

Abstract
On April 29, 2015, Son and colleagues published an article entitled “Granulocyte macrophage colony-stimulating factor (GM-CSF) is required for aortic dissection/intramural haematoma” in Nature Communications. The authors observed that the heterozygous Kruppel-like transcription factor 6 (KLF6) deficiency or absence of myeloid-specific KLF6 led to upregulation of macrophage GM-CSF expression, promoted the development of aortic hematoma/dissection, and stimulated abdominal aortic aneurysm (AAA) formation when the vessel wall was subjected to an inflammatory stimulus. The additional findings of increased adventitial fibrotic deposition, marked infiltration of macrophages, and increased expression of matrix metalloprotease-9 (MMP-9) and IL-6 were blocked with neutralizing GM-CSF antibodies, or recapitulated in normal mice with excess GM-CSF administration. The authors concluded that GM-CSF is a key regulatory molecule in the development of AAA and further suggested that activation of GM-CSF is independent of the transforming growth factor β (TGFβ)-Smad pathway associated with the Marfan aortic pathology. In this perspective, we expand on this mechanism, drawing from previous studies implicating a similar essential role for IL-6 signaling in macrophage activation, Th17 expansion and aortic dissections. We propose a sequential “two-hit” model of vascular inflammation involving initial vascular injury followed by recruitment of Ly6Chi macrophages. Aided by fibroblast interactions inflammatory macrophages produce amplification of IL-6 and GM-CSF expression that converge on a common, pathogenic Janus kinase (JAK)-signal transducers and activations of transcription 3 (STAT3) signaling pathway. This pathway stimulates effector functions of macrophages, promotes differentiation of Th17 lymphocytes and enhances matrix metalloproteinase expression, ultimately resulting in deterioration of vascular wall structural integrity. Further research evaluating the impact of interventions modulating this common JAK-STAT3 pathway may yield new therapeutic interventions for late stages of vascular expansion in inflammation driven aortic disease.
Keywords: Angiotensin II, Janus kinase (JAK), granulocyte macrophage colony-stimulating factor (GM-CSF), IL-6, signal transducers and activations of transcription (STAT), Kruppel-like transcription factor 6 (KLF6)
Human aortic aneurysmal disease
An aneurysm is a permanent dilation of the blood vessel that is lethal if it progresses to dissection and rupture. Aneurysms of the aortic wall are subdivided anatomically into those occurring above or below the diaphragm—thoracic aortic aneurysm (TAA) and abdominal aortic aneurysm (AAA), respectively. AAAs are associated with advanced age, male gender, smoking, atherosclerosis and hypertension (1,2). The pathology of AAA is characterized by atheromatous plaques, inflammatory cell infiltration in the medial and adventitial layers, loss of vascular smooth muscle cell (VSMC) and extracellular matrix (ECM) destruction (3). In addition, AAAs are not associated with any single gene mutation or genetic locus, suggesting that it is a complex disorder involving numerous genetic and environmental factors. In contrast, TAAs often have a strong hereditary link, but are not closely associated with specific cardiovascular risk factors. Pathologically, TAAs involve destructive matrix remodeling and elastin fragmentation, VSMC proliferation, and relatively less inflammatory infiltrates (4). Genetic syndromes that give rise to TAAs include connective tissue disorders such as Marfan syndrome (MFS) and Ehlers-Danlos syndrome, and disorders of cytoskeleton proteins (familial Thoracic Aortic Aneurysm and Dissection) (4).
Experimental models
Over the past 15 years, multiple chemically induced models of AAA—including elastase perfusion, CaCl2, and angiotensin II (Ang II) infusion—have been developed to explore the pathogenesis of human disease (5). In the Ang II model, Ang II is delivered via mini osmotic pumps into hyperlipidimic (LDLR -/- or ApoE -/-) mice over a period of 7 to 28 days. This model mimics multiple salient features that are found in human AAA including aortic medial degeneration, infiltration of macrophages, T cells and B cells, and thrombus formation. A different model of Ang II infusion into normolipidemic mice produces AAA at a lower frequency. In the Ang II model, monocyte infiltration is associated with extensive intramural hematoma formation; interestingly, these hematomas form in the absence of detectable intimal dissections in the early stages of the disease. Also interesting in this model, dissection of the supra-renal aorta occurs shortly after Ang II infusion before bone fide aneurysmal dilation (6). Periaortic application of CaCl2 also leads to medial degeneration and adventitial remodeling but does not promote thrombus formation or dissection. In the study by Son and colleagues (7), a hybrid model was utilized that involved periaortic CaCl2 application followed by Ang II infusion to induce more consistent aortic pathology.
