

Abstract
The rapid and sensitive detection of sugar alcohol sweeteners was demonstrated using ion mobility spectrometry (IMS). IMS provides a valuable alternative in sensitivity, cost, and analysis speed between the lengthy gold-standard liquid chromatography-mass spectrometry (LC-MS) technique and rapid point-of-measurement disposable colorimetric sensors, for the Food and Nutrition industry’s quality control and other “foodomics” area needs. The IMS response, characteristic signatures, and limits of detection for erythritol, pentaerythritol, xylitol, inositol, sorbitol, mannitol, and maltitol were evaluated using precise inkjet printed samples. IMS system parameters including desorption temperature, scan time, and swipe substrate material were examined and optimized, demonstrating a strong dependence on the physicochemical properties of the respective sugar alcohol. The desorption characteristics of each compound were found to dominate the system response and overall sensitivity. Sugar alcohol components of commercial products – chewing gum and a sweetener packet – were detected and identified using IMS. IMS is demonstrated to be an advantageous field deployable instrument, easily operated by non-technical personnel, and enabling sensitive point-of-measurement quality assurance for sugar alcohols.
Keywords: Ion mobility spectrometry, Sugar alcohols, Alternative sweeteners, Artificial sweeteners, Thermal desorption
Introduction
Artificial and alternative sweeteners are used as additives almost ubiquitously in everyday products such as chewing gum, vitamins, and teeth whitening strips. These include artificial compounds (e.g., aspartame – NutraSweet®, saccharin – Sweet’N’Low®, and sucralose – Splenda®), sugar alcohols (e.g., erythritol, sorbitol, and xylitol), and alternative/natural sweeteners (e.g., stevia extracts – Truvia®, tagatose – Naturlose®, and trehalose), all used to replace calorie rich natural sugars. 1–5 Many of these sweeteners are used as sugar substitutes for diabetics. Artificial/alternative sweeteners are also produced in massive quantities; greater than 1 million metric tons of sugar alcohols are produced annually 5 and the public’s interest in low calorie substitutes will continue to increase demand. The direct detection and identification of these compounds plays an important role in the food industry’s quality assurance and control operations. 6 This can become critical for highly regulated items such as infant food. 7
Numerous analytical techniques have demonstrated the detection of artificial/alternative sweeteners. Robust laboratory based techniques such as high performance liquid chromatography (HPLC) and HPLC mass spectrometry (MS) enable sensitive detection and identification of sweeteners and other compounds. 7–11 However, these methods require costly equipment and can require lengthy analysis times. At the other end of the sensitivity and cost spectrum are colorimetric sensors, which sacrifice a degree of sensitivity to be low-cost, portable, and disposable. 12, 13 In addition, alternative analytical techniques based on electrochemical, 14, 15 fluorescence, 16, 17 spectrophotometric 18, 19, chemoluminescence, 20 and ambient ionization mass spectrometry methods 21–23 have demonstrated the detection of artificial and alternative sweeteners.
Here, we implement ion mobility spectrometry (IMS) 24 for the rapid and sensitive detection of alternative sweeteners, specifically, sugar alcohols. IMS provides a compromise between sensitivity, cost, analysis speed, and field portability and has demonstrated applicability in other areas of “foodomics”. 25, 26 Sample introduction platforms range from electrospray ionization 27 to swipe sampling/thermal desorption. Swipe-based collection, followed by thermal desorption, has enabled IMS to evolve into a robust field deployable method for contraband screening arenas, including trace explosives and narcotics detection. 28, 29 These systems typically include gas-phase ionization with a 63Nickel radiation source, however newer instruments have begun using photoionization or corona discharge chemical ionization. Under the application of an electric field and drift gas, ions are separated based on electric mobility. The commercialization of IMS instruments has led to the development of instruments easily operated by non-technical personnel, making them ideal for rapid point-of-measurement quality assurance of sugar alcohols for the food industry.
In this study, we detect, and identify a range of sugar alcohols (i.e., glycerol, erythritol, pentaerythritol, xylitol, inositol, sorbitol, and mannitol) with IMS, and also measure their reduced mobility values. Characteristic performance was evaluated with limit of detection measurements using the pure compounds prepared by inkjet printed material deposition. Method optimization was conducted, focusing on the desorption temperature, scan time, and swipe material. Variability in the IMS response of these compounds revolved around differences in the physicochemical properties and desorption characteristics. Finally, the collection and detection of sugar alcohols in commercial products (chewing gum and sweetener packets) was demonstrated.
