

Abstract
A comprehensive review on the chemistry of Spongia sp. is here presented, together with the biological activity of the isolated compounds. The compounds are grouped in sesquiterpene quinones, diterpenes, C21 and other linear furanoterpenes, sesterterpenes, sterols (including secosterols), macrolides and miscellaneous compounds. Among other reports we include studies on the intraspecific diversity of a Mediterranean species, compounds isolated from associated sponge and nudibranch and compounds isolated from S. zimocca and the red seaweed Laurentia microcladia. Under biological activity a table of the reported biological activities of the various compounds and the biological screening of extracts are described. The present review covers the literature from 1971 to 2015.
Keywords: Spongia sp., sesquiterpene quinones, diterpenes, C21 furanoterpenes, sesterterpenes, sterols, macrolides, biological activity
1. Introduction
Marine sponges have been considered as a very remarkable field for the discovery of bioactive natural products, being so far the most studied source of marine natural products [1]. Some of these metabolites contribute to the chemical defense against predation in their habitat, overgrowth by fouling organisms or competition for space. Moreover, many of them have been found to possess multiple biological activities, such as antitumor, antiviral, anti-inflammatory, immunosuppressive and antibiotic, among others ([1] and previous reviews, [2]). The genus Spongia, Linnaeus 1759, belongs to the family Spongidae of the order Dictyoceratida. It comprises three subgenus, Australospongia, Heterofibria and Spongia, containing 1, 7 and 81 species, respectively, according to “The world Porifera database” “and the WoRMS (World Register of Marine Species)”. Knowledge of the softness, elasticity and water retention capacity has rendered some of the species of Spongia genus useful as bath sponges [3,4]. As a result of overfishing, habitat degradation and spread of diseases, one of them, S. agaricina, is now considered an endangered species under Annex III of the Bern and Barcelona conventions [5]. It is worth mentioning that the nomenclature S. agaricina Pallas 1766 has been proposed to refer only to Philippine specimens while the Mediterranean ones should be better referred to as S. lamella Schultze 1879 [6]. An interesting study on the potential use of three Spongia sp., specially S. agaricina, as precursors in the production of ceramic based tissue engineered bone scaffolds has been recently published [7].
Many reports on the chemistry of Spongia sp. have been published since 1971 and the work of Fattorusso et al. [8] on the C21 furanoterpenes of S. nitens, the first report on the chemistry of Spongia sp. The C21 furanoterpenes, together with spongian diterpenes and scalarane sesterterpenoids, are one of the more abundant metabolite structures of this genus. Other metabolites comprise sesquiterpene quinones (mainly with a rearranged drimane skeleton), sterols and secosterols (mainly of the 5α-cholest-7-en and 5α-hydroxy-cholest-7-en type), and macrolides. A section with reports on the isolation of previously unreported compounds and the biological activity for each of these metabolite classes is presented including at the end reports on X-ray structures, reports on the isolation of known compounds and isolated biological activity studies (other studies). A description of the structure assignment is only given for new compounds, since the known metabolites were identified, in most cases, by comparison with literature data. Under Other reports we include studies on the intraspecific diversity of a Mediterranean species, the compounds isolated from associated sponge and nudibranchs (which are believed to sequester sponge compounds) and geographically co-occurring sponge and seaweed (where the opposite occurs). A section on biological activity summarizing the described biological activities of the compounds and the biological screening of extracts is also provided at the end of the chapter. This review covers the literature from 1971 to 2015.
2. Sesquiterpene Quinones
Urban and Capon [9] reported the isolation of 5-epi-isospongiaquinone 1, together with 5-epi-homoisospongiaquinone 2 (Figure 1), a possible artifact of isolation procedures, from S. hispida, collected in the south western coast of Australia.
Figure 1.
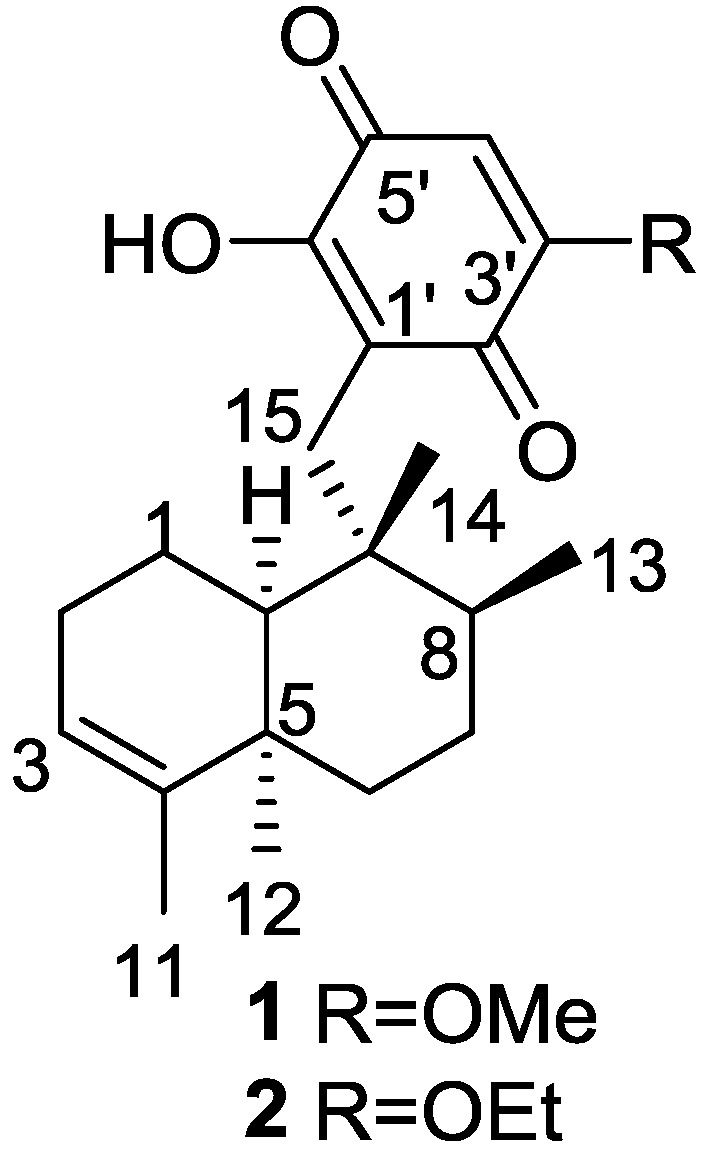
Structures of 5-epi-isospongiaquinone 1 and 5-epi-homoisospongiaquinone 2.
Compound 1 was identified by comparison with the known isospongiaquinone, the C-5 epimer. The fact that the 13C NMR (CDCl3) resonance of C-12 (32.3 ppm) was deshielded compared to that of isospongiaquinone (19.9 ppm) was interpreted by the authors as being diagnostic of a cis, rather than a trans ring junction. Further evidence came from the acid-catalyzed rearrangement of 1 that gave two compounds in all respect identical with the ones obtained from isospongiaquinone. Both 1 and 2 showed antibiotic activity against Staphylococcus aureus (MIC 20 μg/disk and 50 μg/disk, respectively) and Micrococcus sp. (MIC 20 μg/disk and 50 μg/disk, respectively).
Subsequent studies by Capon et al. [10] led to the isolation of the new 3, together with the known dehydrocyclospongiaquinone-1 4 and spongiaquinone 5 from a Spongia sp. collected in the Great Australian Bight (Figure 2). 5 was also isolated as a potassium salt.
Figure 2.

Structures of compound 3, dehydrocyclospongiaquinone-1 4 and spongiaquinone 5.
Compound 3 was identified by 1H and 13C NMR after methylation and comparison with known compounds. For spongiaquinone 5 the previously assigned E configuration of the double bond was confirmed by nOe, and the depicted absolute stereochemistry was established by chemical degradation. X-ray fluorescence spectroscopy confirmed potassium as the main counter ion (sodium was present in trace amounts). The isolated compounds proved to be responsible for the antibiotic activity of the extract against a range of test microorganisms.
From a Spongia sp. collected in Australia, the isolation of the unusual cyclosmenospongine 6 was reported by Utkina et al. [11], together with the already known metabolites smenospongiarine 7, ilimaquinone 8 and smenospongine 9 (Figure 3). It is worth mentioning that ilimaquinone had its structure revised in 1987 [12]. Since the absolute configuration of smenospongine 9 was established by comparison of CD spectra with ilimaquinone 8 [13], the structure here presented is also corrected.
Figure 3.

Structures of cyclosmenospongine 6, smenospongiarine 7, ilimaquinone 8 and smenospongine 9.
For cyclosmenospongine 6 a rearranged drimane skeleton was proposed in the basis of 1H and 13C NMR data, together with the mass spectra fragment at m/z 191. UV and IR spectra indicated the presence of a 1,4-benzoquinone. The bathochromic shift of the absorption maxima observed in the UV spectra together with IR bands confirmed the presence of an amino substituent. A quaternary carbon at δ 88.6 ppm and an IR band at 1244 cm−1 confirmed the presence of an ether linkage. Analysis of 1H–1H COSY, HMQC and HMBC allowed the confirmation of the proposed structure. The relative stereochemistry was ascertained by nOe experiments where irradiation of Me-14 resulted in nOe to H-5 and Me-13. The absolute stereochemistry of 6 was subsequently determined as 5R,8S,9R,10S by chemical correlation [14]. Cyclosmenospongine 6 showed moderate cytotoxic activity against mouse Ehrlich carcinoma cells (IC100 145 μM) and moderate hemolytic activity, inducing 50% hemolysis of mice blood erythrocytes at a concentration of 70 μM in 10 min.
Cao et al. [15] reported the isolation of the new 17-O-isoprenyldictyoceratin-C 10, together with the known dictyoceratin-C 11 (Figure 4), ilimaquinone 8, and a nucleoside 2′-deoxyuridine, from a bioactive extract of Spongia sp. collected in the Philippines. The extract showed inhibitory activity toward the lyase activity of DNA polymerase β at 16.2 μg/mL.
Figure 4.
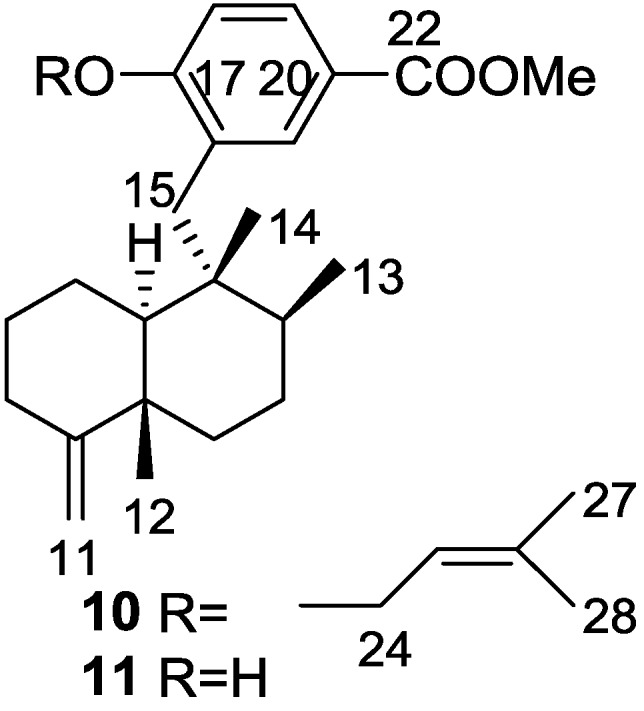
Structures of 17-O-isoprenyldictyoceratin-C 10 and dictyoceratin-C 11.
The structures of 11 and 8 were identified by comparison with literature data and 2′-deoxyuridine with an authentic sample. For 10, the rearranged drimane skeleton was established by the typical 1H NMR signals. The NMR spectra also showed the presence of an 1,2,4-trisubstituted benzene ring (confirmed by UV), and a carbomethoxy moiety (confirmed by IR). The prenyloxy group was identified by the characteristic 1H NMR signals. Comparison of the data with that of 11 confirmed 10 as a O-prenylated derivative, whose location was confirmed by ROESY (correlation from H-24 to H-18). Stereochemistry was assumed on the basis of the value of the optical rotation of both compounds. Further evidence came from conversion of 11 to 10. The absolute stereochemistry was not determined although its co-occurrence with ilimaquinone 8 supports the depicted structure. Purified compounds were used to determine the IC50 values for inhibition of lyase activity of rat DNA polymerase β, as well as for cytotoxicity to A2780 ovarian cancer cells and inhibitory activity toward Cdc25B. Compounds 10, 11 and the nucleoside were inactive in all three assays. 8 showed a IC50 of 45.2 μM as inhibitor of lyase activity of DNA polymerase β. It was also weakly active as an inhibitor of Cdc25B, with an IC50 of 92 μM, and showed moderate toxicity to A2780 cells with an IC50 of 10.9 μM.
Work of Takahashi et al. [16,17,18] allowed the identification of several new metachromins, metachromins J 12 and K 13, L–T, 14–22, together with the known metachromins A 23, C–E 24–26, from a Spongia sp. collected in Okinawa (Figure 5).
Figure 5.

Structures of metachromins J 12 and K 13, L–T, 14–22, A 23, and C–E 24–26.
For metachromin J 12 IR and UV allowed the identification of the carbonyl group and quinone moiety. Comparison of the spectral data with that of metachromins C 24 and E 26 allowed the determination of the proposed structure. NOESY correlations between H-1/Me-13, H-2/Me-14 and H-5/Me-14 allowed the determination of the relative stereochemistry of Me-13 and Me-14 and of a pseudochair conformation for the cyclohexene ring. For metachromin K 13 the hydroxyl group and aromatic ring were identified by IR and UV, respectively. Comparison of the spectral data with that of metachromin C 24, metachromin D 25 and metachromin J 12 allowed the determination of the structure. Both compounds showed weak cytotoxicity against murine lymphoma L1210 cells (IC50 1.0 and 12.6 μg/mL, respectively) and human epidermoid carcinoma KB cells (IC50 9.9 and >20 μg/mL, respectively) in vitro. For metachromin L 14 the presence of OH and/or NH and carboxy groups was established by IR data. A conjugated carbonyl functionality was also present and the UV spectrum suggested the presence of a quinone chromophore. Similarity of the overall NMR data to metachromin A 23 together with the signals corresponding to a glycine residue led to the assignment. Further confirmation was obtained by chemical synthesis of 14 from 23. Comparison of the NMR data of metachromins N 16 and P 18 with that of metachromin L 14 led to the assignment of the former. Again, confirmation of the proposed structures came from their synthesis from metachromin A 23. Metachromins M 15, O 17 and Q 19 were assigned analogously by comparison of the NMR data with that of metachromin C 24. Synthesis from this latter compound confirmed the assigned structures. The structure of metachromin R 20 was assigned by IR, UV and NMR data (including 1H–1H COSY, TOCSY and HMBC). Comparison with the known metachromin G showed that the main differences were the presence of a substituted double bond bearing a methyl group in place of an exomethylene. A phenethylamine unit was inferred from NMR and its connectivity was established by HMBC. The relative stereochemistry of the cyclohexane ring was established by NOESY where cross peaks between H-2β and Me-14 and H-1α and Me-13 were observed. Comparison of the NMR data of metachromin S 21 with that of metachromin R 20, allowed the assignment of the depicted structure. NOESY revealed that the stereochemistry was the same for both compounds. For metachromin T 22 analysis and comparison of the IR, UV and NMR data (including 1H–1H COSY, TOCSY and HMBC) with that of metachromin B allowed the identification of a 6,8-dimethoxy-2-methyl-2Hchromen-5-ol moiety, confirmed by HMBC. Further NMR analysis allowed the identification of the remaining structure, indicating that 22 possessed a cyclohexane ring identical to 20 and 21. The NOESY spectra of 22 indicated that the stereochemistry of the cyclohexane moiety was the same. The absolute configuration at C-9 was deduced as S from CD spectra. For all three compounds 20–22 the absolute stereochemistry at C-5 and C-6 was tentatively assigned as S and R, respectively, since they can be considered to be generated through the same biosynthetic path as metachromin A 23, whose C-6 configuration is R. Metachromins L 14, M 15, S 21 and T 22 showed toxicity against L1210 (IC50 4.0, 3.5, 5.2 and 3.0 μg/mL, respectively) and KB cells (IC50 4.0, 5.4, >10 and 5.6 μg/mL, respectively) in vitro, while metachromins N–Q and R, 16–19 and 20 did not show that activity (IC50 > 10 μg/mL). In a subsequent study [19] metachromin L 14 showed inhibitory activity of EGFR (epidermal growth factor receptor) kinase (IC50 197 μg/mL) and metachromins L–Q, 14–19, showed inhibitory activity of HER2 (human epidermal growth factor receptor 2) kinase (IC50 125, 79, 190, 27, 18 and 22 μg/mL, respectively).
Further investigation of another lot of the same sponge by Takahashi et al. [20] afforded the new dimeric sesquiterpenoid quinones, nakijiquinone E 27 and F 28, together with the known dictyoceratins A–C, 29, 30 and 11, isospongiaquinone 31, 6′-hydroxy-4′-methoxyavarone 32, neoavarol 33, nakijiquinones A–D 34–37, and an endo olefin isomer at C-3 of smenospongine 38 (Figure 6).
Figure 6.

Structures of nakijiquinone E 27 and F 28, dictyoceratins A–C, 29, 30, isospongiaquinone 31, 6′-hydroxy-4′-methoxyavarone 32, neoavarol 33, nakijiquinones A–D 34–37, and an endo olefin isomer at C-3 of smenospongine 38.
For 27 IR data implied the presence of OH and/or NH, carboxy and conjugated carbonyl functionalities. UV suggested the presence of the quinone chromophore. HRESIMS and 1H and 13C NMR data suggested a dimeric sesquiterpenoid quinone. Further analysis of NMR spectra, including 1H–1H COSY and HMBC, identified a tetramethyl decalin with an endo olefin, a trimethyl decalin with an exomethylene, a 2-amino-5-hydroxy-benzoquinone and a methyl 3-amino-2, 4-dihydroxybenzoate. Connection of these moieties was confirmed by HMBC. The relative stereochemistry of the two decaline was established by NOESY. The α-configuration of H-10 and β-configurations of Me-12, Me-13 and Me-14 were deduced from the correlations H-8/H-10, H-10/CH2-15 and Me-12/Me-14. Correlations H-8′/H-10′, H-10′/CH2-15′ and Me-12′/Me-14′ revealed the same orientation for H-10′ and Me-12′, Me-13′ and Me-14′. 28 possessed similar spectral data to that of 27 the difference being the absence of the exomethylene; NMR analysis established its structure. The relative stereochemistry was assigned by NOESY. Both compounds did not show cytotoxicity against P388 and L1210, and KB cells (IC50 > 10 μg/mL).
Other Studies
Utkina and Denisenko [21] reported the isolation of the already known smenoquinone 39 (Figure 7), together with smenospongiarine 7 and ilimaquinone 8, from a Spongia sp. collected in the Vietnam sea.
Figure 7.
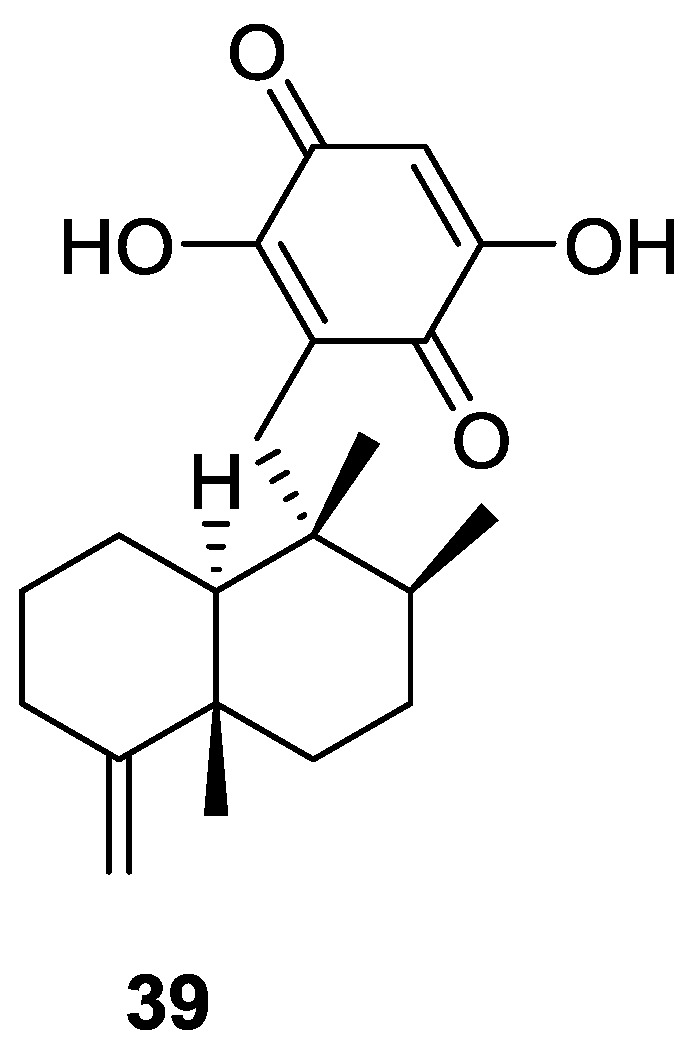
Structure of smenoquinone 39.
The antioxidant activity of 39 was tested using bleaching of solutions of DPPH radical (2,2-diphenyl-1-picrylhydrazyl radical) and ABTS•+ ([2.22-azinobis(3-ethylbenzothiazolin-6-sulfonic acid)]). Compound 39 showed moderate activity from trapping DPPH radicals (IC50 3.7 × 10−4 M, comparable to that of ionol, IC50 3.6 × 10−4 M). The antioxidant activity for reduction of ABTS•+ radical cations corresponded to 0.15 mmol/L of trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) equivalents. Compounds 7 and 8 were inactive.
An independent study by Kittiwisut et al. [22] investigated the antiproliferative activity of several sesquiterpene quinones in a SRB assay. Ilimaquinone 8 showed an IC50 of 7.6 μM against HeLa cells and initiated toxicity, in addition to the already mentioned activity.
3. Diterpenes
Cimino et al. [23] reported the isolation of the first spongian diterpene, isoagatholactone 40 (Figure 8) from a S. officinalis collected in Naples.
Figure 8.
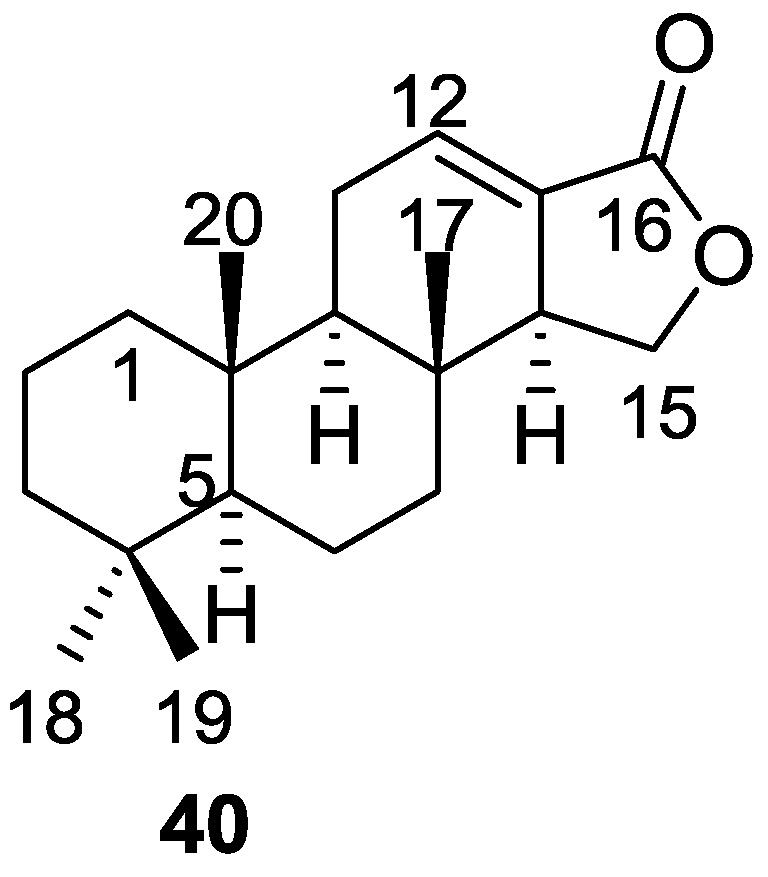
Structure of isoagatholactone 40.
The UV and IR absorptions indicated the presence of a α,β-exounsaturated γ-lactone. 1H NMR showed the presence of four tertiary methyls, an olefinic hydrogen coupled with an allylic methylene and an allylic methine (confirmed by decoupling experiments), and an oxygenated methylene coupled to the allylic methine (confirmed by decoupling experiments). MS data where the base peak at m/z 192 originating from a retro Diels-Alder process was observed, confirmed the position of the double bond in ring C. Chemical correlation with grindelic acid confirmed the structure and the depicted stereochemistry.
Capelle et al. [24] reported the isolation of the new spongia-13(16),14-dien-19-oic acid 41, spongia-13(16),14-dien-19-al 42 and spongia-13(16),14-diene 43 from a S. officinalis collected in Papua-New Guinea (Figure 9).
Figure 9.
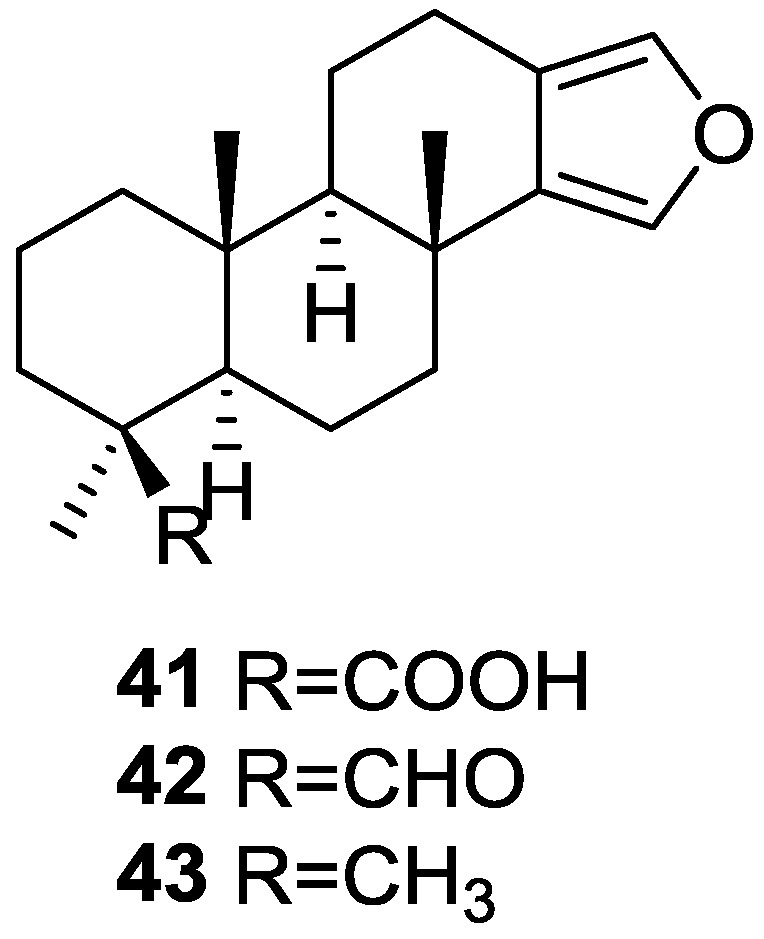
Structures of spongia-13(16),14-dien-19-oic acid 41, spongia-13(16),14-dien-19-al 42 and spongia-13(16),14-diene 43.
UV, IR, 1H NMR and 13C NMR data of 41 allowed the identification of a β,β-disubstituted furan moiety, a carboxylic acid and three tertiary methyl groups, compatible with a tetracyclic diterpene with a furan ring (confirmed by comparison with literature compounds). Location of the COOH group was established by pyridine induced shifts in 1H NMR. The spectral data of 42 was very similar to that of 41, with the characteristic signals of the COOH giving rise to an aldehyde function. Reduction of both compounds to the corresponding alcohol confirmed the assignment. For 43 the appearance of a fourth methyl group and absence of the COOH or CHO functions revealed its structure. Confirmation came from chemical correlation with the alcohol obtained by reduction of 41 and 42.
Cimino et al. [25] reported the isolation of the new 15α,16α-diacetoxyspongian 44, ent-isocopal-12-en-15,16-dial 45, 14-iso-ent-isocopal-12-en-15,16-dial 46 and 15-acetoxy-ent-isocopal-12-en-16-al 47 from a S. officinalis (Figure 10).
Figure 10.