Granulocyte macrophage colony-stimulating factor in the pathogenesis of Ang II + CaCl2-induced aneurysms
Son and colleagues provide evidence that granulocyte macrophage colony-stimulating factor (GM-CSF) plays an essential role in the pathogenesis of aortic wall dilation, intramural hematoma formation, and aortic wall dissection induced by stimuli that induce vessel wall inflammation. In this study, heterozygous Kruppel-like transcription factor 6 (KLF6)-deficient mice developed an exacerbated aortic dilation in response to the CaCl2 + Ang II challenge. Additional findings included increased adventitial fibrotic deposition, marked infiltration of macrophages, and increased expression of matrix metalloprotease-9 (MMP-9) and IL-6, a well-recognized cytokine independently shown to be necessary for Ang II-induced aortic dissections (8). This present study provided compelling genetic data to support the functional implication of KLF6 as an inhibitor of both GM-CSF and IL-6 expression in activated aortic macrophages.
KLF6 is a zinc finger transcription factor belonging to a 17-member family of DNA binding transcriptional regulators initially described as a tumor suppressor gene (9-12), but currently recognized as having multiple, diverse roles during differentiation and development with the capacity to act both as an activator and a repressor of transcription. KLF6 is essential for early hematopoiesis and vasculogenesis since whole-body KLF6 deletion leads to failure of both events and death in utero (13). Identified target genes of KLF6 include collagen 1 (14), urokinase plasminogen activator (15), TGFβ1 and TGFβ1 type I and type II receptors (16). KLF6 directly activates the E-cadherin promoter (17), and has been reported to promote adipocyte differentiation by interaction with histone deacetylase 3 (HDAC3) repressing the Delta-Like 1 Homolog gene (18). HDAC3 is an enzyme that de-acetylates chromatin-associated histones that regulate gene expression. This association perhaps indicates that KLF6 controls epigenetic regulation of cytokine networks in the vessel wall. These authors further demonstrated that myeloid-specific deletion of KLF6 generated the same phenotype of aortic aneurysm and vessel wall inflammation as the whole body heterozygous deletion, but interestingly, also included suprarenal aortic aneurysms. Elevated aortic wall and blood levels of IL-6 and inflammatory monocytes (CD11b + Ly6Chi cells) were observed in the myeloid KLF6-deletion mice, clearly demonstrating that this experimental model was associated with an amplification loop of inflammatory monocytes driven, in part, by GM-CSF.
Aortic macrophages harvested from the myeloid KLF6-deletion mice, treated with CaCl2 + Ang II, expressed 8-fold higher level of GM-CSF mRNA. GM-CSF is a monomeric glycoprotein that is a chemotactic and pro-inflammatory cytokine inducing activation and maturation of macrophages and dendritic cells (19). Previous work has shown that GM-CSF is a highly inducible chemokine locally produced in the aorta by endothelial cells, fibroblasts, smooth muscle cells and macrophages (20). GM-CSF is upregulated by IL-1, TNFα and LPS and its transcription is controlled by activating transcription factors, NF-κB and activating protein 1. Consequently, GM-CSF levels increase in plasma associated with atherosclerosis and is enriched in the aortic sinuses of atherogenic-prone ApoE-deficient mice (21). GM-CSF binds to a heteromeric GM-CSF receptor, composed of α and βc chains; βc is common to GM-CSF, IL-3 and IL-5 receptors. Granulocyte macrophage colony-stimulating factor receptor (GM-CSFR) lacks intrinsic kinase activity but constitutively associates with Janus kinase 2 (JAK2). JAK2 autophosphorylation triggers signaling through STAT3, STAT5 and MAPK (Figure 1). Interestingly, through a direct interaction of GM-CSFRα with the IκB kinase, GM-CSF also potentiates pro-inflammatory NF-κB signaling (22).
Figure 1.
