

Abstract
A systems model was developed to describe the metabolism and disposition of ursodeoxycholic acid (UDCA) and its conjugates in healthy subjects based on pharmacokinetic (PK) data from published studies in order to study the distribution of oral UDCA and potential interactions influencing therapeutic effects upon interruption of its enterohepatic recirculation. The base model was empirically adapted to patients with primary biliary cirrhosis (PBC) based on current understanding of disease pathophysiology and clinical measurements. Simulations were performed for patients with PBC under two competing hypotheses: one for inhibition of ileal absorption of both UDCA and conjugates and the other only of conjugates. The simulations predicted distinctly different bile acid distribution patterns in plasma and bile. The UDCA model adapted to patients with PBC provides a platform to investigate a complex therapeutic drug interaction among UDCA, UDCA conjugates, and inhibition of ileal bile acid transport in this rare disease population.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ PBC is a rare disease characterized by blockage in the bile duct, which leads to accumulation of bile acids in the liver and subsequently permanent damage. Oral UDCA is currently the standard of care for this disease. However, its metabolism in the PBC population has not been extensively characterized.
• WHAT QUESTION DID THIS STUDY ADDRESS?
☑ In this study, we present a systems model that characterizes the metabolism of oral UDCA and its conjugated metabolites in healthy subjects and adapt the model to fit patients with PBC data. The PBC model was then used to investigate the effects of inhibiting ileal reabsorption of UDCA and conjugates on metabolism and distribution.
• WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ To our knowledge, our study provided the first systems model for the metabolism of UDCA and its conjugates in this rare disease population. It facilitated the understanding of disease pathophysiology, the metabolism of oral UDCA, and the potential drug‐drug interactions following coadministration of UDCA with a drug that inhibited enterohepatic recirculation.
• HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS
☑ The UDCA metabolism model for PBC provided a platform to investigate a potentially important therapeutic drug interaction between UDCA, its conjugates, and inhibition of ileal bile acid transport in a rare disease population.
Primary biliary cirrhosis (PBC) is a rare autoimmune disease that largely affects middle‐aged women (30–65 years) and is characterized by destruction of intrahepatic bile ducts, resulting in chronic cholestasis.1 As a result, components of bile, which assist the breakdown of cholesterol and absorption of fatty acids following a meal, accumulate in the liver with toxic effects that may lead to irreversible scarring of liver tissue (cirrhosis) requiring liver transplantation.1 Ursodeoxycholic acid (3α, 7β‐dihydroxy‐5β‐cholanic acid; UDCA) is a minor constituent in human bile2 and generally represents <1–5% of total biliary bile acids in a number of vertebrates.3, 4, 5 In contrast, UDCA is a major bile acid found in a bear's bile5 and has been used in traditional Chinese medicine for liver disease treatment. Currently the only treatment approved by the US Food and Drug Administration for PBC, oral UDCA was first investigated to treat patients with liver diseases in Japan in 19615 and later in patients with PBC in a 1987 open‐label trial that demonstrated a dramatic improvement in liver biochemical markers.6 Poupon et al.7 further confirmed UDCA efficacy for treatment of PBC based on the combined analysis of three large randomized clinical trials that demonstrated improvement in survival free of liver transplantation.7, 8 However, UDCA is known to have little effect on characteristic symptoms of PBC, such as pruritus and fatigue9, 10 and is associated with poor biochemical response that limits its effects on treatment.11, 12 With the unmet needs, there is an increasing interest to investigate second‐line therapy for PBC.13 Recently, human apical sodium bile‐acid transporter (hASBT) inhibitors in the gastrointestinal tract, such as GSK2330672,14 LUM001,15 and A425016 are currently under investigation for treatment of PBC symptoms.
Although the pharmacokinetics (PK) of UDCA have been studied since its approval for use in PBC,17 limited work has been published on modeling the metabolism of UDCA and its two major conjugates, a glycine conjugate, glycoursodeoxycholic acid (GUDCA), and a taurine conjugate, tauroursodeoxycholic acid (TUDCA). Some mechanistic modeling was performed in pediatric patients taking oral UDCA18 but little work was completed in adults. The relative lack of modeling for UDCA and conjugates likely reflects the fact that UDCA and conjugates were not easily separated using traditional assay methods. As measurement techniques for conjugated bile acids have improved, more detailed information on the PK of UDCA conjugates is available to support further understanding based on model development. Hofmann et al.19 published a series of articles modeling metabolism of naturally occurring primary bile acids such as cholic acid,19 chenodeoxycholic acid,20 and deoxycholic acid in humans.21 Due to great complexity, the models were only limited to describing endogenous bile acid distribution patterns but have never been calibrated against the response to oral UDCA treatment. Nonetheless, the publications provided a foundation for the model described herein.
This work described a model that was calibrated and validated against PK data in healthy individuals taking oral UDCA and then adapted to PBC. Besides its descriptive feature for UDCA metabolism and distribution, it also served as a useful experimental platform to explore new interventions for PBC. An example showed in this work is using the model to study effects of inhibiting hASBT on the metabolism of oral UDCA in patients with PBC.
METHODS
Model development
Given that metabolic mechanisms are generally shared among bile acids, the model for UDCA and conjugates was initially based on the model structure proposed by Hofmann et al.19 in 1983. Because of its complexity (9 tissue organs, 27 equations, and over 60 parameters) and the challenge involved with calibrating the model to human UDCA data reported in plasma and bile, the model was simplified by lumping sinusoidal with portal circulations, bile ducts with gallbladder, and among intestinal compartments. Another simplification involved initially fixing all rate constants associated with TUDCA pathway to the values used in GUDCA, except for separate conjugation (KCONJ,2) and deconjugation (KDECONJ,2) rate constants to describe much slower processes for TUDCA compared to GUDCA (KCONJ,1 and KDECONJ,1).19 Later, during model development, adjustments to the initial assumptions were required during parameter estimation to better match model predictions to literature data. Rates for fluxes between portal circulation and liver compartments (KPL,2 and KLP,2) were introduced to describe different affinities of TUDCA for the Na+‐taurocholate co‐transporting polypeptide transporter largely responsible for hepatocyte uptake of the two conjugates.22 Finally, because UDCA is metabolized from chenodeoxycholic acid by intestinal bacteria and hepatic enzymes23 and maintains relatively negligible concentrations in various human tissues as compared to cholic acid and chenodeoxycholic acid,2 endogenous formation of UDCA was excluded from the model.