Structures of 15α,16α-diacetoxyspongian 44, ent-isocopal-12-en-15,16-dial 45, 14-iso-ent-isocopal-12-en-15,16-dial 46 and 15-acetoxy-ent-isocopal-12-en-16-al 47.
For 44 1H NMR identified the two acetyl groups and corresponding oxygenated methines, and the four tertiary methyls. Comparison of 13C NMR data with the known aplysillin (the 12-acetyl analogous, whose relative stereochemistry has been established by X-ray) confirmed the proposed structure. Comparison of the coupling values for H-15 and H-16 in 1H NMR in both compounds supports the depicted stereochemistry. Further evidence came from transformation of 44 into the corresponding furan derivative (already known in the literature) by heating in benzene in the presence of catalytic amounts of silica gel. For 45 four methyl group resonances in 1H NMR spectra suggested a diterpene skeleton. Other signals include an olefinic proton and two aldehyde groups. Assignment of 13C NMR data was accomplished by comparison with literature compounds. Reduction of 45 afforded the diol of known absolute stereochemistry. NMR data of 46 was very similar to that of 45, pointing to a C-14 epimer. Isomerization of 45 afforded 46, confirming the proposed structure of the latter. For 47 an α,β-unsaturated aldehyde, an ester function and an oxygenated methylene could be identified. Reduction with LAH afforded the corresponding diol, confirming structure and absolute stereochemistry.
Gonzalez et al. [26], from the active methanol extract of S. officinalis L. from Tenerife, reported the isolation of the active 11β-hydroxyspongi-12-en-16-one 48 and 11β-acetoxyspongi-12-en-16-one 49, the inactive and already known isoagatholactone 40 and aplysillin 50 (respectively spongia-12-en-16-one and 12α,15α,16α-triacetoxyspongian), and a mixture of the new 7β,11β-dihydroxyspongi-12-en-16-one 51 and 7β,11α-dihydroxyspongi-12-en-16-one 52 (Figure 11), for which no testing was performed. The extract showed antimicrobial activity against Staphylococcus aureus, Pseudomonas aeruginosa and Bacillus sphaericus, and inhibited HeLa cells with values of ID50 1–5 μg/mL.
Figure 11.

Structures of 11β-hydroxyspongi-12-en-16-one 48, 11β-acetoxyspongi-12-en-16-one 49, aplysillin 50, 7β,11β-dihydroxyspongi-12-en-16-one 51, and 7β,11α-dihydroxyspongi-12-en-16-one 52.
For 48 and 49, a spongian skeleton was inferred from 13C NMR data. The presence of an oxygenated substituent at C-11 was established by chemical transformation: acetylation of 48 gave 49; treatment of 48 with TosCl/Py gave the 9,13-diene as a result of concomitant dehydration; and oxidation of 48 with Jones reagent gave the 11-ketolactone. The stereochemistry at C-11 in 48 and 49 was obtained by 1H NMR analysis of hydrogenated derivatives of 49 (cis and trans ring C/D junction) and confirmed by X-ray analysis of a pyrazine derivative obtained by treatment of 49 with diazomethane in ether. The structures of 51 and 52 are proposed on the basis of 1H NMR spectra analysis of the pyrazine derivatives obtained from the acetylated natural products.
Kohmoto et al. [27] reported the isolation of the new 2α,19-dihydroxyspongia-13(16),14-dien-3-one (isospongiadiol) 53, together with the known 54 (epispongiadiol) and 55 (spongiadiol) from a Spongia sp. collected in the Bahamas (Figure 12). Previous biological screening of the extract showed activity against HSV-1 (herpes simplex virus type 1) and P388 murine leukemia cells.
Figure 12.

Structures of 2α,19-dihydroxyspongia-13(16),14-dien-3-one (isospongiadiol) 53, 54 (epispongiadiol) and 55 (spongiadiol).
Compounds 54 and 55 were identified by comparison with literature data. For 53 1H NMR showed the presence of the furan ring, three methyl singlets and an oxygenated methylene. Comparison of the remaining NMR data with that of 54 and 55 suggested a different oxidation pattern in ring A. Further NMR analysis, including C–H correlations, COSY and nOe established the structure. nOe between H-2 and Me-20, and Me-17 and Me-20 suggested a 1,3 diaxial relationship between these substituents and a chair conformation for ring A. The ring A oxidation pattern and absolute configuration was confirmed by comparison of the 1H NMR spectrum and optical rotation of the reduction products of all three compounds. From in vitro assays against P388 cells 53, 54 and 55 yielded IC50 values of 5, 8, and 0. 5 μg/mL, respectively (the value for vinblastine is 0.01 μg/mL). Against HSV-1 the IC50 values for 53, 54 and 55 were 2, 12.5, and 0.25 μg/mL, respectively (the values for ara-A and acyclovir are 50 and 0.5 μg/mL, respectively).
Hirsch and Cashman [28] reported the isolation of the new spongialactone A 56 and 19-acetoxy-3α-hydroxyspongia-13(16),14-dien-2-one 57 from S. officinalis var. arabica collected in gulf of Eilat, together with the known metabolites 3α-17,19-trihydroxyspongia-13(16),14-dien-2-one 58 and 3β,17,19-trihydroxyspongia-13(16),14-dien-2-one 59 (Figure 13). 57 is the acetate of the already known diol 3α,19-dihydroxyspongia-13(16),14-dien-2-one
Figure 13.

Structures of spongialactone A 56, 19-acetoxy-3α-hydroxyspongia-13(16),14-dien-2-one 57, 3α-17,19-trihydroxyspongia-13(16),14-dien-2-one 58, and 3β,17,19-trihydroxyspongia-13(16),14-dien-2-one 59.
For compounds 57, 58 and 59 comparison with literature data confirmed their structures. Furthermore, hydrolysis of 57 gave the known parent diol. Comparison of the NMR data of 56 with that of 57 indicated that these compounds only differed in ring A. The carbonyl of 57 was replaced with two new carbonyls, one belonging to an acid group (proved by methylation and acetylation), and the other to a lactone ring (proved by analysis of IR data). The confirmation of the proposed structure for 56, together with the stereochemistry at C-4 (Me-18 and COOH-19) and C-5 was obtained by NMR experiments (COSY 45, RELAY and NOEDS). The relative configuration of the remaining chiral centers was established by comparison of the NMR data with reference compounds.
Gunasekera and Schimtz [29] reported the isolation of four new metabolites 2β,3β,17,19-tetrahydroxyspongia-13(16),14-diene 60, 2-oxa-17,19-dihydroxyspongia 13(16),14-dien-3-one 61, 17-hydroxy-4-epi-spongialactone A 62 and 19-nor-3-hydroxyspongia-3,13(16),14-trien-2-one 63 (Figure 14), together with the known 3β,17,19-trihydroxyspongia-13(16),14-dien-2-one 59, from an unidentified Spongia sp., collected in Dalton Reef, Australia.
Figure 14.

Structures of 2β,3β,17,19-tetrahydroxyspongia-13(16),14-diene 60, 2-oxa-17,19-dihydroxyspongia 13(16),14-dien-3-one 61, 17-hydroxy-4-epi-spongialactone A 62, and 19-nor-3-hydroxyspongia-3,13(16),14-trien-2-one 63.
The identification of 59 was accomplished by comparison with literature data, although some comments were made. Confirmation of the β-orientation of C-3-OH (inferred from the 13C shift value) came from irradiation of the signal of H-3 that sharpened one of the signals of H-19, showing that H-3 and the oxymethylene group are diaxially disposed. For the remaining compounds the spongiane skeleton, rings B, C and D, two quaternary methyls and the C-17 substituent were established by comparison of the 1H and 13C NMR data with that of 59. For 60 the absence of the carbonyl in IR suggested a reduction derivative of 59. nOe established the β-orientation of C-17, C-19, Me-20 (irradiation of Me-20) and α-orientation of H-3 (irradiation of Me-18). The latter was confirmed by the upfield shift of α-H-1, excluding a 1,3-diaxial relationship with C-3-OH. The configuration at C-2 was inferred from the J coupling values of H-2. For 61 NMR data analysis and nOe (enhancement of CH2-17, CH2-19 and H-1 upon irradiation of Me-20) led to the proposed structure. The authors suggest the presence of an intramolecular hydrogen bond between the C-19-OH and the carbonyl group, based on the low frequency observed for the latter in IR (1702 cm−1). Compound 62 was purified and identified after esterification with diazomethane and acetylation of C-17-OH. Decoupling experiments and nOe (enhancement of Me-19 and CH2-17 upon irradiation of Me-20, and enhancement of CH2-3 upon irradiation of Me-19) confirmed the structure. For 63, confirmation of the structure came from coupling of the vinyl methyl with H-5. Compound 61 showed marginal cytotoxicity to murine leukemia cells (P388), E50 = 3.5 μg/mL, and the other compounds were inactive.
Searle and Molinski [30] reported, among others, the isolation of 5 new diterpenes, 3β,17-dihydroxyspongia-13(16),14-dien-2-one 64, 3α,17-dihydroxyspongia-13(16),14-dien-2-one 65, 2α,17-dihydroxyspongia-13(16),14-dien-3-one 66, 2β,17-dihydroxyspongia-13(16),14-dien-3-one 67 and 3α-hydroxyspongia-13(16),14-dien-3-one 68 (Figure 15), together with the known spongia-13(16),14-diene 43, from a Spongia sp. collected in Australia.
Figure 15.

Structures of 3β,17-dihydroxyspongia-13(16),14-dien-2-one 64, 3α,17-dihydroxyspongia-13(16),14-dien-2-one 65, 2α,17-dihydroxyspongia-13(16),14-dien-3-one 66, 2β,17-dihydroxyspongia-13(16),14-dien-3-one 67, and 3α-hydroxyspongia-13(16),14-dien-3-one 68.
43 was identified by comparison with literature data. For 64, analysis of NMR data, including COSY, HETCOR and COLOC spectra established an oxidized tetracyclic spongian diterpene skeleton and allowed structural identification. Comparison of the furan 13C chemical shifts with those of reported compounds allowed the hydroxymethylene group to be placed at C-17, supported by COSY and COLOC experiments. A chair conformation for A ring with equatorial C-3-OH and Me-18 was established by NOEDS spectra. NMR analysis of 65 identified it as a C-3 epimer of 64. Strong nOe observed between H-3 and Me-20 established a boat conformation for ring A, with C-3-OH in a pseudo-equatorial “prow” position. NMR analysis of 66 and 67 and comparison with 64 and 65 established their structures and allowed their identification as C-2 epimers. NOESY experiments established the relative configuration of 66 and suggested a chair conformation for ring A, with the C-2-OH in an equatorial position. Both 66 and 67 proved to be rather unstable, which prevented full characterization. For 68, analysis of the NMR data and comparison with 64, 65 and literature compounds established its structure. The absolute configuration of 64 was established as 2S,3R by CD studies of the tribenzoate derivative obtained by esterification of the 2β,3β,17β-triol obtained after reduction of the C-2-carbonyl group. This configuration corresponds to the normal “5α,10β” absolute configuration common to all sterols and most polycyclic diterpenes with the exception of ent kaurenes, and is consistent with the findings for earlier spongian derivatives.
Zubía et al. [31] reported the isolation of four new metabolites 12-deacetyl-aplysillin 69, 15,16-diacetoxy-11-oxo-ent-isocopal-12ene 70, 15-hydroxy-ent-isocopal-12-en-16-al 71, 15,17-diacetoxy-ent-isocopal-12-en-16-al 72, and seven already known structures 40, 47, 49, 44, 73, 46 and 45, from a Mediterranean sponge, S. zimoca, Schmidt 1862, collected in the channel of Sicily (Figure 16).
Figure 16.

Structures of 12-deacetyl-aplysillin 69, 15,16-diacetoxy-11-oxo-ent-isocopal-12ene 70, 15-hydroxy-ent-isocopal-12-en-16-al 71, 15,17-diacetoxy-ent-isocopal-12-en-16-al 72, and compound 73.
The known compounds were identified by comparison with literature data while the new compounds were identified by comparison of the NMR spectra with those of 50, 72 and 47. Acetylation of 69 afforded a compound in all respects identical with 50 (including optical rotation). The absolute stereochemistry at C-12 was ascertained by applying a modified Mosher’s method. Attempts to acetylate 71 to obtain 47 failed, probably because of the existence of an intramolecular hydrogen bond between C-15-OH and the aldehyde group. Alternatively methanolysis of 47 with Na2CO3/MeOH (anhydrous) afforded 71. Analysis and comparison of the NMR data of 72 with that of 47 allowed the identification of the former, where the chemical shift of C-7 was diagnostic to localize an acetoxy group at C-17. Compound 70 was identified by comparison with 72 and NMR data analysis. The multiplicity of the olefinic proton allowed the correct localization of all the functionalities. The relative stereochemistry at C-14 was supported by comparison of the 13C NMR δ value for C-7, similar to that reported for 47. The authors suggest that most probably all the metabolites have the same absolute stereochemistry as 40. This suggestion has been proven for 69 (Mosher’s method) and is supported by the fact that 45, 47 and 72 show CD curves opposite to the curves of known compounds (polydiglyal, scalaradial and 12-deacetoxy-scalaradial) supporting the ent-isocopalane skeletons.
Li et al. [32] reported the isolation of two new metabolites 74 and 75 (Figure 17) together with the previously reported 53, 54, 63 and a furanoterpene, from the sponge S. matamata de Laubenfels, 1954, collected in Yap island, Micronesia. This specimen was later reclassified as S. zimocca sensu de Laubenfels by the same authors in a subsequent study [33].
Figure 17.
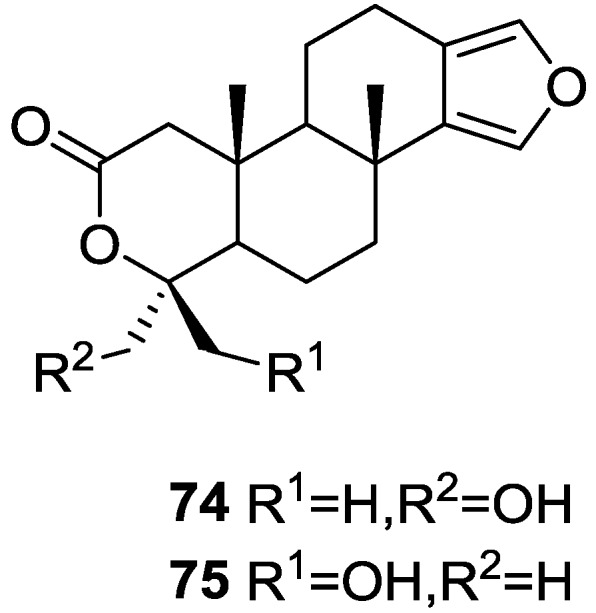
Strutures of furanoterpenes 74 and 75.
The known compounds were identified by comparison of the spectral data with the literature. The new compounds were identified by 1H, 13C, HMQC, HMBC and nOe experiments and comparison with 54, 53 and 63, that showed that they only differed in ring A. The A/B ring trans fusion of 74 was established by comparison of the 13C NMR δ value of Me-20 with those of 54 and 53 and by the coupling constant of H-5. The α-configuration of the oxymethylene was determined on the basis of the nOe enhancement of the oxymethylene proton signals after irradiation of H-5. The β-orientation of the oxymethylene in 75 was confirmed by the nOe enhancement of its signal upon irradiation of Me-20. Analysis of the 1H NMR signals of the these protons before and after proton exchange pointed to the existence of a restricted conformation in which there is significant coupling between only one of the methylene protons and the hydroxyl proton. The authors suggest this is due to a hydrogen bond between the hydroxyl and one of the lactone oxygens. The brine shrimp lethality test was carried out for the purified compounds: 74 was inactive, and the remaining compounds showed mild toxicity with LC50 values of approximately 50–100 μg/mL.
In the subsequent study of S. matamata de Laubenfels collected in Yap island, Micronesia, from the same authors [33] six new terpenoids, 16β-methoxy-15-oxospongi-13-en-19-oic-acid 76, 16α-methoxy-15-oxospongi-13-en-19-oic-acid 77, 15-oxospongi-13-en-19-oic acid 78, 15α-methoxy-16-oxospongi-13-en-19-oic-acid 79, 16-oxospongi-13-en-19-oic acid 80, 13β,14α-dihydroxy-15α,16ξ-dimethoxyspongian-19-oic-acid 81 (Figure 18) and the known spongia-13(16),14-dien-19-oic acid 41 were isolated.
Figure 18.

Strutctures of 16β-methoxy-15-oxospongi-13-en-19-oic-acid 76, 16α-methoxy-15-oxospongi-13-en-19-oic-acid 77, 15-oxospongi-13-en-19-oic acid 78, 15α-methoxy-16-oxospongi-13-en-19-oic-acid 79, 16-oxospongi-13-en-19-oic acid 80, and 13β,14α-dihydroxy-15α,16ξ-dimethoxyspongian-19-oic-acid 81.
The known metabolite 41 was identified by comparison with literature data. Comparison of 1H and 13C NMR data of the new compounds with 41 showed that all compounds had identically substituted rings A and B. Confirmation of these rings stereochemistry was obtained from pyridine-induced solvent shifts. For 76 the NMR data indicated the presence of a carbonyl group, a tetrasubstituted double bond, methoxyl and acetal functions. An α,β-unsaturated-γ-lactone was identified by UV and IR and confirmed by HMBC. The stereochemistry at C-16 of 76 was established on the basis of NOESY of H-16 with H-12α. Although a similar study could not be performed with 77 due to overlapping of the two allylic protons at C-12, the similarity of spectral data allowed its identification as an epimer of 76. For 79 a α,β-unsaturated-γ-lactone with acetal function was confirmed by HMBC. Irradiation of Me-17 in NOESY studies caused an enhancement of H-15, confirming the α-orientation of the methoxyl group. The structure of 81 was established by HMBC data. NOESY experiments confirmed the α-orientation of the C-14-OH and C-15-OMe groups. The deshielding effect on Me-17 on running the NMR spectra in pyridine proved that C-13-OH is β-oriented. The configuration at C-16 could not be resolved. NMR analysis of 78 and 80 indicated the presence of an α,β-unsaturated-γ-lactone (confirmed by UV and IR data) in ring D. The downfield shift of C-14 in 80 when compared to 78 confirmed that this was the β-carbon of the an α,β-unsaturated-γ-lactone and made possible the distinction of both compounds. The brine shrimp lethality test was carried out for all the purified compounds but 76. Only 41 showed mild toxicity, with an LC50 value 10–100 μg/mL.
Mitchell et al. [34] reported the isolation of four new diterpenes, spongiabutenolides A–D, 82–85 (Figure 19), together with the known spongia-13(16),14-dien-19-oic acid 41, from a sample of Spongia sp. collected in the Philippines.
Figure 19.

Structures of spongiabutenolides A–D, 82–85.
Each of the new structures consisted of an inseparable mixture of stereoisomers at the hemiacetal carbon. Furthermore, structures 82 and 83 had to be separated as their methyl esters in order to be identified. The natural products were eventually separated and their spectral data was obtained. The structures were identified by 1H, 13C, HMQC, HMBC and 1D-TOCSY NMR spectra. The relative stereochemistry of the 82 (and 83) was established by ROESY correlations of 82 and its methyl ester. Correlations were seen between Me-20/Me-17/COOMe and Me-18/H-5. The relative stereochemistry of 84 was established by ROESY spectra, while that of 85 was assumed. 82 and 83 were synthesized by singlet oxygen oxidation starting from 41. All the compounds were tested for anti-cancer activity in a 25 cell-line panel but none showed significant cytotoxicity.
Zeng et al. [35] and Su et al. [36] reported the isolation of the new zimoclactone A 86, zimoclactone B 87 and zimoclactone C 88 from S. zimocca subspecies irregularia (Figure 20).
Figure 20.

Structures of zimoclactone A 86, zimoclactone B 87, and zimoclactone C 88.
The structures were determined by 1D and 2D NMR and X-ray diffraction analysis. Zimoclactone A 86 was isolated with 7-dehydrocholesterol and showed moderate cytotoxic activity against P388 cells.
Ponomarenko et al. [37], isolated five new diterpenes, 19-acetoxyspongia-13(16),14-dien-3-one 89, 3β,19-diacetoxyspongia-13(16),14-diene 90, 3β-acetoxyspongia-13(16),14-diene 91, 3α-acetoxyspongia-13(16),14-diene 92 and 2(R),3(S),4(S)-3,18-methylene-2α-acetoxyspongia-13(16),14-diene 93, together with the known 19-acetoxyspongia-13(16),14-diene 94, from S. Heterofibria collected in Northern Cook Islands (Figure 21).
Figure 21.