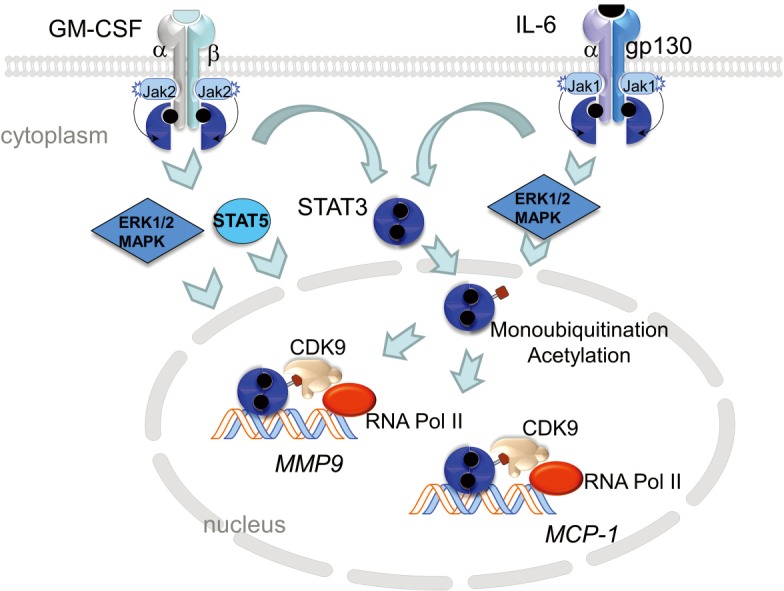
Model of GM-CSF and IL-6 signaling. GM-CSF binding to its receptor, a heterodimer receptor consisting of low-affinity α chain and a longer, signal-transducing β chain leads to conformation changes in the receptor complex and phosphorylation of Janus kinase 2 (JAK2). JAK2 transphosphorylates the β receptor which allows for docking of STAT5 (signal transducer and activator of transcription 5) and STAT3. STAT5 and STAT3 are phosphorylated by JAK2, which promotes their homo-dimerization and translocation to the nucleus to initiate gene transcription. GM-CSFR activation also initiates MAPK signaling involving ERK1/2. Similarly, binding of IL-6 to IL-6Rα leads to dimerization with the signal-transducing membrane protein gp130 and phosphorylation of JAK1. JAK1 transphosphorylates the receptor leading to the recruitment and phosphorylation STAT3. This allows STAT3 to homodimerize and locate to the nucleus. Within the nucleus, phospho-STAT3 is acetylated by p300/CREB-binding protein (CBP) which stabilizes STAT3/p300/CBP complex and promotes enhancesome formation. In addition, STAT3-monoubiquitination on Lys97 promotes binding to BRD4, part of the positive transcription elongation factor (pTEFb) complex that includes CDK9, and promotes transcription elongation of target genes. Both GM-CSF and IL-6 converge on STAT3 signaling suggesting that this common pathway may be a prime target for inhibiting vascular inflammation. GM-CSF, granulocyte macrophage colony-stimulating factor.
To further demonstrate a link between GM-CSF and aortic dilation and dissection, the authors blocked the induced aortic pathology using neutralizing antibodies directed against GM-CSF, and reproduced the pathology in normal mice by administering GM-CSF together with CaCl2 + Ang II. Results of this study provide important new information for our understanding of the role that both systemic and localized tissue inflammation play in these life threatening pathologies of the aortic vessel wall. In addition, results of this and other studies (23) point to significant differences in mechanisms of aortic dissection linked to genetic diseases such as Marfan syndrome, in which TGFβ and its downstream intracellular signaling molecules Smad2/3 and ERK1/2, have important, causal roles in the vessel wall dissections (24,25).
While these authors provide data clearly suggesting that GM-CSF may be a key regulatory molecule influencing the development of aortic dilation and dissection in the presence of an inflammatory stimulus, and further suggest that activation of GM-CSF is independent of the TGFβ-Smad pathway associated with the Marfan aortopathy, they do not develop a mechanistic hypothesis for how GM-CSF and inflammation interact to generate an amplification loop of more inflammation that ultimately results in the aortic wall aneurysms and dissections clearly associated with the mortality of this cardiovascular disease. In the remainder of this Perspective, we will integrate other evidence for the pathogenic mechanism of aortic cytokines and propose an integrative model.