Experimental Methods
Materials
Sugar alcohols, including, glycerol (CAS number 56-81-5), erythritol (CAS no. 149-32-6), pentaerythritol (CAS no. 115-77-5), xylitol (CAS no. 87-99-0), myo-inositol (CAS no. 87-89-8), sorbitol (CAS no. 50-70-4), D-mannitol (CAS no. 69-65-8), and maltitol (CAS no. 585-88-6), were purchased from Sigma Aldrich (St. Louis, MO, USA) at 98 % purity or greater. Solutions of these compounds were prepared gravimetrically (AT21 comparator balance, Mettler Toledo, Columbus, OH, USA) and dissolved in Chromasolv grade water (Sigma Aldrich) to a nominal concentration of 1 mg mL−1. Samples were prepared by inkjet printing precise amounts of the compound (50 ng unless otherwise stated) onto the swipe surface of interest. Inkjet printing (Jetlab 4 XL-B, MicroFab Technologies, Inc., Plano, TX, USA)*, in which a piezo-electric print head was used to precisely control the size and number of drops deposited onto substrates, allowed for reduction in variability from sample to sample, in comparison to pipetting a solution onto a surface. This technique allowed for the deposition of samples in the same spot on each swipe substrate with a mass precision of less than 0.5 % RSD (relative standard deviation). Details on the inkjet printing technique for sample test material production can be found elsewhere in the literature. 30–33 Samples were deposited onto commercial Nomex® swipes (Manual Swab-6821201-B, Smiths Detection, Edgewood, MD, USA) for all parts of this work. In addition, polytetrafluoroethylene (PTFE, Teflon) coated fiberglass weave (Sample Traps-ST1318P, DSA Detection, LLC, Andover, MA, USA) and muslin (Sample Swabs-SSW5883P, DSA Detection) swipes were also used in the examination of swipe substrates.
Instrumentation
The analysis presented here was completed on a Smiths IONSCAN 400B instrument (Smiths Detection). The 400B IMS uses a 63Ni source for ionization and a faraday plate for detection. A list of the base case parameters is shown in Table 1. A doped reactant gas, hexachloroethane, was continuously bled into the ionization region, generating product ions for directed adduct formation. Multiple blank Nomex swipes were run between each sample to reduce carry-over. Raw signals were digitally processed by proprietary algorithms within the instrument firmware, which included a baseline correction. A custom MATLAB-based code (MATLAB R2015a, Mathworks, Inc., Natick, MA, USA) was used to batch post-process series of data files. IMS response measurements were based on the cumulative amplitude of a specified peak (drift time) across all scans (full sampling time). These signal intensities were given in a generic unit of “counts”. Peak areas were calculated for limits of detection and parametric investigations by integrating the cumulative spectra between bracketing points around the peak maximum for which the first derivative cross the y-axis (zero slope).
Table 1.
Parameters for the IMS operation, unless otherwise stated.
| IMS Parameter | Value | |
|---|---|---|
|
| ||
| Desorber temperature (°C) | 225 | |
| Detection/tube temperature (°C) | 111 | |
| Drift flow (cm3 min−1) | 350 | |
| Drift gas | Dry Air | |
| Sampling time (s) | 20 | |
* Certain commercial products are identified in order to adequately specify the procedure; this does not imply endorsement or recommendation by NIST, nor does it imply that such products are necessarily the best available for the purpose.
Results and Discussion
Reduced Mobility Values
IMS drift times were measured for each analyte for base case instrument parameters (Table 2). Drift time measurements are a direct function of the IMS instrument’s drift tube environmental conditions and parameters, specifically, temperature, pressure, humidity, tube length, and applied electric field. Therefore, drift times are routinely normalized to a reduced mobility value, K0(td)=L/tdE(PTo/PoT), where td is the drift time, P and T are the drift tube pressure and temperature, Po and To are standard pressure and temperature, L is drift tube length, and E is the electric field applied across the drift tube. 34, 35 Often the reduced mobility value is experimentally measured through calibration by a proportional relationship with an assigned calibrant mobility and measured drift time. 34, 35 However, the commercial firmware used here measured the analyte drift time and internally calculated the respective reduced mobility values based on system parameters.
Table 2.
Analyte properties including structure, vapor pressure (VP; at 25 °C), relative sweetness to sucrose, drift times, and reduced mobilities for 50 ng deposits of specified sugar alcohols at base case instrument conditions (Table 1). Drift times and reduced mobility values represent the preliminarily assigned chloride adducts. Vapor pressures were estimated using EPI Suite v4.11, US EPA, 2014.
| Compound | Structure | Molecular Formula | Molecular Weight (g mol−1) | Relative Sweetness to Sucrose (%) 37, 38 | VP (kPa) | Drift Time (ms) | K0 (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|
| Glycerol |
|
C3H8O3 | 92.09 | 80 | 1.06×10−5 | 10.5 | 1.761 |
| Erythritol |
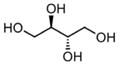
|
C4H10O4 | 112.12 | 70 | 6.99×10−8 | 11.4 | 1.624 |
| Pentaerythritol |
|
C5H12O4 | 136.15 | - | 3.37×10−9 | 12.0 | 1.545 |
| Xylitol |
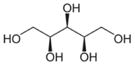
|
C5H12O5 | 152.15 | 100 | 2.61×10−4 | 12.3 | 1.499 |
| Inositol |
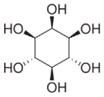
|
C6H12O6 | 180.16 | 50 | 1.77×10−11 | 13.1 | 1.417 |
| Sorbitol |
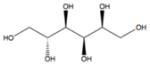
|
C6H14O6 | 182.17 | 50–60 | 1.75×10−10 | 13.2 | 1.405 |
| Mannitol |
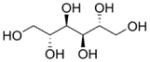
|
C6H14O6 | 182.17 | 50–70 | 1.75×10−10 | 13.1 | 1.410 |
| Maltitol |
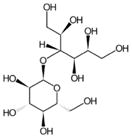
|
C12H24O11 | 344.13 | 90 | 2.98×10−18 | - | - |
Figure 1 displays representative IMS spectra for 50 ng of select sugar alcohols, including straight chain and ringed monosaccharides (glycerol, erythritol, pentaerythritol, xylitol, inositol, and sorbitol); the spectra demonstrate the cumulative intensity across the sampling time. Characteristic peaks for each analyte and the reactant ion peak (RIP) were identified and labeled. The RIP peaks, which come from the dopant gas, hexachloroethane, have previously been identified as Cl− and Cl2− using tandem IMS - mass spectrometry (IMS-MS). 33, 36 One to three distinct peaks were observed for the sugar alcohols investigated here. Cursory peak assignments were based on knowledge of common product ions generated by the dopant gas and past experience. The three potential ions expected for this system were a deprotonated ion [M-H]− formed by proton abstraction from an adduct species; a chloride adduct, [M+Cl]−, generated through dopant product ions; and a nitrate adduct, [M+NO3]−, formed through ionization of the ambient atmosphere (‘M’ represents the respective sugar alcohol). All of these adducts were singly charged and expected to exhibit increased drift time in order of size: [M-H]−, [M+Cl]−, [M+NO3]−. Given the continuously introduced dopant gas and ion chemistry, the dominant peaks were preliminarily assigned to the chloride adduct (Table 2). Future work must be completed using a tandem IMS-MS system to verify adduct ion assignments of observed peaks. 36
Figure 1.
