory behavior, or skin ulceration (both associated with infiltrating tumor cells in surrounding tissues) (Figure S3A). Mammary tumors from Wcre; PtenF/+mice displayed diverse histological phenotypes, including solid carcinomas, squamous metaplastic carcinomas, and EMT-like tumors, while Wcre; PtenF/F mice solely developed squamous metaplastic carcinomas (Figures 3B and S3A), suggesting that somatic PTEN loss, on its own, preferentially drives the development of this particular tumor type.
Notably, additional inactivation of E-cadherin completely shifted mammary tumor morphology. About half (57%) of mammary tumors in Wcre;Cdh1F/F;PtenF/+ mice and nearly all (96%) tumors in Wcre;Cdh1F/F;PtenF/F mice displayed infiltrative patterns of single files of tumor cells into the surrounding stroma (Figures 3B and 3C). IHC staining of tumor sections confirmed the absence of E-cadherin and PTEN protein in CK8-marked tumor cells, whereas surrounding stromal cells retained PTEN expression (Figure 3C). Moreover, IHC staining revealed elevated levels of phosphorylated (p)-AKT, p-4ebp1, and p-S6 in tumor cells, confirming the activation of PI3K signaling due to loss of PTEN (Figure S3B). Given that this phenotype comprises key characteristics of human CLC, we designated these tumors as mouse CLC (mCLC). Interestingly, mCLCs from Wcre;Cdh1F/F;PtenF/+ mice completely lost PTEN protein expression as analyzed by IHC, indicating that ILC formation requires complete loss of PTEN expression (Figure S3E). The remaining mammary tumors from Wcre;Cdh1F/F;PtenF/+ mice were classified as solid ILC, squamous metaplastic carcinomas, or EMT-like tumors that possibly developed due to incomplete loss of PTEN expression (Figure 3B).
Since the majority of human CLCs are estrogen receptor (ER) positive, we analyzed ER expression in mCLCs from Wcre;Cdh1F/F;PtenF/F mice, revealing a mean of 26% ER-positive tumor cells (Figures 3C, 3D, and S3C). In contrast, a mean of only 0.1% ER-positive cells was observed in Brca1Δ/Δ;Trp53Δ/Δ mouse mammary tumors, which are known to be ER negative (Liu et al., 2007). Also, the expression of Esr1 mRNA, encoding ERα, was found to be increased in Cdh1Δ/Δ;PtenΔ/Δ mCLCs, as compared to Brca1Δ/Δ;Trp53Δ/Δ mouse mammary tumors (Figure S3F). Expression of Esr1 mRNA was also observed in FACS (fluorescence-activated cell sorting)-sorted EpCAM (epithelial cell adhesion molecule)-positive tumor cells, confirming the ER positivity of tumor cells in mCLC (Figures S3C and S3F).
Based on their gene expression profiles, human CLCs are classified as luminal-A tumors (Ciriello et al., 2015, Iorfida et al., 2012). To assess the molecular subtype of mCLCs from Wcre;Cdh1F/F;PtenF/F mice, hierarchical clustering of RNA-seq data from 956 human breast cancers and 42 mouse mammary tumors was performed using the PAM50 gene set, which allows classification of breast tumors into luminal, basal-like, HER2-positive and normal-like subgroups (Parker et al., 2009). Whereas the 22 Brca1Δ/Δ;Trp53Δ/Δ mammary tumors clustered together with human basal-like breast cancers, all 20 Cdh1Δ/Δ;PtenΔ/Δ mCLCs were classified as luminal-A tumors, indicating that these mouse tumors closely resemble the molecular subtype of human CLC (Figure 3E). In summary, the combined loss of E-cadherin and PTEN drives a rapid onset of mCLCs that recapitulate the morphology, ER positivity, and molecular subtype of human CLC.
Mammary Gland Reconstitution Exposes Slow and Linear Growth of De Novo mCLCs
Human CLCs are typically slow-growing tumors with a low-to-intermediate mitotic index. Unfortunately, mCLC growth kinetics could not be measured in Wcre;Cdh1F/F;PtenF/F mice, which display multifocal tumor formation in all mammary glands. To assess the growth kinetics of mCLCs, we performed mammary gland reconstitutions by orthotopic transplantation of small fragments of tumor-free mammary gland tissue from Wcre;Cdh1F/F;PtenF/F mice into cleared mammary fat pads of wild-type mice. In 82 out of 104 reconstituted mammary glands, a palpable tumor was detected after a median latency period of 102 days, which is similar to the median latency of 109 days in the spontaneous mouse model (Figure 4A). Since tumor development after mammary gland reconstitution occurred at a single location, it allowed reliable assessment of tumor size. Up to 250 days after tumor detection, which is more than twice the age the spontaneous mouse model reaches, a linear and slow growth rate was observed for all 82 tumors to an average size of only 8.6 mm2 (Figures 4B, 4C, and S4A). These linear growth kinetics are unique for mouse tumors, which generally exhibit exponential tumor growth, and are very comparable to the slow growth kinetics of human CLC. The fact that mCLCs display constant and linear growth kinetics from the time of detection onward suggests that no additional mutations are required for CLC formation besides loss of E-cadherin and PTEN.