Actions of GM-CSF in tissue inflammation driven by activated macrophages
GM-CSF is a chemotactic cytokine involved in macrophage maturation that is secreted locally by aortic fibroblasts (26), and infiltrating monocyte/macrophages and Th17 cells (27). The actions of GM-CSF have been most intensively investigated in models of lung inflammation, where it induces macrophage activation including expression of cell adhesion molecules (CD11a and CD11c), promotes macrophage polarization and acquisition of phagocytic activity (28). In the vasculature, GM-CSF signaling produces intimal proliferation and macrophage priming. Its function in macrophage recruitment appears to be its major initial effect, based on findings that GM-CSF knockout mice have lower tissue macrophage populations (29), and conversely, GM-CSF overexpression in tissues leads to macrophage expansion and tissue inflammation, followed later by fibrosis (30). GM-CSF has the interesting properties that it is cell-surface bound and associated with extracellular matrix (31). These latter properties may account for how GM-CSF has local inflammatory effects in tissues where it is overexpressed, rather than activating systemic inflammation.
GM-CSF in vascular disease
An increase in GM-CSF production has been observed by multiple groups in models of aortic inflammation and aneurysm formation. In a rat elastase perfusion model of AAA, macrophage density in media and adventitia correlated with elevated gene expression of GM-CSF in the aortic wall (32). A different model of Smad3 deficiency that leads to aneurysm-osteoarthritic syndrome (AOS) is associated with an increase in GM-CSF and IL-6 secretion from CD4+ T cells (33). The pathogenic role of GM-CSF in this model was demonstrated by antibody neutralization, which ameliorated aortic inflammation and aortic root enlargement. Furthermore, a surgical aortic specimen from a patient with AOS demonstrated increased inflammatory infiltrates and GM-CSF immunostaining. Our group has found increased GM-CSF secretion and phospho-STAT3, the downstream signaling molecule, in addition to IL-6 and MCP-1, in aortas from a hypomorphic fibrillin mouse model of MFS (34). Collectively, these data suggest that GM-CSF is involved in the development of aortic inflammation and likely exacerbates destruction of aortic architecture.
Effects of GM-CSF on leukocyte effector functions
Multiple cell types of myeloid lineage are capable of responding to GM-CSF including mature granulocytes, monocytes, macrophages and dendritic cell lineages (19). In vitro stimulation of monocytes and macrophages elicits a robust production of cytokines including IL-6, IL-8, TNFα and IL-1, and specific patterns of macrophage polarization (35). The study of Son et al. implicated aortic macrophages as primary cells secreting GM-CSF, suggesting that its autocrine and paracrine actions probably result in an exaggerated inflammatory response in the vessel wall. More insight into the role of GM-CSF on monocytes is derived from experiments involving conditional deletion of the GM-CSFR βc from CCR2 + Ly6Chi monocytes either before or after onset of experimental autoimmune encephalitis (EAE) (36). Removal of βc receptor on Ly6Chi monocytes before onset blocked EAE and removal after onset reduced autoimmune tissue damage. Furthermore, βc receptor seemed dispensable on resident macrophages, neutrophils and conventional dendritic cells, suggesting βc receptor is most important for monocyte-mediated inflammation. The authors further demonstrated that βc-receptor-deficient, monocyte-derived dendritic cells are not able to produce sufficient IL-1β—a key cytokine needed for expansion of pathogenic CD4+ Th cells—due to downregulation of apoptosis-associated speck like protein containing a CARD (ASC) component of the NLRP3 inflammasome. In Ang II infused mice, inflammasome activation via increased mitochondrial reactive oxygen species (ROS) generation was observed in aortic adventitial macrophages and bone marrow-derived macrophages (37). Deletion of inflammasome components decreased aortic IL-1β production, leukocyte recruitment, and AAA formation. Taken together, these studies suggest that GM-CSF/GM-CSFR interaction on recruited monocytes is upstream of enhanced cytokine secretion, T cell polarization, and leukocyte recruitment to the aortic wall.