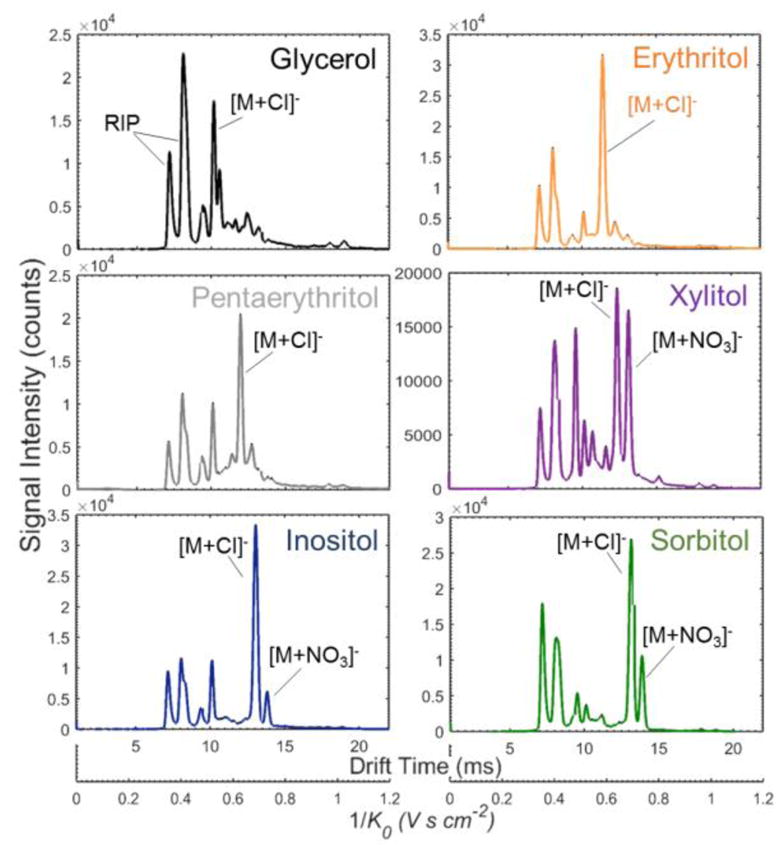
Representative IMS spectra (cumulative across sampling time) for 50 ng deposits of select sugar alcohols.
Analyte drift times and reduced mobility values were determined from ten replicate measurements of eight sugar alcohols under base case instrument parameters (Table 1). Seven of the sugar alcohols demonstrated characteristic peaks attributable to the sugar alcohol, when compared to blank Nomex control measurements. Table 2 displays the reduced mobility and drift time values for these identified peaks along with relevant analyte parameters. A discernable signature for the disaccharide, maltitol, was unobtainable. Efforts to obtain a signature through varying system parameters were also unsuccessful. These difficulties were attributed to maltitol’s low volatility and presumed inability to effectively vaporize/thermally desorb during the 20 s sampling time. Glycerol was found to be readily detected in the background signature of most swipes, and was traced to at least one possible source (nitrile gloves). Because of the high level of background, glycerol was not studied further. Also, because sorbitol and mannitol are isomers, it was assumed that they would behave similarly in the IMS, and therefore, only sorbitol was analyzed in the additional studies.
Limits of Detection
Following characterization of the IMS signatures for the select sugar alcohols, baseline performance of the instrument for their detection was evaluated. Limits of detection (LOD) were calculated by inkjet printing a series of incrementally lower masses onto Nomex swipes and analyzing them under the base case parameters (Table 1). Ten replicates of each of five levels (1 ng, 5 ng, 10 ng, 25 ng, and 50 ng) were analyzed in addition to ten blanks. Cumulative peak area of the base sugar alcohol peak in the plasmagram was then calculated for each replicate (see Experimental Methods for details). Calculations of the limits of detection were completed in accordance with ASTM E2677 following ISO-IUPAC guidelines 39 and using the ASTM E2677 Limit of Detection Web Portal. 40 A 90 % confidence level (LOD90) was used for all measurements. Table 3 highlights the limits of detection for select sugar alcohols in addition to the 90 % upper confidence limit for the limit of detection. Limits of detection were less than 1 ng with the exception of erythritol. The lower sensitivity for erythritol, when compared to other compounds, was likely due to its increased thermal lability and the elevated level of background noise for drift times below 12 ms. The background noise levels observed decreased for increasing drift times, enhancing the detection limits for the longer drift time compounds. While IMS achieved sensitive detection of sugar alcohols (Table 3), the technique demonstrated only an approximately single order-of-magnitude linear range, limiting quantification capabilities.