The simplified model was composed of 19 ordinary differential equations, with 7 for UDCA and 12 for its conjugates (Figure 1 , Supplemental Model Code). Based on mass conservation law, each equation represented the amount (in millimole) of analyte in a tissue compartment at a given time and was derived from the sum of fluxes coming into and leaving the compartment, described either as a constant (e.g., oral UDCA dose) or a linear rate constant (unit of hour−1).
Figure 1.
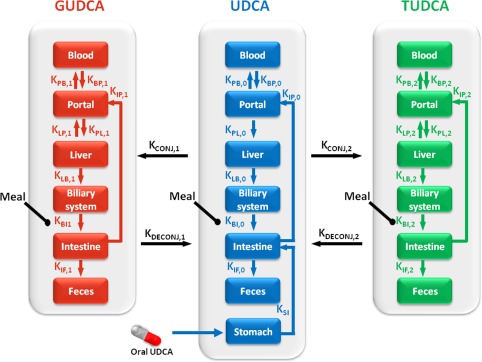
Schematic representation of the metabolism in humans for ursodeoxycholic acid (UDCA) and its conjugates, glycoursodeoxycholic acid (GUDCA), and tauroursodeoxycholic acid (TUDCA). The arrows represent the fluxes of UDCA and conjugates with rate constant near each arrow.
As a common approximate approach to characterize drug dissolution in the stomach over time, various forms of absorption models were explored, such as square wave (step function) and PK‐type functions. Eventually, a square wave with duration of 0.5 hour and the area under the curve (AUC) being the dose strength in millimole was deemed most appropriate for its adequacy and simplicity. UDCA tablets were reported to have decreasing fraction of absorption into recirculation as the dose increased.24 We assumed that fractional absorption only applied when the drug first entered into the intestine through the stomach, but not during recirculation. Fractional absorption was taken from literature (0.31) for 1,000 mg UDCA tablet2 and estimated (0.66) for 150 mg UDCA tablet based upon extrapolation from linear regression based on reported fractional absorption values from 200 to 2,000 mg25 and corresponding logarithmic UDCA doses in milligram (R2 = 0.99). UDCA and conjugates were assumed to exit the circulating bile acid pool once they entered the colon, which represented removal of UDCA and the conjugate mass from the system either through fecal excretion or metabolism to lithocholic acid or sulfated metabolites during the treatment period.26 Due to the lack of information on the rate of UDCA and conjugate metabolism in the colon, only fecal output was modeled and its value during a target time window was calculated as the difference of amounts between the ending and starting times in the fecal compartment.
Gallbladder contraction significantly increases bile acid flux into the intestine following food ingestion.27 In addition, it has been demonstrated that meal composition is a major determinant of gastric emptying and bile acid secretion from the gallbladder.28 Both meals (lunch and dinner) and a snack were involved in the data used for model calibration, however, the detailed composition was not published. To simplify the model, food effects on gallbladder contraction were modeled by applying two different constant scaling factors Emeal and Esnack on rate constants for fluxes from the biliary system to the intestines (KBS,0 and KBS,1) for a duration of time. The duration of food effects on these rate constants were empirically and manually calibrated with best outcome achieved as 1 hour for a meal and 0.5 hour for a snack. Lunch and dinner were modeled with the same scaling factor Emeal in both studies. Effects of snacks on gallbladder contraction were also assumed to be the same (Esnack) for both studies.
During model fitting to literature data, predicted bile acids in bile and plasma were converted from amount in millimole to concentration in μmol/L after being scaled by the estimated volumes of distribution for UDCA and conjugates in the bile (75 mL) and plasma (2,500 mL), respectively.19 For simplicity, the volumes of distribution for UDCA and its conjugates were assumed to be the same in each tissue compartment.
Data for calibration
Two primary sources of data were selected to calibrate the model for healthy subjects. The first dataset, from Xiang et al.,29 consisted of mean serial concentration‐time profiles of UDCA and its conjugates following a single 150 mg oral dose of UDCA in 27 healthy volunteers, with a postdose warm meal consumed at 4 hours and two snacks given at 7 and 10 hours, respectively. The second set of data was reported by Dilger et al.2 for daily oral administration of UDCA to both healthy (n = 11) and PBC subjects (n = 11) at a dose of 15 mg/kg body weight for 3 weeks. Meals, including lunch, a light snack, and dinner, were given at 4, 7, and 10 hours following the morning dose of UDCA, respectively. Reported mean values of peak concentration (Cmax) in plasma on day 22, baseline (predose) concentration in duodenal bile at steady state (day 22), and derived steady state trough in duodenal bile from reported peak‐trough‐fluctuation values for UDCA and its conjugates for healthy subjects were used in model calibration. It is noted that the standard deviation (SD) for the reported values are generally large, sometimes greater than the corresponding mean values, which suggests significant between‐subject variability. Due to the absence of individual doses and PK parameters, 1,000 mg was used in the model fitting for this study as an average dose estimated based on the mean body weight reported for healthy subjects in the study.
Parameter optimization
The ordinary differential equations system for the model was written in MATLAB (The Mathwords, Natick, MA). The simultaneous second and third order Runge‐Kutta algorithm (ode23) was used for parameter estimation and model simulations. Parameter estimation was performed using the Optimization Toolbox within MATLAB. The initial estimates for the parameters during optimization were partially taken from the bile acid model discussed in Hofmann et al.19 and partially by empirical manual handling. UDCA and its conjugates were expected to have negligible presence in most of the organs during fasting except the gallbladder, biliary systems, and intestine.30 Therefore, the actual reported mean baseline values for concentration of UDCA and conjugates in plasma and duodenal bile from both studies were used as initial conditions for the bile acid level in plasma and the biliary system, whereas baseline values in other tissues were set to be 0 due to the lack of information in the literature.