IL-6/MCP-1 amplification in vascular inflammation
Earlier studies using the Ang II infusion model have shown time-dependent increases in IL-6 and MCP-1 secretion in the aortic wall (38). Here, IL-6 was also independently demonstrated to be necessary for aortic dissection mediated by recruitment of circulating monocytes expressing the MCP-1 chemokine receptor, CCR2. Genetic deficiency of IL-6 resulted in the inhibition of monocyte recruitment and MMP-9 expression, preventing the rapid appearance of Ang II-induced aortic dissection and aneurysm. Similarly, inhibition of monocyte recruitment with CCR2 deficiency decreased aortic IL-6 and MCP-1 secretion and significantly decreased aortic dissection, whereas infusion of CCR2+/+ monocytes into CCR2-/- mice restored aortic IL-6 and MCP-1 levels along with an increased incidence of dissections. In vitro co-culture of aortic fibroblasts and monocytes, without direct contact, led to multi-fold increases in IL-6 and MCP-1, suggesting that intercellular communication between these two cells in the adventitia promotes amplified cytokine secretion. It is interesting that although IL-6 promotes macrophage maturation, it does not have independent chemotactic activity, unlike that of MCP-1 and GM-CSF.
This property suggests that the initial increase in GM-CSF and MCP-1 secretion in the initial phases of vascular response to Ang II infusion likely leads to recruitment of circulating monocytes to the adventitia where they interact with fibroblasts and amplify cytokine production. Localized aortic cytokine production has systemic secondary effects. For example, we have observed that IL-6-STAT3 signaling leads to Th17 lymphocyte differentiation and a 4-fold increase in splenic Th17 population; this expansion is mitigated by a STAT3-peptide inhibitor (39). Th17 cells producing IL-17 are recruited to the aorta and are known to promote aneurysm formation (38-40). The function of the spleen is important because this organ serves as a reservoir of CD11b + Ly6Chi inflammatory monocytes. Interestingly, splenectomy partially protects from Ang II-induced AAA formation, probably through this mechanism (41). Data from Ang II infusion in IL-6-/- and KLF6-/- mice demonstrate that both IL-6 and GM-CSF are necessary for the full manifestation of aortic pathology as these cytokines amplify aortic cytokine production and promote leukocyte recruitment and maturation.
IL-6 promotes macrophage effector functions
IL-6 promotes monocyte differentiation to macrophages in vitro by increasing cell size, developing cytoplasmic vacuoles and acquiring surface adherence (8). Macrophage cell surface markers such as F4/80 and macrophage-colony stimulating factor (M-CSF) receptor are also increased by IL-6 (42). Surprisingly, IL-6 deficiency does not alter mature numbers of macrophages in vivo, but does make IL-6-/- mice succumb to infections by Listeria monocytogenes—a bacterium that survives and proliferates in macrophages (43). This suggests that loss of IL-6 affects macrophage effector function in vivo. In AAA, the MCP-1/CCR2 axis is important for monocyte recruitment to the aortic wall where their secretion of MMPs leads to elastolysis (38,44). IL-6 has been demonstrated to increase transcription of MCP-1 (45) and MMP-9 (46), linking it to monocyte recruitment and matrix remodeling. In addition, IL-6, in concert with TGFβ, promotes differentiation of naïve T cells into IL-17 producing Th17 lymphocytes (47). IL-17 is a strong inducer of MCP-1 and therefore leukocyte recruitment; IL-17A-/- mice are resistant to Ang II-induced aortic monocyte accumulation and AAA formation (39).
Mechanistically, IL-6 binding to IL-6Rα leads to oligomerization with the transmembrane gp130 β-subunit. This event promotes association of JAK1 with the receptor complex leading to autophosphorylation and recruitment of STAT3 (Figure 1). Tyrosine phosphorylation of STAT3 leads to dimerization of the transcription factor and translocation to the nucleus. Nuclear STAT3 is modified via acetylation and mono-ubiquitination, events necessary for strong interaction with enhancer protein complex p300/CBP (48) and BRD4/CDK9 (49), respectively. Activated STAT3/BRD4/CDK9 complex phosphorylates RNA polymerase II on the C-terminus initiating transcription elongation of target genes (49,50) The STAT3 signaling pathway is a shared pathway triggered by both GM-CSF and IL-6 making its exploration in the development of AAA of substantial importance.