Figure 4.

Mammary Gland Transplantation Reveals Linear Growth Kinetics of mCLCs
(A) Mammary-tumor-free survival in Wcre;Cdh1F/F;PtenF/F mice and wild-type mice transplanted with small precancerous mammary gland fragments from Wcre;Cdh1F/F;PtenF/F mice.
(B) Average size of tumors in mice transplanted with precancerous Wcre;Cdh1F/F;PtenF/F mammary gland fragments (n = 82).
(C) Pie chart showing distribution of tumor phenotypes from mice transplanted with precancerous Wcre;Cdh1F/F;PtenF/F mammary gland fragments (n = 58).
(D) A fraction (n = 17) of mice transplanted with precancerous Wcre;Cdh1F/F;PtenF/F mammary gland fragments showed a sudden transition from linear to exponential growth 250–450 days after tumor onset.
(E) Overview of oncogenic mutations identified in exponentially growing solid ILCs and EMT tumors.
(F) Number of genomic aberrations in linear-growing mCLCs (n = 14) or exponential-growing solid ILCs and EMT tumors (n = 7).
Interestingly, 17 out of 82 mCLCs showed a sudden transition from linear to exponential growth kinetics between 8 and 21 months after tumor detection (Figure 4D). These exponentially growing tumors displayed non-CLC phenotypes, including solid ILC and EMT-like tumors, which were also observed in a small fraction of the spontaneous tumors from Wcre;Cdh1F/F;PtenF/F mice (Figures 3B and S3B). The sudden and stochastic transition from linear to exponential growth kinetics suggests the acquisition of additional mutations in these tumors over time. Indeed, whole-exome sequencing of seven randomly selected exponentially growing tumors and 14 mCLCs revealed that all exponentially growing tumors harbored mutations in at least one cancer-related gene, while none of the mCLCs showed additional mutations (Figure 4E). Similarly, DNA copy-number variation (CNV) analysis showed that mCLCs displayed relatively stable genomic profiles, whereas exponentially growing tumors contained multiple DNA copy-number aberrations (Figures 4F and S4B).
In summary, we successfully developed a mammary gland reconstitution model for CLC, highlighting a linear growth rate of these tumors and indicating that combined E-cadherin and PTEN loss is sufficient and, thus, causal for CLC development.
Metastatic Spectrum of mCLCs Mimics that of Human CLC
Human CLC is characterized by its invasive and metastatic behavior. Similarly, mCLCs displayed tumor invasion, as detected by the examination of tumor sections, revealing irregular tumor edges with files of tumor cells invading surrounding tissues such as fat, muscle, and nerves (Figures 5A and 5B). In line with previous observations (Cheung et al., 2013), we observed a mixture of CK8 and cytokeratin-14 (CK14) double-positive tumor cells and CK14 single-positive tumor cells at the invasive front (Figure S5A). We also investigated metastatic behavior for each tumor genotype. In Wcre;PtenF/+ and Wcre;PtenF/F mice, no microscopic metastases could be detected in any organ, possibly due to the presence of intact adherens junctions (Figure 5C). In contrast, microscopic or macroscopic metastases were observed in 34% and 5% of tumor-bearing Wcre;Cdh1F/F;PtenF/+ and Wcre;Cdh1F/F;PtenF/F mice, respectively (Figure 5C). These metastases were detected at various distant sites that are also typical for human CLC, including lymph nodes, lungs, pleura, peritoneum, and abdominal viscera (Figures 5C and 5D). The distribution of the primary tumor phenotypes in mice that developed metastases was similar to the overall distribution, with the majority of primary tumors displaying an mCLC phenotype (Figures 3B and S5B). A plausible explanation for the lower number of metastases in Wcre;Cdh1F/F;PtenF/F mice lies in the fact that these mice were sacrificed at a relatively young age (median, 210 days), as compared to Wcre;Cdh1F/F;PtenF/+ mice, which were sacrificed at a median age of 634 days (Figure S5C). Therefore, Wcre;Cdh1F/F;PtenF/F mice might have been too young at the time of sacrifice to permit detection of (slow-growing) metastases. This notion is supported by the fact that the Wcre;Cdh1F/F;PtenF/F mice with detectable metastases were the oldest of all mice analyzed (Figure S5D). However, no metastatic lesions were observed in lungs from ten aged mice that had been reconstituted with mammary gland tissue from Wcre;Cdh1F/F;PtenF/F mice as described earlier (Figure 4B). Therefore, it cannot be excluded that the late onset of tumor formation in Wcre;Cdh1F/F;PtenF/+ mice might favor the development of distinct ILCs with increased metastatic capacity.