Following fitting to the mean observations, a bootstrap analysis was carried out such that over 100 sets of dataset were randomly generated from the reported mean and SD values at each observation for healthy subjects from both studies.2, 29 The model was then fitted to each dataset and the best‐fit parameter set obtained from each run eventually constructed a range for each model parameters, from which 95% confidence interval (CI; 2.5–97.5th quantiles) was summarized.
Model validation
Following parameter optimization, the model was validated against data from a separate source31 for chronic oral UDCA treatment (900 mg twice daily for 21 days, fractional absorption was assumed to be 0.31 in healthy subjects). Model simulations based on the parameter sets from bootstrap analysis were compared to the reported PK parameters in the plasma on the last day of dosing (day 21) and integrated fecal output (solid and aqueous forms) averaged over the last 3 days of dosing. Given the limitation of the model to differentiate on the form of fecal output, model simulations were compared to the sum of the two forms reported in the literature.
Sensitivity analysis
A sensitivity analysis was conducted to evaluate potential model redundancy and the influence of individual parameters to the AUC0‐24h of UDCA and conjugates in all tissues at steady state following chronic once daily dosing of 1 gram UDCA. Given the complexity of the model and the challenge of obtaining a physiological range for every parameter, a one‐at‐a‐time approach was used for the sensitivity evaluation,32, 33 such that the parameter Pi was moved one at a time between their respective extreme values, Pi,min (0.1×) and Pi,max (10×), from the best‐fit value, while all the other parameters were fixed at their best‐fit values. A sensitivity index (SIj,i) was defined as steady state fluctuation for the jth tissue part with respect to parameter Pi:
| (3) |
where AUCj (.) was the AUC0‐24h for UDCA and conjugates evaluated with a parameter set in the jth tissue at steady state, max(AUCj) was the maximum value of AUCj achieved during the variations around Pi.
Model for PBC
PBC is known to be associated with accumulation of bile acids in the liver and plasma, reduced endogenous bile acid synthesis, and diminished intestinal excretion.34 Dilger et al.2 have also suggested that UDCA and conjugate concentrations did not change substantially in the duodenal bile compared with healthy subjects. This information, together with results of sensitivity analyses, guided the choice of reducing rate constants for fluxes of UDCA and conjugates from the liver to the biliary system (KLB,0, KLB,1) during adaptation of healthy model to PBC model. During the comparison, it appeared that the model for PBC may require a separate rate constant for TUDCA biliary uptake (KLB,2) to account for the different changes in GUDCA and TUDCA from healthy state. Therefore, it was tuned along with the other two parameters against the PBC data.2
Inhibition of the ileal reabsorption of bile acids
The in vitro experiments suggest parent bile acids are reabsorbed across ileal membrane predominantly through passive transport, whereas their conjugates mostly through active transport,22 but the extent of passive transport is unknown. Therefore, two scenarios of hASBT inhibition were hypothesized using the PBC model: (1) inhibition of ileal reabsorption of both UDCA and conjugates (KIP,0, KIP,1, and KIP,2); (2) only inhibition of conjugate reabsorption (KIP,1 and KIP,2). For simplicity, inhibition was assumed to be constant over the entire treatment interval of oral UDCA (i.e., 24 hours). In the event of inhibition, the corresponding rate constants were reduced by 50% and 90% and compared with the control state (no inhibition). Simulated steady state AUC0‐24h in the plasma and bile were summarized for both parent and conjugated bile acids for analysis.
RESULTS
Model calibration
Comparisons of predictions from the best‐fit model to the observed data from healthy volunteers are displayed in Figure 2 and model parameters are listed in Table 1. The majority of predicted results fell within reported measurement variability, indicating a reasonable overall fit of both single and chronic dosing regimens within the constraints of datasets from two separate studies. For chronic dosing, predictions for plasma concentrations at steady state fell between the baseline (predose) and Cmax values and predictions for accumulation in duodenal bile matched reported values. The predicted plasma Cmax of TUDCA was slightly higher than the observed mean plus one SD range. Given the relatively large variability in the reported value (mean ± SD of 0.2 ± 0.2 μmol/L) and the constraints from single dose profile for TUDCA, and the relative magnitude of TUDCA vs. UDCA and GUDCA, the fitting discrepancy seems acceptable.
Figure 2.
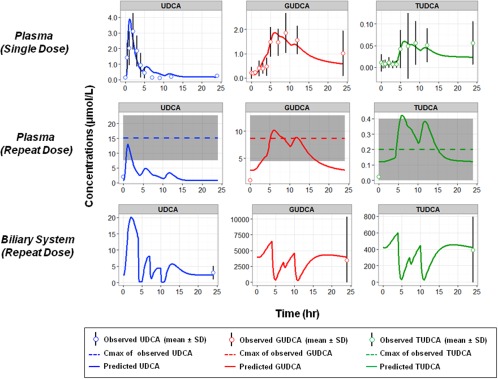
Comparison of model prediction (solid lines) of ursodeoxycholic acid (UDCA) and conjugates to literature data (circles for mean and error bars for SD) following single tablet dose of UDCA 150 mg (first row) and chronic dosing of UDCA 1 g/day (second and third rows). Observed plasma Cmax values from repeated dose were plotted as mean (dashed lines) with SD (gray shaded area) due to the lack of information on the time the Cmax was achieved (tmax).
Table 1.
