

Abstract
The MAG gene encodes myelin‐associated glycoprotein (MAG), an abundant protein involved in axon–glial interactions and myelination during nerve regeneration. Several members of a consanguineous family with a clinical syndrome reminiscent of Pelizaeus–Merzbacher disease and demyelinating leukodystrophy on brain MRI were recently found to harbor a homozygous missense p.Ser133Arg MAG mutation. Here, we report two brothers from a nonconsanguineous family afflicted with progressive cognitive impairment, neuropathy, ataxia, nystagmus, and gait disorder. Exome sequencing revealed the homozygous missense mutation p.Arg118His in MAG. This Arg118 residue in immunoglobulin domain 1 is critical for sialic acid binding, providing a compelling mechanistic basis for disease pathogenesis.
Introduction
Myelin‐associated glycoprotein (MAG; OMIM #159460) is a well‐known protein involved in glial–axon maintenance. It belongs to the SIGLEC superfamily of lectins that are able to bind sialic acid.1 MAG contains a large extracellular domain with five immunoglobulin (Ig) domains, a transmembrane domain, and a smaller cytoplasmic domain (Fig. 1A). It is enriched at periaxonal regions of myelin sheaths in peripheral as well as central nervous systems.1 Alternatively spliced mRNA transcripts generate long and short isoforms that differ in the size of the cytoplasmic region.2 MAG has been associated with at least two different activities: (1) as an inhibitor of axon growth in the CNS after injury3, 4 and (2) as an enhancer of axon radial growth at nodes of myelinated axons via phosphorylation of neurofilaments.5, 6, 7, 8 MAG interacts directly with axons of nerve cells through at least two different sites. One is centered within Ig domain 1, which is also critical for sialic acid binding, and the other is within Ig domains 4 and 5.9, 10, 11
Figure 1.
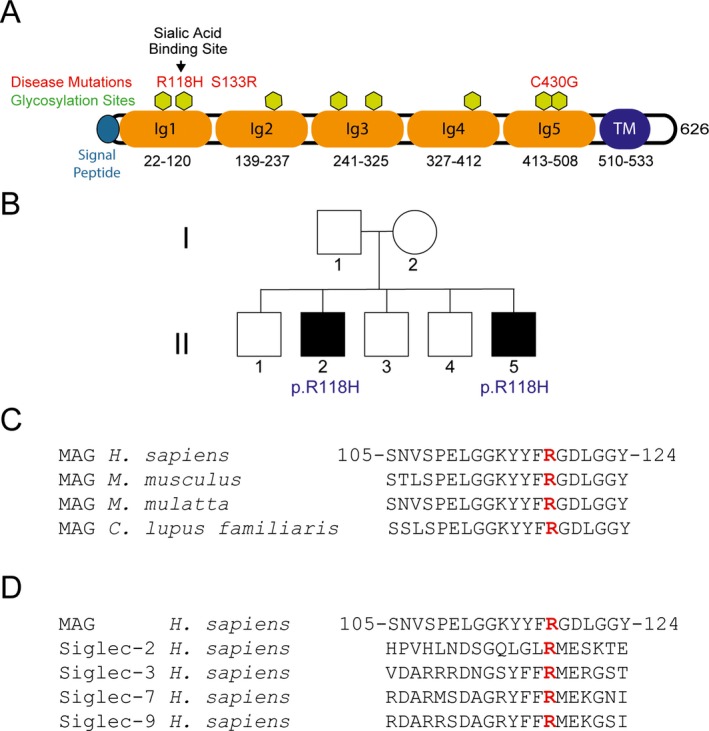
Domain organization and disease mutations of the associated glycoprotein. (A) Structural domains of MAG and mutations in our patients as well as previously reported mutations. Amino acid residue numbers are indicated (single letter amino acid code). (B) Family pedigree, with affected individuals harboring homozygous p.Arg118His mutations identified by black‐filled squares. Alignments of the residues surrounding Arg118, showing conservation among species (C) and across other human sialoproteins (D). Amino acid residue numbers are indicated for human MAG.
Antibodies against MAG lead to a prevalent, adult‐onset, autoimmune neuropathy associated with IgM‐type monoclonal antibodies (M‐protein). Nerve biopsies typically show signs of myelin loss, and electrophysiological studies demonstrate nerve conduction slowing and increased distal latencies, consistent with demyelination.12, 13, 14, 15 Recently, whole exome evaluation of a large cohort of patients with genetically undiagnosed hereditary spastic paraplegia (HSP) raised the possibility that a homozygous c.1288T>G (p.Cys430Gly) mutation in MAG might lead to HSP.16 A very recent report has supported this assignment, describing the clinical features of three siblings with a homozygous missense c.399C>G (p.Ser133Arg) mutation in MAG.17 In both studies, patients were spastic, and syndromes with mutations in MAG leading to spasticity have been designated SPG75 (OMIM #616680). Notably, in the latter report patients also had hyporeflexia.