Structures of 19-acetoxyspongia-13(16),14-dien-3-one 89, 3β,19-diacetoxyspongia-13(16),14-diene 90, 3β-acetoxyspongia-13(16),14-diene 91, 3α-acetoxyspongia-13(16),14-diene 92, 2(R),3(S),4(S)-3,18-methylene-2α-acetoxyspongia-13(16),14-diene 93, and 19-acetoxyspongia-13(16),14-diene 94.
Structure of 89 was established on the basis of 1H, 13C, COSY, HSQC and HMBC and a single crystal X-ray diffraction study, followed by CD spectroscopy—the conformations of A and B rings are chairs, while that of ring C is a half chair. The CD spectrum of 89 showed positive Cotton effects, and application of the octant rule established the depicted stereochemistry (4S,5R,8R,9R,10R), in accordance with other spongians diterpenoids. Alkaline hydrolysis afforded the known 19-hydroxyspongia-13(16),14-dien-3-one, although the observed and reported values for optical rotation where somewhat different. Comparison of the NMR data of the remaining compounds with that of 89 established the spongian-based furanoditerpene skeleton. It was suggested that all these metabolites shared with 89 the same absolute configurations in their polycyclic structures. The nature and orientation of the substituents was established by NMR data analysis (including 1H–1H COSY, HSQC, HMBC, and NOESY spectra, and irradiation experiments). For 90 the configuration at C-3 came from the J coupling value of H-3 and its NOESY with H-5 and Me-18. NOESY of CH2-19 with Me-20 established the β-orientation of the former. For 91 and 92 the orientation of the acetoxy group was inferred from the J coupling values of H-3, and NOESY with H-5 and Me-18 in the case of 91. The unusual cyclopropane ring in 93 was identified by the high field 1H NMR signals and its location in C-3, C-4 and C-18 was established on the basis of the HMBC spectra. Its α-orientation was established by nOe of one H-18 with H-5. The α-orientation of the acetoxy group at C-2 was established by the J coupling observed for H-2 and its nOe with Me-20. Compounds 90 and 94 were tested for immunomodulatory properties by the methods reported in the literature and demonstrated a slight lysosomal activation (about 130% of control) of mice spleenocytes at concentrations of 100 μg/mL.
Carroll et al. [38] reported the isolation of four new spongian diterpenes 20-acetoxy-19-hydroxyspongia-13(16),14-diene 95, 19-acetoxy-20-hydroxyspongia-13(16),14-diene 96, 19,20-diacetoxyspongia-13(16),14-diene 97 and 19,20-dihydroxyspongia-13(16),14-diene 98 (Figure 22) together with the known spongia-13(16),14-diene 43, from an extract of Spongia sp. collected in Wreck Reef, Coral Sea, that showed TRH-R2 binding affinity.
Figure 22.
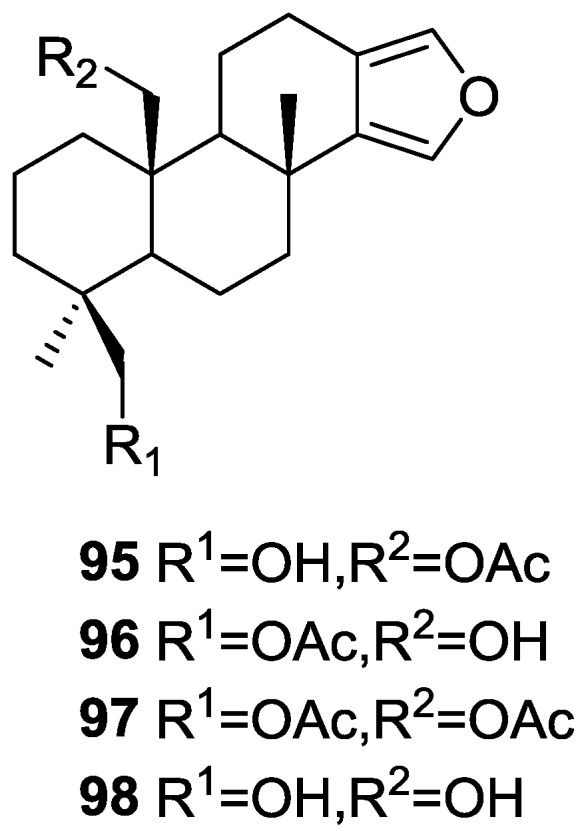
Structures of 20-acetoxy-19-hydroxyspongia-13(16),14-diene 95, 19-acetoxy-20-hydroxyspongia-13(16),14-diene 96, 19,20-diacetoxyspongia-13(16),14-diene 97, and 19,20-dihydroxyspongia-13(16),14-diene 98.
The known compound was identified by 2D NMR data analysis and comparison with literature data. For the remaining compounds analysis of the 1H, 13C, gCOSY, gHMQC, HMBC and ROESY spectra allowed their full characterization. All four compounds showed positive Cotton effects in their CD spectra, confirming the 4S,5R,8R,9R,10S configurations. For 95 Me-18 was established as equatorial on basis of the chemical shift. ROESY correlations between CH2-19 and CH2-20 confirmed a 1,3-diaxial relationship. For 96, comparison with 95 revealed an isomeric relationship. HMBC confirmed the structure. For 97 the comparison with 95 and 96 led to the proposed structure. For 98 the lack of the acetate band in IR, of the corresponding methyl signal in 1H NMR and the upfield shifts of CH2-20 when compared to 95 led to the proposed structure. TRH is a tripeptide that has been proposed to play an important role in neurotransmitter signaling. Two subtypes of the TRH receptor, TRH-R1 and TRH-R2 are found in rat brain tissues. Agonists and antagonist of TRH binding show potential therapeutic value in regulating endocrine function, in controlling pain, and in the treatment of spinal cord injury. Compound 95 was the most active of the five compounds in the TRH-R2 receptor binding assay, exhibiting an IC50 of 23 μM. Compounds 96, 97, 98 and 43 were only weakly active, displaying IC50’s of 70 μM, 400 μM, 600 μM and 1 mM, respectively. The reference compound TRH had an IC50 of 23 nM.
Ponomarenko et al. [39] reported the isolation of the new 19-norspongia-13(16),14-dien-3-one 99, together with the known 93, 102, 100, 91, 92, 94, 89, 90 and 101 from a Spongia ssp. (subgenus Heterofibria) collected in Northern Cook Islands. From a Spongia ssp. (subgenus Heterofibria) collected in Vietnam the known 43 and 91 were isolated. Compound 99 had previously been synthesized (Figure 23).
Figure 23.

Structures of 19-norspongia-13(16),14-dien-3-one 99, and compounds 100–102.
MS and 13C NMR data for 99 suggested a norditerpenoid structure. In the NMR spectra, signals corresponding to the furan ring, two methyls at quaternary carbons, one methyl group at a tertiary carbon and a carbonyl group were observed. HMBC confirmed that only one methyl was attached to C-4. nOe enhancements of H-4 and Me-17 upon irradiation of Me-20 proved that H-4 is β-oriented. The effects of 89, 90, 91, 99, 100, 101 and 102 on the biosynthesis of nucleic acids and embryonic development of the sea urchin Strongylocentrotus intermedius were studied. All the compounds inhibited sea urchin embryo development at concentrations of 20 μg/mL and above and DNA biosynthesis at the dose of 10 μg/mL. The inhibitory effect of these diterpenoids may partly be explained by the inhibition of thymidine kinase activity. The same compounds stimulated RNA synthesis in the developing sea urchin embryos.
Parrish et al. [40] reported the isolation of three new diterpenes 18-nor-3,17-dihydroxyspongia-3,13(16),14-trien-2-one 103, 18-nor-3,5,17-trihydroxyspongia-3,13(16),14-trien-2-one 104 and spongiapyridine 105 (Figure 24) together with the known 62, from an unidentified Spongia sp. collected in Sulawesi, Indonesia.
Figure 24.

Structures of 18-nor-3,17-dihydroxyspongia-3,13(16),14-trien-2-one 103, 18-nor-3,5,17-trihydroxyspongia-3,13(16),14-trien-2-one 104, and spongiapyridine 105.
Structure of compound 103 was established on the basis of 1H, 13C, HMBC and COSY NMR spectra. The relative configuration was ascertained by ROESY spectra where correlations between Me-20 and CH2-17 indicated they were syn diaxial. Correlation between H-9 and H-5 (axial, on the basis of J coupling values with H-6) identified H-9 as axial. The presence of the 5-OH substituent in 104 was suggested by the downfield shift of C-5 when compared to 103, and confirmed by HMBC. Of the four stereocenters of 104 only two could be determined by NOESY: Me-20 and CH2-17 in a syn diaxial relationship. The coupling constant of H-9 indicated it was axial as well. C-5 could not be determined due to rapid exchange of the alcoholic proton in aprotic solvents, and at lower temperatures. For compound 105 comparison with 103 showed identical rings A and B. 1H and 13C NMR data analysis, including 1JC–H values for H-16, were consistent with the presence of a pyridine ring, which was confirmed by 1H–15N HMBC. Additional structural features were deduced based on HMBC correlations that connected the pyridine ring to ring B, and indicated that the carbonyl was at C-12. The relative configuration were established by NOESY: again a correlation between Me-20 and CH2-18 confirmed both substituents as syn diaxial; 1H J values for H-5 and H-9 suggested both to be axial oriented. For 62, NMR data analysis was in agreement with the known structure, including the configuration of C-4. Although this is not a new structure full 1H and 13C NMR data are presented since the former characterization was for the 17-acetyl methyl ester derivative [29]. Since all the spongian diterpenes for which absolute configurations were determined belong to the same enantiomeric series, the authors suggest that all the compounds in this study have the 5R,8R,9R,10R configuration. The authors also propose a biosynthetic route to compounds 103, 104 and 105 (Figure 25).
Figure 25.

Proposed biosynthesis route for compounds 103, 104 and 105.
Several bioactivity tests were performed in search of the chemopreventive capacity of the isolated compounds. Modest inhibition of TNF-α-activated NF-κB activity was observed for 62, 103, 104 and 105 with ED50 values around 50 μM. No significant activity was observed for inhibition of iNOS activity in LPS-induced RAW 264.7 murine macrophage cells, and no significant induction occurred in a retinoic X receptor response element luciferase reporter gene assay. Compound 104 inhibited aromatase in a dose-dependent manner with an IC50 value of 34.4 μM. The other compounds did not achieve 50% inhibition at a concentration of 50 μM. 104 was also tested as an QR1 (NAD(P)H: quinone reductase 1) inducer. With cultured Hepa 1c1c7 cells 104 showed a CD (concentration required to double the specific activity) value of 11.2 μM, which is similar to the CD value of resveratrol (21 mM), a weak QR1 inducer. None of the compounds showed any significant activity towards the aspartic protease BACE1 (<100 μM).
Pham et al. [41] reported the isolation of an unusual nitrogenous spongian metabolite, haumanamide 106, together with the known spongia-13(16),14-dien-19-oic acid 41 from a Spongia sp. collected in Pohnpei, Micronesia (Figure 26).
Figure 26.

Structure of haumanamide 106.
The structure of 106 was established by comparison of the 13C NMR data with that of 41. The α,β-unsaturated-γ-lactam in D ring was confirmed by the chemical shift of C-15, an IR band at 1665 cm−1, and HMBC spectrum analysis. Difference nOe measurements confirmed that the relative stereochemistry of both compounds is the same. 106 showed activity against KB (MIC 5 μg/mL) and LoVo (MIC 10 μg/mL) cancer cells.
De Marino et al. [42] reported the isolation of the new spongidines A–D 107–110 from a Spongia sp. collected in Vanuatu Islands, Australia (Figure 27).
Figure 27.

Structures of spongidines A–D 107–110.
For 107, mass spectrum, IR and 13C NMR data indicated the presence of a carboxyl group. The 13C NMR data also revealed a tricarbocyclic skeleton with geminal dimethyl groups at C-4, and two methyl groups at the ring junctions C-8 and C-10. A disubstituted pyridinium salt was also inferred from 13C NMR, confirmed by UV and IR absorptions typical of alkylpyridinium salts. COSY and HMBC allowed the proposal of the structure. For 108 the comparison with 107 allowed the identification of the acetoxymethyl group, located at C-4 by the downfield shift observed at C-4 and upfield shift observed at C-3. The stereochemistry at C-4 was determined by ROESY (intense cross peaks between CH2-17 and Me-19). For 109, 1H and 13C NMR, COSY and HMBC data, together with comparison with 107 and 108 allowed the determination of the proposed structure. For 110 the comparison with 107 and the differences observed for the pyridine salt moiety, together with COSY and IR data allowed the determination of the taurine residue. HMBC established its location. Inhibition of specific PLA2 enzymes constitutes a potentially useful approach for treating a great variety of inflammatory disorders. Compounds 107, 108, 109 and 110 were tested as inhibitors of sPLA2 (secretory phospholipase A2) enzymes belonging to the groups I (Naja naja venom and porcine pancreatic enzymes), II (human synovial recombinant and rat air pouch secretory enzymes) and III (bee venom enzymes). All compounds inhibited human synovial PLA2 at 10 μM, compound 110, containing a sulfonic acid group, being the most interesting inhibitor. In this regard these compounds can offer new structural requirements for further studies about mechanistic interactions between PLA2 enzymes and inhibitors. All compounds were inactive to cPLA2. The results are summarized in Table 1.
Table 1.
Effect of compounds 107–110 on different sPLA2 activities a.
| Compound | N. naja Venom %I (10 μM) | Pancreas %I (10 μM) | Human Synovial %I (10 μM) IC50 (μM) | RAPb + Zymosan %I (10 μM) | Bee Venom %I (10 μM) IC50 (μM) |
|---|---|---|---|---|---|
| 107 | 0.5 ± 0.5 | 18.0 ± 8.1 | 40.1 ± 7.7 d | 17.1 ± 4.6 | 33.1 ± 6.0 c |
| 108 | 0.4 ± 0.4 | 14.2 ± 5.1 | 34.6 ± 5.8 d | 17.9 ± 4.2 | 32.2 ± 6.0 c |
| 109 | 3.1 ± 2.2 | 9.1 ± 3.5 | 40.4 ± 5.7 d | 30.9 ± 5.3 c | 36.2 ± 5.4 d |
| 110 | 0.0 ± 0.0 | 7.6 ± 4.0 | 48.2 ± 3.8 d | 19.6 ± 5.4 | 37.6 ± 6.5 c |
| manoalide | 17.0 ± 1.7 c | 32.3 ± 2.7 d | 93.2 ± 0.2 d 3.9 | 38.4 ± 0.5 d | 62.5 ± 3.8 d 7.5 |
a Results show percentages of inhibition at 10 μM and IC50 (μM) values determined only for those compounds that reach 50% of inhibition. Mean ± S.E.M. (n = 6); b RAP: Rat air pouch PLA2; c p < 0.05; d p < 0.01.
Mori et al. [43] reported the isolation of spongolactams A–C, 111–113 (Figure 28), together with the known spongia-13(16),14-dien-19-oic acid 41, from a Spongia sp. collected in Okinawa, Japan, whose extract showed a 70% inhibition of FTase (Farnesyl transferase) at 20 μg/mL, in a new assay described by the authors.
Figure 28.

Structures of spongolactams A–C, 111–113.
Structure elucidation for all compounds was based on 1H, 13C, HMQC, HMBC and DQF-COSY spectra. For compounds 111 and 112 the carboxyl group was identified by its δ value in 13C NMR and IR bands; its location was determined by HMBC. Similar process led to identification and localization of the tertiary amide. The 5-imidazolyl ring connected to the C-22 methylene group was established by its typical chemical shifts and HMBC data. The relative stereochemistry of 111 was inferred from NOESY spectra where correlation between H-12/H-16 specified the direction of the lactam group. For 112 the reversal of the chemical shifts of C-15 and C-16, together with NOESY between H-7/H-15 confirmed the structure. Compound 113 was identified by comparison with 111: significant differences in the spectra were the absence of the imidazole moiety and the replacement of the C-22 methylene by a carbonyl group. The structure and absolute stereochemistry of 111 and 112 were confirmed by synthesis from 41. The structure of 113 was also confirmed by synthesis from the same precursor. The synthesis of other spongolactam related compounds are also presented. FTase inhibitors are believed to be candidates for novel chemotherapeutic drugs. In the FTase inhibition assays the synthetic sample of 111 showed an IC50 23 mM (natural sample 22 mM). The activity of spongolactams B and C was determined only with synthetic samples due to inadequate amounts of natural material (130 μM and >260 μM, respectively). Cytotoxicity of these compounds against a human vulval-derived epidermoid carcinoma cell line, A431, was also evaluated and apparently some correlation exists between the two assays. The authors suggest that FTase could be a molecular target in the expression of spongolactam cytotoxicity.
Other Studies
Kazlauskas et al. [44] reported the isolation of 3α,19-dihydroxyspongia-13(16),14-dien-2-one 55, 3β,19-dihydroxyspongia-13(16),14-dien-2-one 54, 3α,17,19-trihydroxyspongia-13(16),14-dien-2-one 58 and, 3β,17,19-trihydroxyspongia-13(16),14-dien-2-one 59, together with their acetyl derivatives 3α,19-diacetoxyspongia-13(16),14-dien-2-one 114, 3β,19-diacetoxyspongia-13(16),14-dien-2-one 115, 3α,17,19-triacetoxyspongia-13(16),14-dien-2-one 102 and 3β,17,19-triacetoxyspongia-13(16),14-dien-2-one 116 from several Spongia sp. collected in the Great Barrier Reef (Figure 29). These specimens were subsequently reclassified as Rhopaloeides odorabile [45].
Figure 29.

Structures of 3α,19-diacetoxyspongia-13(16),14-dien-2-one 114, 3β,19-diacetoxyspongia-13(16),14-dien-2-one 115, and 3β,17,19-triacetoxyspongia-13(16),14-dien-2-one 116.
For 114 fragment ions in mass spectra indicated successive losses of CH3, AcOH and 2xAcOH. The 1H NMR spectrum indicated three quaternary methyls, two acetoxy methyls, an oxygenated methylene and methine and two furan protons. For 102 mass spectra showed the successive losses of CH2OAc and AcOH, which suggested that a quaternary methyl group had been replaced by an acetoxymethyl. This was confirmed by 1H NMR, where the remaining signals were very similar in both compounds. IR and 13C NMR showed the presence of a ketone group for both compounds. Analysis of the 13C NMR spectra of both compounds allowed the identification of the furan ring and establishment of the functionality at C-17. 1H NMR analysis together with biogenetic considerations established ring A. The position of an acetoxymethyl group at C-4 was assigned for 114 by an 1H NMR study in the presence of Eu(fod)3. Definite proof of stereochemistry came from a single crystal X-ray diffraction study of 102 where ring A was shown to be present as a boat conformation with atoms C-1, C-2, C-4 and C-5 coplanar, ring B formed a chair, ring C a distorted half-chair and ring D was practically flat. CD and ORD established the absolute configuration of 102. Acetylation of 59 produced 116 identified as a C-3 epimer of 102. Acetylation of 54 produced 115, the C-3 epimer of 114. Compound 58 was also acetylated to give 102. Acetylation of a mixture of 55 and 54 gave 114 and 115.
Puliti and Matia [46] determined the relative configuration of ent-isocopal-12-en-15,16-dial 45 as 5S*,8R*,9R*,10S*,14S* by X-ray analysis. A trans fused tricyclic system with four methyl substituents, three of which are axially β-oriented (at C-4, C-8, and C-10) was determined. The β-orientation of the aldehyde substituent at C-14 was confirmed.
An independent study by Yong et al. [47] determined the absolute configurations and conformations of 100, 54 and 55 by X-ray analysis. For 100 a twisted-boat ring A, chair B and C rings and a planar furan ring was determined, with an absolute stereochemistry of 5R,8R,9R,10R. In 54 ring A adopts a chair conformation and the hydroxymethylene group donates an intramolecular hydrogen bond to the C-3-OH. An absolute stereochemistry of 3R,4S,5R,8R,9R,10R was determined. For 55 a disordered ring A with a dominant chair conformer with the C-3-OH in an axial position was observed. The minor contribution was a distorted-boat conformer where the hydroxyl group adopts an equatorial position. An absolute stereochemistry of 3S,4S,5R,8R,9R,10R was determined. The authors point out that all the literature spongian diterpenes for which absolute stereochemistry had been reported belonged to the same enantiomeric series, even though some configurations had been assigned by Mosher esters analysis or CD data (as is the case of 64, 89, 95 and 102).
An independent study by Betancur-Galvis et al. [48] tested several spongian diterpenes for their activity against herpes simplex virus type 2 and cytotoxic effect on tumor cells. Compound 40 showed low cytotoxicity and 43 was poorly active against HSV-2. Compound 100 showed no anti viral activity but was cytotoxic to HeLa (human cervix epithelioid carcinoma-CC100 30 μg/mL), Hep-2 (human larynx epidermoid carcinoma-CC100 40 μg/mL), CHO (Cricetulus griseus Chinese hamster ovary cells ATCC CCL-61-CC100 30 μg/mL) and Bon-Fib (primary culture of bovine ear subcutaneous fibroblasts-CC100 40 μg/mL) cells.
4. C21 and Other Linear Furanoterpenes
Work of Fattorusso et al. [8] and Cimino et al. [49,50,51] on the chemistry of S. nitens and S. officinalis, both from the Mediterranean, allowed the isolation and characterization of the furanoterpenes, nitenin 117 and dihydronitenin 118 (both from S. nitens), furospongin-1 119, anhydrofurospongin-1 120, furospongin-2 121, isofurospongin-2 122, dihydrofurospongin-2 123, tetrahydrofurospongin-2 124, furospongin-3 125 and furospongin-4 126 (from S. officinalis) (Figure 30).
Figure 30.

Structures of nitenin 117, dihydronitenin 118, furospongin-1 119, anhydrofurospongin-1 120, furospongin-2 121, isofurospongin-2 122, dihydrofurospongin-2 123, tetrahydrofurospongin-2 124, furospongin-3 125, and furospongin-4 126.
The structures were identified on the basis of UV, IR, 1H NMR data with double irradiation experiments, mass spectra, and chemical transformation and degradation. Typical of the furan moiety seem to be a positive Ehrlich test, a λmax ca. 220 nm in UV (cyclohexane), characteristic IR bands at 3140, 1570, 1500, 875 and 780 cm−1, 1H NMR (CCl4) signals at δ 7.26–7.15 ppm and δ 7.14–7.05 ppm for the α-protons of both rings (usually equivalent), one proton signal at δ 6.16–6.14 ppm for the β-protons of both rings (usually equivalent), and mass fragments at m/z 67, 81 and 95. The isoprene unit is usually recognized by a methyl singlet at δ 1.58 ppm, broadened by long range coupling to the trans olefinic proton. For nitenin 117 and dihydronitenin 118 the configuration of C-11 was assigned as R, by applying Horeau’s method to the C-7 unsaturated and saturated diols, respectively, obtained after LAH reduction. This assignment was confirmed by 1H NMR analysis of the Mosher’s esters in a subsequent study by Fontana et al. [52] that isolated both compounds from S. agaricina from NE Spain. These authors also determine a R absolute stereochemistry for C-8 of 118 on the basis of nOe spectra. For furospongin-1 119 the absolute configuration at C-11 was established as S by applying Horeau’s method. Subsequent studies by Kobayashi et al. [53] corrected this assignment to R by applying Mosher’s method, further supported by nOe studies and pyridine induced shift. Although the authors [49] assigned a R configuration to C-13 on the basis of chemical degradation of the dehydrated derivative, this was later corrected to S in a subsequent paper [50].
The same correction is applied to the configuration of C-13 of 123. For 121 UV and IR indicated an α,β-unsaturated ketone, confirmed by 1H NMR with irradiation experiments of the signals of the corresponding H-12 and vinylic methyl. The low field resonance of the vinylic methyl at C-13 suggested it was cis to the carbonyl group. Further 1H NMR analysis led to the proposal of the structure. 122 showed UV, IR and mass spectra identical to 121. The only difference in the 1H NMR spectrum was the upfield shift of the methyl at C-13 that led the authors to assume a different configuration of the ∆12,13 bond. For 123 the presence of the ketone was inferred from IR, and mass fragments corresponding to the cleavage of the C-10/C-11 bond, together with 1H NMR analysis, led to the proposal of the structure. For 124 the ketone group identified by IR, 1H NMR data analysis, the fact that it presented no optical rotation, and that in the mass spectrum only one fragment for α-cleavage of the carbonyl group was observed, led the authors to propose the meso compound depicted. 125 and 126 were isolated as a mixture resistant to separation. IR spectra indicated the presence of conjugated ester and carboxylic acid substituents that justify the intensity of the UV absorption observed, further confirmed by 1H NMR shifts of the corresponding olefinic protons. The location of the carboxylic acid was inferred from mass spectra and the trans orientation of the carboxyl substituents to the corresponding olefinic proton was established by spin decoupling experiments.
Further work by the same authors [54] on the same S. officinalis led to the isolation of eight new structures (isolated as mixtures) related to furospongin-1 119, with γ-hydroxy-α,β-butenolide and β,γ-epoxybutenolide rings, 127–134 (Figure 31).
Figure 31.

Structures of furospongin-1 119 related compounds with γ-hydroxy-α,β-butenolide and β,γ-epoxybutenolide rings, 127–134.
The structures were identified as mixtures on the basis of UV, IR, 1H NMR data, mass spectra and comparison with furospongin-1 119. The mixtures of 127 and 128, and 129 and 130 were also identified by chemical correlation with 119. Since the structure of the latter was reviewed after the publication of this study, the structures here presented are also corrected. The mixture of 131–134 readily underwent decomposition to 127–130. The fact that the authors were unable to identify any of the metabolites after exposure of a methanolic solution of furospongin-1 119 to the light, reinforces the nature of the β,γ-epoxybutenolides as natural products.
Kazlauskas et al. [55] reported the isolation of tetradehydrofurospongin-1 135 from an Australian Spongia sp (Figure 32). The proposed structure was subsequentially corrected.
Figure 32.

Structure of tetradehydrofurospongin-1 135.
The structure was identified on the basis of IR, 1H NMR data with double irradiation experiments, mass spectra and chemical transformation. Capon et al. [56] in a subsequent study in 1982 assigned 13C data, established the E configuration of the double bonds based on the 13C NMR resonances of the vinylic methyls and J coupling values of H-6 and H-7, and an R configuration at C-11 (Horeau’s method). In the already mentioned study of Fontana et al. [52] of 1996 the enantiomer of (−)-untenospongine B is isolated from S. virgultosa from NE Spain. The NMR data of this new compound 136 (Figure 33) was assigned by 1D and 2D experiments and an R configuration at C-11 was confirmed by 1H NMR analysis of the Mosher’s esters. Comparison of the obtained NMR data with that of the reported for 135 led the authors to reassign the structure of 136 to tetradehydrofurospongin-1.
Figure 33.