Conclusions
Based on these data, we know the progression of aortic disease from initial injury to dissection or dilation is a complex interaction between innate inflammation of resident stromal cells with recruitment of circulating macrophages and lymphocytes, in much as the same way in the development of complex atherosclerotic plaques (51). We propose a sequential two-hit model for aortic disease, beginning with a local injury, followed by adventitial cytokine amplification. This initial injury induces production of monocyte chemotactic factors (MCP-1, GM-CSF) by resident vascular cells (endothelium, fibroblasts, smooth muscle cells), responsible for recruitment of Ly6Chi monocytes via CCR2 engagement (Figure 2). Once Ly6Chi monocytes are recruited, IL-6 and GM-CSF stimulate production of ROS and acquisition of phagocytic activity. As the disease progresses, infiltrating Ly6Chi monocytes promote dramatic increases in cytokine production, MMP secretion, Th17 lymphocyte recruitment and fibrotic adventitial expansion. These events occur concurrently with intimal dissection and medial rupture causing aneurysmal dilation (Figure 2). The work of Son et al. has shown that both IL-6 and GM-CSF are under tonic inhibition by KLF6; the known interaction of KLF6s with HDAC3 may provide mechanistic information for how IL-6/GM-CSF are tonically suppressed, perhaps through the epigenetic actions of HDAC3. Importantly, these studies indicate an intricate interplay between the vessel wall, innate and adaptive immune cells and adventitial fibrosis. More work will be required to establish whether targeting JAK-STAT pathway will block both IL-6- and GM-CSF-mediated inflammation and inhibit AAA formation.
Figure 2.

Two hit model for Ang II-induced vascular inflammation and aneurysm formation. 1, after an initial insult of Ang II and CaCl2, aortic fibroblasts and resident macrophages are activated leading to secretion of chemokines GM-CSF and MCP-1 in addition to secretion of pro-fibrotic cytokine IL-6; 2, circulating Ly6Chi monocytes are recruited to the adventitia where they interact with fibroblasts and amplify cytokine/chemokine secretion. GM-CSF secreted by aortic monocyte/macrophages acts in an autocrine and paracrine manner stimulating the production of IL-1β and IL-6 and their downstream target MCP-1. In addition, IL-6 promotes MMP-9 production leading to aortic wall remodeling and aortic dilation. Systemic effects of aortic IL-6 include Th17 lymphocyte polarization; Th17 home to the abdominal aorta where they compound inflammation by producing more GM-CSF and IL-17. Excessive activation of MMPs and destabilization of extracellular matrix leads to intramural hematomas and eventually to aortic dissection. GM-CSF, granulocyte macrophage colony-stimulating factor; MMP, matrix metalloprotease.
Acknowledgements
None.
Footnotes
Provenance: This is an invited Perspective commissioned by the Section Editor Lei Zhang (Department of Vascular Surgery, Changhai Hospital, Second Military Medical University, Shanghai, China).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Lederle FA, Johnson GR, Wilson SE, et al. The Aneurysm Detection and Management (ADAM) Veterans Affairs Cooperative Study Investigators . J Vasc Surg 1997;26:595-601. 10.1016/S0741-5214(97)70057-0 [DOI] [PubMed] [Google Scholar]
- 2.Lu H, Rateri DL, Bruemmer D, et al. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin Sci (Lond) 2012;123:531-43. 10.1042/CS20120097 [DOI] [PubMed] [Google Scholar]
- 3.Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol 2015;35:1746-55. 10.1161/ATVBAHA.115.305269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindsay ME, Dietz HC. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 2011;473:308-16. 10.1038/nature10145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2004;24:429-34. 10.1161/01.ATV.0000118013.72016.ea [DOI] [PubMed] [Google Scholar]
- 6.Saraff K, Babamusta F, Cassis LA, et al. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2003;23:1621-6. 10.1161/01.ATV.0000085631.76095.64 [DOI] [PubMed] [Google Scholar]
- 7.Son BK, Sawaki D, Tomida S, et al. Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat Commun 2015;6:6994. 10.1038/ncomms7994 [DOI] [PubMed] [Google Scholar]