Figure 5.

Metastatic Spectrum of mCLCs
(A and B) Representative H&E stainings revealing tumor cell invasion into surrounding muscular tissue (A) and nerve tissue (B) in mammary glands from 12-week-old Wcre;Cdh1F/F;PtenF/F mice (scale bars, 100 μm).
(C) Incidence of distant metastases in Wcre;PtenF/+ (n = 15), Wcre;Cdh1F/F;PtenF/+ (n = 32), Wcre;PtenF/F (n = 26), and Wcre;Cdh1F/F;PtenF/F (n = 62) mice. Colors depict different metastatic sites in metastasis-bearing Wcre;Cdh1F/F;PtenF/+ (n = 11) and Wcre;Cdh1F/F;PtenF/F (n = 3) mice.
(D) Representative H&E stainings of tumor infiltration (marked by asterisks) in lymph node, lung, pleura, peritoneum, and spleen. Scale bars, 200 μm.
(E) Representative FACS plots of GFP-sorted cells obtained from mammary tumors, blood, lungs, and bone marrow of tumor-bearing Wcre;Cdh1F/F;PtenF/F;mTmG mice, revealing dissemination of GFP-marked tumor cells to distant organs.
(F) Fractions of Wcre;Cdh1F/F;PtenF/F;mTmG mice (n = 5) harboring GFP-marked tumor cells in indicated organs.
Next, we used FACS analysis to search for single mCLC cells in the circulation or in common sites of human CLC metastasis, such as bone marrow or lungs. To facilitate the identification of single tumor cells, we used Wcre;Cdh1F/F;PtenF/F;mTmG mice in which GFP expression marks Wcre-mediated loss of E-cadherin and PTEN (Muzumdar et al., 2007). IHC staining of mammary gland sections indeed confirmed the formation of GFP-positive mCLCs in Wcre;Cdh1F/F;PtenF/F;mTmG mice (Figure S5E). FACS analysis of primary tumor tissue from five Wcre;Cdh1F/F;PtenF/F;mTmG mice revealed 10%–15% GFP-positive cells, implying that the majority of cells in mCLC are non-tumor cells (Figure 5E). As expected, FACS analysis of blood samples revealed low numbers of circulating tumor cells (CTCs) in all Wcre;Cdh1F/F;PtenF/F;mTmG mice analyzed, suggesting that tumor cells are able to shed from the primary tumor and intravasate into the blood circulation (Figures 5E and 5F). Furthermore, low numbers of GFP-positive cells were detected in bone marrow and lung tissue, indicating infiltration of disseminated tumor cells into these organs (Figures 5E and 5F). The low number of GFP-marked tumor cells might underestimate the actual number of disseminated tumor cells, as the presence of Tomato and GFP double-positive CTCs in the blood revealed incomplete switching of the homozygous mTmG reporter alleles. Nevertheless, these results show that mCLCs exhibit an invasive and metastatic behavior that strongly resembles that of human CLC.
Early Stromal Recruitment in CLC
Histological and FACS analysis indicated mCLCs to contain a large proportion of non-tumor cells (Figures 3C and 5E). Therefore, we performed a detailed histological characterization of the tumor microenvironment of mCLCs and compared it to that of human CLCs. Besides H&E staining, we included Masson’s trichrome staining to visualize collagen fibers, a major component of the extracellular matrix, and IHC stainings to detect fibroblasts (PDGFRβ), T cells as a major component of the adaptive immune system (CD3), and macrophages as a major component of the innate immune system (F4/80 or CD68).