Structure of tetradehydrofurospongin-1 136.
Another study of Australian Spongia sp. by Kazlauskas et al. [57] led to the isolation of two new compounds, furospongenol 137 and furospongenone 138 (Figure 34).
Figure 34.

Structures of furospongenol 137 and furospongenone 138.
The structures were identified on the basis of IR, UV, 1H NMR data with irradiation experiments, mass spectra and chemical transformation. No absolute configuration was assigned to 137.
Walker et al. [58] reported the isolation of the new idiadione 139 and the known furospinulosin-1 140 from S. idia de Laubenfels collected in San Diego, California (Figure 35).
Figure 35.

Structures of idiadione 139 and furospinulosin-1 140.
140 was identified by comparison with literature data. 139 was identified by IR, 1H and 13C NMR, and chemical transformation and degradation. The position of the ketone groups was established by mass fragmentation of the diketone-tetrahydrofuran obtained after full hydrogenation; NMR analysis of the products synthesized by reduction and acetylation, followed by ozonolysis and hydrogenation of the ozonides, confirmed the position of the double bonds; their geometry was determined by the chemical shifts of the vinylic methyls. 139 was toxic to the sea star Pisaster giganteus at a concentration of 5 mg/L, immobilized the larvae of the red abalone Haliotis rufescens at 1 mg/L in sea water, and was toxic to the ectoproct Membranipora membranacea at 10 mg/L. Both compounds were toxic to brine shrimp Artemia sp. at 10 mg/L.
From anWestern Australian Spongia sp., Capon et al. [56] isolated a new C-21 furanoterpene, 141 (Figure 36).
Figure 36.

Structure of C-21 furanoterpene 141.
The structure was elucidated on the basis of UV, IR, 1H and 13C NMR, and mass spectra. The presence of a tertiary carbinol system came from IR, 1H and 13C NMR data. Significant downfield shifts were observed for the ∆6,7 vinylic protons and the methyl singlet upon recording the 1H NMR spectrum in the presence of tris[3-(trifluoromethylhydroxymethylene-d-camphorato]europium(III). Confirmation of the proposed structure came from ozonolysis. The E configuration of the double bond at C-11 came from JH,H coupling analysis of H-11 and H-12; the same configuration was assigned to the double bond at C-13 on the basis of the high field resonance observed for Me-14 (shielded by cis-allylic methylene group). Analysis of the JH,H coupling of H-6 and H-7 obtained by spectral simulation allowed the assignment of the E configuration to the ∆6,7 double bond.
Subsequent work of Capon et al. [59] on Spirastrella papilosa led to the reisolation of the compound, and revision of the proposed structure to 142 (Figure 37), for which the name of (−)-isotetradehydrofurospongin-1 is proposed.
Figure 37.

Structure of (−)-isotetradehydrofurospongin-1 142.
The revision was based on 2D NMR data. The E configuration of ∆5,6 was confirmed by J coupling analysis and the chemical shift of Me-14 in 13C NMR; the E configuration of ∆10,11 is confirmed by NOESY. Ozonolysis was repeated and (R)-dimethyl citramalate was recovered: its assignment was confirmed by 1H NMR, [α]D and chiral HPLC comparison with authentic samples of both the R and S enantiomers.
Tanaka and Higa [60] reported the isolation of the new kurospongin 143 from a Spongia sp. collected in Miyako Island, Japan (Figure 38).
Figure 38.

Structure of kurospongin 143.
The structure was identified by mass spectrometry, IR, 1H and 13C NMR and irradiation experiments. These allowed the identification of the furan rings, the α,β-unsaturated γ-lactone, a vinyl methyl, a trans di-substituted double bond and a tri-substituted double bond. The geometry of the latter was assigned as E by the value of the 13C chemical shift of the vinylic methyl. The absolute stereochemistry at C-11 was assigned as S by applying Horeau’s partial resolution method to the diol obtained after treatment with ethylmagnesiumbromide. Compound 143 was ichthyotoxic, killing goldfish at the concentration of 5 μg/mL within 4 h. In feeding experiments using the omnivorous fish Tilapia mosambica 143 impregnated in feed completely deterred its consumption at the concentration level of 0.3%.
De Giulio et al. [61] reported the isolation of furospongin-2 121, together with its three new isomers 144–146 (Figure 39), from a S. officinalis L. collected in northern Adriatic, whose extract showed cytotoxic activity (LD50 45 μg/mL) in the brine shrimp assay.
Figure 39.

Structures of furospongin-2 121 isomers 144–146.
For all compounds, the presence of an α,β-unsaturated ketone was established by UV and IR and confirmed by 1H and 13C NMR spectra. The use of COSY and HETCOR spectra allowed the assignment of all resonances for 144. For this compound the fact that the 13C NMR spectra only showed 11 signals led to the conclusion that it was symmetrical. Comparison of its data with the remaining compounds led to identification of the latter. The stereochemistry of the double bonds of all compounds were assigned on the basis of the chemical shifts in 1H and 13C spectra for the vinylic methyls and allylic methylenes. 13C NMR data for 121 is assigned based on COSY and HETCOR. All compounds showed high activity (LD50 0.09–1.6 μg/mL) in the Artemia salina shrimp bioassay, an in-house substitute for 9 KB and 9 PS cytotoxicities.
Lumsdon et al. [62] reported the isolation of the new tetronic acid 147 from a Spongia sp. collected in Australia (Figure 40). The crude ethanol extract evoked a large triphasic contraction of smooth muscle in the isolated guinea-pig ileum. It also appeared to inhibit contractions elicited by different drugs (acetylcholine, 5-hydroxytryptamine and histamine) of the isolated guinea-pig ileum, and inhibited the growth of several bacteria (Staphylococcus aureus, Micrococcus sp. and Serrata sp.) in a standard antibiotic disk assay.
Figure 40.

Structure of tetronic acid 147.
Compound 147 was identified by NMR, where resonances for a β-substituted furan, three substituted double bonds with vinylic methyls and a tetronic acid moiety were observed. The presence of this latter feature and confirmation of the structure came by comparison with palominin, its known geometrical isomer. The observed chemical shifts for the olefinic methyl resonances of 147 confirmed the E geometry of all double bonds. CD data supported a 21R stereochemistry. The antibiotic activity of the extract was attributed entirely to 147. Preliminary testing suggested that this compound was also responsible for the inhibitory activity detected in the crude ethanol extract. This compound reversibly blocked contractions, evoked by acetylcholine, 5-hydroxytryptamine and histamine, of isolated guinea-pig ileum, and electrical stimulation of intrinsic nerves. Purification of the extract appeared to remove the contracting substance detected, due to its loss or to a synergistic activity between the isolated compounds.
Urban and Capon [63] reported the isolation of the new cometins A–C 148–150 (Figure 41), together with the known furospinosulin-1 140, from a Spongia sp. collected in the Great Australian Bight.
Figure 41.

Structures of cometins A–C 148–150.
Compound 148 was identified by mass spectrometry and 1H and 13C NMR where resonances for the difuran and tetronic acid moieties were identified. Confirmation came from comparison with literature compounds. The 1,1,4-trisubstituted 1,3 diene functionality was further identified by NMR. The geometry of the ∆16,17 double bond was determined as E by the J coupling value observed for the olefinic protons. Comparison of the δ value in 13C NMR for the olefinic methyl with reference compounds allowed the determination of the E configuration for the trisubstituted double bond. Although stereochemistry at C-18 was not determined, a R configuration is proposed on the basis of CD data. Comparison of the NMR data of 149 with that of 148 allowed the replacement of the tetronic acid moiety by a conjugated γ-butenolide. Confirmation of the structure came from COSY spectra and nOe. The geometry of the double bond was determined as E by the 13C NMR shift of the olefinic methyl. Stereochemistry at C-18 was not determined. Comparison of the data of 150 with that of 149, together with analysis of the COSY spectra, allowed the proposal of the structure for the former. The geometry of the ∆12,13 double bond was determined as E by the 13C chemical shift of Me-14, and the presence and orientation of the butenolide fragment were confirmed by MS (observation of the fragment derived by allylic fragmentation) and nOe (enhancement of H-20 and Me-25 upon irradiation of H-22). Biological testing of the pure compounds against Staphylococcus aureus and a Serratia sp. confirmed that only 148 was active. The minimum concentrations for these activities were determined as 3–5 μg/disk and 5 μg/disk, respectively.
From the already mentioned study of Searle et al. [30] of a Spongia sp. from Western Australia, the known ambliofuran 151 together with the new (S)-12-hydroxyambliofuran 152, (S)-12-acetoxyambliofuran 153, and 154 were identified (Figure 42).
Figure 42.

Structures of ambliofuran 151, (S)-12-hydroxyambliofuran 152, (S)-12-acetoxyambliofuran 153, and compound 154.
Compound 151 was identified by comparison of 1H and 13C NMR data with the literature. For compound 153 the location of the acetoxy substituent (confirmed by IR and 1H signal of the corresponding methine) was determined by COSY, HETCOR and HMBC data. Compound 152 was assigned by comparison with 153. Hydrolysis of the latter allowed confirmation of the proposed structure. The absolute configuration of 152 and 153 (after hydrolysis) was determined by a modified Mosher’s method. For both compounds a 3:1 mixture of enantiomers was determined, with excess of the 12S enantiomer. This ratio was confirmed by the 1H NMR spectra of 153 in the presence of Eu((+)-hfc)3. The authors suggest the existence of two different enzymes, one with the S specificity and other with the R that would oxidize 151 to 152; further acetylation would produce 153 in the same ratio. For 154, the presence of a trisubstituted epoxide was suggested by 13C NMR. COSY, HETCOR, COLOC and 2D-INADEQUATE spectra allowed confirmation of the proposed structure. Although the absolute stereochemistry of 154 was not determined, a trans epoxide is proposed on the basis of high field signal of the attached methyl group in 13C NMR (due to steric compression by the syn methylene group) and comparison with the observed values for the corresponding methyl group in trans geraniol-2,3-epoxide and cis nerol-2,3-epoxide. The E configuration of the double bonds was determined by the 13C chemical shift values of the vinylic methyls.
Lenis et al. [64] reported the isolation of the new isonitenin 155 (Figure 43) and the known nitenin 117 and dihydronitenin 118 from S. officinalis collected in the Galician coast.
Figure 43.

Structure of isonitenin 155.
The known compounds were identified by comparison with literature data. The structure of 155 was established by NMR (including 1H–1H COSY and HMQC). Comparison of the data with that of 117 showed an downfield shift of H-7 and upfield shift of CH2-6, consistent with an E stereochemistry of the ∆7,8 double bond. This geometry was further confirmed by the upfield shift observed for C-10 due to the cis arrangement of C-10 and C-6. Since optical rotation and CD spectrum of 155 where almost identical to those of 117 an R stereochemistry at C-11 is proposed. For 118 an 8R stereochemistry is proposed on the basis of nOe observed between H-8 and H-11. The E stereochemistry of the ∆12,13 double bond was confirmed by nOe between CH2-15 and H-12.
Garrido et al. [65] reported the isolation of the new furospongin-5 156, cyclofurospongin-2 157 and demethylfurospongin-4 158 (Figure 44), together with the known, 121–124, 126, and 144–146, from a S. officinalis L. collected in Cádiz, Spain.
Figure 44.

Structures of furospongin-5 156, cyclofurospongin-2 157, and demethylfurospongin-4 158.
Compound 156 was identified by comparison with 121 and analysis of the IR (non conjugated ketone) and NMR data. The stereochemistry of the double bonds was determined on the basis of the 13C chemical shifts of the vinylic methyls. Confirmation of the proposed structure and double bond stereochemistry came from nOe difference spectroscopy, where irradiation of H-7 caused enhancement of CH2-10, and irradiation of H-15 enhanced Me-14. For 157 IR, 1H and 13C data showed it was an isomer of 156 with a (E)-furylmethylpentenyl fragment linked to a central ketone. An extra α,β-disubstituted furan ring with a fused methylated cyclohexene and an isolated methylene were established by 1H and 13C NMR. Acid treatment of 121 yielded (±)-cyclofurospongine-2 as expected. Since the natural product is optically active occurrence of the cyclisation process during isolation is excluded. The structure of 158 was established by IR, 1H and 13C NMR data, and comparison with the data of a mixture of 125 and 126, and 154. 13C NMR data is presented for all compounds. The new compounds were tested against P-388, A-549, HT-29 and MEL-28. Compounds 156–158 showed low cytotoxicity with E97 values over 10 μg/mL in all cases with the exception of 156 that showed mild cytotoxicity against P-388 cell line (ED50 5 μg/mL).
Manzo et al. [66] reported the isolation of the new 7,8-epoxy-furospongin-1 159 and isofurospongin-4 160 together with the known 119, 120, 123, 124, 126 and 161, from S. officinalis L. collected in Sicily (Figure 45).
Figure 45.

Structures of 7,8-epoxy-furospongin-1 159, isofurospongin-4 160, and compound 161.
Compound 159 was identified by NMR data and comparison to furospongin-1 119. The presence of an epoxide ring was identified by 1H and 13C NMR and the proposed structure was confirmed by 2D NMR. The relative configuration was determined by nOe difference experiments where effects where observed between CH2-6 and Me-9, and between H-7 and CH2-10. The absolute configuration at C-11 was established by applying a modified Mosher’s method. Based on biogenetic considerations the absolute configuration at C-13 was assigned as in furospongin-1 119. The NMR data of 160 closely resembled that of furospongin-4 126. Analysis of 1H–1H COSY, HSQC and HMBC spectra showed that both compounds differed in the esterification site. Further confirmation of the structure came from comparison of the dimethyl ester obtained from both compounds. Compounds 119, 120, 123, 124, 126, 160 and 161 were tested for antibacterial and antifungal activity against E. coli, Staphylococcus aureus and Candida albicans. Only 126 showed weak activity against S. aureus at 100 μg/mL. For compounds 123 and 124 an interesting biofilm induction activity of E. coli PHL628 was observed, this activity being more efficient by an increase in the concentration of 124. The authors suggest that this activity is related to the symbiosis that marine organisms are able to form with strains of bacteria that do not allow biofouling stratification on their surfaces.
Other Studies
Li et al. [32] reported the isolation of the known 162 (Figure 46) from the already mentioned study of on a S. matamata de Laubenfels collected in Yap Island, Micronesia, a specimen reclassified as S. zimocca sensu de Laubenfels by the same authors in a subsequent study [33].
Figure 46.

Structure of compound 162.
Purified 162 showed mild toxicity, with LC50 values of approximately 50–100 μg/mL, in the brine shrimp lethality test.
Rueda et al. [67] reported the isolation of the known 117, 118, 120, 140, and 155 from S. agaricina collected in Cádiz, Spain.
5. Sesterterpenes
Cimino et al. [68] reported the isolation of deoxoscalarin 163 from S. officinalis, collected in Naples. The C-19 stereochemistry of this structure was revised in a subsequent publication [69] where the known scalarin 164 was isolated from S. virgultosa and the new 12-epi-deoxoscalarin 165 and 12-epi-scalarin 166 were isolated from the Mediterranean S. nitens, whose extracts had already afforded furanoterpenes [8] (Figure 47).
Figure 47.

Structures of deoxoscalarin 163, scalarin 164, 12-epi-deoxoscalarin 165, and 12-epi-scalarin 166.
Compound 163 was identified by 1H NMR with spin decoupling, mass spectrometry, comparison with 164 and chemical correlation between both compounds. For all compounds the mass fragments corresponding to the rupture of ring D at m/z 258 (retro Diels Alder and concomitant loss of acetic acid), ring C at m/z 205 and m/z 191 (cleavages of the 8,14 and 11,12, and 8,14 and 9,11 bonds, respectively, with associated loss of hydrogen from the charge retaining rings A and B) and fragmentation of ring B at m/z 137 and m/z 123 (cleavage of the 6,7 and 9,10, and 5,6 and 9,10 bonds, respectively) are identified. Comparison of the 1H NMR data showed main differences in the shape of the H-12 signal and J analysis established its orientation. Further evidence came from the fact that scalarin type compounds (12-OH axial) are more readily oxidized by Jones’ reagent than epi-scalarin compounds (12-OH equatorial). Chemical correlation of 165 and 166 with 164 confirmed their structures. Analysis of several derivatives (NMR, CD and shifts induce by Eu([2H9]fod)3) allowed the determination of a trans-transoid-trans-skeleton with an H-18-α absolute configuration and an axial orientation of the Me-24, Me-25, H-9 and H-14. Stereochemistry at C-19 was again inferred from chemical studies of derivatives. 13C NMR data is presented for 164, 165 and 166.
Further studies on the extracts of the same S. nitens [70] led to the isolation of 12-epi-scalaradial 167 and 12,18-di-epi-scalaradial 168 (Figure 48).
Figure 48.
Structures of 12-epi-scalaradial 167 and 12,18-di-epi-scalaradial 168.
Compound 167 was assigned on basis of the UV, IR, 1H and 13C NMR data and comparison with the known scalaradial. Orientation at C-12 was based on the shape of the H-12 signal in 1H NMR. Full confirmation, including absolute stereochemistry, came from correlation with 12-epi-deoxoscalarin 165. 13C NMR data comparison of 167 with that of scalaradial allowed the confirmation of its structure and determination of an all trans-anti-trans configuration. Compound 168 was established by comparison of the obtained data with that of 167. 13C NMR data allowed confirmation of its structure and determination of the stereochemistry.
From the already mentioned study of Walker et al. [58] 12-deacetyl-12,18-di-epi-scalaradial 169, scalarafuran 170 and scalarolide 171, together with the known heteronemin 172 and 165, were isolated from S. idia de Laubenfels, collected in San Diego, California (Figure 49).
Figure 49.

Structures of 12-deacetyl-12,18-di-epi-scalaradial 169, scalarafuran 170, scalarolide 171, and 172.
Compounds 165 and 172 were identified by comparison with literature values. 169 was converted into the known 12,18-di-epi-scalaradial 168. Compound 170 was obtained from 172 by controlled pyrolysis. For 171 a scalarin skeleton with an equatorial hydroxyl at C-12, an olefin at C-17(18), and lactone were inferred from 1H and 13C data. The position of the lactone at C-19 was established by chemical transformation and confirmed by chemical correlation to 172. Compound 169 was toxic to the sea star Pisaster giganteus at a concentration of 5 mg/L, immobilized the larvae of the red abalone Haliotis rufescens at 1 mg/L in seawater and was toxic to the hydroid Bougainvilla sp. at 10 mg/L. Compounds 165, 169 and 172 were toxic to brine shrimp Artemia sp. at 10 mg/L. Compound 172 immobilized the larvae of the red abalone Haliotis rufescens at 1 mg/L in seawater and was toxic to the gametes of the giant kelp Macrocystis pyrifera at 10 mg/L.
Subsequent work by Cimino et al. [71] on S. nitens allowed the isolation of scalarolbutenolide 173 (Figure 50).
Figure 50.
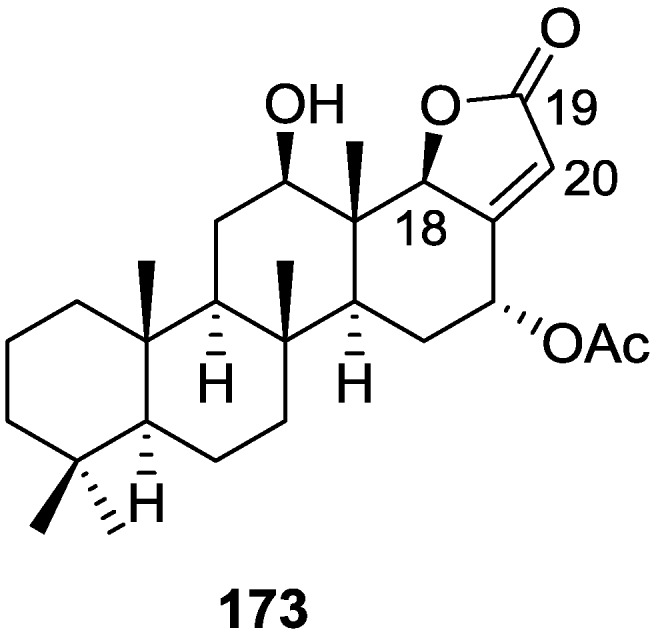
Structure of scalarolbutenolide 173.
A tetracyclic scalarin type skeleton with an acetoxy group and a hydroxyl at C-12 was inferred from mass spectra. This latter assignment is confirmed by the corresponding H-12 signal in 1H NMR. Comparison of 13C data (including decoupling experiments) with those of known compounds confirmed the proposed structure. The upfield shifts observed upon acetylation indicated the presence of a hydrogen bond between C-12-OH and the lactone oxygen. The localization of the acetoxy group in C-16 was suggested by the ease of elimination observed in mass spectra. Its orientation was based on the upfield shift of H-14 when compared with a known compound, justifiable by a γ-gauche interaction with an axially oriented group. The upfield resonance of C-25 indicated an α-orientation of C-18. The β,γ-disubstituted-α,β-butenolide ring suggested by UV, IR and 1H NMR data was confirmed by chemical transformation. The relative stereochemistry is in agreement to previous findings for scalarin type compounds. Application of Horeau’s method determined an R absolute configuration at C-12.
From the already mentioned study of De Giulio et al. [61] 16-deacetoxy-12-epi-scalarafuran acetate 174 and deoxoscalarin acetate 175, together with (−)-12-epi-deoxoscalarin 176 (Figure 51), were isolated from a S. officinalis L. collected in northern Adriatic, whose extract showed cytotoxic activity (LD50 45 μg/mL) in the brine shrimp assay.
Figure 51.

Structures of 16-deacetoxy-12-epi-scalarafuran acetate 174, deoxoscalarin acetate 175, and (−)-12-epi-deoxoscalarin 176.
For all compounds 1H and 13C NMR data comparison with literature compounds supported a scalarane skeleton. For 174 the presence of an acetyl group was inferred from IR data and confirmed by 1H NMR. This spectra also showed the typical signals of a β,β-disubstituted furano ring. This data suggested that 174 was identical to the furan obtained in the reaction of acetylation of deoxoscalarin 163 [68]. The relative stereochemistry at C-12 was assigned by the chemical shift and multiplicity observed in 1H NMR. Compound 175 was identified by IR and 1H NMR data that showed it to be the acetyl derivative of deoxoscalarin 163. 175 was unstable and readily transformed into 174, what suggested that 174 was an artifact. The spectral data of 176 (including COSY and NOESY) were in agreement with those of 165. The fact that it presented a similar melting point and an optical rotation of the same magnitude but opposite sign indicated it was the enantiomer of 165. All compounds were tested in the Artemia salina shrimp bioassay, an in-house substitute for 9 KB and 9 PS cytotoxicities, (LD50 180–200 mg/mL). The authors point out that the co-occurrence of two related compounds (175 and 176) as epimers at C-12 shows that the enzymatic pathways for oxidation at C-12 for scalarane sesterterpenes are non stereoselective.
Davis and Capon [72] reported the isolation of the new isoscalarafuran A 177 and isoscalarafuran B 178, together with the known hyrtiosal 179, from a S. hispida collected in the Great Australian Bight (Figure 52).
Figure 52.

Structures of isoscalarafuran A 177, isoscalarafuran B 178, and hyrtiosal 179.
Compound 179 was identified by comparison with literature data. For 177 NMR data identified five tertiary methyls, two furan α-protons, acetoxy and hydroxy functions, and five sp2 carbons that led the authors to propose a tetracyclic sesterterpene. Comparison of the data with the known scalarafuran 170 and other literature compounds confirmed the localization of the hydroxy and acetoxy functions. Confirmation of the α-orientation at C-12 was obtained by the 1H chemical shift and multiplicity of H-12. Stereochemistry at C-16 was determined by molecular modelling. For 178 the main difference in respect to the 1H NMR data of 177 was the H-16 resonance, consistent with 178 being the C-16 epimer. Both compounds underwent decomposition before absolute stereochemistry, optical rotation and biological activity could be determined.
He et al. [73] reported the isolation of the new spongianolides A–F 180–185 (Figure 53) from a Spongia sp. collected in Florida in a bioguided study of protein kinase C (PKC) inhibitors.
Figure 53.