Table 3.
Limits of detection (LOD90) and 90 % Upper Confidence limit for the LOD for the sugar alcohols examined.
| Compound | LOD90 (ng) | 90% Upper Limit (ng) |
|---|---|---|
| Erythritol | 3.92 | 7.57 |
| Pentaerythritol | 0.43 | 0.81 |
| Xylitol | 0.51 | 0.97 |
| Inositol | 0.89 | 2.15 |
| Sorbitol | 0.55 | 1.14 |
Desorption Temperature
Next, we conducted a number of parametric investigations into the IMS response as a function of the desorption temperature, scan time, and swipe/collection material. Thermal desorption is the primary method used for analyte vaporization in a large number of commercial IMS instruments and was a primary factor in the response optimization for sugar alcohols with a wide range of volatilities (used here to qualitatively describe sublimation rates, intermolecular forces, and surface adsorption). Here, 50 ng deposits of five sugars (sorbitol, erythritol, pentaerythritol, xylitol, and inositol) were analyzed at temperatures ranging from 75 °C to 300 °C, varied in 25 °C increments. Figure 2 shows the IMS response for each sugar alcohol as a function of desorber temperature for 10 replicates. The data displayed in Figure 2 represents the cumulative peak area of the base peak across the 20 s collection time (segmented into 40 individual scans). Higher vapor pressure sugar alcohols were desorbed rapidly during the scan duration (Figure 3(a)), while those with lower vapor pressure were desorbed slower, resulting in a longer duration of signal (Figure 3(b)). The select sugar alcohols (erythritol, pentaerythritol, xylitol, inositol, and sorbitol) investigated here demonstrated optimal temperatures in the range of 125 °C to 200 °C. At low temperatures, near 75 °C, the heat provided by the desorber was not sufficient enough to volatilize the sugar alcohols, resulting in lower signal. On the other extreme, elevated temperatures reduced signal either through flash heating or thermal decomposition. Optimal desorber temperature was found to correlate with molecule size (erythritol: 125 °C, pentaerythritol: 125–150 °C, xylitol: 150 °C, inositol: 200 °C, and sorbitol: 200 °C) and roughly inversely with vapor pressure (Figure 2). Practically, these trends could be implemented to assign an optimal desorber temperatures for each target analyte individually or chose a desorber temperature to achieve overall optimal performance for the complete range of analytes, e.g., ≈200 °C.
Figure 2.
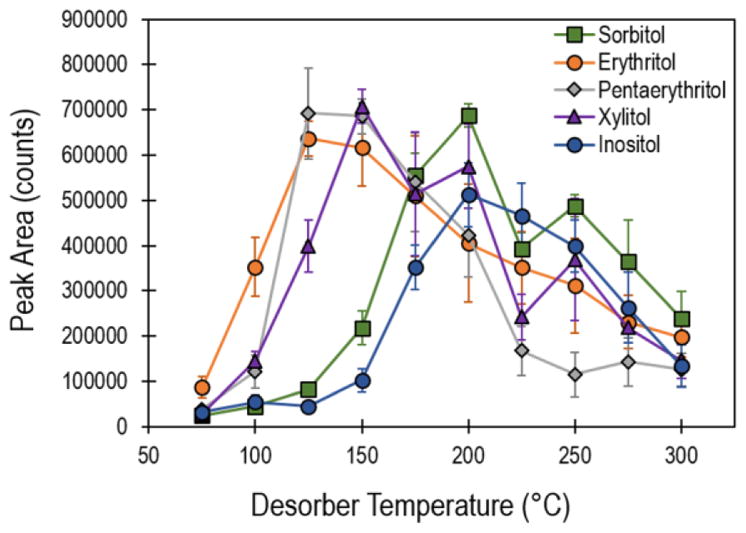
Response (cumulative peak area of base peak) of representative sugar alcohols as a function of desorber temperature. Data points and uncertainty represent the average and standard deviation of 10 replicate samples.
Figure 3.

Representative IMS spectra for (a) erythritol and (b) inositol displaying signal as a function of drift time and scan number. (c) represents the signal intensity at the drift times for erythritol and inositol as a function of scan number for the IMS spectra in (a) and (b). Measurements were taken for base case instrument parameters. Relative signal intensity increases from dark blue to red color.