We analyzed both early lesions and more advanced tumors in 6-week-old and 25-week-old wild-type, Wcre;Cdh1F/F, Wcre;PtenF/F, and Wcre;Cdh1F/F;PtenF/F mice, respectively (Figure 6A). In 6-week-old wild-type and Wcre;PtenF/F mice (Figures 6A, S6A, and S6B) and 25-week-old wild-type mice (data not shown), mammary ducts were surrounded by a thin layer of blue-colored collagen fibers and PDGFRβ-positive fibroblasts, containing few CD3-positive T cells and F4/80-positive macrophages. In contrast, mammary ducts of both Wcre;Cdh1F/F and Wcre;Cdh1F/F;PtenF/F mice clearly showed an altered microenvironment. In Wcre;Cdh1F/F mammary glands, a relatively normal epithelial layer was surrounded by an thickened layer of collagen fibers and fibroblasts, showing slightly increased numbers of macrophages, but no notable changes in T cell numbers (Figure 6A). Even more profound changes in the microenvironment of mammary ducts were observed in Wcre;Cdh1F/F;PtenF/F mice. Early lesions of 6-week-old Wcre;Cdh1F/F;PtenF/F mice showed a clear increase in collagen and numbers of fibroblasts and displayed increased numbers of infiltrating macrophages and T cells, especially at the periphery (Figure 6A). Similar phenotypes were observed in more advanced tumors of 25-week-old Wcre;Cdh1F/F;PtenF/F mice, indicating that stromal recruitment is a continuous process with preservation of the composition (Figure 6A).
Figure 6.

Early Stromal Recruitment in Mouse and Human CLC
(A and B) In (A), H&E and Masson’s trichrome staining (revealing collagen) and IHC of PDGFRβ (fibroblasts), CD3 (T cells), and F4/80 (macrophages) in tumor-free mammary glands and mCLCs from wild-type, Wcre;Cdh1F/F, Wcre;PtenF/F, and Wcre;Cdh1F/F;PtenF/F mice of indicated age were used to examine stromal compositions in each indicated genotype, which were subsequently compared to (B) the microenvironment of healthy human mammary glands and the tumor microenvironment of human CLCs.
To investigate whether the stromal composition of mCLC resembles that of human CLC, we analyzed breast tissue samples from five healthy individuals and tumor tissue from 12 CLC patients. Analysis of normal breast tissue yielded similar results as those from wild-type mice, showing little collagen and low numbers of fibroblasts, T cells, or macrophages surrounding mammary ducts (Figure 6B). Similar to mCLCs, human CLCs showed abundant collagen fibers and fibroblasts, infiltrating macrophages, and increased numbers of T cells that were mainly located at the tumor periphery (Figure 6B). In summary, both human CLCs and mCLCs arising in Wcre;Cdh1F/F;PtenF/F mice are heterotypic tumors characterized by a rich stromal compartment consisting mainly of collagen and fibroblasts.
Inhibition of PI3K Signaling Has Antitumor Activity in Wcre;Cdh1F/F;PtenF/F Mice
As previously mentioned, the PI3K signaling pathway is frequently mutated in human ILC. In line with the role of PTEN as a negative regulator of PI3K signaling, CLCs from our Wcre;Cdh1F/F;PtenF/F mouse model showed activated PI3K signaling, as evidenced by elevated levels of p-AKT and p-S6 (Figure 7A). Given that improved treatments for human CLC are highly desirable, we asked whether the PI3K signaling pathway might be a potential therapeutic target for CLC. To this end, we performed a preclinical intervention study in Wcre;Cdh1F/F;PtenF/F female mice with the dual PI3K/mTOR (mammalian target of rapamycin) inhibitor BEZ235 (Liu et al., 2009). Eight-week-old littermates were treated with BEZ235 or vehicle for a period of 28 days. Target inhibition of the PI3K signaling pathway by BEZ235 was confirmed by IHC and western blotting, showing decreased phosphorylation of both AKT and S6 in CLCs from mice treated with BEZ235 (Figures 7A and 7B). As expected, vehicle-treated Wcre;Cdh1F/F;PtenF/F female mice showed, at the end of treatment, more and larger lesions than age-matched Wcre;Cdh1F/F;PtenF/F mice did at the start of treatment (t = 0) (Figures 7C and 7D). Significantly fewer and smaller CLC lesions were observed in the mammary glands of BEZ235-treated littermates compared to t = 0 and vehicle-treated Wcre;Cdh1F/F;PtenF/F mice, indicating that this PI3K/mTOR inhibitor strongly inhibited tumor formation in our mCLC model (Figures 7C and 7D). Together, these findings indicate that pharmacological inhibition of PI3K signaling may be a promising therapeutic strategy to target CLCs that harbor activating mutations in this pathway.
Figure 7.

Inhibition of PI3K Signaling Has Antitumor Activity in Wcre;Cdh1F/F;PtenF/F Mice
(A) Representative IHC of p-AKT and p-S6, reflecting PI3K signaling activity in mCLCs from Wcre;Cdh1F/F;PtenF/F mice treated with either vehicle or BEZ235.
(B) Western blot analysis of p-AKT/AKT and p-S6/S6 expression, reflecting PI3K signaling activity in mCLCs from vehicle-treated and BEZ235-treated Wcre;Cdh1F/F;PtenF/F mice.
(C) H&E staining of mammary glands from Wcre;Cdh1F/F;PtenF/F mice treated with vehicle or BEZ235. Lesions are indicated by arrows.