The model parameter values from the best fitting to clinical data based on healthy subjects and precision (95% CI) derived from bootstrap analysis, displayed by analytes (UDCA, GUDCA, and TUDCA) and physiological processes they are associated with
| Physiological processes | UDCA | GUDCA | TUDCA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter [hr−1] | Value | Precision (95% CI) | Parameter [hr−1] | Value | Precision (95% CI) | Parameter [hr−1] | Value | Precision (95% CI) | |
| Stomach → intestine | KSI | 16.61 | (9.9–21.5) | – | – | – | – | ||
| Intestine → portal | KIP,0 | 2.70 | (1.22–3.03) | KIP,1 | 0.32 | (0.16–0.54) | KIP,2 | Same as KIP,1 | – |
| Portal → blood | KPB,0 | 0.61 | (0.49–0.80) | KPB,1 | 0.36 | (0.18–0.45) | KPB,2 | Same as KPB,1 | – |
| Blood →portal | KBP,0 | 6.94 | (5.52–9.21) | KBP,1 | 0.66 | (0.48–0.84) | KBP,2 | Same as KBP,1 | – |
| Portal → liver | KPL,0 | 0.82 | (0.73–1.03) | KPL,1 | 1.68 | (1.34–2.41) | KPL,2 | 3.98 | (2.55–4.40) |
| Liver → portal | – | – | KLP,1 | 0.10 | (0.04–0.19) | KLP,2 | 0.03 | (0–0.05) | |
| Liver → biliary system | KLB,0 | 0.33 | (0.19–0.50) | KLB,1 | 0.32 | (0.22–0.46) | KLB,2 | Same as KLB,1 | – |
| Biliary system → intestine | KBI,0 | 0.64 | (0.41–0.79) | KBI,1 | 0.13 | (0.04–0.25) | KBI,2 | Same as KBI,1 | – |
| Intestine → feces | KIF,0 | 0.79 | (0.64–1.23) | KIF,1 | 0.21 | (0.15–0.41) | KIF,2 | Same as KIF,1 | – |
| UDCA→ GUDCA | – | – | KCONJ,1 | 54.61 | (24.12–67.52) | – | – | – | |
| GUDCA→ UDCA | – | – | KDECONJ,1 | 0.76 | (0.49–1.00) | – | – | – | |
| UDCA→ TUDCA | – | – | – | – | – | KCONJ,2 | 3.83 | (1.08–7.68) | |
| TUDCA→ UDCA | – | – | – | – | – | KDECONJ,2 | 0.17 | (0.06–0.28) | |
| Meal effect on KBI,0 and KBI,1 | Emeal,0 | 35.33 | (25.37–42.24) | Emeal,1 | Same as Emeal,0 | – | Emeal,2 | Same as Emeal,0 | – |
| Snack effect on KBI,0 and KBI,1 | Esnack,0 | 9.53 | (3.77–14.56) | Esnack,1 | Same as Esnack,0 | – | Esnack,2 | Same as Esnack,0 | – |
CI, confidence interval; GUDCA, glycoursodeoxycholic acid; TUDCA, tauroursodeoxycholic acid; UDCA, ursodeoxycholic acid.
Model validation
Simulations from the best‐fit model for healthy subjects according to study design from Hess et al.31 compared favorably with the reported values of postprandial peak for UDCA concentration (Cmax), AUC0‐4h average concentration during the first 4 hours of dosing (Cavg), and the average fecal output of UDCA at steady state (Table 2). Most of the ranges of simulated exposure values (mean ± SD) fell within reported ranges, whereas the mean of AUC0‐4h was ∼30% lower than the observed mean with one third of the range overlapping with the observed range. The underprediction of AUC0‐4h may be due to the lack of representation for liquid meal (given before the last UDCA dose) in the model, which was not part of the calibration dataset. Nonetheless, the reasonable prediction for plasma exposure and fecal output, which was not available in the calibration datasets, had improved the confidence level of the model.
Table 2.
Comparison of model prediction for exposure of UDCA in plasma and fecal output to literature reference (Hess et al.31) on day 21 following chronic dosing of 900 mg oral UDCA daily for 21 days
| Parameters | Reported values in Hess et al.31 , a | Predictions from best model and bootstrapa | |
|---|---|---|---|
| Plasma (postprandial) | Cmax (μg/mL) | 5.61 ± 6.55 | 5.4 ± 2.6 |
| Cavg (μg/mL) | 4.63 ± 2.05 | 3.0 ± 2.0 | |
| AUC0‐4h (μg.min/mL) | 1105 ± 287 | 731 ± 469 | |
| Feces | Aqueous phase (mg/day) | 13.5 ± 20.3 | 147 ± 26 |
| Solid phase (mg/day) | 136 ± 126 | ||
AUC, area under the curve; Cavg, average concentration during the first 4 hours of dosing; UDCA, ursodeoxycholic acid.
aMean ± SD.
Sensitivity analysis
Table 3 provides a heat map illustrating the values for the sensitivity index. The top two most influential parameters to steady state AUC0‐24h were the rate constants for ileal absorption of UDCA (KIP,0) and its fecal output rate (KIF,0). This could be predicted, as they served as the input rate (KIP ,0 due to oral dose of UDCA) and one of the output rates (KIF,0) for the physiologic system. Two additional parameters, KCONJ,1 and KCONJ,2, had greater impact on tissue content of the conjugates than other parameters. Describing the rate‐limiting process for the formation of GUDCA and TUDCA, the importance of these two parameters was anticipated. Furthermore, large changes in these parameters had opposite effects on the distribution of GUDCA and TUDCA, because a large increase in the formation of one metabolite was counterbalanced by a large decline in the other.
Table 3.