Scan Time/Scan Number
The vapor pressure of these compounds varied widely, corresponding to variability in the temporal delay observed for the optimal desorption of material (Figure 3). The number of independent scans (and thus total sample analysis time) was shortened and extended to investigate signal improvements and optimization. Generally, the strongest signal was observed as the analyte desorbs/vaporizes off the swipe in the first few seconds of scanning (40 scans in a 20 s sampling period), followed by a rapid or gradual decay with time depending on the analyte properties. For example, erythritol and inositol display very different vapor pressure and volatilities, which led to observed differences in their desorption profiles (Figure 3). Erythritol, the more volatile of these two example compounds, rapidly reached maximum signal (in ≈2 s (scan no. 4)) that then quickly decayed (Figure 3(a) and 3(c)). Because of its rapid desorption, shorter sampling times would be appropriate for volatile compounds, such as erythritol, at elevated temperatures. Alternatively, the lower volatility inositol displayed a more gradual signal rise to maximum (in ≈5 s), achieving a broad (in time) high intensity period from approximately 3 s to 8 s, followed by a steady decay (Figure 3(b) and 3(c)). In a low volatility case such as this, increased sampling time led to an enhancement in IMS response based on cumulative signal (e.g., area under the curve in Figure 3(c)). However, these compounds would demonstrate comparable detection of alarm algorithms based on maximum signal intensity across a scan period (Figure 3(c)).
As demonstrated, both the desorber temperature and sampling time played significant and coupled roles in the IMS response. Figure 4 displays the IMS response as a function of the desorber temperature and scan number (40 scans in a 20 s sampling period) for erythritol and inositol. This visual representation of the data clearly demonstrates the coupling of desorber temperature and scan number, identifying differences in optimal parameters for compounds with differing chemical properties. The trade-off between increasing the scan time or increasing desorption temperature was deduced from these plots. As demonstrated in Figure 2, erythritol experienced optimal IMS response for lower desorption temperatures. Figure 4 further demonstrates that as the desorption temperature was increased, shorter sampling times were appropriate. This contrasted noticeably with inositol, which displayed very poor performance at low desorption temperatures and benefited from longer sampling times at its optimal temperature. This configuration can allow the appropriate combination of optimal scan time and desorption temperature for a specific compound to be chosen, which could further enhance the sensitivity of the instrument for that compound. As might be expected based on chemical properties, the compounds with lower vapor pressures benefited from elevated temperatures and increased scan times.
Figure 4.
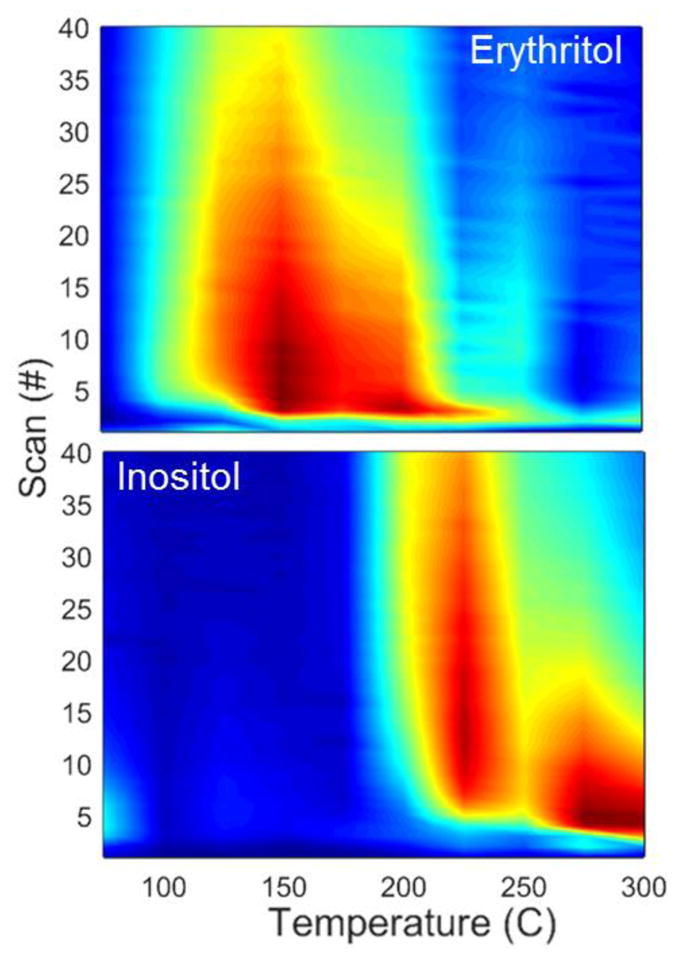
IMS response as a function of desorber temperature and scan number (directly related to sampling time) for representative erythritol and inositol data. Relative signal intensity increases from dark blue to red color. Maximum intensities (dark red) correspond to 2.17 × 104 counts per scan for erythritol and 1.78 × 104 counts per scan for inositol.