(D) Quantification of lesion formation by measuring total lesion area over total mammary gland tissue area for vehicle-treated (n = 6) and BEZ235-treated (n = 6) Wcre;Cdh1F/F;PtenF/F littermates, compared to age-matched Wcre;Cdh1F/F;PtenF/F mice (n = 9) at start of treatment (t = 0).
Discussion
In this study, we show that concomitant inactivation of E-cadherin and PTEN leads to rapid formation of mouse mammary tumors that faithfully recapitulate the histopathology, molecular phenotype, ER status, growth kinetics, metastatic behavior, and tumor microenvironment of human CLC. Inactivating mutations of E-cadherin and PTEN co-occur in a substantial fraction (13%) of human ILCs (Ciriello et al., 2015). In line with this, our data revealed potent synergism and causality between E-cadherin and PTEN inactivation in CLC formation. Previously, we reported that inactivation of E-cadherin and P53 in mammary epithelium results in the development of pleomorphic ILC, a relatively rare subtype of ILC (Derksen et al., 2006, Derksen et al., 2011). In contrast, inactivation of PTEN alters the tumor outcome of E-cadherin-deficient cells toward CLC, which is the most common subtype of ILC (Iorfida et al., 2012). Although E-cadherin inactivation alone is insufficient to induce tumor formation (Boussadia et al., 2002, Derksen et al., 2006, Derksen et al., 2011), it appears to be required for ILC formation by dictating tumor outcome of P53- and PTEN-deficient mammary epithelial cells toward pleomorphic ILC and CLC, respectively. Thus, besides being recognized as a hallmark of ILC (Zschiesche et al., 1997), E-cadherin inactivation is also required for ILC formation.
In apparent contrast to its tumor-suppressor role, E-cadherin inactivation limits mammary epithelial cell survival in vivo as E-cadherin-deficient luminal epithelial cells were found to accumulate in the lumen of mammary ducts and undergo apoptosis, which is most likely due to reduced junctional integrity caused by E-cadherin loss (Shamir et al., 2014). Previously, E-cadherin inactivation in the mammary gland was found to limit the survival of alveolar cells during the onset of lactation (Boussadia et al., 2002), supporting a role for E-cadherin in maintaining the survival of luminal mammary epithelial cells. E-cadherin-deficient mammary epithelium was also unable to expand in vitro and required concomitant inactivation of PTEN for outgrowth and invasion. PTEN inactivation also enabled the survival of E-cadherin-deficient mammary epithelial cells in vivo, permitting the formation of CLC. Importantly, as tumor onset in female Wcre;Cdh1F/F;PtenF/F mice was extremely rapid, and no additional mutations were detected in the ensuing mCLCs, the combined inactivation of E-cadherin and PTEN appears to be sufficient and, thus, causal for CLC formation.
PTEN is an important negative regulator of the PI3K signaling pathway, which controls cell survival (Cully et al., 2006). It is, therefore, likely that activation of PI3K signaling due to loss of PTEN promotes the survival of E-cadherin-deficient mammary epithelial cells and subsequent formation of CLC. Indeed, mCLCs from our Wcre;Cdh1F/F;PtenF/F mouse model showed activated PI3K signaling, as evidenced by expression of p-AKT and p-S6. Also, human ILCs were found to show frequent activation of PI3K signaling, as evidenced by increased AKT phosphorylation, reduced PTEN expression, and a high incidence of somatic mutations leading to PI3K pathway activation (Ciriello et al., 2015). Together, these data suggest that PI3K pathway inhibitors might effectively target CLC. In line with this, mCLC formation in female Wcre;Cdh1F/F;PtenF/F mice was significantly decreased by treatment with the dual PI3K/mTOR inhibitor BEZ235, indicating that the PI3K signaling pathway is an important Achilles’ heel of CLC. At present, BEZ235 is under evaluation in phase I/II clinical trials (Bendell et al., 2015), and future studies are needed to investigate its efficacy in patients with CLC. However, it should be noted that antitumoral efficacy of PI3K pathway inhibitors might be reduced, in case CLCs acquire additional activating mutations in oncoproteins, such as KRAS, or loss-of-function mutations in tumor suppressors, such as PTPN11/SHP2 and p53, which we identified to alter tumor outcome and growth kinetics of mCLCs.