Structures of spongianolides A–F 180–185.
For 180 the sesterterpenoid skeleton was suggested by four methyl resonances in 1H and 13C NMR spectra where additional resonances could be assigned to rings A and B of spongiane type diterpenes. A γ-hydroxide butenolide moiety was also identified by NMR and UV indicated conjugation with a trans double bond. The full structure was assigned by HMBC. The A/B and B/C ring trans junctions were supported by nOe (H-9 to H-5, and H-9 to H-14). The β-configuration of the substituents at C-13 and C-14 was based on nOe between Me-23 and one CH2-24, and Me-23 and H-15. For 181, comparison with 180 showed the replacement of the acetyl group by a γ-hydroxybutyryl group. Comparison of 182 and 183 with 180 revealed that the olefinic protons were replaced by a methylene and an oxygenated methine, consistent with a tetrahydrofuranyl ring confirmed by COSY spectra. The authors suggest that biosynthetically this ring is formed by addition of C-13-OH to C-16 in 180. The diastereomeric relationship of 182 and 183 at C-16 was established by the J coupling values of H-16 with the diastereotopic H-15. The configuration of this center in both compounds was determined by nOe (H-18/H-24 for 182 and H-16/H-24, H-16/H-15β and Me-23/H-24 for 183). The measured coupling constant of CH2-15 and H-16 were very close to the corresponding values obtained from molecular mechanics calculations. Compounds 184 and 185 possessed a γ-hydroxybutyryl ester side chain instead of the acetyl group of 182 and 183. Lactones 180–184 inhibited PKC at IC50 20–30 μM and did not inhibit the human 85 kD phospholipase A2. Compounds 180–183 potently inhibited (IC50 0.5–1.4 μM) the proliferation of the mammary tumor cell line MCF-7. After completion of the manuscript compounds 182 and 183 were isolated from another sponge and designated as lintenolides A and B [74]. However, they were characterized only after acetylation and only partial spectroscopic data were reported for the natural products.
Lu and Faulkner [75] reported the isolation of 12α-acetoxy-19β-hydroxyscalara-15,17-dien-20,19-olide 186 and 12α,16β-diacetoxyscalarolbutenolide 187, together with the known scalarin 164 and 12α-acetoxy-16β-hydroxyscalarolbutenolide 188, from S. matamata de Laubenfels 1954, collected in a marine lake at Palau, Western Caroline Islands (Figure 54).
Figure 54.

Structures of 12α-acetoxy-19β-hydroxyscalara-15,17-dien-20,19-olide 186, 12α,16β-diacetoxyscalarolbutenolide 187, and 12α-acetoxy-16β-hydroxyscalarolbutenolide 188.
For 186 the presence of five methyl signals and an acetate group in 1H NMR indicated a pentacyclic scalarane skeleton. UV (conjugated diene), IR, 1H and 13C NMR, HMQC, HMBC, DQCOSY and NOEDS spectra allowed identification: coupling of the olefinic protons to a methine placed the conjugated diene on ring D; H-12 was assigned by HMBC correlations with Me-25 and nOe showed that it is β-equatorial (irradiation produced nOe on H-19 and Me-25); nOe also defined the regiochemistry of the butenolide ring showing that H-19 (which overlapped with H-15) is α-oriented (irradiation caused enhancement on the acetate methyl signal). 187 showed spectral data very similar to 188, except for the presence of an extra acetyl group. The significant downfield shift of H-16 confirmed its location. Further confirmation came from acetylation of 188. This compound had been reported in the abstract of a paper but not on the main text [76]. Its full characterization is reported and its assignment is based on spectral data analysis and comparison to 16-O-deacetyl-16-epi-scalarolbutenolide. The stereochemistry at C-12 was deduced from the small coupling constants observed.
From the already mentioned study of Rueda et al. [67] on S. agaricina, from Cádiz, Spain, 12,16-di-epi-12-O-deacetyl-16-O-acetylfuroscalarol 189 and 16-epi-scalarolbutenolide 190 (Figure 55), together with the known 12-epi-scalaradial 167, 12,18-di-epi-scalaradial 168, 12-epi-deoxoscalarin 165, 12-epi-scalarin 166, and scalarolbutenolide 173 were isolated.
Figure 55.

Structures of 12,16-di-epi-12-O-deacetyl-16-O-acetylfuroscalarol 189 and 16-epi-scalarolbutenolide 190.
For 189 the signals in 1H NMR for the five methyl groups and the furan moiety established the furoscalarol skeleton. IR and 1H and 13C NMR identified the hydroxyl and acetyl groups. Comparison with the known furoscalarol indicated substantial differences on the signals of protons geminal to the oxygenated functionalities. Thus, H-12 was assigned as axial and H-16 as equatorial. 190 was assigned a scalarane type skeleton on the basis of the 1H and 13C NMR signals of the five methyl groups. IR and 13C NMR established the α,β-unsaturated-γ-lactone. Comparison with the data of scalarolbutenolide 173 indicated the same functionality and stereochemistry at C-12 and an opposite orientation of the acetoxy group at C-16. Compound 189 showed cytotoxicity against P-388, A-549, HT-29 and MEL-28 tumor cell lines with IC50 values of 1 μg/mL and 190 showed weaker activity with IC50 of 5 μg/mL.
Tsukamoto et al. [77] described the bioassay guided isolation of 12-O-deacetylscalafuran 191, 12-O-deacetyl-12-epi-scalarin 192 and 12-O-acetyl-16-O-methylhyrtiolide 193 (Figure 56) together with the known 12-O-deacetyl-12-epi-19-deoxyscalarin 194, 12-epi-deoxoscalarin 165 and 12-epi-scalarin 166, from a Spongia sp. collected in the sea of Japan.
Figure 56.

Structures of 12-O-deacetylscalafuran 191, 12-O-deacetyl-12-epi-scalarin 192, 12-O-acetyl-16-O-methylhyrtiolide 193, and 12-O-deacetyl-12-epi-19-deoxyscalarin 194.
Compound 191 was assigned on the basis of 1H and 13C NMR, HMQC and HMBC data. The pentacyclic sesterterpene skeleton was inferred from the 13C NMR resonances of the five methyl groups and the presence of a furan ring was established by HMBC. The C-12-OH was suggested by IR and H-12 was established as α-axial on the basis of J couplings. Although this compound had previously been reported as a product of the hydrogenolysis of scalarafuran 170, this was its first occurrence as a natural product. Compound 192 was assigned by NMR data. The C-12-OH was established as α-axial on the basis of J couplings of H-12. nOe correlation with H-18 implied that the latter was also α-oriented. Stereochemistry at C-19 could not be resolved. Further confirmation came from the transformation into 12-epi-scalarin 166 by acetylation. For 193 the γ-hydroxy-butenolide ring was suggested by the deshielded methine signals, two quaternary olefinic carbons and a carbonyl resonance. Comparison of the data with that of hyrtiolide showed the extra O-acetyl and O-methyl groups, whose location came from HMBC. J couplings showed that H-12 was α-axial and that H-16 was β-equatorial. nOe showed that H-19 was β-oriented (correlation with Me-25). All six compounds were tested against L1210, HeLa, A549 and KB cell lines. The results are in Table 2.
Table 2.
Cytotoxicity of compounds against tumor cell lines [77].
| IC50 (μg/mL) | ||||
|---|---|---|---|---|
| Compound | L1210 | HeLa | A549 | KB |
| 165 | 2.1 | 22.5 | 29.4 | 16.2 |
| 166 | 13.2 | 26.0 | 23.7 | 18.5 |
| 191 | >50 | 19.5 | >50 | >50 |
| 192 | 2.3 | 15.0 | 14.8 | 14.3 |
| 193 | 2.2 | 5.3 | 5.3 | 15.6 |
| 194 | 1.6 | 16.5 | 16.5 | 17.1 |
| Mitomycin C | 0.020 | 0.015 | 0.020 | 0.015 |
Compounds 192, 165 and 194 were also tested for in vivo mean survival times (MST) and increases of life spans (ILS) in sarcoma-180-implanted mice. 194 showed significant ILS: 50.3% of ILS at 5 mg/Kg intraperitonial administration, and this is more potent than a positive control 5-fluoroacil (5-FU; 32.9%) at the same dose. Compound 165 also showed comparable ILS (28%) to 5-FU at 10 mg/kg, and 192 was inactive at 5 mg/kg.
Tokue et al. [78] reported the isolation of the new deacetoxy scalarin 195, together with the known 166, 192, 194, 196 and 197 from a Spongia sp. collected in Toyama Bay, Japan sea (Figure 57).
Figure 57.

Structures of deacetoxy scalarin 195, and compounds 196 and 197.
The known compounds were identified by comparison with literature data. For 195, IR, 1H and 13C NMR identified a carbonyl, an hydroxyl, an olefin, an oxygenated methine and five methyl groups. Further NMR data analysis (including COSY, HMBC and nOe) and comparison with 192 established the structure. nOe correlations between H-19 and Me-25 established the β-orientation of the former. Neurotrophic activity was tested using PC-12 (pheochromocytoma) cells. At the concentration of 50 μg/mL 196, 194, 166, 192 and 197 induced neurite outgrowth in PC-12 cells (68, 65, 58, 50 and 24% of the cells underwent outgrowth), but 195 was inactive.
Nam et al. [79] reported the isolation of three new sesterterpenes 12,24-diacetoxy-deoxoscalarin 198, 12-O-deacetoxyl-24-hydroxyl-deoxoscalarin 199 and 12-O-deacetoxyl-19-O-methyldeoxoscalarin 200, together with the known 201 and 165, from a Spongia sp. collected in Korea (Figure 58).
Figure 58.

Structures of 12,24-diacetoxy-deoxoscalarin 198, 12-O-deacetoxyl-24-hydroxyl-deoxoscalarin 199, and 12-O-deacetoxyl-19-O-methyldeoxoscalarin 200, and compound 201.
The structure of 198 was established by IR, 1H and 13C NMR, DEPT, COSY, HSQC, HMBC and NOESY, where olefin, acetal and acetate groups were revealed. A scalarane type skeleton with acetyl groups at C-12 and C-24 was thus assigned. The relative stereochemistry was established by NOESY where Me-23, C-24, Me-25 and H-19 showed the same orientation. H-18 was shown to be trans to H-19 by the J coupling observed and correlation with H-12 showed that both have a cis relationship (confirmed by J coupling value of the latter). Comparison of 199 with 198 showed that both have the same skeleton, the only difference being that two hydroxyl groups of the former are replaced by acetyl groups in the latter, as evidenced by the upfield shifts of H-12 and H-24. Synthesis of the triacetate of both compounds confirmed the assignment. The spectral data of 200 showed, when compared to 199, extra methoxyl and methyl groups and the loss of the downfield C-24 methylene. HMBC confirmed the location of the methoxyl group. All compounds were tested against FXR (nuclear hormone receptor, farnesoid X-activated receptor, a promising drug target to treat hypercholesterolemia in humans) transactivation (Table 3). The authors suggest that acetyl groups at C-12 and C-24 are critical for FXR antagonistic activity.
Table 3.
Activity against FXR [79].
| Compound | Inhibition FXR Transactivation IC50 (μM) | Cytotoxicity IC50 (μM) (CV-1 cell) |
|---|---|---|
| 165 | 81.1 | 98.5 |
| 198 | 8.1 | 32.7 |
| 199 | 64.5 | >100 |
| 200 | 24.8 | 86.9 |
| 201 | 25.3 | 29.2 |
| Z-Guggulsterone | 10.0 | Not determined |
Further studies of Nam et al. [80] on the activity of extracts as FXR transactivation inhibitors led to the isolation of the new 12-O-deacetyl-12-epi-19-deoxy-21-hydroxyscalarin 202, 12-O-deacetyl-12-epi-19-deoxy-22-hydroxyscalarin 203 and 12-O-deacetyl-12-epi-19-O-methylscalarin 204 (Figure 59), together with the known 12-O-deacetyl-12-epi-scalarin 192, 12-epi-scalarin 166 and 12-O-deacetyl-12-epi-19-deoxyscalarin 194, from a Spongia sp. collected in the South sea of Korea.
Figure 59.

Structures of 12-O-deacetyl-12-epi-19-deoxy-21-hydroxyscalarin 202, 12-O-deacetyl-12-epi-19-deoxy-22-hydroxyscalarin 203, and 12-O-deacetyl-12-epi-19-O-methylscalarin 204.
Compound 202 was assigned by IR, 1H and 13C NMR, COSY, HSQC, HMBC and ROESY: IR and 13C data indicated an hydroxyl and α,β-exounsaturated-γ-lactone; an oxygenated methylene at C-19 was assigned by COSY (coupling to H-18) and HMBC, and an oxygenated methylene at C-21 was assigned by HMBC. The relative stereochemistry was assigned by J coupling analysis and ROESY that showed that Me-22, Me-23, Me-24, Me-25 were axial, and were on the same plane as C-19. The magnitude of the J coupling of H-12 indicated also an axial orientation. The spectral data for 203 showed an identical structure to 202 except for ROESY correlations that established it was a diastereomer. The spectral data for 204 indicated a scalarane skeleton with a methoxy group at C-19. The location of the acetal, the γ-position of the lactone and the position of the double bond were determined by HMBC. The relative stereochemistry was assigned by J coupling and NOESY data that established H-12 as axial (correlation with H-18). The α-configuration of C-19-OMe was established by a NOESY correlation between H-19 and Me-25. All compounds were tested against FXR transactivation (Table 4). The stereochemistry at C-4 seems to be critical for biological activity since compounds 203, 194, 192 and 166 showed almost no activity below the IC50 value of cytotoxicity.
Table 4.
Activity against FXR [80].
| Compound | Inhibition FXR Transactivation IC50 (μM) | Cytotoxicity IC50 (μM) (CV-1 Cell) |
|---|---|---|
| 166 | 60.4 | 75.1 |
| 192 | 75.0 | 77.2 |
| 194 | 31.6 | 41.4 |
| 202 | 2.4 | 49.4 |
| 203 | >100 | >100 |
| 204 | 24.0 | 48.0 |
| E-Guggulsterone | 4.1 | Not determined |
Direct binding of scalarins 202–204, 194, 192 and 166 to the ligand binding domain (LBD) of FXR was monitored by using surface plasmon resonance (SPR) spectroscopy using a BIAcore system. They decreased the affinity of FXR LBD for SRC-1 peptide, which was facilitated by CDCA (chenodeoxycholic acid, a natural ligand for FXR). 202, which showed the most potent antagonistic activity against FXR in the cell-based cotransfection assay, was only a weak inhibitor or no inhibitor at all of the specific interaction between FXR and SRC-1 peptide. This result suggested that 202 should inhibit FXR transactivation by an indirect mechanism or by interaction with one of the other cofactors such as SRC-2or-3 in cells, which was not tested. Compounds 204, 192, 166 and 194 showed very strong direct interactions with FXR, although they were not especially potent in the cell based assay. This may be caused by the fact that FXR controls target gene expression in a ligand- and promoter-specific fashion. They may interact with FXR very well as on a natural promoter such as a bile salt export pump or cholesterol 7α-hydrolase, a well known target for FXR, while they activate the luciferase reporter gene poorly on a nonmammalian promoter, ecdysone receptor response element.
From the already mentioned study of De Marino et al. [42] on a Spongia sp. collected in Vanuatu Islands, Australia, 21-hydroxy petrosaspongiolide K 205 and 21-hydroxy petrosaspongiolide P 206, together with the known petrosapongiolides D 207 and G 208 were isolated (Figure 60). Compounds 205 and 206 are the 21-hydroxy derivatives of the known petrosaspongiolides K and P, respectively.
Figure 60.

Structures of 21-hydroxy petrosaspongiolide K 205, 21-hydroxy petrosaspongiolide P 206, petrosapongiolides D 207, and G 208.
For 205 comparison with the know petrosaspongiolides K led to the proposed structure. The localization of the hydroxyl group was confirmed by HMBC. For 206 the comparison with petrosaspongiolide P led to the proposed structure. The upfield shift for C-3 and the presence of an oxygenated methylene carbon were consistent with the proposed structure. For 205 mass spectrum, IR and 13C NMR data indicated the presence of a carboxyl group. The 13C NMR data also revealed a tricarbocyclic skeleton with geminal dimethyl groups at C-4, and two methyl groups at the ring junctions C-8 and C-10. A disubstituted pyridinium salt was also inferred from 13C NMR, confirmed by UV and IR absorptions typical of alkylpyridinium salts. COSY and HMBC allowed the proposal of the structure. For 206 the comparison with 205 allowed the identification of the acetoxymethyl group, located at C-4 by the downfield shift observed at C-4 and upfield shift observed at C-3. The stereochemistry at C-4 was determined by ROESY (intense cross peaks between CH2-17 and Me-19). For 207 1H and 13C NMR, COSY and HMBC data, together with comparison with 205 and 206 allowed the determination of the proposed structure. For 208 the comparison with 205 and the differences observed for the pyridine salt moiety, together with COSY and IR data allowed the determination of the taurine residue. HMBC established its location. . Inhibition of specific PLA2 enzymes constitutes a potentially useful approach for treating a great variety of inflammatory disorders. Compounds 205 and 206 were tested as inhibitors of sPLA2 (secretory phospholipase A2) enzymes belonging to the groups I (Naja naja venom and porcine pancreatic enzymes), II (human synovial recombinant and rat air pouch secretory enzymes) and III (bee venom enzymes). Compound 206 inhibited preferentially human synovial PLA2 in the μM range, showing a slightly lower potency towards this enzyme that that of the reference inhibitor, manoalide. None of the compounds was active against cPLA2 (cytosolic phospholipase A2 from macrophage cell line RAW 264.7), although this enzyme was partially inhibited by manoalide at 10 μM. The results are shown in Table 5.
Table 5.
Effect of compounds 205 and 206 on different sPLA2 activities a.
| Compound | N. naja Venom %I (10 μM) | Pancreas %I (10 μM) | Human Synovial %I (10 μM) IC50 (μM) | RAPb + Zymosan %I (10 μM) | Bee Venom %I (10 μM) IC50 (μM) |
|---|---|---|---|---|---|
| 205 | 1.3 ± 0.8 | 14.3 ± 6.8 | 34.4 ± 6.5 d | 18.8 ± 3.2 | 37.1 ± 6.3 d |
| 206 | 8.7 ± 3.9 | 19.5 ± 3.6 d | 87.2 ± 2.1 d 5.8 | 25.6 ± 1.9 d | 5.4 ± 2.1 |
| manoalide | 17.0 ± 1.7 c | 32.3 ± 2.7 d | 93.2 ± 0.2 d 3.9 | 38.4 ± 0.5 d | 62.5 ± 3.8 d 7.5 |
a Results show percentages of inhibition at 10 μM and IC50 (μM) values determined only for those compounds that reach 50% of inhibition. Mean ± S.E.M. (n = 6); b RAP: Rat air pouch PLA2; c p < 0.05; d p < 0.01.
Carr et al. [81] reported the isolation of a new nitrogen containing sesterterpenoid irregularasulfate 209, together with the known hipposulfate C 210, halisulfate-7 211 and igernellin 212 (Figure 61), from the bioassay guided fractioning of extracts of S. irregularis Ledenfeld from Papua New Guinea.
Figure 61.

Strutures of irregularasulfate 209, hipposulfate C 210, halisulfate-7 211, and igernellin 212.
Compound 212 was isolated from an inactive fraction. Compounds 210, 211 and 212 were identified by comparison with literature data. For 209, comparison of the NMR data (1H, 13C, DEPT, COSY, HSQC, HMBC) with that of halisulfate-7 211 showed the molecules were closely related. HMBC correlations indicated the attachment of C-16 to the α-carbon of an α,β-unsaturated-γ-lactam. COSY and HMBC identified the remaining isopentenyl fragment, whose attachment to nitrogen was confirmed by HMBC. The near identity of the carbon and proton shifts of the decalin system of 209 and 211 showed that the relative configurations at C-5, C-8 and C-9 were identical. The relative configuration at C-13 remained to be assigned in both compounds. Calcineurin (a serine/threonine protein phosphatase) is the indirect cellular target of two important immunosuppressive natural product drugs. Irregularasulfate 209 inhibited calcineurin in vitro with IC50 of 59 μM, while hipposulfate C 210 and halisulfate 211 showed calcineurin inhibition with IC50’s of 66 and 69 μM, respectively. To test for selectivity, 210 and 211 were also tested against pure preparations of the catalytic subunits of protein phosphatases PP-1 and PP-2A. They both showed similar potency against PP-1 (IC50’s 71 and 64 μM, respectively) as they did against calcineurin, but were less active to PP-2A (IC50’s of 130 and 140 μM, respectively). It seemed possible that the sulfated C-24 hydroxymethyl fragment in 209, 210 and 211 mimicked a phosphorylated serine residue in the natural substracts for calcineurin, PP-1 and PP-2A. In order to test this further, thiophosphate and phosphate analogues of the major S. irregularis metabolites hipposulfate C 210 and halisulfate-7 211 were prepared in an attempt to increase their potency as calcineurin inhibitors. The phosphate analogue of 211 was significantly less active than 211 against calcineurin (IC50 36 μM) but was more active against PP-1c (IC50 36 μM). A possible explanation was that the analogue acted as substrate and was being converted to the inactive alcohol during the assay. The thiophosphate analogue of 210 showed comparable activity (IC50 70 μM) against calcineurin to that of 210. The sulfate functionality seems to be essential for phosphatase inhibition.
Other Studies
Petrosaspongiolides A 213, B 214 and I 215 were isolated from a Spongia sp. collected in Vanuatu Islands, Australia [82] (Figure 62).
Figure 62.

Structures of petrosaspongiolides A 213, B 214, and I 215.
Deoxoscalarin 163, 16-deacetoxy-12-epi-scalarafuran acetate 174 and scalaradial 216 (Figure 63), were isolated from the already mentioned study on S. officinalis L. from Sicily of Manzo et al. [66]
Figure 63.
Structure of scalaradial 216.
6. Sterols
Aiello et al. [83] reported the isolation of three new 3β,5α-dihydroxy-6β-methoxycholest-7-enes from S. agaricina collected in Naples (Figure 64).
Figure 64.

Structures of 3β,5α-dihydroxy-6β-methoxycholest-7-enes 217, 218, and 219.
Compound 217 was identified by 1H and 13C NMR were the steroid skeleton was recognized by the five methyl resonances. The high field position of Me-18 in 13C NMR was in good agreement with a ∆7 sterol. Further evidence came from spin-decoupling experiments. The multiplicity of H-4 axial suggested that C-5 was not protonated. This fact, together with the downfield shift of H-3 relative to 5α-cholestan-3β-ol and the downfield shift observed for H-3 when the spectra was run in pyridine, led to assignment of the 5α-hydroxyl group. Spin decoupling experiments led to the oxidation pattern of ring B. Selective esterification of C-3-OH confirmed that the remaining hydroxyl group was located at C-5. The configuration at C-6 was established by J coupling analysis and was consistent with the lower than normal resonances of Me-19 and axial H-4. For 218 the comparison with 217 led to the assignment. The presence of a double bond in the side chain was inferred from mass spectrum and 1H NMR aided by spin decoupling experiments. The E configuration was established by the large coupling constant of H-22 and H-23. For 219 the comparison with 217 and 218 led to the assignment. The possibility that the compounds were formed by methanolysis of the allylic alcohol during extraction was excluded by repeating the extraction with EtOH.
Madaio et al. [84] reported the isolation of the known 220–228 together with the new 229–234 3β,5α,6β-trihydroxycholest-7-enes from S. officinalis collected in Naples (Figure 65).
Figure 65.

Structures of 3β,5α,6β-trihydroxycholest-7-enes 220–234.
The 3β,5α,6β-trihydroxy sterol nucleus was identified by the chemical shift values of H-3, H-4, H-6, H-7, Me-18 and Me-19. In addition the signals of H-3, axial H-4 and Me-19 showed the typical pyridine induced deshielding due to the 1,3-diaxial interaction with C-5 and C-6 hydroxyl groups. The 13C NMR shifts of C-3, C-5, C-6, C-7 and C-8 confirmed the assignment. For all the compounds, a [M+] could not be observed, the highest peak in mass spectra being at m/z values corresponding to [M − H2O]+. The new compounds were all isolated as mixtures (229 and 230, 231 and 232, 233 and 234). For 229 and 230 analysis of the mass spectra allowed the identification of a saturated C9H19 side chain. The chemical shifts of the side chain signals were consistent with those of authentic samples of campesterol and 24-epi-campesterol. For 231 and 232 the mass spectra allowed the identification of a C10H19 side chain containing a double bond; 1H–1H COSY 45 and irradiation allowed the assignment of the 1H NMR shifts. Comparison of the methyl region of the 1H NMR spectra with that of a 1:1 mixture of stigmasterol and poriferasterol revealed the presence of both epimers. For 233 and 234 the mass spectra allowed the identification of a C10H19 side chain containing a double bond and 1H decoupling experiments confirmed the presence of the ethylidene group at C-24. The side chain chemical shifts were consistent with those of fucosterol and 28-iso-fucosterol; the stereochemistry of the 24(28) double bond was determined by the chemical shift value of H-25, in accordance with the values reported for the known compounds.
Work of Migliuolo et al. [85] allowed the isolation of six new tetrahydroxylated sterols, 5α-cholest-7-ene-3β,5,6β,9-tetraol 235, (22E)-5α-cholest-7,22-diene-3β,5,6β,9-tetraol 236, (22E,24S)-24-methyl-5α-cholest-7,22-diene-3β,5,6β,9-tetraol 237, 24-methylene-5α-cholest-7-ene-3β,5,6β,9-tetraol 238, (24S)-24-ethyl-5α-cholest-7-ene-3β,5,6β,9-tetraol 239 and (24R)-24-ethyl-5α-cholest-7-ene-3β,5,6β,9-tetraol 240 from S. officinalis collected in Naples (Figure 66).
Figure 66.