Case Report
Two brothers were referred to the NIH Neurogenetics Branch for progressive ataxia. They provided informed consent to participate in a clinical research protocol (00‐N‐0043) approved by the NIH Combined Neuroscience Institutional Review Board. There are no other known affected family members in previous or later generations (Fig. 1B).
Patient II.5 was clumsy as a child. However, during his teens and 20s he was an avid runner, until he noted difficulty with his legs and began tripping and falling frequently. In his 40s, he had to stop driving because of difficulty with his vision, and he started using a hearing aid for hearing loss. At age 48, he first came to our attention. On examination, his cognition was normal. He had circular nystagmus, worse in his right eye and less in downgaze. In addition, he exhibited saccadic dysmetria and saccadic pursuits. He had no optic neuropathy or maculopathy. He was diffusely hyporeflexive, with mild bilateral hip‐flexor weakness and severe dorsiflexor weakness. Tone was normal, but Babinski sign was present bilaterally. Vibratory sensation was absent at the knees, and there was a distal, gradient impairment of pinprick sensation in the legs and feet. There was mild truncal ataxia, and he had a waddling gait.
Patient II.2 was 54 years old upon initial evaluation at NIH. By history, he walked late (about 2 years of age), had frequent falls as a child, and was comparatively slow in physical activities as compared to his brothers. He was diagnosed with cerebral palsy at 14 years of age. He also suffered from mild learning difficulties in school. In his late 20s, he developed nystagmus and oscillopsia. By his late 40s, he was legally blind. He also suffered from a spastic bladder. Neuropsychologic testing revealed prominent impairments in attention and working memory. Ophthalmologic examination revealed central scotomas as well as bilateral optic atrophy (Fig. 2A), circular nystagmus, and severe vision loss (OD: Snellen: 20/500, OS: Snellen: 20/400). He was mildly dysarthric and mildly spastic, with preserved strength in the upper extremities but weakness in hip flexion and dorsiflexion more so than plantar flexion in the lower extremities. He was diffusely hyporeflexive throughout, but Babinski sign was present bilaterally. Romberg sign was present. Vibratory sensation was absent at the great toes, and there was truncal more so than appendicular dysmetria. The subject used a wheelchair, and gait was not tested.
Figure 2.
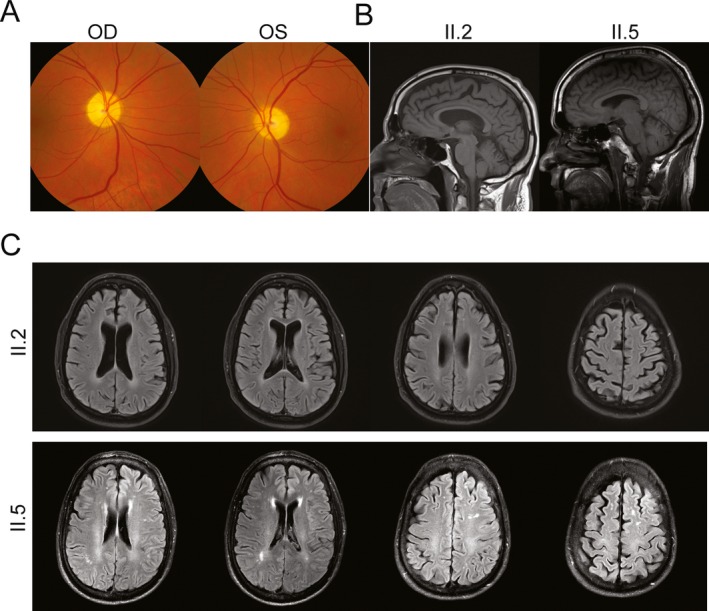
Brain and retinal imaging of two subjects with homozygous p.Arg118His MAG mutation. (A) Retinal images show optic disk atrophy in II.2. (B) Sagittal brain T1‐weighted MRI images reveal diffuse atrophy in both patients, but more prominently in II.2. (C) Transverse brain MRI FLAIR images for II.2 and II.5 demonstrate global atrophy as well as white matter abnormalities in II.5.