Other strategies to target CLC might involve targeting the tumor microenvironment, which can promote growth, metastasis, and therapy resistance of breast cancer (Boelens et al., 2014, Giussani et al., 2015, Unsworth et al., 2014). Furthermore, the massive deposition of collagen in the tumor microenvironment of both mouse and human CLC suggests that collagen production is important for CLC development and progression. Collagen has been implicated in the induction of tumor cell invasion, as it was found to accumulate at invasive edges in breast tumors (Egeblad et al., 2010, Provenzano et al., 2006) and to induce in vitro invasion of cultured breast tumor cells (Nguyen-Ngoc et al., 2012). Similarly, tumor cells in mCLCs align with collagen as they invade the surrounding tissue. Both mouse and human CLC showed massive infiltration of fibroblasts, which are, in general, the main source of collagen deposition. Future studies will be required to investigate the importance of these cancer-associated fibroblasts (CAFs) for mCLC development and progression and whether targeting CAFs might provide a useful strategy for treatment of human CLC.
In conclusion, in this study we presented concomitant inactivation of E-cadherin and PTEN in mammary epithelium to drive the formation of mCLC, thus developing a pertinent mouse model for human CLC. This mouse model provides a powerful in vivo tool to develop novel therapeutic strategies for CLC patients, which may extend the relative limited treatment options that are currently available in the clinic.
Experimental Procedures
Generation of Mice
We intercrossed Wcre;Cdh1F/F mice (Derksen et al., 2011, Derksen et al., 2006) with PtenF/F mice (Ma et al., 2005) and mTmG reporter mice (Muzumdar et al., 2007). Mice were bred onto an FVB/N background and genotyped as described previously (Derksen et al., 2006, Ma et al., 2005); (Muzumdar et al., 2007). All animal experiments were approved by the Animal Ethical Committee and were conducted in compliance with the Netherlands Cancer Institute and Dutch Animal Welfare guidelines.
Isolation of Primary Mammary Epithelium for Organoid Generation
Purified primary mammary epithelial fragments were obtained from isolated mammary glands by mechanical disruption, collagenase/trypsin digestion, and differential centrifugation to generate organoids as previously described (Ewald et al., 2008). For additional details, see the Supplemental Information.
Immunoblotting, IF, and Histopathology
Immunoblotting, IF, and histopathological analysis was performed as described in the Supplemental Information.
Mammary Gland Reconstitution
Mammary gland reconstitutions were performed based on previously described mouse mammary epithelial cell transplantation (Evers et al., 2010). In brief, the fourth mammary glands of 18- to 21-day-old wild-type or mTmG mice on FVB/N background were cleared by surgical removal of the epithelium-containing part, including the lymph node, and were engrafted with small fragments (1-2 mm in diameter) of mammary gland tissue from 5- to 6-week-old Wcre;Cdh1F/F;PtenF/F mice. Transplanted mice were checked biweekly for the development of tumors.
Genomic Analysis
RNA sequencing, whole-exome sequencing, and CNV analysis was performed as described in the Supplemental Information.
Flow Cytometry
Blood samples were collected in tubes containing heparin. Tumors and lungs were mechanically chopped using a McIlwain tissue chopper (Mickle Laboratory Engineering), followed by digestion for 1 hr at 37°C in 3 mg/ml collagenase type A (Roche) and 25 μg/ml DNase (Sigma-Aldrich) in serum-free DMEM medium. Enzyme activity was neutralized by the addition of cold DMEM/10% fetal bovine serum (FBS), and suspensions were dispersed through a 70-μm cell strainer. Bone marrow was isolated through the flushing of two femurs using PBS. All single-cell suspensions were treated with erythrocyte lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.5 M EDTA) and collected in PBS + 1% BSA. DAPI (1:20) was added to exclude dead cells. GFP-positive cells were analyzed using a Beckman Coulter CyAn ADP flow cytometer and FlowJo software.
BEZ235 Intervention Study
Eight-week-old littermates were orally dosed on a daily basis with 30 mg/kg BEZ235 dissolved in 10% N-methyl-2-pyrrolidone/90% PEG 400 (polyethylene glycol 400) (Sigma-Aldrich) or vehicle for a period of 28 days. Two hours after the last dosing, mice were sacrificed, and mammary glands were harvested for further analysis. Antitumor activity was assessed by calculating the ratio between the sum of all CLC regions and the total mammary gland area using ImageJ.
Statistics
The following statistics were used for the indicated figures: for Figures 1C, 1E, 2D, S2C, and S2D, an unpaired t test was used; for Figures 3A and S3A, we used a log-rank Mantel-Cox test; for Figure 3D, a Mann-Whitney test was used; for Figure S3C, differential expression was assessed using limma (Ritchie et al., 2015); and a one-tailed Mann-Whitney U test was used for Figure 4E. For Figure 7D, a paired t test was performed on these ratios between BEZ235- and vehicle-treated littermates.
Author Contributions
M.C.B., M.N., S.K., R.v.A., and J.J. conceived experiments and analyzed data. M.C.B., M.N., S.K., E.S., N.B., A.L.Z., J.R.d.R., E.W., and E.v.d.B. conducted experiments. L.W. provided expertise and feedback. M.C.B., M.N., and J.J. wrote the manuscript.