Sensitivity index calculated for steady state AUC0‐24h of UDCA (U) and its conjugates (G and T) in various tissue compartments for each parameter in the model for healthy subjects.
| Intestine | Portal | Blood | Liver | Biliary | Feces | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | U | G | T | U | G | T | U | G | T | U | G | T | U | G | T | U | G | T |
| KSI | 0.0 | −0.1 | −0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 4.3 | −0.8 | −0.6 | 0.1 | 0.1 | 0.2 |
| KIP,0 | −89.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 91.1 | 92.0 | 92.3 | 91.8 | −89.5 | 90.4 | 90.4 |
| KPL,0 | 0.0 | −0.5 | −0.2 | −99.0 | 0.0 | 0.0 | −99.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 2.4 | −10.2 | −6.3 | 3.7 | 6.6 | 6.7 |
| KLB,0 | 2.0 | −2.7 | −2.7 | 2.3 | −2.7 | −2.7 | 2.3 | −2.7 | −2.7 | −2.8 | −2.7 | −2.7 | 99.0 | −2.7 | −2.7 | 2.4 | −2.7 | −2.7 |
| KBI,0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | −97.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| KIF,0 | −91.1 | −91.2 | −91.1 | −91.1 | −91.1 | −91.1 | −91.1 | −91.1 | −91.1 | −91.1 | −91.1 | −91.1 | −91.0 | −92.5 | −92.0 | 89.6 | −90.3 | −90.3 |
| KPB,0 | 0.0 | 0.2 | 0.1 | 0.0 | 0.0 | 0.0 | 99.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | −3.6 | 2.6 | 1.7 | −0.3 | −0.6 | −0.6 |
| KBP,0 | 0.0 | −0.1 | 0.0 | 0.0 | 0.0 | 0.0 | −99.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.3 | −2.6 | −1.6 | 0.3 | 0.6 | 0.6 |
| KCONJ,1 | 25.8 | 69.4 | −96.7 | 25.7 | 69.3 | −96.7 | 25.7 | 69.3 | −96.7 | −96.7 | 69.3 | −96.7 | −96.7 | 70.6 | −96.7 | 24.6 | 68.9 | −96.8 |
| KDECONJ,1 | 60.6 | −92.2 | 61.8 | 61.7 | −66.7 | 61.7 | 61.7 | −66.6 | 61.7 | 61.7 | −13.5 | 61.7 | 60.9 | −15.4 | 65.1 | 60.5 | −90.4 | 60.3 |
| KIP,1 | −4.2 | −17.4 | −21.4 | −2.5 | 95.3 | 98.2 | −2.5 | 95.4 | 98.3 | −2.5 | 85.5 | 92.9 | −2.1 | 87.5 | 93.8 | −15.1 | −25.8 | −37.6 |
| KPL,1 | 0.0 | 0.2 | 0.0 | 0.0 | −99.0 | 0.0 | 0.0 | −99.0 | 0.0 | 0.0 | 0.0 | 0.0 | 1.1 | −1.7 | 0.6 | 2.7 | 5.0 | 2.8 |
| KLP,1 | 0.0 | 0.1 | 0.0 | 0.0 | 87.8 | 0.0 | 0.0 | 87.8 | 0.0 | 0.0 | 0.0 | 0.0 | −0.2 | 4.1 | −0.4 | −1.2 | −2.3 | −1.3 |
| KLB,1 | 2.1 | 2.9 | 5.2 | 2.1 | −87.4 | −58.4 | 2.1 | −87.3 | −58.1 | 2.1 | −99.0 | −98.9 | 0.9 | −13.6 | −7.8 | 14.9 | 26.0 | 31.2 |
| KBI,1 | 0.3 | −1.0 | 0.7 | 0.7 | 0.9 | 1.2 | 0.8 | 1.1 | 1.9 | 0.8 | 1.0 | 1.5 | 3.8 | −97.4 | −97.4 | 10.8 | 18.7 | 23.7 |
| KIF,1 | −68.4 | −92.0 | −97.5 | −68.5 | −86.2 | −96.6 | −68.5 | −86.2 | −96.5 | −68.5 | −78.8 | −90.6 | −69.1 | −84.3 | −93.3 | −64.9 | 89.8 | 61.7 |
| KPB,1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 99.0 | 99.0 | 0.0 | 0.0 | 0.0 | −0.7 | 0.9 | 0.2 | −1.0 | −1.8 | −1.3 |
| KBP,1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | −99.0 | −99.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.5 | 0.2 | 1.0 | 1.8 | 1.3 |
| EMeal | −2.2 | −13.5 | −8.6 | 0.0 | −1.0 | −3.9 | 0.0 | 0.0 | −0.1 | 0.0 | 0.0 | 0.0 | −11.5 | −41.1 | −39.5 | 1.2 | 2.0 | 2.6 |
| ESnack | −0.3 | −2.0 | −1.3 | 0.0 | −0.1 | −0.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | −10.9 | −13.6 | −13.8 | 0.3 | 0.4 | 0.6 |
| KCONJ,2 | −26.7 | −69.0 | 96.8 | −26.9 | −69.0 | 96.8 | −26.9 | −69.0 | 96.8 | −69.0 | −69.0 | 96.8 | −68.9 | −70.3 | 96.7 | −25.8 | −68.5 | 96.8 |
| KDECONJ,2 | 16.7 | 16.9 | −86.9 | 16.8 | 16.8 | −83.1 | 16.8 | 16.8 | −82.9 | 16.8 | 16.8 | −58.5 | 16.9 | 18.7 | −62.1 | 15.9 | 15.8 | −85.6 |
| KPL,2 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | −99.0 | 0.0 | 0.0 | −99.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | −1.7 | 0.1 | 0.1 | 0.9 |
| KLP,2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 60.5 | 0.0 | 0.0 | 60.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.0 | 0.0 | −0.1 |
AUC, area under the curve; UDCA, ursodeoxycholic acid.
The color represents the relative magnitude of the Sensitivity index (SI) values with positive extreme value in dark red and negative extreme value in dark blue. Values in between are colored with lighter colors transitioning from the two extreme colors.
Among all the parameters, the effects of the meal (Emeal) and snack (Esnack) seem to be comparatively less influential to steady state AUC. As food effects were introduced primarily to account for the meal‐induced peaks caused by gallbladder contraction in the plasma PK profiles of GUDCA and TUDCA following a single UDCA dose, these two parameters were required in the model to characterize meal to meal variations. No other parameters were considered to be redundant based on the sensitivity analysis and all remained in the final model.