In both patients, electrophysiological studies revealed an axonal sensorimotor polyneuropathy. There were also demyelinating features, with prolonged F‐wave and distal latencies (see Data S1). Exome analysis (see Data S2) of both affected subjects revealed a homozygous c.353G>A (p.Arg118His) missense mutation in MAG; these results were confirmed through Sanger sequencing. This variant has been noted only once in 118,740 alleles, and in no homozygotes, in the database of the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) (December 2015). Exon‐level oligo array CGH did not detect any deletions or duplications of the MAG gene in either brother (GeneDx, Gaithersburg, MD). This further suggests that they are homozygous for this allele, and that the inheritance is recessive despite the lack of consanguinity; we were unable to test either parent for additional confirmation. This missense mutation is within a highly conserved domain present in numerous sialoproteins as well as across various species for MAG itself (Fig. 1C and D); it is predicted to be damaging, with a score of 1.00 by PolyPhen 2.18 Prior commercial genetic testing including for mutations in SYNE1 and ATM (Prevention Genetics, Marshfield, WI) did not reveal any pathogenic mutations. An additional screen for recessive causes of ataxia including ADCK3, AFG3L2, ANO10, APTX, ATM, FLVCR1, FXN, GRM1, MRE11A, MTPAP, POLG, SACS, SETX, SIL1, SYNE1, SYT14, TDP1, and TTPA did not reveal any pathogenic mutations (Athena Diagnostics, Marlborough, MA).
Brain MRI of both patients showed global atrophy, without any obvious preference for the cerebellum (Fig. 2B and C). In patient II.5, discrete white matter lesions were evident on FLAIR sequences, reminiscent of those previously reported in patients with the p.Ser133Arg mutation in MAG.17 Skin biopsies performed to evaluate epidermal nerve fibers for morphological abnormalities revealed only nonspecific axonal swellings (data not shown).
Discussion
We identified two brothers with the same homozygous p.Arg118His mutation in Ig domain 1 of MAG. This is a highly conserved residue critical for sialic acid binding.8, 11 Although antibodies against MAG have been widely described as causing a well‐characterized auto‐immune neuropathy, there have only been two reports of patients with homozygous MAG mutations.16, 17 The two patients described here appear similar in presentation to those described previously. Their predominant symptoms are ataxia, nystagmus, mild distal weakness and, in one affected brother, cognitive difficulties.
The results here and those of Lossos et al.17 suggest that homozygous p.Arg118His and p.Ser133Arg mutations both lead to a loss of MAG protein function in peripheral nervous tissue. Sural nerve biopsy in one patient with the p.Ser133Arg mutation did not reveal significant morphological alterations, other than rare, ill‐formed onion‐bulb structures, but there was a significant decrease in MAG levels, and the p.Ser133Arg mutation decreased protein stability.17 We similarly did not note significant morphological abnormalities other than axonal swellings in epidermal nerve biopsies from our patients. Previous studies have shown that mutating Arg118 to either Ala or Asp abolishes its sialic acid binding activity.11 We suspect that this loss of function could underlie the symptoms in our patients, though we cannot rule out the possibility of decreased MAG protein stability as well.
Clinical findings, global brain atrophy and FLAIR abnormalities on MRI, and optic nerve atrophy on fundoscopy are also suggestive of central nervous system disease. Interestingly, myelination still occurs in the absence of MAG in mice.19, 20 However, while compaction of myelin is normal, the organization of the peri‐axonal spaces is not,19 and these abnormalities extend to the optic nerves of examined mice.20 Patient II.2 had significant optic atrophy on examination (Fig. 2A), suggesting that there is some correlation between Mag null mouse models and patients with loss‐of‐function MAG mutations. Similarly, a late‐onset axonal neuropathy is seen in aged Mag‐deficient mice, reminiscent of that seen in our patients.6
To date, mutations in Ig 1 and Ig 5 domains as well as between Ig 1 and Ig 2 have been identified. Although there are not enough mutations to determine whether hot spots exist, it is intriguing that Ig 1 and Ig 4,5 domains in fact serve different functions.8 Studies in mice have also demonstrated an increase in N‐CAM (another adhesion molecule) mRNA expression in the absence of MAG, suggesting the presence of a compensatory mechanism that might ameliorate what could otherwise be a more severe presentation. However, a more complete pathological assessment of the effects of these mutations in central glia is still required. Recessive MAG mutations should be considered as a possible cause of any unsolved ataxia and neuropathy syndromes, and new cases should shed further light on pathogenic mechanisms.