Structures of 5α-cholest-7-ene-3β,5,6β,9-tetraol 235, (22E)-5α-cholest-7,22-diene-3β,5,6β,9-tetraol 236, (22E,24S)-24-methyl-5α-cholest-7,22-diene-3β,5,6β,9-tetraol 237, 24-methylene-5α-cholest-7-ene-3β,5,6β,9-tetraol 238, (24S)-24-ethyl-5α-cholest-7-ene-3β,5,6β,9-tetraol 239, and (24R)-24-ethyl-5α-cholest-7-ene-3β,5,6β,9-tetraol 240.
The mass spectra, 1H and 13C NMR data of all compounds allowed the identification of a common skeleton, thoroughly determined for 235 by NMR analysis of the 1H spectra run in CD3OD and pyridine, together with 13C NMR, DEPT, COSY 45, irradiation experiments and homo-decoupling spectral measurements. Five methyl signals of the cholestane skeleton were observed and the signal of H-3 showed the normal complexity of an α-oriented proton for an A/B trans steroid; its low field chemical shift was typical of 3β-hydroxysteroids with a 5α-hydroxyl group. The downfield shifts observed in pyridine for H-1 axial, H-3 axial, H-4 axial and Me-19 led to the placement of two hydroxyl groups in the 6β and 9α positions. The side chain structure was established by comparison of the 13C NMR data with that of cholesterol. nOe difference experiments confirmed the proposed structure and allowed the assignment of the overall relative stereochemistry: irradiation of 9α-OH enhanced axial H-1 and H-14; irradiation of H-3 enhanced equatorial H-2 and equatorial H-4; irradiation of Me-19 caused nOe on 6β-OH confirming its orientation. Chirality at C-17 was also determined by nOe that proved Me-18 and Me-21 are in proximity. For 236 and 237, analysis of the mass spectra and NMR data allowed the identification of the side chain. The E stereochemistry of both double bonds was established on the basis of the J coupling observed for H-22/H-23. For 237 the assignment of the configuration at C-24 was performed by comparison with an epimeric mixture of brassicasterol. For 238 the identity of the side chain was established by mass spectrometry. NMR data analysis, together with comparison of the 13C NMR shifts with those of 24-methylene-5α-cholest-7-ene-3β,6β-diol allowed its confirmation. Compounds 239 and 240 could not be separated, and NMR data analysis was difficult on account of signal overlapping. The chemical shifts of the side chain carbons and methyl protons, as well as the absolute configuration at C-24, were established by comparison with those of sitosterol and clionasterol. The authors also point out that, analogously to what had been stated by other authors on the biosynthesis of another organism, the co-occurrence in the same sponge of the isolated metabolites together with ∆7-3β,5α,6β-trihydroxysterols and the corresponding ∆5,7-3β-hydroxysterols, may indicate that the latter may be the biosynthetic precursors of both the above mentioned ∆7-tri and ∆7-tetrahydroxysterols.
Subsequent work by Migliuolo et al. [86] on a Mediterranean S. officinalis led to the isolation of the diacetate derivatives of six new sterols, 5α,6α-epoxycholest-8(14)-ene-3β,7α-diol 3,7-diacetate 241, (22E,24ξ)-5α,6α-epoxy-24-methylcholesta-8(14),22-diene-3β,7α-diol 3,7-diacetate 242, 5α,6α-epoxy-24-methylcholesta-8(14),24(28)-diene-3β,7α-diol 3,7-diacetate 243, 5α,6α-epoxy-cholest-8-ene-3β,7α-diol 3,7-diacetate 244, (22E,24ξ)-5α,6α-epoxy-24-methylcholesta-8,22-diene-3β,7α-diol 3,7-diacetate 245 and 5α,6α-epoxy-24-methylcholesta-8,24(28)-diene-3β,7α-diol 3,7-diacetate 246 (Figure 67).
Figure 67.

Structures of 5α,6α-epoxycholest-8(14)-ene-3β,7α-diol 3,7-diacetate 241, (22E,24ξ)-5α,6α-epoxy-24-methylcholesta-8(14),22-diene-3β,7α-diol 3,7-diacetate 242, 5α,6α-epoxy-24-methylcholesta-8(14),24(28)-diene-3β,7α-diol 3,7-diacetate 243, 5α,6α-epoxy-cholest-8-ene-3β,7α-diol 3,7-diacetate 244, (22E,24ξ)-5α,6α-epoxy-24-methylcholesta-8,22-diene-3β,7α-diol 3,7-diacetate 245, and 5α,6α-epoxy-24-methylcholesta-8,24(28)-diene-3β,7α-diol 3,7-diacetate 246.
Isolation of the compounds in pure form proved very difficult and they could only be separated after acetylation. Previous NMR analysis indicated the absence of natural acetyl groups. For 241 mass analysis after hydrolysis established the molecular formula and the presence of two hydroxyl groups in the natural compound. NMR analysis of 241 allowed the identification of the structure. The ∆8,(14) double bond was established by the chemical shift values of Me-18, C-8 and C-14. The chemical shift and shape of the signal of the 3α proton was in accordance with an A/B trans 3β-acetoxysteroid having a oxygenated function at C-5 with an α-orientation, confirmed by COSY spectra. This function was identified as an epoxide ring (1H and 13C NMR resonances). The second acetoxy group was located at C-7 since H-7 correlated with H-6. The cholestane type side chain was indicated by NMR and mass data. nOe experiments failed to establish the stereochemistry at C-7. The proposed structure was confirmed by X-ray analysis. Comparison of 242 and 243 with 241 showed that these compounds only differed in the side chain. These were established by MS data and NMR, including decoupling experiments. For 242 the configuration at C-24 remained unassigned. For 244 the mass spectrum and NMR data were very similar to 241. The positioning of all functionalities in the steroid nucleus was aided by 1H–1H COSY-60 and decoupling experiments. The α-orientation of the acetate group at C-7 was indicated by nOe between H-7 and Me-19. The double bond could only be located at the ∆8 position. Again comparison of 245 and 246 with 244 showed that these compounds only differed in the side chain. These were established by mass data and decoupling experiments. For 245 the configuration at C-24 remained unassigned.
Aoki et al. [87] reported the bioassay-guided isolation of agosterol A 247 from a Spongia sp. collected in Mie Prefecture. The ethyl acetate soluble portion of the acetone extract showed strong growth inhibition at 10 μg/mL against P-gp (P-glycoprotein) overexpressing MDR tumor cells (KB-C2) in the presence of 0.1 μg/mL of colchicine, while it exhibited little cytotoxicity against parental KB-3-1 cells at 10 μg/mL. Further work [88] led to the isolation of agosterol B 248, C 249, A4 250, D2 251, A5 252 and C6 253 together with agosterol A 247 (Figure 68).
Figure 68.

Structures of agosterol A 247, B 248, C 249, A4 250, D2 251, A5 252 and C6 253.
Compound 247 was identified on the basis of 1H and 13C NMR, COSY, TOCSY and HMBC spectral data. The relative stereochemistry was assigned on the basis of ROESY spectra and J couplings. The absolute configurations at C-11 and C-22 were established as R by a modified Mosher’s method. The configuration at C-20 was determined as S by comparison of the CD data of the 22-keto derivative (obtained by treatment of 247 with pyridinium dichromate) to that of (20S)-22-ketocholesterol.
The compounds 248–253 were analyzed by 2D NMR (COSY, HMQC, HOHAHA, HMBC and NOESY), compared to agosterol A 247, and their structures were elucidated. Compound 248 was very similar to 247, the difference being the lack of an acetyl group at C-6 (indicated by the higher field resonances for H-6 and C-6). For 249 the lack of the 3-O-acetyl group and higher field resonances of protons and carbons at C-3 and C-11 led to the proposed structure. For 250 the exomethylene group was located in C-24 by HMBC; the absolute configuration at C-22 was assigned as R by a modified Mosher’s method. The 20S configuration was presumed on the basis of the similarity of the C-21 shift with that of 247. For 251, treatment with 2,2-dimethoxypropane and PPTS afforded the acetonide derivative. Comparison of the Me-21 1H chemical shifts of both compounds established an 20R,22R configuration. The absolute configuration at C-22 was confirmed by a modified Mosher’s method. For 252 the E geometry of the ∆22 double bond was assigned by NOESY (correlation between H-22, H-24 and Me-28). The absolute stereochemistry at C-20 and C-24 were not determined. For 253 the side chain was identified by 1H NMR and HMBC. The absolute stereochemistry at C-20, C-22 and C-24 were not determined. Agosterol A 247 completely reversed the resistance to colchicine in KB-C2 cells (P-gp overexpressing strain) at 3 μg/mL and also the resistance of vincristine in KB-CV60 cells (which overexpress multidrug resistance-associated protein MRP), at 1 μg/mL. Compound 247 was not cytotoxic even at 10 μg/mL. The reversing activity of the analogous 248–253 and other derivatives were further examined. The results also showed that three acetyl groups in rings AB seem to be crucial for reversing activity. The C-11 and C-22 hydroxyl groups also seem to be crucial. Further studies on the mechanism by which agosterol A 247 reverses MRP I-mediated drug resistance through the investigation of the interaction between agosterols and MRP I in MRP I-over expressing human KB carcinoma cells were undertaken [89].
Migliuolo et al. [90] reported the isolation of the new 9,11-secosterol, 3β,6α-dihydroxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al 254, from S. officinalis L. collected in Naples (Figure 69).
Figure 69.

Structure of 9,11-secosterol, 3β,6α-dihydroxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al 254.
The structure of 254 was elucidated with mass spectrometry, UV, IR, 1H and 13C NMR, 1H–1H COSY, proton double quantum 2D NMR, 1H–13C heterocorrelation and decoupling experiments. The conjugated ketone was identified by UV and IR. Five methyl resonances indicated a steroid nucleus. The H-3 signal indicated a 3β-hydroxysteroid with A/B trans rings and the stereochemistry at C-6 was inferred from the J coupling values; the fact that C-6-OH was equatorial was indicated by the small downfield shift observed for Me-19, and the more substantial downfield shift on H-4 equatorial, when the proton spectrum was run in pyridine-d5. The localization of the CH2CHO fragment came from nOe that also fully supported the seco structure for ring C: irradiation of Me-18 caused effects on Me-19 and Me-21, and a strong effect on the H-7 signal, implying that the ring-D-containing portion of the molecule was slightly rotated around the C-8/C-14 bond. Mass spectra identified the side chain, confirmed by 13C NMR. Further proof of the proposed structure came from its synthesis from 7-dehydrocholesterol, the first synthesis of a natural ring-C secosterol.
Further studies of the same sponge [91] led to the isolation of the new 9,11-seco-3β,6α,11-trihydroxy-5α-cholest-7-en-9-one 255 and 9,11-seco-3β,6α,11-trihydroxy-24-methylene-5α-cholest-7-en-9-one 256 (Figure 70).
Figure 70.

Structures of 9,11-seco-3β,6α,11-trihydroxy-5α-cholest-7-en-9-one 255, and 9,11-seco-3β,6α,11-trihydroxy-24-methylene-5α-cholest-7-en-9-one 256.
For 255 the conjugated ketone was identified by UV and IR and confirmed by 13C NMR. Five methyl resonances indicated a steroid nucleus. The unsaturation equivalents deduced from the molecular formula indicated a secosterol structure. MS data and NMR, including 1H–1H COSY-45 indicated that the structure contained the same 9,11-seco-3β,6α-dihydroxy-7-en-9-one steroidal structure of 254 with an hydroxyl group at C-11 instead of the aldehyde group. The side chain structure was supported by MS and 13C NMR. Further confirmation came by synthesis from 254 by aldehyde selective reduction with tetra-n-butylammonium triacetoxyborohydride. Comparison of 256 with 255 showed that these compounds only differed in the side chain, which was identified by MS analysis and COSY-45. Assignment of the chemical shifts for the side chain carbons in the 13C NMR spectrum was based on comparison with the known values for 24-methylene-5α-cholest-7-en-3β,6α-diol.
Further investigation of the same sponge [92], led to the isolation of 3β-acetoxy-5,6β-dihydroxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al 257 (Figure 71).
Figure 71.

Structure of 3β-acetoxy-5,6β-dihydroxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al 257.
Compound 257 was elucidated with mass spectrum, UV, IR, and NMR with decoupling experiments, and comparison with 254. IR indicated an ester, α,β-unsaturated-ketone and hydroxyl functions. The enone moiety was confirmed by UV and 13C NMR. Four methyl resonances (one integrating for six protons) suggested a steroid of the cholestane series. The unusually high chemical shift resonance of H-3 indicated, apart from acetylation, an α-oriented hydroxyl group at C-5, which was confirmed by the multiplicity of both H-4, and the strong pyridine induced shifts experienced by the axial H-1 and H-3. The β-orientation of C-6-OH was also determined on the basis of the pyridine downfield shifts observed for Me-19 and H-4 axial. 1H–1H COSY and 1H–13C heterocorrelation fully elucidated rings A and B. nOe experiments established the location of the CH2CHO group and a ring-C seco structure (strong enhancement of H-7 upon irradiation of Me-18). The sidechain was identified by 1H NMR and mass fragments. Further confirmation of the structure came from its synthesis from 7-dehydrocholesterolacetate. The authors suggest that both 257 and 254 derive from a common 5,7,9(11)-triene sterol through oxidation at C-5 and C-6, in the case of 257, or only at C-6 in the case of 254, with concomitant oxidative cleavage of the 9,11 double bond.
From the already mentioned study of Lu and Faulkner [75] on S. matamata de Laubenfels 1954, collected in a marine lake at Palau, Western Caroline Islands, the new 3β-hydroxy-5α,6α-epoxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al 258 was isolated (Figure 72).
Figure 72.

Structure of 3β-hydroxy-5α,6α-epoxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al 258.
Compound 258 was identified by comparison with the known luffasterol A, the acetyl derivative. Synthesis of this compound by acetylation of 258 confirmed the structure.
From the already mentioned study of Rueda et al. [67] on S. agaricina, from Cádiz, Spain, the new 3-O-deacetylluffasterol B 259 and 3-O-deacetyl-22,23-dihydro-24,28-dehydroluffasterol B 260 (Figure 73), together with the known 258, were isolated.
Figure 73.

Structures of 3-O-deacetylluffasterol B 259 and 3-O-deacetyl-22,23-dihydro-24,28-dehydroluffasterol B 260.
Compound 259 was identified by spectral data and comparison with the known luffasterol B, the acetyl derivative. Synthesis of this compound by acetylation of 259 confirmed the structure. Compound 260 was identified by comparison with 259, where the replacement of Me-28 and H-22 and H-23 proton signals by two exomethylene signals, was observed. The authors also propose that 259, 260 and 258 derive from the same 5,7,9(11)-trienesterol precursor proposed by Adinolfi et al. [92]. Compounds 259 and 260 showed cytotoxicity against P-388, A-549, HT-29 and MEL-28 tumor cell lines with IC50 values of 1 μg/mL.
7. Macrolides
Quiñoà et al. [93] reported the isolation of two cytotoxic macrocyclic lactones, fijianolides A 261 and B 262, from S. mycofijiensis, collected in Vanuatu Islands, Australia (Figure 74).
Figure 74.

Structures of fijianolides A 261 and B 262.
Both metabolites were identified by mass spectrometry and extensive NMR analysis. For 261 the unsaturated structures were assigned as follows: the but-2(Z)-enoyl array (as a lactone) was recognized by IR and NMR shifts; the dihydropyran ring A with vinylic methyl and equatorial (E)-ethenyl was identified by NMR resonances and the large J value at H-23; the second dihydropyran (ring C) with a disubstituted double bond was deduced from the value of J6,7 and the 1,3 C-ring substituents were assigned as trans by J analysis of H-5 and H-9; 1H–1H and 1H–13C COSY data allowed the proposal of a 2-methyl-4-methylenepentanyl group and the confirmation of the connections showed in 261 for the contiguous carbons. A vicinal diol was recognized by conversion to the diacetate and dioxolane and analysis of the H-15 and H-16 J values. Ring B was identified by 1H NMR chemical shifts and 1H–1H COSY spectra revealed its location. Its stereochemistry was confirmed by comparison to furanose rings and J value analysis. 2D nOe experiments confirmed the cis arrangement of H-19 and H-20. The relative H-16/H-17 erythro stereochemistry was assigned by comparison of J values with model compounds. A relative stereochemistry of 15S*,16S*,17R*,19S*,20S* is proposed for 261. Compound 262 was identified by comparison of the NMR data with those of 261 that showed that the structure and the stereochemistry were the same except for the region of C-15 to C-20. Conversion to the diacetyl derivative allowed further identification. The trans epoxide ring was identified by the characteristic 1H and 13C NMR shifts and the value of J16,17. The authors suggest that both compounds are biogenetically related by an SN2 transposition with inversion at C-17 and that their stereochemistry is the same at the remaining centers. As such, 262 is assigned a 15S*,16S*,17S*,19S*,20S* relative configuration. Moderate in vitro cytotoxicity was shown by 261 and 262 acetate against HT-29 (IC50 11 μg/mL and 0.5 μg/mL, respectively) and P388 (IC50 9 μg/mL and 6 μg/mL, respectively).
Work of Pettit et al. [94,95,96] led to the isolation of spongistatins 1–3 263–265 and dictyostatin 1 266 from a Spongia sp. collected in the Eastern Indian Ocean, and its recollection in the Republic of the Maldives (Figure 75).
Figure 75.

Structures of spongistatins 1–3 263–265 and dictyostatin 1 266.
All compounds were isolated from P388 lymphocytic leukemia bioassay active fractions. The structure of 263 was identified on the basis of extensive NMR data analysis in three solvents, at 400 and 500 MHz (including APT, 1H–1H and 1H–13C COSY, HMBC and nOe). Selective acylation experiments were also employed. Due to the small amount available and difficulties in crystallization, the relative and absolute stereochemistry was not established. Compounds 264 and 265 were identified by extensive NMR analysis and comparison with 263. For 264 the presence of a ABX spin system in the 1H NMR spectra confirmed that the difference between both compounds was at C-50, confirmed by shifts observed at C-51, C-50, C-49, C-48 and C-47. For 265 the main difference relative to 263 was the absence of an acetyl group at C-5, established by the upfield shift of the corresponding H-5. The structure of 266 was identified on the basis of extensive NMR data analysis at 400 and 500 MHz (including APT, 1H–1H COSY, HMQC, HMBC and nOe). An ABX spin system in 1H NMR indicated a terminal unit. A broad singlet of an oxygenated methine correlating to a carbonyl in HMBC suggested a macrolide. The coupling relationships of signals corresponding to H-2, H-3, H-4 and H-5 were established and extended to H-13. Coupling of H-13 to H-19 was also established and extended to H-23 and H-26. The absence of a dihydropyran ring between C-9 and C-13 was established by nOe experiments and mass spectrometry. The geometry of the double bonds was inferred from J coupling values. The definitive relative and absolute stereochemistry were not assigned. At the same time, the isolation of spongistatin 1 263 was reported, Kobayashi [97] and Fusetani [98] reported the isolation of the same metabolite from other marine sources (altohyrtin A and cinachyrolide A, respectively). The proposed configuration for spongistatin 1 263 [96,99] was in disagreement with the proposed configuration of the other compounds [98,100]. The absolute configurations of spongistatin 1 263 here depicted were only determined by synthesis in subsequent studies by other authors [101,102]. The authors suggest that the depicted configurations are common to all the members of this class of spongipyran natural products. Compound 263 was found to be extremely potent (GI50 typically 2.5–3.5 × 10−11 M) against a subset of highly chemoresistant tumor types (e.g., HL-60, SR leukemias; NCI-H226, NCI H23, NCI H460, NCI H522 non-small cancer lung; DMS 114 and DMS 273 small cell lung; HCT-116, HT29, KM12, KM 20L2 and SW-620, colon; SF-539, U251 brain; SK-MEL-5 melanoma; OVCAR-3 ovarian; and RXF-393 renal cancers) comprising the NCI panel of 60 human cancer cell lines. Cell lines derived from human melanoma and lung, colon, and brain cancers were found to be especially sensitive to spongistatin 1 263. The distinctive pattern of relative cellular sensitivity to spongistatin 1 263 was analyzed by computerized pattern-recognition algorithms and found to be closely related with the important general mechanistic class of microtubule interactive antimitotics. Spongistatins 2 264 and 3 265 showed a diminished overall potency when compared to 263 in the NCI 60 cell line in vitro screening panel. Compounds 263, 264 and 265 remain however among the most potent of substances tested in the NCI screen.
Bai et al. [103] showed that spongistatin 1 263 inhibited mitosis, microtubule assembly and the binding of vinblastine to tubulin. Subsequent studies [104] on the interaction of macrocyclic lactone polyethers with tubulin showed that spongistatin 3 265 inhibits the formation of the same intrachain cross-link in tubulin as is inhibited by vinblastine. Unlike vinblastine, 265 has no effect on the exposure of either sulfhydryl groups or hydrophobic areas on the tubulin molecule.
From the already mentioned study of Grassia et al. [82] on a Spongia sp. collected in Vanuatu Islands, Australia, the new spongidepsin 267 was isolated by bioassay guided fractionation (Figure 76).
Figure 76.
Structure of spongidepsin 267.
Compound 267 was identified through extensive NMR analysis including HSQC, HMBC, COSY and TOCSY. These spectra allowed the identification of the 9-hydroxy-2,4,7-trimethyltetradeca-14-ynoic acid. Further confirmation of the alkyne function came from 13C NMR resonances and HMBC data. The remaining NMR data suggested the presence of a N-methylphenylalanine residue. Further HMBC analysis allowed the closing of the 13-membered macrocyclic ring and determination of the carbon framework. The l-series was assigned to the N-methylphenylalanine residue through the application of Marfey HPLC method on the acidic hydrolysate of 267. The relative configuration at C-2 and C-4 was investigated using an NMR approach described by Murata that relies on 3JH,H and 2,3JCH measurements in combination with ROESY responses, which allows the identification of the predominant conformers among the six staggered rotamers with threo and erythro relative configurations. Stereochemistry at C-7 and C-9 could not be assigned due to signal overlap that precluded J analysis. Subsequent work by two other authors [105,106] led to the determination of the absolute configuration spongidepsin 267 by total synthesis. Thus, (2R,4R,7R,9R,16S)-spongidepsin can be assigned to structure 268 (Figure 77).
Figure 77.
Structure of (2R,4R,7R,9R,16S)-spongidepsin 268.
Antiproliferative assays for spongidepsin 268 and of the control (6-mercaptopurine) are shown in Table 6.
Table 6.
In vitro antiproliferative activity of spongidepsin 268 [82].
| Cell Lines | 268 IC50 (μM) | 6-Mercaptopurine IC50 (μM) |
|---|---|---|
| J774.A1 | 0.56 | 0.003 |
| HEK-293 | 0.66 | 0.007 |
| WEHI-164 | 0.42 | 0.017 |
8. Miscellaneous Compounds
From the already mentioned study of Lumsdon et al. [62] on a Spongia sp. collected in Australia, the new p-quinol 269 was isolated (Figure 78).
Figure 78.

Structure of p-quinol 269.
NMR analysis identified a 1,2,4-trisubstituted aromatic ring, four internal trisubstituted double bonds and one terminal trisubstituted double bond of a pentaprenyl side chain with the corresponding methyl groups. The 13C NMR shifts of the vinylic methyls established an all E geometry of the double bonds while the doublet multiplicity of the benzylic methylene due to coupling to the olefinic proton allowed the double bond regiochemistry to be assigned. The substituents on the aromatic ring were supported by MS data and IR absorptions. Comparison with literature compounds supported the aromatic pattern shown. Although the crude extract evoked a large triphasic contraction of smooth muscle in the isolated guinea-pig ileum, appeared to inhibit contractions elicited by different drugs (acetylcholine, 5-hydroxytryptamine and histamine) of the isolated guinea-pig ileum, and inhibited the growth of several bacteria (Staphylococcus aureus, Micrococcus sp. and Serrata sp.) in a standard antibiotic disk assay, 269 showed no activity.
Kalidindi et al. [107] reported the isolation of pokepola ester 270 from S. oceania collected in Maui, Hawaii (Figure 79).
Figure 79.

Structure of pokepola ester 270.
The structure was identified by mass spectrometry, IR and NMR (including HMBC and COSY), where a homoserine moiety connected as an amide to C12 carboxylic acid terminated by a furan ring, and an esterified 2-methylhexanol residue were identified. 13C NMR, where coupling to phosphorus was observed, together with 31P NMR indicated the presence of a phosphate group. The geometry of the double bond was established as E by nOe experiments where effects were observed between H-3′ and H-5′ and CH2-2′ and Me-8′. The absolute configuration of homoserine was determined to be D((R)-2-amino-4-hydroxybutyric acid) by Marfey’s method. This absolute configuration suggests a microbial origin whereas the C12 carboxylic acid appears to be a trisnorsesquiterpene. Compound 270 showed mild anti HIV activity at a concentration of 0.2 μg/mL without showing any cytotoxicity.
Pettit et al. [108] reported the isolation of spongilipid 271 from S. cf. hispida collected in the Republic of Singapore (Figure 80).
Figure 80.

Structure of spongilipid 271.
IR data indicated the presence of hydroxyl, ester and ether groups and preliminary 1H NMR analysis showed a long chain aliphatic unit; mass spectrometry indicated a C16 saturated aliphatic ester (palmitoyl group). Conversion to the pentaacetate allowed simplification of the spectra, identification of a β-linked sugar and of a glycerol unit. APT NMR experiment for carbon hybridization combined with COSY, HMQC and HMBC led to the assigned structures of both the pentaacetate and 271. The structure was unequivocally confirmed and the absolute chemistry assigned by X-ray analysis. Compound 271 was active against Enterococcus faecalis. Under standard P388 techniques, the activity of 271 ranged from 2 to >100 mg/mL, presumably owing to difficulties caused by severe solubility problems and suspension formation in the cell media.
Xu et al. [109] reported the isolation of three new alkaloids 272–274 from S. obligue collected in the South China Sea (Figure 81).
Figure 81.