Sensitivity analysis guided the selection of the most appropriate parameters to vary when the healthy model was adapted to the PBC population. Structurally, the small bile duct destruction associated with PBC can affect fluxes both from liver to bile (KLB,0, KLB,1, KLB,2) and from bile to intestine (KBI,0, KBI,1, KBI,2). Table 3 suggested that altering KLB,0, KLB,1, and KLB,2 resulted in greater increase in bile acid levels in the plasma and, more importantly, smaller changes in the bile concentrations for patients with PBC as compared with healthy subjects, both of which were consistent with what was observed in patients with PBC.2 Thus, KLB,0, KLB,1, and KLB,2 were varied to adapt the healthy model to PBC.
Model for PBC
As illustrated in Supplemental Figure S1, simulations following 90% reduction on KLB,0, 70% on KLB,1, and 90% on KLB,2 indicated significant changes in the conjugate levels in the liver, portal vein, and plasma, which is consistent with literature.34 The simulation suggested a slight reduction in Cmax of GUDCA and TUDCA in both intestinal and biliary systems, which may be associated with diminished excretion in PBC.34 While comparing with observed data from Dilger et al.2 for PBC subjects (Figure 3), the simulations in plasma and bile are reasonably consistent with mean observations. In addition, the relative ratios among the three analytes (i.e., TUDCA ∼15–30× lower than UDCA and GUDCA) and the relative changes in their plasma levels (i.e., no change in UDCA and 2–4× increase in conjugates) as compared to healthy state is also consistent with the reported values.
Figure 3.
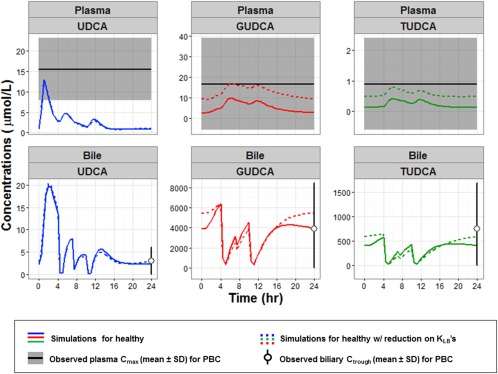
Simulation of ursodeoxycholic acid (UDCA; blue), glycoursodeoxycholic acid (GUDCA; red) and tauroursodeoxycholic acid (TUDCA; green) in plasma (top row) and biliary system (bottom row) at steady state (day 14) following the chronic dosing of 1,000 mg/day using the best‐fit model with no change (solid lines) and when reduction (dotted lines) was applied to KLB,0 (90%), KLB,1 (70%), and KLB,2 (90%). The observed data from patients with primary biliary cirrhosis (PBC; Dilger et al.2), Cmax in plasma (mean ± SD) are represented in broad dashed lines surrounded by shaded areas and biliary trough concentrations (mean ± SD) are in open circle and error bar.
Inhibition of ileal reabsorption of bile acids
Simulations based on the two proposed hypotheses revealed substantial decline in plasma (Figure 4 a) and biliary levels (Figure 4 c) as inhibition of ileal absorption for all three analytes increased from 0% (“no inh”) to 90%, which suggested potential interaction with UDCA dosing. However, when inhibition was only exerted on reabsorption of conjugates, there was a negligible difference in UDCA regardless of inhibition levels and much smaller reductions in conjugate levels as inhibition increased (Figures 4b and 4d). The discrepancy between the two hypotheses can be explained by the fact that when UDCA absorption was inhibited in hypothesis #1, the source of conjugated bile acids was blocked from entering the system, which resulted in reduction in all three analytes. In hypothesis #2, they could be regenerated through enterohepatic recirculation, which includes the steps of deconjugation into UDCA, reabsorption through ileal enterocytes, and reconjugation to form GUDCA and TUDCA in the liver.
Figure 4.
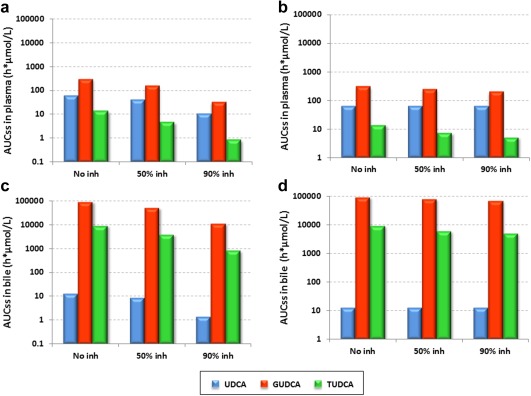
Simulations of steady state area under the curve (AUCss,0‐24h) in the plasma (a, b) and biliary system (c, d) based on ursodeoxycholic acid (UDCA) model for patients with primary biliary cirrhosis (PBC) based on two inhibition scenarios with inhibition on ileal reabsorption of both UDCA and its conjugates (a, c) and inhibition only on conjugates' ileal reabsorption (b, d).
DISCUSSION
The purpose of this work was to study the absorption, distribution, metabolism, and elimination of oral UDCA and its conjugated metabolites and to build a predictive systems model for both healthy subjects and patients with PBC. As a rare disease, clinical trials on PBC are faced with challenges in patient enrolment despite the great needs to slow disease progression and improve disease symptoms. Modeling and simulation could serve as a useful tool to facilitate the understanding of disease pathophysiology, the metabolism of current standards of care (UDCA), as well as the potential effects following a coadministration of UDCA with a drug that interacts with its metabolism.