- 8.Hou T, Tieu BC, Ray S, et al. Roles of IL-6-gp130 Signaling in Vascular Inflammation. Curr Cardiol Rev 2008;4:179-92. 10.2174/157340308785160570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andreoli V, Gehrau RC, Bocco JL. Biology of Krüppel-like factor 6 transcriptional regulator in cell life and death. IUBMB Life 2010;62:896-905. 10.1002/iub.396 [DOI] [PubMed] [Google Scholar]
- 10.Ito G, Uchiyama M, Kondo M, et al. Krüppel-like factor 6 is frequently down-regulated and induces apoptosis in non-small cell lung cancer cells. Cancer Res 2004;64:3838-43. 10.1158/0008-5472.CAN-04-0185 [DOI] [PubMed] [Google Scholar]
- 11.Narla G, Heath KE, Reeves HL, et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science 2001;294:2563-6. 10.1126/science.1066326 [DOI] [PubMed] [Google Scholar]
- 12.Reeves HL, Narla G, Ogunbiyi O, et al. Kruppel-like factor 6 (KLF6) is a tumor-suppressor gene frequently inactivated in colorectal cancer. Gastroenterology 2004;126:1090-103. 10.1053/j.gastro.2004.01.005 [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto N, Kubo A, Liu H, et al. Developmental regulation of yolk sac hematopoiesis by Kruppel-like factor 6. Blood 2006;107:1357-65. 10.1182/blood-2005-05-1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratziu V, Lalazar A, Wong L, et al. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis. Proc Natl Acad Sci U S A 1998;95:9500-5. 10.1073/pnas.95.16.9500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kojima S, Hayashi S, Shimokado K, et al. Transcriptional activation of urokinase by the Krüppel-like factor Zf9/COPEB activates latent TGF-beta1 in vascular endothelial cells. Blood 2000;95:1309-16. [PubMed] [Google Scholar]
- 16.Kim Y, Ratziu V, Choi SG, et al. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem 1998;273:33750-8. 10.1074/jbc.273.50.33750 [DOI] [PubMed] [Google Scholar]
- 17.DiFeo A, Narla G, Camacho-Vanegas O, et al. E-cadherin is a novel transcriptional target of the KLF6 tumor suppressor. Oncogene 2006;25:6026-31. 10.1038/sj.onc.1209611 [DOI] [PubMed] [Google Scholar]
- 18.Li D, Yea S, Laborda J, et al. KLF6 promotes adipocyte differentiation through histone deacetylase 3 (HDAC3)-dependent repression of Dlk1. Gastroenterology 2003;124:A69-A70. 10.1016/S0016-5085(03)80343-8 [DOI] [Google Scholar]
- 19.Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol 2016;12:37-48. 10.1038/nrrheum.2015.161 [DOI] [PubMed] [Google Scholar]
- 20.Plenz G, Reichenberg S, Koenig C, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) modulates the expression of type VIII collagen mRNA in vascular smooth muscle cells and both are codistributed during atherogenesis. Arterioscler Thromb Vasc Biol 1999;19:1658-68. 10.1161/01.ATV.19.7.1658 [DOI] [PubMed] [Google Scholar]
- 21.Ditiatkovski M, Toh BH, Bobik A. GM-CSF deficiency reduces macrophage PPAR-gamma expression and aggravates atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 2006;26:2337-44. 10.1161/01.ATV.0000238357.60338.90 [DOI] [PubMed] [Google Scholar]
- 22.Ebner K, Bandion A, Binder BR, et al. GMCSF activates NF-kappaB via direct interaction of the GMCSF receptor with IkappaB kinase beta. Blood 2003;102:192-9. 10.1182/blood-2002-12-3753 [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Ait-Oufella H, Herbin O, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest 2010;120:422-32. 10.1172/JCI38136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006;312:117-21. 10.1126/science.1124287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cook JR, Clayton NP, Carta L, et al. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol 2015;35:911-7. 10.1161/ATVBAHA.114.305150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tieu BC, Ju X, Lee C, et al. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J Vasc Res 2011;48:261-72. 10.1159/000320358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Codarri L, Gyülvészi G, Tosevski V, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 2011;12:560-7. 10.1038/ni.2027 [DOI] [PubMed] [Google Scholar]
- 28.Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol 2005;25:405-28. 10.1615/CritRevImmunol.v25.i5.50 [DOI] [PubMed] [Google Scholar]
- 29.Paine R, 3rd, Morris SB, Jin H, et al. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. Am J Physiol Lung Cell Mol Physiol 2001;281:L1210-8. [DOI] [PubMed] [Google Scholar]