Acknowledgments
We are grateful to Anne Paulien Drenth, Ute Boon, and Renske de Korte-Grimmerink for help with the mammary gland reconstitutions and to the NKI Molecular Pathology & Biobanking Facility for supplying human tumor material and lab support. We thank the NKI animal facility, animal pathology facility, flow cytometry facility, and genomics core facility for technical support. This work was supported by grants from the Dutch Cancer Society (NKI 2015-7589); the Netherlands Organization for Scientific Research ([VENI 016156012, NGI-Zenith 93512009, and VICI 91814643], Cancer Genomics Netherlands [CGCNL], Cancer Systems Biology Center [CSBC], and a National Roadmap grant for the Mouse Clinic for Cancer and Aging research [MCCA]); the European Union Seventh Framework Programme (EurocanPlatform project 260791 and Infrafrontier-I3 project 312325); and the European Research Council (ERC Synergy project CombatCancer).
Published: August 11, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.059.
Accession Numbers
The accession number for the raw data of RNA sequencing and exome sequencing reported in this paper is European Nucleotide Archive (ENA): PRJEB14147.
Supplemental Information
References
- Bendell J.C., Kurkjian C., Infante J.R., Bauer T.M., Burris H.A., 3rd, Greco F.A., Shih K.C., Thompson D.S., Lane C.M., Finney L.H., Jones S.F. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Invest. New Drugs. 2015;33:463–471. doi: 10.1007/s10637-015-0218-6. [DOI] [PubMed] [Google Scholar]
- Boelens M.C., Wu T.J., Nabet B.Y., Xu B., Qiu Y., Yoon T., Azzam D.J., Twyman-Saint Victor C., Wiemann B.Z., Ishwaran H. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159:499–513. doi: 10.1016/j.cell.2014.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussadia O., Kutsch S., Hierholzer A., Delmas V., Kemler R. E-cadherin is a survival factor for the lactating mouse mammary gland. Mech. Dev. 2002;115:53–62. doi: 10.1016/s0925-4773(02)00090-4. [DOI] [PubMed] [Google Scholar]
- Buttitta F., Felicioni L., Barassi F., Martella C., Paolizzi D., Fresu G., Salvatore S., Cuccurullo F., Mezzetti A., Campani D., Marchetti A. PIK3CA mutation and histological type in breast carcinoma: high frequency of mutations in lobular carcinoma. J. Pathol. 2006;208:350–355. doi: 10.1002/path.1908. [DOI] [PubMed] [Google Scholar]
- Cheung K.J., Gabrielson E., Werb Z., Ewald A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C., TCGA Research Network Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cully M., You H., Levine A.J., Mak T.W. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer. 2006;6:184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- Derksen P.W.B., Liu X., Saridin F., van der Gulden H., Zevenhoven J., Evers B., van Beijnum J.R., Griffioen A.W., Vink J., Krimpenfort P. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–449. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Derksen P.W.B., Braumuller T.M., van der Burg E., Hornsveld M., Mesman E., Wesseling J., Krimpenfort P., Jonkers J. Mammary-specific inactivation of E-cadherin and p53 impairs functional gland development and leads to pleomorphic invasive lobular carcinoma in mice. Dis. Model. Mech. 2011;4:347–358. doi: 10.1242/dmm.006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M., Nakasone E.S., Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers B., Speksnijder E.N., Schut E., Ciampricotti M., Smalley M.J., Derksen P.W.B., Jonkers J., de Visser K.E. A tissue reconstitution model to study cancer cell-intrinsic and -extrinsic factors in mammary tumourigenesis. J. Pathol. 2010;220:34–44. doi: 10.1002/path.2655. [DOI] [PubMed] [Google Scholar]
- Ewald A.J., Brenot A., Duong M., Chan B.S., Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani M., Merlino G., Cappelletti V., Tagliabue E., Daidone M.G. Tumor-extracellular matrix interactions: Identification of tools associated with breast cancer progression. Semin. Cancer Biol. 2015;35:3–10. doi: 10.1016/j.semcancer.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Houssami N., Irwig L., Simpson J.M., McKessar M., Blome S., Noakes J. The contribution of work-up or additional views to the accuracy of diagnostic mammography. Breast. 2003;12:270–275. doi: 10.1016/s0960-9776(03)00098-5. [DOI] [PubMed] [Google Scholar]