To our knowledge, this model is the first to feature UDCA and metabolite metabolism in adult human with oral UDCA dose built into the model and with adaption to the patients with PBC population. In contrast to the limited predictive power of a published bile acid model,12 this UDCA model, which was calibrated against a training set of literature data for healthy subjects from both single and chronic UDCA dosing, also accurately predicted the plasma and fecal output reported in a separate healthy volunteer study31 during external validation. It was adapted from healthy to PBC populations, and simulated results were consistent with clinical observations.2
One advantage of a systems model is the knowledge gained from model predictions that describe behaviors in tissues or organs that are remote, technically challenging, or cost prohibitive to measure directly. Model simulations for patients with PBC (Supplementary Figure S1) suggested that following chronic oral UDCA dosing, most of the parent drug was in the intestine and portal vein, while the conjugates were primarily concentrated in the liver and biliary system, with plasma containing only a minor amount of the drug. Therefore, for UDCA and conjugates, measurement of plasma levels only in a clinical setting will not provide enough information to properly study the metabolism and enterohepatic circulation of UDCA. For example, samples from duodenal bile greatly elucidated the change in bile composition of UDCA in patients with PBC.2 The sensitivity analysis performed on the model for the healthy population not only provided insights on how a few parameters significantly influence the steady state exposure for UDCA and conjugates in various tissues, but also the efficient way to perturb the model for healthy subjects to achieve the anticipated adaption to PBC populations.
It is important to note some limitations of the work. The use of mean literature data from multiple resources for model calibration only allowed an average estimate of parameters for the healthy population. Despite intensive PK sampling in plasma, the sparse PK samples for duodenal bile posed a challenge for characterizing parameters related to the biliary system. Additionally, the linearity built into the transport processes and inhibition of hASBT can be both time‐variant and saturable theoretically at higher concentrations or amounts of drug or conjugates. However, it can be adjusted to be time‐variant and saturable depending on physiological situations, such as genetic mutation or pharmacological property of hASBT. Nonetheless, this work illustrates differences between two hypotheses describing the mechanism hASBT inhibition.
Model validation and parameter identification are common concerns with large systems models. To address the first concern, the model was constructed based on known physiology based on a more complex published model19 and then simplified by lumping parameters. Some challenge was experienced when using the initial estimates for parameter optimization based on physiological values from Hofmann et al.,19 which possibly came from lumping and potential differences for the model in describing endogenous metabolism vs. state with exogenous input (oral UDCA). Therefore, preliminary wide random searching for the initial estimates was also performed even outside the previously assumed physiological range for endogenous state to facilitate the parameter optimization as well as to prevent the possibility of reaching local minimum for parameters. Although direct comparison with literature for the parameters were not carried out due to the aforementioned challenges, bootstrap analysis integrating the observed variability in clinical data and cross‐validation case for plasma and fecal output of bile acids were thought to provide sufficient confidence to address the concerns.
Besides studying the hASBT inhibitor in patients with PBC, it seems possible that this model can be usefully adapted to other bile acid species and with perturbation on various components to characterize diverse hepatic disease states not just limited to PBC.
Supporting information
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://www.wileyonlinelibrary.com/psp4).
Supporting Information
Supporting Information
Acknowledgment
The authors acknowledge the constructive feedback provided by Dr. Alan Hofmann during the model development.
Conflict of Interest
P.Z., R.L.O‐S., and M.A.Y. are employees of PAREXEL International and R.L.D. is an employee of Cempra. This work began while all authors were employed by GlaxoSmithKline. They hold GlaxoSmithKline stocks and/or stock options.
Author Contributions
P.Z., R.L.D., R.L.O‐S., and M.A.Y. wrote the manuscript. P.Z., R.L.D., R.L.O‐S., and M.A.Y. designed the research. P.Z. and R.L.D. performed the research. P.Z. analyzed the data.
References
- 1. Imam, M.H. & Lindor, K.D. The natural history of primary biliary cirrhosis. Semin. Liver Dis. 34, 329–333 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Dilger, K. et al Effect of ursodeoxycholic acid on bile acid profiles and intestinal detoxification machinery in primary biliary cirrhosis and health. J. Hepatol. 57, 133–140 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Tammar, A.R. A comparative study of steroids with special reference to bile salts (Ph.D. Diss., University of London, London, 1970). [Google Scholar]
- 4. Haslewood, G.A.D. The Biological Utility of Bile Salts (North Holland Publishing Co, Amsterdam, UK, 1978). [Google Scholar]
- 5. Hagey, L.R. , Crombie, D.L. , Espinosa, E. , Carey, M.C. , Igimi, H. & Hofmann, A.F. Ursodeoxycholic acid in the Ursidae: biliary bile acids of bears, pandas, and related carnivores. J. Lipid Res. 34, 1911–1917 (1993). [PubMed] [Google Scholar]
- 6. Poupon, R. , Chrétien, Y. , Poupon, R.E. , Ballet, F. , Calmus, Y. & Darnis, F. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet 1, 834–836 (1987). [DOI] [PubMed] [Google Scholar]
- 7. Poupon, R.E. , Lindor, K.D. , Cauch‐Dudek, K. , Dickson, E.R. , Poupon, R. & Heathcote, E.J. Combined analysis of randomized trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 113, 884–890 (1997). [DOI] [PubMed] [Google Scholar]
- 8. Carey E.J. & Lindor K.D., eds. Cholestatic liver disease. Second ed. Chapter: treatment. 40 (Springer, New York, NY, 2014). [Google Scholar]
- 9. Mells, G.F. et al Impact of primary biliary cirrhosis on perceived quality of life: the UK‐PBC national study. Hepatology 58, 273–283 (2013). [DOI] [PubMed] [Google Scholar]
- 10. Carbone, M. et al Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastorenterology 144, 560–569 (2013). [DOI] [PubMed] [Google Scholar]
- 11. Corpechot, C. et al Biochemical response to ursodeoxycholic acid and long‐term prognosis in primary biliary cirrhosis. Hepatology 48, 871–877 (2008). [DOI] [PubMed] [Google Scholar]
- 12. Parés, A. , Caballería, L. & Rodés, J. Excellent long‐term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic acid. Gastroenterology 130, 715–720 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Dyson, J.K. et al Novel therapeutic targets in primary biliary cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 12, 147–158 (2015). [DOI] [PubMed] [Google Scholar]
- 14. Clinicaltrials.gov. A study to evaluate the safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of repeat doses of GSK2330672 administration in subjects with primary biliary cirrhosis (PBC) and symptoms of pruritus (2014). US National Library of Medicine <https://clinicaltrials.gov/ct2/show/NCT01899703>. Accessed 9 February 2016.