- 30.Lang RA, Metcalf D, Cuthbertson RA, et al. Transgenic mice expressing a hemopoietic growth factor gene (GM-CSF) develop accumulations of macrophages, blindness, and a fatal syndrome of tissue damage. Cell 1987;51:675-86. 10.1016/0092-8674(87)90136-X [DOI] [PubMed] [Google Scholar]
- 31.Farrar WL, Brini AT, Harel-Bellan A, et al. Hematopoietic growth-factor signal transduction and regulation of gene expression. Immunol Ser 1990;49:379-410. [PubMed] [Google Scholar]
- 32.Sho E, Sho M, Hoshina K, et al. Hemodynamic forces regulate mural macrophage infiltration in experimental aortic aneurysms. Exp Mol Pathol 2004;76:108-16. 10.1016/j.yexmp.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 33.Ye P, Chen W, Wu J, et al. GM-CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest 2013;123:2317-31. 10.1172/JCI67356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ju X, Ijaz T, Sun H, et al. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J Am Heart Assoc 2014;3:e000476. 10.1161/JAHA.113.000476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Däbritz J, Weinhage T, Varga G, et al. Reprogramming of monocytes by GM-CSF contributes to regulatory immune functions during intestinal inflammation. J Immunol 2015;194:2424-38. 10.4049/jimmunol.1401482 [DOI] [PubMed] [Google Scholar]
- 36.Croxford AL, Lanzinger M, Hartmann FJ, et al. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 2015;43:502-14. 10.1016/j.immuni.2015.08.010 [DOI] [PubMed] [Google Scholar]
- 37.Usui F, Shirasuna K, Kimura H, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol 2015;35:127-36. 10.1161/ATVBAHA.114.303763 [DOI] [PubMed] [Google Scholar]
- 38.Tieu BC, Lee C, Sun H, et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest 2009;119:3637-51. 10.1172/JCI38308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ju X, Ijaz T, Sun H, et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler Thromb Vasc Biol 2013;33:1612-21. 10.1161/ATVBAHA.112.301049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Z, Wang Y, Zhang K, et al. Inhibiting the Th17/IL-17A-related inflammatory responses with digoxin confers protection against experimental abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 2014;34:2429-38. 10.1161/ATVBAHA.114.304435 [DOI] [PubMed] [Google Scholar]
- 41.Mellak S, Ait-Oufella H, Esposito B, et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe-/- mice. Arterioscler Thromb Vasc Biol 2015;35:378-88. 10.1161/ATVBAHA.114.304389 [DOI] [PubMed] [Google Scholar]
- 42.Chomarat P, Banchereau J, Davoust J, et al. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol 2000;1:510-4. 10.1038/82763 [DOI] [PubMed] [Google Scholar]
- 43.Kopf M, Baumann H, Freer G, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994;368:339-42. 10.1038/368339a0 [DOI] [PubMed] [Google Scholar]
- 44.Daugherty A, Rateri DL, Charo IF, et al. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin Sci (Lond) 2010;118:681-9. 10.1042/CS20090372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biswas P, Delfanti F, Bernasconi S, et al. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood 1998;91:258-65. [PubMed] [Google Scholar]
- 46.Kothari P, Pestana R, Mesraoua R, et al. IL-6-mediated induction of matrix metalloproteinase-9 is modulated by JAK-dependent IL-10 expression in macrophages. J Immunol 2014;192:349-57. 10.4049/jimmunol.1301906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kimura A, Naka T, Kishimoto T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci U S A 2007;104:12099-104. 10.1073/pnas.0705268104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hou T, Ray S, Lee C, et al. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem 2008;283:30725-34. 10.1074/jbc.M805941200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ray S, Zhao Y, Jamaluddin M3, et al. Inducible STAT3 NH2 terminal mono-ubiquitination promotes BRD4 complex formation to regulate apoptosis. Cell Signal 2014;26:1445-55. 10.1016/j.cellsig.2014.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou T, Ray S, Brasier AR. The functional role of an interleukin 6-inducible CDK9.STAT3 complex in human gamma-fibrinogen gene expression. J Biol Chem 2007;282:37091-102. 10.1074/jbc.M706458200 [DOI] [PubMed] [Google Scholar]
- 51.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011;473:317-25. 10.1038/nature10146 [DOI] [PubMed] [Google Scholar]