Conflict of Interest
None declared.
Supporting information
Data S1. Description of methods used for exome analysis and list of genes screened.
Data S2. Electromyography and nerve conduction data.
Acknowledgments
We thank Elizabeth Hartnett for help with patient scheduling and the Johns Hopkins Hospital Cutaneous Nerve Laboratory for performing the biopsy analysis. We also thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison; a list of contributing groups is available at http://exac.broadinstitute.org/about.
References
- 1. Quarles RH. Myelin‐associated glycoprotein (MAG): past, present and beyond. J Neurochem 2007;100:1431–1448. [DOI] [PubMed] [Google Scholar]
- 2. Salzer JL, Holmes WP, Colman DR. The amino acid sequences of the myelin‐associated glycoproteins: homology to the immunoglobulin gene superfamily. J Cell Biol 1987;104:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mukhopadhyay G, Doherty P, Walsh FS, et al. A novel role for myelin‐associated glycoprotein as an inhibitor of axonal regeneration. Neuron 1994;13:757–767. [DOI] [PubMed] [Google Scholar]
- 4. McKerracher L, David S, Jackson DL, et al. Identification of myelin‐associated glycoprotein as a major myelin‐derived inhibitor of neurite growth. Neuron 1994;13:805–811. [DOI] [PubMed] [Google Scholar]
- 5. Yin X, Crawford TO, Griffin JW, et al. Myelin‐associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 1998;18:1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weiss MD, Luciano CA, Quarles RH. Nerve conduction abnormalities in aging mice deficient for myelin‐associated glycoprotein. Muscle Nerve 2001;24:1380–1387. [DOI] [PubMed] [Google Scholar]
- 7. Garcia ML, Lobsiger CS, Shah SB, et al. NF‐M is an essential target for the myelin‐directed “outside‐in” signaling cascade that mediates radial axonal growth. J Cell Biol 2003;163:1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nguyen T, Mehta NR, Conant K, et al. Axonal protective effects of the myelin‐associated glycoprotein. J Neurosci 2009;29:630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kelm S, Pelz A, Schauer R, et al. Sialoadhesin, myelin‐associated glycoprotein and CD22 define a new family of sialic acid‐dependent adhesion molecules of the immunoglobulin superfamily. Curr Biol 1994;4:965–972. [DOI] [PubMed] [Google Scholar]
- 10. Cao Z, Qiu J, Domeniconi M, et al. The inhibition site on myelin‐associated glycoprotein is within Ig‐domain 5 and is distinct from the sialic acid binding site. J Neurosci 2007;27:9146–9154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang S, Shen YJ, DeBellard ME, et al. Myelin‐associated glycoprotein interacts with neurons via a sialic acid binding site at ARG118 and a distinct neurite inhibition site. J Cell Biol 1997;138:1355–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steck AJ, Murray N, Meier C, et al. Demyelinating neuropathy and monoclonal IgM antibody to myelin‐associated glycoprotein. Neurology 1983;33:19–23. [DOI] [PubMed] [Google Scholar]
- 13. Steck AJ, Murray N, Dellagi K, et al. Peripheral neuropathy associated with monoclonal IgM autoantibody. Ann Neurol 1987;22:764–767. [DOI] [PubMed] [Google Scholar]
- 14. Steck AJ, Stalder AK, Renaud S. Anti‐myelin‐associated glycoprotein neuropathy. Curr Opin Neurol 2006;19:458–463. [DOI] [PubMed] [Google Scholar]
- 15. Kaku DA, England JD, Sumner AJ. Distal accentuation of conduction slowing in polyneuropathy associated with antibodies to myelin‐associated glycoprotein and sulphated glucuronyl paragloboside. Brain 1994;117:941–947. [DOI] [PubMed] [Google Scholar]
- 16. Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lossos A, Elazar N, Lerer I, et al. Myelin‐associated glycoprotein gene mutation causes Pelizaeus‐Merzbacher disease‐like disorder. Brain 2015;138:2521–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li C, Tropak MB, Gerlai R, et al. Myelination in the absence of myelin‐associated glycoprotein. Nature 1994;369:747–750. [DOI] [PubMed] [Google Scholar]
- 20. Montag D, Giese KP, Bartsch U, et al. Mice deficient for the myelin‐associated glycoprotein show subtle abnormalities in myelin. Neuron 1994;13:229–246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Description of methods used for exome analysis and list of genes screened.
Data S2. Electromyography and nerve conduction data.