Swipe Material
In addition to desorption temperature and sampling time, swipe material was investigated for improving sensitivity for sugar alcohols. A number of commercial swab/swipe materials that exist on the market and have been previously investigated for collection efficiency of particles 41 and reuse robustness 42 were considered to evaluate this effect. The response of select sugar alcohols (erythritol, pentaerythritol, xylitol, sorbitol, and inositol) was characterized for Nomex, muslin, and PTFE-coated fiberglass weave swipe materials under base case system parameters. Figure 5 displays the IMS response for 10 replicates of each sugar alcohol. The results clearly demonstrate superior performance was achieved by Nomex and the PTFE-coated fiberglass weave substrates over the muslin. The absorbent nature of the thicker muslin material may have resulted in a gradient of inkjet printed material wicked into the fabric, reducing the efficiency of the thermal desorption step. Alternatively, the hydrophobic nature of the PTFE-coated fiberglass weave likely led to discrete surface distributions of the analytes, in turn leading to efficient desorption. However, the topographical nature of the weave (interlacing 500 μm threads – see literature for scanning electron microscope images 42 ) may have been the driving factor behind the large standard deviations for this substrate. Analytes may be on an elevated weave peak or depressed trough, thereby changing the surrounding material (air vs PTFE-coated fiberglass) and thermal conductivity properties. In addition, the thermal properties of the swab substrate material affected the thermal desorption process. 21 The rate at which the substrate heated directly affected the desorption process. As demonstrated above, for the base case system parameters, the volatile compounds (erythritol, pentaerythritol, xylitol) desorbed rapidly and had a very short (in time) signal duration on Nomex swabs. The thicker (larger thermal mass) and lower thermal conductivity PTFE-coated fiberglass weave substrates heated much slower (thermal conductivity of fiberglass is approximately an order of magnitude lower than Nomex), increasing the signal duration for these compounds, thereby increasing the cumulative signal (Figure 5). However, this thermal effect also resulted in sub-optimal thermal desorption of the low(er) volatility compounds – inositol and sorbitol.
Figure 5.
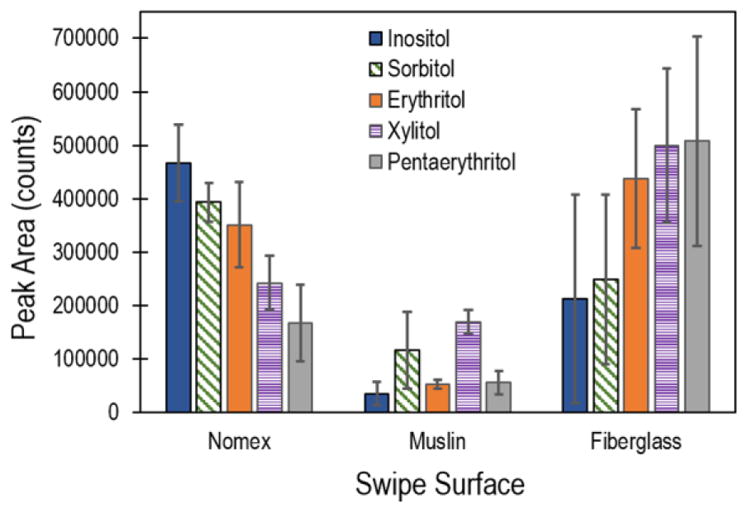
IMS response of select sugar alcohols as a function of swipe material – Nomex, PTFE-coated fiberglass, and muslin. Data points and uncertainty represent the average and standard deviation of 10 replicate samples.
Complex Mixtures
Finally, the detection and identification of sugar alcohols from commercial products was demonstrated. First, a stick of 5® sugar-free gum (spearmint rain, Wm. Wrigley Jr Company, Peoria, IL, USA) was swiped with a Nomex swab in a single continuous motion. Figure 6(a) displays the IMS spectrum obtained under the base case system parameters. The nutrition facts list the following ingredients: sorbitol, gum base, glycerol, and natural and artificial flavors. IMS clearly detected the sorbitol sugar alcohol sweetener (13.2 ms), the principal ingredient. In addition, the peaks observed at 8.4 ms and 10.5 ms were attributed to the reactant ion and glycerol, respectively. Gum base components of chewing gum vary by brand and may include elastomers, resins, waxes, fats, and emulsifiers as a delivery system for the sweeteners and flavors. A peak at 16.8 ms was also observed, which may be attributed to gum base components, however IMS-MS capabilities would be required for proper identification. Next, a single crystal of the natural sweetener, Truvia® (Cargill, Inc., Minneapolis, MN, USA) was collected on a Nomex swab and analyzed (Figure 6(b)). Truvia’s® natural sweetness is extracted from the stevia leaf, however, erythritol remains the main component/ingredient (0.857 g erythritol per 1 g Truvia®) used to disperse and dilute the strong stevia sweetness (200× sweeter than sugar). The IMS spectra for Truvia® was dominated by the signal for erythritol (11.4 ms), with a very minor peak for the reactant ion. Samples were also extracted into solvents (acetone and water) and solution deposited onto swabs for analysis, demonstrating analogous IMS spectra.
Figure 6.