Structure of the alkaloids 272–274.
The structures were established by UV, IR, MS, 1D and 2D NMR and elemental analysis.
Kobayashi et al. [110] reported the isolation of spongiacysteine 275 from a Spongia sp. (Figure 82).
Figure 82.
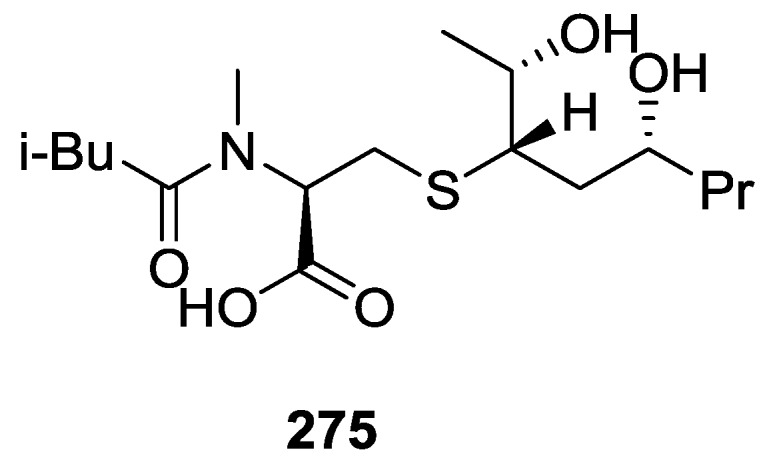
Structure of spongiacysteine 275.
The structure was elucidated by spectroscopic analysis and the absolute stereostructure was established by total synthesis. 275 showed moderate antimicrobial activity against the rice blast fungus Pyricularia oryzae.
Lin et al. [111] reported the isolation of a new ceramide 276, together with 277 and the guanidine acetic salt 278 from S. zimocca subspecies irregularia (Ledenfeld) (Figure 83).
Figure 83.

Structures of ceramide 276, compound 277 and the guanidine acetic salt 278.
The structures were determined by spectroscopic methods.
Xu and Yang [112] reported the isolation of three new ceramides 2-hydroxy-N-(1,3,4-trihydroxy-17-methyloctadecan-2-yl)-18-methylarachidamide 279, 2-hydroxy-N-(1,3,4-trihydroxy-17-methyloctadecan-2-yl)-19-methyl-henicosanamide 280 and 2-hydroxy-N-(1,3,4-trihydroxy-17-methyloctadecan-2-yl)-20-methyl-behenamide 281 from S. suriganensis collected in the South China Sea (Figure 84).
Figure 84.

Structures of the ceramides 2-hydroxy-N-(1,3,4-trihydroxy-17-methyloctadecan-2-yl)-18-methylarachidamide 279, 2-hydroxy-N-(1,3,4-trihydroxy-17-methyloctadecan-2-yl)-19-methyl-henicosanamide 280, and 2-hydroxy-N-(1,3,4-trihydroxy-17-methyloctadecan-2-yl)-20-methyl-behenamide 281.
The structures were confirmed by spectroscopic analysis and hydrolysis.
Guan and Zeng [113] reported the isolation of a ceramide, N-palmitoyl-heptacosane-1,3,5-triol 282, from Spongia sp. collected in Hainan Province, China (Figure 85). The isolation of this compound was also reported in a Chinese Journal [114], where the presence of several other compounds (including sterols) is mentioned.
Figure 85.

Structures of N-palmitoyl-heptacosane-1,3,5-triol 282.
The structure was identified by IR, 1H, 13C and DEPT, together with FAB-MS data. The FAB-MS fragmentation showed peaks corresponding to fragments G and T, according to sphingolipid mass fragmentation patterns, which allowed the identification of the proposed structure. Further confirmation came from hydrolysis of 282: the hexane extract furnished methyl palmitate as sole product.
Salim et al. [115] reported the isolation of heterofibrins A1 283, A2 284, A3 285, B1 286, B2 287 and B3 288 from Spongia (heterofibria) sp. collected in the Great Australian Bight (Figure 86).
Figure 86.

Structures of heterofibrins A1 283, A2 284, A3 285, B1 286, B2 287 and B3 288.
For 283 UV indicated a diyne-ene moiety. The NMR indicated a carboxylic acid, two fully substituted acetylenes, an E-1,2-disubstituted olefin, a primary methyl and ten methylene carbons suggestive of an acyclic unbranched fatty acid. Analysis of 2D-NMR revealed a diagnostic correlation sequence from the carboxylic acid terminus, to the four acetylenic carbons, and from there to the olefinic methines, and on through the methylenes C-11, C-12 and C-13. The structure was thus assigned. Comparison of the data of 286 with that of 283 allowed the determination of the proposed structure. For 284, comparison with 283 allowed the determination of an extra lactyl ester and a diastereotopic nature for both H-2 hydrogens. The 2′S absolute stereochemistry was assigned on the basis of the comparison of the value of the optical rotation with that of the synthesized (2′S)-lactyl linoleoate (the (2′R) isomer was also prepared). For 287, comparison with 284 allowed the identification of the structure. The 2′S stereochemistry was assigned on the basis of the comparison of the optical rotation with those of 284 and (2′S)-lactyl linoleoate. Both 285 and 288 proved hard to purify. They were isolated as mixtures with 13-methylmyristic acid and palmitic acid, respectively. The ratios and the structure elucidation of each heterofibrin and associated fatty acid were confirmed by spectroscopic analysis and comparisons with model compounds (all the possible isomers of dilactyl linoleate). Only 283 and 286 inhibited the formation of lipid droplets (LD) in A431 fibroblast cells (up to 60% at 10 μM). All of the metabolites were non cytotoxic to the A431 fibroblast cells in the LD bioassay, or mammalian HeLa (cervical) and MDA-MB-231 (mammary epithelium) cancer cell lines (up to 30 μM). Likewise they were all non-cytotoxic to Candida albicans, Pseudomonas aeruginosa or Escherichia coli (IC50 values > 50 μM). They did display weak antibacterial activity against Gram positive bacterium Bacillus subtilis (10 < IC90 < 60 μM), while 283 and 286 displayed weak activity against Staphylococcus aureus (4 < IC90 < 45 μM). Further studies [116] on the effect of heterofibrin A1 283 on the cellular storage of neutral lipids were undertaken [116]. Inhibition of LD biogenesis by heterofibrin A1 283 was observed for A431 cells and AML12 hepatocytes. The activity was dose dependent with 20 μM inhibiting LD formation and triglyceride accumulation by 50% in the presence of 50 μM oleic acid. Compound 283 significantly reduced the intracellular accumulation of fatty acids and resulted in the formation of distinct fatty acid metabolites in both cultured cells and in embryos of the zebrafish Danio rerio.
From the already mentioned study of Manzo et al. [66] on S. officinalis collected in Sicily, officinoic acid A 289 and officinoic acid B 290 were isolated (Figure 87).
Figure 87.

Structures of officinoic acid A 289 and officinoic acid B 290.
For 289 IR and 13C NMR allowed the identification of a carboxylic acid and an α,β-unsaturated ester. The 13C NMR spectrum also showed signals due to a trisubstituted double bond and 16 sp3 resonances including a signal due to an oxygenated methylene, which were consistent with an acyclic carbon skeleton. The 1H NMR spectrum showed, among others, four methyl signals. Analysis of 1H–1H COSY and HMBC identified the structure. The geometry of the double bond was established as Z by the value of the vinyl methyl carbon signal and nOe between Me-10′ and H-2′. The configurations at C-3 and C-7 were unassigned. The 2D NMR data of the methyl derivative was in accordance with the proposed structure. This derivative was submitted to methanolysis and the reaction mixture was analyzed by GC-MS where diagnostic molecular ion peaks for the two methyl esters were observed. Compound 290 was identified by NMR data analysis and comparison to 289. The geometries of the double bonds were established as Z by the chemical shift values of the vinyl methyls and nOe (between Me-11 and H-4, and Me-11′ and H-2′). The configuration at C-9 was unassigned. Compound 290 was also converted to the methyl ester which was characterized by 2D NMR. Methanolysis of the derivative gave a reaction mixture analyzed by GC-MS, where again diagnostic molecular ion peaks for the two methyl esters were observed.
From the already mentioned study of Ponomarenko et al. [39] on a Spongia ssp. (subgenus Heterofibria) collected in Northern Cook Island 291 was isolated (Figure 88).
Figure 88.
Structure of compound 291.
The effects of 291 on the biosynthesis of nucleic acids and on the embryonic development of the sea urchin Strongylocentrotus intermedius were studied. The compound inhibited sea urchin embryo development at concentrations of 20 μg/mL and above and DNA biosynthesis at the dose of 10 μg/mL. The inhibitory effect of this compound may partly be explained by the inhibition of thymidine kinase activity. Compound 291 stimulated RNA synthesis in the developing sea urchin embryos.
Carballeira et al. [117] reported the total phospholipid fatty acid composition of S. tampa, collected in Puerto Rico. The most prominent phospholipid fatty acids were 5,9-pentacosadienoic acid and 5,9-hexacosadienoic acid. 9- and 16-pentacosenoic acids and 9-, 15- and 16-hexacosenoic acids were identified as trace amounts. The purified fraction of phosphatidylethanolamines was shown to contain C14–C22 fatty acids. The principal saturated fatty acids were tetradecanoic acid, hexadecanoic acid and octadecanoic acid. The principal fatty acids in the phosphatidylethanolamines were the monosaturated 9-octadecenoic acid and 13-docosenoic acid. 9-hexadecenoic acid and 11-octadecenoic acid were also found. The only dienoic acid present was 9,12-octadecadienoic acid. The positional distribution of the C14–C22 fatty acids in the phosphatidylethanolamines was examined. 9,12-octadecadienoic acid was shown to have no preference for the sn-1 or sn-2 positions in the phosphatidylethanolamines. The other saturated and unsaturated fatty acids with chains between 14 and 22 carbons long were also found to be equally distributed between the sn-1 and sn-2 positions of the phosphatidylethanolamines of this sponge.
Junqua et al. [118] reported the isolation and partial characterization of glycoconjugates from S. officinalis collected near Marseille. Two types of glycoconjugates were distinguished with respect to the size of their sugar moiety: type 1 contained 4%–7% carbohydrate, showed affinity for lectins and represent about 63% of total glycoconjugates; type 2 contained about 30% carbohydrate and some sulphate (about 2.7%) and were not bound to lectins. Type 1 glycoproteins had mol. wt. of 10, 16, 21 and 32 K. Glucuronic acid was present in all fractions together with galactose, mannose, glucose, fucose, arabinose and N-acetylglucosamine. Type 2 glycoconjugates had a much higher mol. wt. N-acetylglucosamine, N-acetylgalactosamine, galactose, mannose, fucose, arabinose and glucuronic acid were present. The aminoacid and carbohydrate composition revealed a striking similarity between sponge glycoproteins and structural glycoproteins isolated from vertebrate tissues.
9. Other Reports
Noyer et al. [119] reported the intraspecific diversity of the Mediterranean S. lamella. The chemical profiles of seven populations spreading over 1200 km in the Western Mediterranean were obtained by SPE-HPLC-DAD-ELSD. Nitenin 117, isonitenin 155, dihydronitenin 118, 12-episcalarin 166, 12-epi-deoxoscalarin 165, 12-epi-scalaradial 167, 12,18-di-epi-scalaradial 168 and ergosteryl myristate 292 (Figure 89) were identified by comparison of the HPLC-MS spectra and 1H NMR with literature data.
Figure 89.

Structure of ergosteryl myristate 292.
Terem and Scheuer [120] reported the isolation of the known scalaradial 216 and 12-deacetylscalaradial 293 from an associated S. oceania and Chromodoris youngbleuthi, collected in O’ahu, Hawaii. From the nudibranch 216 was absent and 293, and the new 12-deacetyl-12-epi-scalaradial 294 and 12-deacetyl-18-epi-12-oxoscalaradial 295 were isolated (Figure 90).
Figure 90.

Structures of 12-deacetylscalaradial 293, 12-deacetyl-12-epi-scalaradial 294, and 12-deacetyl-18-epi-12-oxoscalaradial 295.
216 and 293 were identified by comparison of their spectral data with literature compounds. For 295 the chemical shift of both H-11 suggested a 12-oxo functionality, confirmed by 13C NMR. Stereochemistry at C-18 was inferred by the J coupling value of the C-19 aldehyde proton, and confirmed by nOe, where irradiation of Me-25 enhanced the equatorial H-18. Analysis of the NMR data of 294 indicated a diastereomer of 293. nOe studies were inconclusive for the assignment of the stereochemistry that had to be assigned by comparison of the spectral data of the acetylated derivative with literature compounds. The relative amounts of the 293, 294 and 295 suggest that 293 is converted into the other metabolites by the nudibranch, which is supported by the absence of 294 and 295 in the sponge. Both 293 and 294 tasted more bitter than 295. Fish feeding studies on Tilapia sp. in freshwater failed to quantify this finding although preliminary studies on filefishes showed on the basis of frequency counts that 293 was rejected more frequently than 295.
Kakou and Crews [121] reported the isolation of dendrolasin 296 and latrunculin A 297 from an association of S. mycofijijensis and the nudibranch Chromodoris lochi (Figure 91).
Figure 91.

Structures of dendrolasin 296 and latrunculin A 297.
Collections were from two distinct locations in the Fiji Islands. Although both organisms are associated separate extracts were performed and the content of each of the metabolites determined by 13C NMR. Compound 297 was completely toxic to Hep-2 and MA-104 cells at 0.072 μg/mL and 0.23 μg/mL, respectively. Compound 296 was completely toxic to Hep-2 at 24 μg/mL but inactive against MA-104 cells.
Further studies [122] led to the recolection of the same pair of organisms in Vanuatu Islands, Australia. From the sponge, mycothiazole 298 was isolated (Figure 92).
Figure 92.

Structure of mycothiazole 298.
It is noteworthy that 13C analysis of the extract showed the absence of dendrolasin A 296. Mycothiazole 298 was identified by mass spectrometry and extensive NMR analysis (including spectra in three different solvents, 1H–1H COSY and 1H–13C COSY). For the determination of the connection site of the thiazole a study of the substituent increment shifts in 13C NMR and of the JCH coupling values in substituted thiazole rings was performed, using literature data. A biosynthetic relation for mycothiazole 298 and lantrunculin B is proposed. Compound 298 was completely active at 50 μg/mL in an antihelminthic (in vitro) assay against Nippostrongylus braziliensis. It was deadly to mice at 10 mg/kg when injected intraperitonially, but no toxicity was seen by this route at 3 mg/kg.
Three reports by Guella et al. [123,124,125] focused on the metabolites of S. zimocca and of the seaweed Laurentia microcladia. Both species grow in a narrow area off torrent of Il Rogiolo, south of Livorno. From the sponge the new rogiolol acetate 299, rogiolenyne B 300 and rogiolenyne C 301 were isolated, together with the known isopimarane 302 (semisynthetic), chamigrene 4E 303, chamigrene 4Z 304, bromosphaerol 305, sphaerococcenol A 306 and furospongin-1 119 (Figure 93).
Figure 93.