- 15. Clinicaltrials.gov. Phase 2 study to evaluate LUM001 in combination with ursodeoxycholic acid in patients with primary biliary cirrhosis (CLARITY) (2015). US National Library of Medicine <https://clinicaltrials.gov/ct2/show/NCT01904058>. Accessed 9 February 2016.
- 16. Clinicaltrials.gov. An exploratory, phase IIa study to demonstrate the safety and efficacy of A4250 in patients with primary biliary cirrhosis and cholestatic pruritus (2015). US National Library of Medicine <https://clinicaltrials.gov/ct2/show/NCT02360852? term=NCT02360852&rank=1>. Accessed 9 February 2016.
- 17. Crosignani, A. , Setchell, K.D. , Invernizzi, P. , Larghi, A. , Rodrigues, C.M. & Podda, M. Clinical pharmacokinetics of therapeutic bile acids. Clin . Pharmacokinet. 30, 333–358 (1996). [DOI] [PubMed] [Google Scholar]
- 18. Baillie, R. , Gordi, T. , Vuong, L. , Bosley, J. & Blood, A. Pediatric PK modeling using AMS data and PhysioPDTM modeling of ursodiol in a neonate clinical trial. <http://www.rosaandco.com/posters/rosaAAPS2011abstract.pdf> (2011).
- 19. Hofmann, A.F. , Molino, G. , Milanese, M. & Belforte, G. Description and simulation of a physiological pharmacokinetic model for the metabolism and enterohepatic circulation of bile acids in man. Cholic acid in healthy man. J. Clin. Invest. 71, 1003–1022 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Molino, G. , Hofmann, A.F. , Cravetto, C. , Belforte, G. & Bona, B. Simulation of the metabolism and enterohepatic circulation of endogenous chenodeoxycholic acid in man using a physiological pharmacokinetic model. Eur. J. Clin. Invest. 16, 397–414 (1986). [DOI] [PubMed] [Google Scholar]
- 21. Hofmann, A.F. , Cravetto, C. , Molino, G. , Belforte, G. & Bona, B. Simulation of the metabolism and enterohepatic circulation of endogenous deoxycholic acid in humans using a physiologic pharmacokinetic model for bile acid metabolism. Gastroenterology 93, 693–709 (1987). [DOI] [PubMed] [Google Scholar]
- 22. Balakrishnan, A. , Wring, S.A. & Polli, J.E. Interaction of native bile acids with human apical sodium‐dependent bile acid transporter (hASBT): influence of steroidal hydroxylation pattern and C‐24 conjugation. Pharm. Res. 23, 1451–1459 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ellis, E. et al Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology 38, 930–938 (2003). [DOI] [PubMed] [Google Scholar]
- 24. Walker, S. , Rudolph, G. , Raedsch, R. & Stiehl, A. Intestinal absorption of ursodeoxycholic acid in patients with extrahepatic biliary obstruction and bile drainage. Gastroenterology 102, 810–815 (1992). [DOI] [PubMed] [Google Scholar]
- 25. Crosignani, A. et al Effects of ursodeoxycholic acid on serum liver enzymes and bile acid metabolism in chronic active hepatitis: a dose‐response study. Hepatology 13, 339–344 (1991). [PubMed] [Google Scholar]
- 26. Hagey, L.R. & Krasowski, M.D. Microbial biotransformations of bile acids as detected by electrospray mass spectrometry. Adv. Nutr. 4, 29–35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher, R.S. , Rock, E. & Malmud, L.S. Effects of meal composition on gallbladder and gastric emptying in man. Dig. Dis. Sci. 32, 1337–1344 (1987). [DOI] [PubMed] [Google Scholar]
- 28. Brunner, H. , Northfield, T.C. , Hofmann, A.F. , Go, V.L. & Summerskill, W.H. Gastric emptying and secretion of bile acids, cholesterol, and pancreatic enzymes during digestion. Duodenal perfusion studies in healthy subjects. Mayo Clin. Proc. 49, 851–860 (1974). [PubMed] [Google Scholar]
- 29. Xiang, X. , Vakkilainen, J. , Backman, J.T. , Neuvonen, P.J. & Niemi, M. No significant effect of the SLCO1B1 polymorphism on the pharmacokinetics of ursodeoxycholic acid. Eur. J. Clin. Pharmacol. 67, 1159–1167 (2011). [DOI] [PubMed] [Google Scholar]
- 30. Milani, M. Ursodeoxycholic acid (UDCA) in biliary diseases: a clinical review. Br. J. Med. Med. Res. 4, 1783–1790 (2014). [Google Scholar]
- 31. Hess, L.M. et al Results of a phase I multiple‐dose clinical study of ursodeoxycholic acid. Cancer Epidemiol. Biomarkers Prev. 13, 861–867 (2004). [PubMed] [Google Scholar]
- 32. Fassò, A. & Perri, P.F. Sensitivity analysis. Encyclopedia of Environmetrics 4, 1968–1982 (2006). [Google Scholar]
- 33. Hamby, D.M. A review of techniques for parameter sensitivity analysis of environmental models. Environ. Monit. Assess. 32, 135–154 (1994). [DOI] [PubMed] [Google Scholar]
- 34. Salen, G. & Batta, A.K. Bile acid abnormalities in cholestatic liver diseases. Gastroenterol. Clin. North. Am. 28, 173–193 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information accompanies this paper on the CPT: Pharmacometrics & Systems Pharmacology website (http://www.wileyonlinelibrary.com/psp4).
Supporting Information
Supporting Information