- Iorfida M., Maiorano E., Orvieto E., Maisonneuve P., Bottiglieri L., Rotmensz N., Montagna E., Dellapasqua S., Veronesi P., Galimberti V. Invasive lobular breast cancer: subtypes and outcome. Breast Cancer Res. Treat. 2012;133:713–723. doi: 10.1007/s10549-012-2002-z. [DOI] [PubMed] [Google Scholar]
- Li G., Robinson G.W., Lesche R., Martinez-Diaz H., Jiang Z., Rozengurt N., Wagner K.-U., Wu D.-C., Lane T.F., Liu X. Conditional loss of PTEN leads to precocious development and neoplasia in the mammary gland. Development. 2002;129:4159–4170. doi: 10.1242/dev.129.17.4159. [DOI] [PubMed] [Google Scholar]
- Li C.I., Malone K.E., Porter P.L., Weiss N.S., Tang M.-T.C., Daling J.R. Reproductive and anthropometric factors in relation to the risk of lobular and ductal breast carcinoma among women 65-79 years of age. Int. J. Cancer. 2003;107:647–651. doi: 10.1002/ijc.11465. [DOI] [PubMed] [Google Scholar]
- Liu X., Holstege H., van der Gulden H., Treur-Mulder M., Zevenhoven J., Velds A., Kerkhoven R.M., van Vliet M.H., Wessels L.F.A., Peterse J.L. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. USA. 2007;104:12111–12116. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.-J., Koul D., LaFortune T., Tiao N., Shen R.J., Maira S.-M., Garcia-Echevrria C., Yung W.K.A. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009;8:2204–2210. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Ziel-van der Made A.C., Autar B., van der Korput H.A., Vermeij M., van Duijn P., Cleutjens K.B., de Krijger R., Krimpenfort P., Berns A. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 2005;65:5730–5739. doi: 10.1158/0008-5472.CAN-04-4519. [DOI] [PubMed] [Google Scholar]
- Michaut M., Chin S.-F., Majewski I., Severson T.M., Bismeijer T., de Koning L., Peeters J.K., Schouten P.C., Rueda O.M., Bosma A.J. Integration of genomic, transcriptomic and proteomic data identifies two biologically distinct subtypes of invasive lobular breast cancer. Sci. Rep. 2016;6:18517. doi: 10.1038/srep18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A., Di Cristofano C., Lami A., Collecchi P., Caligo A., Decarli N., Leopizzi M., Aretini P., Bertacca G., Porta R.P. PIK3CA in breast carcinoma: a mutational analysis of sporadic and hereditary cases. Diagn. Mol. Pathol. 2009;18:200–205. doi: 10.1097/PDM.0b013e31818e5fa4. [DOI] [PubMed] [Google Scholar]
- Moll R., Mitze M., Frixen U.H., Birchmeier W. Differential loss of E-cadherin expression in infiltrating ductal and lobular breast carcinomas. Am. J. Pathol. 1993;143:1731–1742. [PMC free article] [PubMed] [Google Scholar]
- Muzumdar M.D., Tasic B., Miyamichi K., Li L., Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nguyen-Ngoc K.-V., Cheung K.J., Brenot A., Shamir E.R., Gray R.S., Hines W.C., Yaswen P., Werb Z., Ewald A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA. 2012;109:E2595–E2604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Mullins M., Cheang M.C.U., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestalozzi B.C., Zahrieh D., Mallon E., Gusterson B.A., Price K.N., Gelber R.D., Holmberg S.B., Lindtner J., Snyder R., Thürlimann B., International Breast Cancer Study Group Distinct clinical and prognostic features of infiltrating lobular carcinoma of the breast: combined results of 15 International Breast Cancer Study Group clinical trials. J. Clin. Oncol. 2008;26:3006–3014. doi: 10.1200/JCO.2007.14.9336. [DOI] [PubMed] [Google Scholar]
- Provenzano P.P., Eliceiri K.W., Campbell J.M., Inman D.R., White J.G., Keely P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamir E.R., Pappalardo E., Jorgens D.M., Coutinho K., Tsai W.-T., Aziz K., Auer M., Tran P.T., Bader J.S., Ewald A.J. Twist1-induced dissemination preserves epithelial identity and requires E-cadherin. J. Cell Biol. 2014;204:839–856. doi: 10.1083/jcb.201306088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsworth A., Anderson R., Britt K. Stromal fibroblasts and the immune microenvironment: partners in mammary gland biology and pathology? J. Mammary Gland Biol. Neoplasia. 2014;19:169–182. doi: 10.1007/s10911-014-9326-8. [DOI] [PubMed] [Google Scholar]
- Zschiesche W., Schönborn I., Behrens J., Herrenknecht K., Hartveit F., Lilleng P., Birchmeier W. Expression of E-cadherin and catenins in invasive mammary carcinomas. Anticancer Res. 1997;17(1B):561–567. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.