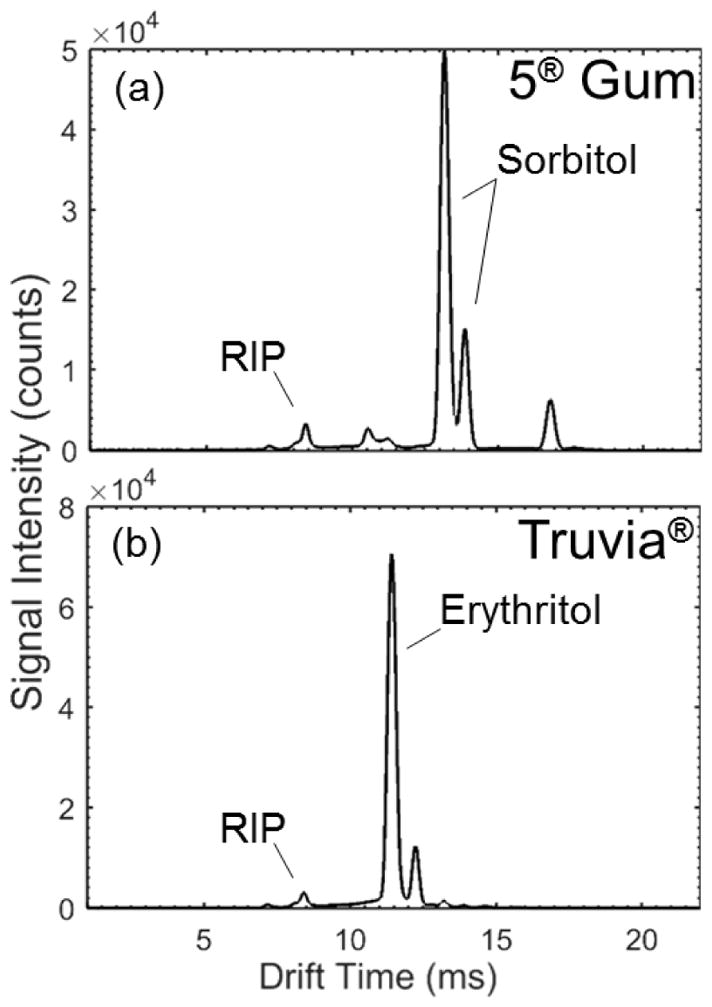
Representative IMS spectra (cumulative across sampling time) for commercial products containing sugar alcohols: (a) 5® gum and (b) Truvia® natural sweetener.
Conclusions
The demand for zero or low-calorie artificial and/or alternative sweeteners has drastically increased for their use in a wide range of commercial products. One category of alternative sweeteners, sugar alcohols, are produced in excess of 1 million metric tons annually. Here, we employed ion mobility spectrometry (IMS) as a low cost, rapid, sensitive, and field deployable alternative for quality assurance/quality control detection and identification of sugar alcohols. Nanogram to sub-nanogram limits of detection were demonstrated for a range of sugar alcohols, including erythritol, pentaerythritol, xylitol, inositol, and sorbitol. System optimization was investigated as a function of desorption temperature, scan time, and swipe material. These investigations identified the physicochemical properties and desorption characteristics of each sugar alcohol as the dominant factor in IMS performance. Overall, volatility – qualitatively encompassing intermolecular forces, sublimation and melting rates, and surface adsorption – dictated the rate of desorption and signal duration, which led to control over optimal operation through desorption temperature and scan time parameters. Sugar alcohols were also directly detected from commercial products demonstrating the ability of IMS to provide a sensitive point-of-measurement tool for the Food and Nutrition industry.
Acknowledgments
The authors thank Jeff Lawrence of the National Institute of Standards and Technology for inkjet printing the samples for this study. The NIST SURF program provided support for CAB.
Footnotes
Official contribution of the National Institute of Standards and Technology; not subject to copyright in the United States
References
- 1.Gardner C, Wylie-Rosett J, Gidding SS, Steffen LM, Johnson RK, Reader D, Lichtenstein AH. Diabetes Care. 2012;35:1798–1808. doi: 10.2337/dc12-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deis RC. Cereal Foods World. 2000;45:418–421. [Google Scholar]
- 3.Fedor WS, Millar J, Accola AJ. Industrial & Engineering Chemistry. 1960;52:282–286. [Google Scholar]
- 4.Tomasik P. Chemical and Functional Properties of Food Saccharides. CRC Press; 2003. [Google Scholar]
- 5.Tundo P, Perosa A, Zecchini F. Methods and Reagants for Green Chemistry: An Introduction. John Wiley & Sons, Inc; 2007. [Google Scholar]
- 6.Dorne JLCM, Dorne JLCM, Bordajandi LR, Amzal B, Ferrari P, Verger P. TrAC Trends in Analytical Chemistry. 2009;28:695–707. [Google Scholar]
- 7.Yang D-j, Chen B. Journal of Agricultural and Food Chemistry. 2009;57:3022–3027. doi: 10.1021/jf803988u. [DOI] [PubMed] [Google Scholar]
- 8.Buchgraber M, Wasik A. Journal of AOAC International. 2008;92:208–222. [PubMed] [Google Scholar]
- 9.Chen QC, Wang J. Journal of Chromatography A. 2001;937:57–64. doi: 10.1016/s0021-9673(01)01306-1. [DOI] [PubMed] [Google Scholar]
- 10.Wasik A, McCourt J, Buchgraber M. Journal of Chromatography A. 2007;1157:187–196. doi: 10.1016/j.chroma.2007.04.068. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Guo Y, Ye M, James FS. Journal of Chromatography A. 2005;1085:143–146. doi: 10.1016/j.chroma.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 12.Musto CJ, Lim SH, Suslick KS. Analytical Chemistry. 2009;81:6526–6533. doi: 10.1021/ac901019g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim SH, Musto CJ, Park E, Zhong W, Suslick KS. Organic letters. 2008;10:4405–4408. doi: 10.1021/ol801459k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filho JC, Santini AO, Nasser ALM, Pezza HR, Eduardo de Oliveira J, Melios CB, Pezza L. Food Chemistry. 2003;83:297–301. [Google Scholar]
- 15.Nikolelis DP, Pantoulias S, Krull UJ, Zeng J. Electrochimica Acta. 2001;46:1025–1031. [Google Scholar]
- 16.Zhang Y, He Z, Li G. Talanta. 2010;81:591–596. doi: 10.1016/j.talanta.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 17.Li G, Jiang T, Zhang X, Wang Q, Li G. A new fluorescent vesicular sensor for saccharides based on boronic acid-diol recognition on the interfaces of vesicles. 2007 [Google Scholar]
- 18.Capitán-Vallvey LF, Valencia MC, Nicolás EA. Food Additives & Contaminants. 2004;21:32–41. doi: 10.1080/0265203032000158283. [DOI] [PubMed] [Google Scholar]
- 19.Capitán-Vallvey LF, Valencia MC, Arana Nicolás E, García-Jiménez JF. Analytical and Bioanalytical Chemistry. 2006;385:385–391. doi: 10.1007/s00216-006-0346-0. [DOI] [PubMed] [Google Scholar]
- 20.Niu W, Kong H, Wang H, Zhang Y, Zhang S, Zhang X. Analytical and Bioanalytical Chemistry. 2011;402:389–395. doi: 10.1007/s00216-011-5305-8. [DOI] [PubMed] [Google Scholar]
- 21.Sisco E, Forbes TP. Analyst. 2015;140:2785–2796. doi: 10.1039/c4an02347a. [DOI] [PubMed] [Google Scholar]
- 22.Nielen MWF, Hooijerink H, Zomer P, Mol JGJ. TrAC Trends in Analytical Chemistry. 2011;30:165–180. [Google Scholar]
- 23.Hajslova J, Cajka T, Vaclavik L. TrAC Trends in Analytical Chemistry. 2011;30:204–218. [Google Scholar]
- 24.Eiceman GA, Karpas Z, Hill HH., Jr . Ion Mobility Spectrometry. CRC Press; Boca Raton, FL: 2013. [Google Scholar]
- 25.García-Cañas V, Simó C, Herrero M, Ibáñez E, Cifuentes A. Analytical Chemistry. 2012;84:10150–10159. doi: 10.1021/ac301680q. [DOI] [PubMed] [Google Scholar]
- 26.Karpas Z. Food Research International. 2013;54:1146–1151. [Google Scholar]
- 27.Midey AJ, Camacho A, Sampathkumaran J, Krueger CA, Osgood MA, Wu C. Analytica Chimica Acta. 2013;804:197–206. doi: 10.1016/j.aca.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Fetterolf DD. Advances in Analysis and Detection of Explosives: Proceedings of the 4th International Symposium on Analysis and Detection of Explosives. In: Yinon J, editor. September 7–10, 1992; Jerusalem, Israel. Netherlands, Dordrecht: Springer; 1993. pp. 117–131. [Google Scholar]
- 29.Ewing RG, Atkinson DA, Eiceman GA, Ewing GJ. Talanta. 2001;54:515–529. doi: 10.1016/s0039-9140(00)00565-8. [DOI] [PubMed] [Google Scholar]
- 30.Windsor E, Najarro M, Bloom A, Benner B, Fletcher R, Lareau R, Gillen G. Analytical Chemistry. 2010;82:8519–8524. doi: 10.1021/ac101439r. [DOI] [PubMed] [Google Scholar]
- 31.Verkouteren RM, Verkouteren JR. Analytical Chemistry. 2009;81:8577–8584. doi: 10.1021/ac901563j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verkouteren RM, Verkouteren JR. Langmuir. 2011;27:9644–9653. doi: 10.1021/la201728f. [DOI] [PubMed] [Google Scholar]
- 33.Verkouteren JR, Lawrence J, Klouda GA, Najarro M, Grandner J, Verkouteren RM, York SJ. Analyst. 2014;139:5488–5498. doi: 10.1039/c4an01184h. [DOI] [PubMed] [Google Scholar]
- 34.Eiceman GA, Nazarov EG, Stone JA. Analytica Chimica Acta. 2003;493:185–194. [Google Scholar]
- 35.Kaur-Atwal G, O’Connor G, Aksenov AA, Bocos-Bintintan V, Paul Thomas CL, Creaser CS. Int J Ion Mobil Spec. 2009;12:1–14. [Google Scholar]
- 36.Kozole J, Stairs JR, Cho I, Harper JD, Lukow SR, Lareau RT, DeBono R, Kuja F. Analytical Chemistry. 2011;83:8596–8603. doi: 10.1021/ac201999a. [DOI] [PubMed] [Google Scholar]
- 37.Desai BB. Handbook of nutrition and diet. Marcel Dekker, Inc; New York: 2000. [Google Scholar]
- 38. [Accessed 3/30/2016];Calorie Control Council - Polyols. 2016 www.polyols.org.
- 39.Currie LA. Analytica Chimica Acta. 1999;391:127–134. [Google Scholar]
- 40.ASTM E2677 Limit of Detection Web Portal. 2016 https://www-s.nist.gov/loda/
- 41.Verkouteren JR, Coleman JL, Fletcher RA, Smith WJ, Klouda GA, Gillen G. Measurement Science and Technology. 2008;19:115101. [Google Scholar]
- 42.Staymates JL, Staymates ME, Lawrence J. Int J Ion Mobil Spec. 2015;19:41–49. doi: 10.1007/s12127-015-0185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]