Structures of rogiolol acetate 299, rogiolenyne B 300, rogiolenyne C 301, isopimarane 302, chamigrene 4E 303, chamigrene 4Z 304, bromosphaerol 305, and sphaerococcenol A 306.
From the seaweed L. microcladia the deacetyl derivative of 299 was isolated, together with 300, 303 and 304. The presence of the di-deacetyl derivative of 302 could not be ascertained. The finding that 303 and 304, as well as traces of 300, are present ion both species strongly suggest that these metabolites are obtained by the sponge from the seaweed. A similar suggestion can be made about 305 and 306 since they are produced by the red seaweed Sphaerococcus coronopifolius, which also grows in the same area as S. zimocca. The authors suggest that S. zimocca either engulfs the seaweeds like the action of a solvent, or that it filters solid matrices (maybe algal spores) containing the compounds released by the seaweeds. The authors also suggest that compounds 299, 301 and 302 are acetylated by the sponge. NMR data and MS data of both 299 and the deacetyl derivative, obtained by mild saponification, allowed the determination of the molecular formula and establishment of the presence of halogens. The β-chamigrene structure was deduced from NMR data where the exo methylidene group, the gem-dimethyl group and the spiro center could be identified. The location of the substituents was obtained by comparison with literature compounds and confirmed by COSY and 1H–13C correlation experiments. The relative configuration at the five carbon centers was derived from J coupling values and nOe. Further confirmation came from the addition of Eu(fod)3 to the deacetyl derivative, where all the paramagnetic shifts observed were in accordance with the proposed structure. Chemical transformation of the deacetyl derivative of 299 allowed the proposal of an absolute stereochemistry of (2R,3S,6R,8R,9R) (CD data) confirmed by chemical correlation of the deacetyl derivative with a known compound. For 300 and 301 mass spectrometry established the molecular formula and the presence of the chlorine and bromine atoms. Comparison of the NMR data with rogiolenyne A (epoxide ring in positions C-9/C-10, isolated from L. microcladia) identified both as chlorohydrin and acetyl chlorohydrin derivatives, respectively. Confirmation came from the transformation of 301 into rogiolenyne A by treatment with K2CO3/MeOH at room temperature. The location of the Cl atom at C-9 was inferred from COSY spectra. The pseudo-equatorial positions of H-9 and H-10 were inferred from the W-coupling of H-11/H-9 and H-10/one CH2-8, and the small J coupling values of H-9 and H-10. Confirmation came from nOe (one CH2-8/H-12 and H11/H-12. Eu(fod)3-induced deshielding of the H-12 resonance of 300 established the β-orientation of the OH group while the quasi-insensitive resonance of H-7 further supported the trans-relationship of the side chains at C-7 and C-12. The authors suggest that rogiolenyne A is transferred to the sponge where it undergoes epoxide opening by Cl− anion to give 300 and 301. Compounds 303 and 304 proved to be stable at low temperature (−15 °C) in the dark but to undergo interconversion when exposed to daylight at room temperature.
10. Biological Activity
In Table 7, a summary of the biological activities mentioned in the text is presented.
Table 7.
Biological activities of Spongia sp. Metabolites.
| Activity | Sesquiterpene Quinones | Diterpenes | C21 and Other Furano Terpenes | Sesterterpenes | Sterols | Macrolides | Miscellaneous Compounds |
|---|---|---|---|---|---|---|---|
| Artemia Salina |
53,54,63,74,75 [32] 41 [33] |
139,140 [58] 121,144,145,146 [61] 162 [32] |
165,169,172 [58] | ||||
| Anticancer (cytotoxicity and/or antiproliferative) |
6 [11] 8 [15,22] 12,13 [16] 14,15 [17] 21,22 [18] |
53,54,55 [27] 61 [29] 86 [36] 106 [41] 40,100 [48] |
156,157,158 [65] 296 [121] |
180,181,182,183 [73] 189,190 [67] 165,166,191,192,193,194 [77] |
259,260 [67] |
261,262 [93] 263 [94,95] 267 [82] 297 [121] |
271 [108] 283,284,285,286,287,288 [115] |
| Anticancer (other actions) |
8 [15] 14,15,16,17,18,19 [20] |
111 [43] |
180,181,182, 183,184 [73] 165,192,194 [77] |
263 [103] | |||
| Chemopreventive | 103,104 [40] | ||||||
| DNA repair | 8 [15] | ||||||
| Embryo development | 89,90,91,99,100,101,102 [39] | 291 [39] | |||||
| Immunomodulatory | 90,94 [37] | ||||||
| Immunosuppression | 209,210,211 [81] | ||||||
| P-glycoprotein modulation |
247 [87] 247,248,249,250,251,252,253 [88] |
||||||
| HSV1 or HSV2 |
53,54,55 [27] 43 [48] |
||||||
| HIV | 270 [107] | ||||||
| Microorganisms |
1,2 [9] 3,4,5 [10] |
48,49 [26] |
147 [62] 148 [63] 126 [66] |
271 [108] 275 [110] 283,284,285,286,287,288 [115] |
|||
| Antihelminthic | 298 [122] | ||||||
| Predation/Defense |
139 [58] 143 [60] |
169,172 [58] | |||||
| Biofilm induction of E. coli PHL628 | 123,124 [66] | ||||||
| Hypercholesterolemia |
165,198,199,200,201 [79] 166,192,194,202,204 [80] |
||||||
| Lipid metabolism |
283,286 [115] 283 [116] |
||||||
| Antiinflamatory | 205,206 [42] | ||||||
| Antioxidant | 39 [21] | ||||||
| Hemolytic activity | 6 [11] | ||||||
| Smooth muscle paralyzing agent | 147 [62] | ||||||
| Neurotransmission | 43,95,96,97,98 [38] | ||||||
| Neurotrophic | 166,192,194,196,197 [78] |
Several reports have appeared in the literature concerning the activity of extracts of Spongia sp.
Extracts of S. barbara collected in the Florida Keys significantly inhibited MAPK/ERK1,2 (mitogen-activated protein kinase-extracellular signal regulated kinase) activity (44% of control levels without altering cell survival) in cultured SW-13 human adrenal carcinoma cells. Results showed that ERK2 predominated over ERK1 and that the phosphorylated forms of these isozymes were strongly suppressed by active extracts [126].
Further studies [127] reported the effects of the extracts of the same sponge on cell cycle regulatory protein, cyclin B1; on cell cycle growth phase (sub G1/apoptosis, G1, S, and G2/M); and on cell survival in SW-13 human adrenal carcinoma cultures. A 70%–90% reduction in cyclin B1 levels was observed, together with a 10-fold increase in the percentage of cells entering apoptosis. During the same time the percentage of cells in G2/M was increased by 2 fold. Cell growth/survival studies also indicated a time-dependent decline in the percentage confluence of cell cultures exposed to the extracts.
A screening of the activity of S. magellanica (Thiele, 1905) against genotoxic biomarkers (mitotic index, cell proliferation kinetics and sister chromatid exchanges) was reported, although no activity was found [128].
Reports on the activity of extracts S. officinalis include:
-
-
the screening against clinical isolates of bacteria including multi-drug resistant (MDR) strains and fungi, where activity against MDR strains of Streptococcus pyogenes and Acinetobacter sp. was observed [129];
-
-
the anticonvulsant (using pentylenetetrazole seizure model) and analgesic (using writhing test in mice) activities of the extract (and its fractions) of the defensive secretion on the sponge, where analgesic activity in a dose dependent manner was observed for the extract [130];
-
-
the evaluation of antiproliferative (A549—lung cell carcinoma, HCT15—colon cell carcinoma and MCF7—breast adenocarcinoma) and anti-inflammatory (carrageenan-induced rat paw edema) activities of the extract (and its semi-purified fractions) of the defensive secretion of the sponge [131] and of the sponge methanol extract (and its semi-purified fractions) [132], where significant antiproliferative activities and anti-inflammatory activity were observed, and
-
-
the antiamoebic activity of extracts of S. officinalis var. ceylonensis against Entamoeba histolytica, where the alkaloids xestospongins and araguspongins where identified (by LCMS) as the major components of the active fraction [133].
11. Conclusions
The chemical composition of Spongia species has been studied in specimens obtained from different geographic areas. From these marine sponges a broad array of metabolites has been identified since 1971. The terpenic metabolites (sesqui-, di-, sesterpenes, and sterols) are the most representative compounds of the genera along macrolides, long chain lipid compounds and some alkaloids. More specifically we can verify that sesquiterpene quinones, C21 furanoterpenes, spongian diterpenes, scalarane sesterterpenes, sterols and secosterols are the most abundant skeletons biosynthesized by these marine organisms.
Some of the terpenic structures exhibit action on different cancer cell lines, are anti-HIV, anti-HSV1 and anti-HSV2, and immunomodulators, from a long list of assays described in literature. In general the description of long chain lipid compounds concerns their structure and they may be considered as part of the chemical fingerprint of Spongia genus. No activity assays are described for these compounds.
We highlight the most interesting activity encountered in Spongia metabolites that corresponds to the macrolide structure spongistatin 1 263 that displays an extremely potent action against a broad array of highly chemoresistant tumour types in the range of nanograms. Macrolides are anticancer leads and spongistatin 1 263 has been the subject of intense studies of synthetic strategies and anticancer mechanism of action. However the minute quantities in which this compound occurs from natural source and the complexity of the syntheses limit the progress of clinical studies.
Among the compounds of natural source that were identified as active and whose mechanism of action is somewhat effective on a given pathology there are few that reach the stage of clinical trials and even fewer come on the market for therapeutic purposes. Efforts are continuous to bring new lead structures to reach their role for the sake of human wellbeing. In this scenario marine organisms are an untold source of potent toxins that encourage scientists to break through the barriers to reach efficient therapeutic drugs.
Acknowledgments
We would like to thank the funding programs Promar (31-04-01-FEP-308) and QREN (2014/39985).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2015;32:116–211. doi: 10.1039/C4NP00144C. [DOI] [PubMed] [Google Scholar]
- 2.Perdicaris S., Vlachogianni T., Valavanidis A. Bioactive Natural Substances from Marine Sponges: New Developments and Prospects for Future Pharmaceuticals. Nat. Prod. Chem. Res. 2013;1 doi: 10.4172/2329-6836.1000114. [DOI] [Google Scholar]
- 3.Pronzato R., Manconi R. Mediterranean Commercial Sponges: Over 5000 Years of Natural History and Cultural Heritage. Mar. Ecol. 2008;29:146–166. doi: 10.1111/j.1439-0485.2008.00235.x. [DOI] [Google Scholar]
- 4.Voultsiadou E., Dailianis T., Antoniadou C., Vafidis D., Dounas C., Chintiroglou1 C.C. Aegean Bath Sponges: Historical Data and Current Status. Rev. Fish. Sci. 2011;19:34–51. doi: 10.1080/10641262.2010.531794. [DOI] [Google Scholar]
- 5.Noyer C., Agell G., Pascual M., Becerro M.A. Isolation and Characterization of Microsatellite Loci from the Endangered Mediterranean Sponge Spongia agaricina (Demospongiae: Dictyoceratida) Conserv. Genet. 2009;10:1895–1898. doi: 10.1007/s10592-009-9848-6. [DOI] [Google Scholar]
- 6.Noyer C., Becerro M.A. Relationship between Genetic, Chemical, and Bacterial Diversity in the Atlanto-Mediterranean Bath Sponge Spongia lamella. Hydrobiology. 2012;687:85–99. doi: 10.1007/s10750-011-0884-0. [DOI] [Google Scholar]
- 7.Cunningham E., Dunne N., Walker G., Maggs C., Wilcox R., Buchanan F. Hydroxyapatite Bone Substitutes Developed via Replication of Natural Marine Sponges. J. Mater. Sci. Mater. Med. 2010;21:2255–2261. doi: 10.1007/s10856-009-3961-4. [DOI] [PubMed] [Google Scholar]
- 8.Fattorusso E., Minale L., Sodano G., Trivellone E. Isolation and Structure of Nitenin and Dihydronitenin, New Furanoterpenes from Spongia nitens. Tetrahedron. 1971;27:3909–3917. doi: 10.1016/S0040-4020(01)98252-3. [DOI] [Google Scholar]
- 9.Urban S., Capon R.J. 5-epi-isospongiaquinone, A New Sesquiterpene/Quinone Antibiotic from an Australian Marine Sponge, Spongia Hispida. J. Nat. Prod. 1992;55:1638–1642. doi: 10.1021/np50089a012. [DOI] [PubMed] [Google Scholar]
- 10.Capon R.J., Groves D.R., Urban S., Watson R.G. Spongiaquinone Revisited-Structural and Stereochemical Studies on Marine Sesquiterpene Quinones from a Southern Australian Marine Sponge, Spongia sp. Aust. J. Chem. 1993;46:1245–1253. doi: 10.1071/CH9931245. [DOI] [Google Scholar]
- 11.Utkina N.K., Denisenko V.A., Scholokova O.V., Virovaya M.V., Prokof’eva N.G. Cyclosmenospongine, a New Sesquiterpenoid Aminoquinone from an Australian Marine Sponge Spongia sp. Tetrahedron Lett. 2003;44:101–102. doi: 10.1016/S0040-4039(02)02497-8. [DOI] [Google Scholar]
- 12.Capon R.J., MacLeod J.K. A Revision of the Absolute Stereochemistry of Ilimaquinone. J. Org. Chem. 1987;52:5059–5060. doi: 10.1021/jo00231a051. [DOI] [Google Scholar]
- 13.Kondraki M.-L., Guyot M. Smenospongine: A Cytotoxic and Antimicrobial Aminoquinone Isolated from Smenospongia sp. Tetrahedron Lett. 1987;28:5815–5818. doi: 10.1016/S0040-4039(01)81061-3. [DOI] [Google Scholar]
- 14.Utkina N.K., Denisenko V.A., Scholokova O.V., Makarchenko A.E. Determination of the Absolute Stereochemistry of Cyclosmenospongine. J. Nat. Prod. 2003;66:1263–1265. doi: 10.1021/np030115r. [DOI] [PubMed] [Google Scholar]
- 15.Cao S., Gao Z., Thomas S.J., Hecht S.M., Lazo J.S., Kingston D.G.I. Marine Sesquiterpenoids that Inhibit the Lyase Activity of DNA Polymerase β. J. Nat. Prod. 2004;67:1716–1718. doi: 10.1021/np049849+. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi Y., Tsuda M., Fromont J., Kobayashi J. Metachromins J and K, New Sesquiterpenoids from Marine Sponge Spongia Species. Heterocycles. 2006;67:791–796. doi: 10.1002/chin.200630193. [DOI] [Google Scholar]
- 17.Takahashi Y., Kubota T., Fromontb J., Kobayashi J. Metachromins L–Q, New Sesquiterpenoid Quinones with an Amino Acid Residue from Sponge Spongia sp. Tetrahedron. 2007;63:8770–8773. doi: 10.1016/j.tet.2007.06.031. [DOI] [Google Scholar]
- 18.Takahashi Y., Yamada M., Kubota T., Fromont J., Kobayashi J. Metachromins R–T, New Sesquiterpenoids from Marine Sponge Spongia sp. Chem. Pharm. Bull. 2007;55:1731–1733. doi: 10.1248/cpb.55.1731. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi Y., Kubota T., Yamamoto S., Kobayashi J. Inhibitory Effects of Metachromins L–Q and Its Related Analogs Against Receptor Tyrosine Kinases EGFR and HER2. Bioorg. Med. Chem. Lett. 2013;23:117–118. doi: 10.1016/j.bmcl.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi Y., Kubota T., Kobayashi J. Nakijiquinones E and F, New Dimeric Sesquiterpenoid Quinones from Marine Sponge. Bioorg. Med. Chem. 2009;17:2185–2188. doi: 10.1016/j.bmc.2008.10.080. [DOI] [PubMed] [Google Scholar]
- 21.Utkina N.K., Denisenko V.A. Sesquiterpene Quinones from a Viet Nam Sea Sponge Spongia sp. Chem. Nat. Compd. 2011;47:135–137. doi: 10.1007/s10600-011-9858-8. [DOI] [Google Scholar]
- 22.Kittiwisut S., Yuenyongsawad S., Mooberry S.L., Plubrukarn A. DNA Damage Initiated by Merosesquiterpenes from the Sponge Spongia sp. Planta Med. 2012;78:1147. doi: 10.1055/s-0032-1320698. [DOI] [Google Scholar]
- 23.Cimino G., Derosa D., Destefan S., Minale L. Isoagatholactone, a Diterpene of a New Structural Type from Sponge Spongia officinalis. Tetrahedron. 1974;30:645–649. doi: 10.1016/S0040-4020(01)97059-0. [DOI] [Google Scholar]
- 24.Capelle N., Braekman J.C., Daloze D., Tursch B. Chemical Studies of Marine-Invertebrates .44. 3 New Spongian Diterpenes from Spongia officinalis. Bull. Des Soc. Chim. Belges. 1980;89:399–404. doi: 10.1002/bscb.19800890511. [DOI] [Google Scholar]
- 25.Cimino G., Morrone R., Sodano G. New Diterpenes from Spongia officinalis. Tetrahedron Lett. 1982;23:4139–4142. doi: 10.1016/S0040-4039(00)88369-0. [DOI] [Google Scholar]
- 26.Gonzalez A.G., Estrada D.M., Martin J.D., Martin V.S., Perez C., Perez R. New Antimicrobial Diterpenes from a Sponge Spongia officinalis. Tetrahedron. 1984;40:4109–4113. doi: 10.1016/0040-4020(84)85092-9. [DOI] [Google Scholar]
- 27.Kohmoto S., McConnell O.J., Wright A., Cross S. Isospongiadiol, A Cytotoxic And Antiviral Diterpene From A Caribbean Deep-Water Marine Sponge, Spongia sp. Chem. Lett. 1987;16:1687–1690. doi: 10.1246/cl.1987.1687. [DOI] [Google Scholar]
- 28.Hirsch S., Ashman Y.K. Spongialactone A, a New Spongian Diterpene from Spongia arabica. J. Nat. Prod. 1988;51:1243–1245. doi: 10.1021/np50060a032. [DOI] [Google Scholar]
- 29.Gunasekera S.P., Schmitz F.J. New Spongian Diterpenoids from a Great Barrier Reef Sponge, Spongia sp. J. Org. Chem. 1991;56:1250–1253. doi: 10.1021/jo00003a057. [DOI] [Google Scholar]
- 30.Searle P.A., Molinski T.F. Scalemic 12-Hydroxyambliofuran and 12-Acetoxyambliofuran, Five Furanoditerpenes and a Furanosesterterpene from Spongia sp. Tetrahedron. 1994;50:9893–9908. doi: 10.1016/S0040-4020(01)89605-8. [DOI] [Google Scholar]
- 31.Zubia E., Gavagnin M., Scognamiglio G., Cimino G. Spongiane and Ent.isocopalane Diterpenoids from the Mediterranean Sponge Spongia zimocca. J. Nat. Prod. 1994;57:725–731. doi: 10.1021/np50108a007. [DOI] [Google Scholar]
- 32.Li C.-J., Schmitz F.J., Kelly-Borges M. New Diterpene Lactones from the Sponge Spongia matamata. J. Nat. Prod. 1998;61:546–547. doi: 10.1021/np9705219. [DOI] [PubMed] [Google Scholar]
- 33.Li C.-J., Schmitz F.J., Kelly-Borges M. Six New Spongian Diterpenes from the Sponge Spongia matamata. J. Nat. Prod. 1999;62:287–290. doi: 10.1021/np9803981. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell S.S., Harper M.K., Faulkner D.J. Spongiabutenolides A–D: Minor γ-Hydroxybutenolide Diterpenoids from a Philippines Spongia sp. Tetrahedron. 1999;55:10887–10892. doi: 10.1016/S0040-4020(99)00622-5. [DOI] [Google Scholar]
- 35.Zeng L.M., Guan Z., Su J.Y., Feng X.L., Cai J.W. Two New Spongian Diterpene Lactones. Acta Chim. Sin. 2001;59:1675–1679. [Google Scholar]
- 36.Su J.Y., Lin C.W., Zeng L.M., Yan S.J., Feng X.L., Cai J.W. Separation and Structure Determination of a New 19-nor-spongian Diterpenoid. Chem. J. Chin. Univ. 2003;24:817–819. [Google Scholar]
- 37.Ponomarenko L.P., Kalinovsky A.I., Afiyatullov S.S., Pushilin M.A., Gerasimenko A.V., Krasokhin V.B., Stonik V.A. Spongian Diterpenoids from the Sponge Spongia (Heterofibria) sp. J. Nat. Prod. 2007;70:1110–1113. doi: 10.1021/np070068t. [DOI] [PubMed] [Google Scholar]
- 38.Carroll A.R., Lamb J., Moni R., Hooper J.N.A., Quinn R.J. Spongian Diterpenes with Thyrotropin Releasing Hormone Receptor 2 Binding Affinity from Spongia sp. J. Nat. Prod. 2008;71:884–886. doi: 10.1021/np070658r. [DOI] [PubMed] [Google Scholar]
- 39.Ponomarenko L.P., Terent’eva N.A., Krasokhin V.B., Kalinovsky A.I., Rasskazov V.A. Terpenoid Metabolites from Spongia spp. and Their Effects on Nucleic Acid Biosynthesis in Sea Urchin Eggs. Nat. Prod. Commun. 2011;6:773–776. [PubMed] [Google Scholar]
- 40.Parrish S.M., Yoshida W.Y., Kondratyuk T.P., Park E.-J., Pezzuto J.M., Kelly M., Williams P.G. Spongiapyridine and Related Spongians Isolated from an Indonesian Spongia sp. J. Nat. Prod. 2014;77:1644–1649. doi: 10.1021/np500256w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pham A.T., Carney J.R., Yoshida W.Y., Scheuer P.J. Haumanamide, a Nitrogenous Spongian Derivative from Spongia sp. Tetrahedron Lett. 1992;33:1147–1148. doi: 10.1016/S0040-4039(00)91881-1. [DOI] [Google Scholar]
- 42.Marino S.D., Iorizzi M., Zollo F., Debitus C., Menou J.-L., Ospina L.F., Alcaraz M.J., Payá M. New Pyridinium Alkaloids from a Marine Sponge of the Genus Spongia with a Human Phospholipase A2 Inhibitor Profile. J. Nat. Prod. 2000;63:322–326. doi: 10.1021/np990374+. [DOI] [PubMed] [Google Scholar]
- 43.Mori D., Kimura Y., Kitamura S., Sakagami Y., Yoshioka Y., Shintani T., Okamoto T., Ojika M. Spongolactams, Farnesyl Transferase Inhibitors from a Marine Sponge: Isolation through an LC/MS-Guided Assay, Structures, and Semisyntheses. J. Org. Chem. 2007;72:7190–7198. doi: 10.1021/jo071003y. [DOI] [PubMed] [Google Scholar]
- 44.Kazlauskas R., Murphy P.T., Wells R.J., Noack K., Oberhansli W.E., Schonholzer P. New Series of Diterpenes from Australian Spongia Species. Aust. J. Chem. 1979;32:867–880. doi: 10.1071/CH9790867. [DOI] [Google Scholar]
- 45.Thompson J.E., Murphy P.T., Bergquist P.R., Evans E.A. Environmentally Induced Variation in Diterpene Composition of the Marine Sponge Rhopaloeides Odorabile. Biochem. Syst. Ecol. 1987;15:595–606. doi: 10.1016/0305-1978(87)90111-6. [DOI] [Google Scholar]
- 46.Puliti R., Mattia C.A. Ent-Isocopal-12-ene-15,16-dialdehyde from Spongia officinalis. Acta Crystallogr. 1999;55:2160–2163. [Google Scholar]
- 47.Yong K.W.L., Garson M.J., Bernhardt P.V. Absolute Structures and Conformations of the Spongian Diterpenes Spongia-13(16),14-dien-3-one, Epispongiadiol and Spongiadiol. Acta Crystallogr. Sect. C. 2009;65:O167–O170. doi: 10.1107/S0108270109008816. [DOI] [PubMed] [Google Scholar]
- 48.Betancur-Galvis L., Zuluaga C., Arnó M., González M.A., Zaragozá R.J. Cytotoxic Effect (on Tumor Cells) and in Vitro Antiviral Activity against Herpes Simplex Virus of Synthetic Spongiane Diterpenes. J. Nat. Prod. 2002;65:189–192. doi: 10.1021/np010206t. [DOI] [PubMed] [Google Scholar]
- 49.Cimino G., Stefano S.D., Minale L. Furospongin-1, a New C-21 Furanoterpene from the Sponges Spongia officinalis and Hippospongia communis. Tetrahedron. 1971;27:4673–4679. doi: 10.1016/S0040-4020(01)98174-8. [DOI] [Google Scholar]
- 50.Cimino G., Stefano S.D., Minale L. Minor C-21 Furanoterpenes from the Sponges Spongia officinalis and Hippospongia communis. Tetrahedron. 1972;28:267–273. doi: 10.1016/0040-4020(72)80132-7. [DOI] [Google Scholar]
- 51.Cimino G., Stefano S.D., Minale L. Further Linear Furanoterpenes from Marine Sponges. Tetrahedron. 1972;28:5983–5991. doi: 10.1016/0040-4020(72)88131-6. [DOI] [Google Scholar]
- 52.Fontana A., Albarella L., Scognamiglio G., Uriz M., Cimino G. Structural and Stereochemical Studies of C-21 Terpenoids from Mediterranean Spongiidae Sponges. J. Nat. Prod. 1996;59:869–872. doi: 10.1021/np9600964. [DOI] [Google Scholar]
- 53.Kobayashi M., Chavakula R., Murata O., Sarma N.S. Marine Terpenes and Terpenoids. Part 16. Revised Structure of Marine Furanoterpene (+)-furospongin-1. J. Chem. Res. 1992:366–367. doi: 10.1002/chin.199311253. [DOI] [Google Scholar]
- 54.Cimino G., Stefano S.D., Minale L. Oxidized Furanoterpenes from the Sponge Spongia officinalis. Experientia. 1974;30:18–20. doi: 10.1007/BF01921570. [DOI] [Google Scholar]
- 55.Kazlauskas R., Murphy P.T., Quinn R.J., Wells R.J. Tetradehydrofurospongin-1, a New C21 Furanoterpene from a Sponge. Tetrahedron Lett. 1976;16:1331–1332. doi: 10.1016/S0040-4039(00)78055-5. [DOI] [Google Scholar]
- 56.Capon R.J., Ghisalberti E.L., Jefferies P.R. A New Furanoterpene from a Spongia sp. Experientia. 1982;38:1444–1445. doi: 10.1007/BF01955759. [DOI] [Google Scholar]
- 57.Kazlauskas R., Murphy P.T., Quinn R.J., Wells R.J. Two New Unsymetrically Oxygenated C21 furanoterpenes from a Sponge. Tetrahedron Lett. 1976;16:1333–1334. doi: 10.1016/S0040-4039(00)78056-7. [DOI] [Google Scholar]
- 58.Walker R.P., Thompson J.E., Faulkner D.J. Sesterterpenes from Spongia idia. J. Org. Chem. 1980;45:4976–4979. doi: 10.1021/jo01312a032. [DOI] [Google Scholar]
- 59.Capon R.J., Jenkins A., Rooney F., Ghisalberti E.L. Structure Revision and Assignment of Absolute Stereochemistry of a Marine C21 Bisfuranoterpene. J. Nat. Prod. 2001;64:638–639. doi: 10.1021/np000477o. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka J., Higa T. The Absolute Configuration of Kurospongin, a New Furanoterpene from a Marine Sponge, Spongia sp. Tetrahedron. 1988;44:2805–2810. doi: 10.1016/S0040-4020(88)90016-6. [DOI] [Google Scholar]
- 61.Giulio A.D., Rosa S.D., Vincenzo G.D. Terpenoids from the North Adriatic Sponge Spongia officinalis. J. Nat. Prod. 1989;52:1258–1262. doi: 10.1021/np50066a010. [DOI] [Google Scholar]
- 62.Lumsdon D., Capon R.J., Thomas S.G., Beveridge A.A. A New Sesterterpene Tetronic Acid and a Pentaprenylated Para-Quinol from an Australian Marine Sponge, Spongia sp. Aust. J. Chem. 1992;45:1321–1325. doi: 10.1071/CH9921321. [DOI] [Google Scholar]
- 63.Urban S., Capon R.J. Cometins (A–C), New Furanosesterterpenes from an Australian Marine Sponge, Spongia sp. Aust. J. Chem. 1992;45:1255–1263. doi: 10.1071/CH9921255. [DOI] [Google Scholar]
- 64.Lenis L.A., Nunez L., Jimenez C., Riguera R. Isonitenin and Acetylhomoagmatine New Metabolites from the Sponges Spongia officinalis and Cliona Celata Collected at the Galician Coast (NW Spain) Nat. Prod. Lett. 1996;8:15–23. doi: 10.1080/10575639608043233. [DOI] [Google Scholar]
- 65.Garrido L., Zubía E., Ortega M.J., Salvá J. New Furanoterpenoids from the Sponge Spongia officinalis. J. Nat. Prod. 1997;60:794–797. doi: 10.1021/np970160x. [DOI] [Google Scholar]
- 66.Manzo E., Ciavatta M.L., Villani G., Varcamonti M., Sayem S.M.A., Soest R., Gavagnin M. Bioactive Terpenes from Spongia officinalis. J. Nat. Prod. 2011;74:1241–1247. doi: 10.1021/np200226u. [DOI] [PubMed] [Google Scholar]
- 67.Rueda A., Zubía E., Ortega M.J., Carballo J.L., Salvá J. New Metabolites from the Sponge Spongia agaricina. J. Nat. Prod. 1998;61:258–261. doi: 10.1021/np970390b. [DOI] [PubMed] [Google Scholar]
- 68.Cimino G., Stefano S.D., Minale L. Deoxoscalarin, a Further Sesterterpene with the Unusual Tetracyclic Carbon Skeleton of Scalarin, from Spongia officinalis. Experientia. 1973;29:934–936. doi: 10.1007/BF01930382. [DOI] [Google Scholar]
- 69.Cimino G., Stefano S.D., Minale L., Trivellone E. 12-epi-Scalarin and 12-epi-Deoxoscalarin, Sesterterpenes from the Sponge Spongia nitens. J. Chem. Soc. Perkin Trans. 1977;13:1587–1593. doi: 10.1039/p19770001587. [DOI] [PubMed] [Google Scholar]
- 70.Cimino G., Stefano S.D., Luccia A.D. Further Sesterterpenes from the Sponge Spongia nitens: 12-epi-scalaradial and 12,18-diepi-scalaradial. Experientia. 1979;35:1277–1278. doi: 10.1007/BF01963953. [DOI] [Google Scholar]
- 71.Cimino G., Rosa S.D., Stefano S.D. Scalarolbutenolide, a New Sesterterpenoid from the Marine Sponge Spongia nitens. Experientia. 1981;37:214–216. doi: 10.1007/BF01991615. [DOI] [Google Scholar]
- 72.Davis R., Capon R.J. Two New Scalarane Sesterterpenes-Isoscalarafuran-A and Isoscalarafuran-B, Epimeric Alcohols from a Southern Australian Marine Sponge, Spongia hispida. Aust. J. Chem. 1993;46:1295–1299. doi: 10.1071/CH9931295. [DOI] [Google Scholar]
- 73.He H., Kulanthaivel P., Baker B.J. New Cytotoxic Sesterterpenes from the Marine Sponge Spongia sp. Tetrahedron Lett. 1994;35:7189–7192. doi: 10.1016/0040-4039(94)85357-6. [DOI] [Google Scholar]
- 74.Conte M.R., Fatorrusso E., Lanzotti V., Magno S., Mayol L. Lintenolides, new pentacyclic bioactive sesterterpenes from the caribbean sponge Cacospongia cf. linteiformis. Tetrahedron. 1994;50:849–856. doi: 10.1016/S0040-4020(01)80799-7. [DOI] [Google Scholar]
- 75.Lu Q., Faulkner D.J. Two New Sesterterpenoids and a New 9,11-Secosterol from Spongia matamata. J. Nat. Prod. 1997;60:195–198. doi: 10.1021/np9606411. [DOI] [PubMed] [Google Scholar]
- 76.Sakamoto K., Miyamot T., Amano H., Higuchi R., Komori T., Sasaki T. 34th Tennen Yuki Kagobutsu Toronkai Koen Yoshishu. Volume 34. Organizing Committee of the 34th Symposium on the Chemistry of Natural Products; Tokyo, Japan: 1992. pp. 455–462. Chem. Abstr. 173183. [Google Scholar]
- 77.Tsukamoto S., Miura S., Soest R.W.M.V., Ohta T. Three New Cytotoxic Sesterterpenes from a Marine Sponge Spongia sp. J. Nat. Prod. 2003;66:438–440. doi: 10.1021/np020497l. [DOI] [PubMed] [Google Scholar]
- 78.Tokue T., Miura S., Kato H., Hirota H., Ohta T., Tsukamoto S. Neurotrophic Sesterterpenes Isolated from a Marine Sponge, Spongia sp. Heterocycles. 2006;69:521–526. doi: 10.1002/chin.200723178. [DOI] [Google Scholar]
- 79.Nam S.-J., Ko H., Shin M., Ham J., Chin J., Kim Y., Kim H., Shin K., Choi H., Kang H. Farnesoid X-activated Receptor Antagonists from a Marine Sponge Spongia sp. Bioorg. Med. Chem. Lett. 2006;16:5398–5402. doi: 10.1016/j.bmcl.2006.07.079. [DOI] [PubMed] [Google Scholar]
- 80.Nam S.-J., Ko H., Ju M.K., Hwang H., Chin J., Ham J., Lee B., Lee J., Won D.H., Choi H., et al. Scalarane Sesterterpenes from a Marine Sponge of the Genus Spongia and Their FXR Antagonistic Activity. J. Nat. Prod. 2007;70:1691–1695. doi: 10.1021/np070024k. [DOI] [PubMed] [Google Scholar]
- 81.Carr G., Raszek M., Soest R.V., Matainaho T., Shopik M., Holmes C.F.B., Andersen R.J. Protein Phosphatase Inhibitors Isolated from Spongia irregularis Collected in Papua New Guinea. J. Nat. Prod. 2007;70:1812–1815. doi: 10.1021/np0702887. [DOI] [PubMed] [Google Scholar]
- 82.Grassia A., Bruno I., Debitus C., Marzocco S., Pinto A., Gomez-Paloma L., Riccio R. Spongidepsin, a New Cytotoxic Macrolide from Spongia sp. Tetrahedron. 2001;57:6257–6260. doi: 10.1016/S0040-4020(01)00587-7. [DOI] [Google Scholar]
- 83.Aiello A., Ciminiello P., Fattorusso E., Magno S. 3,5-Dihydroxy-6-methoxycholest-7-enes from the Marine Sponge Spongia agaricina. J. Nat. Prod. 1988;51:999–1002. doi: 10.1021/np50059a036. [DOI] [PubMed] [Google Scholar]
- 84.Madaio A., Piccialli V., Sica D. New Polyhydroxysterols from the Dictyoceratid Sponges Hippospongia communis, Spongia officinalis, Ircinia variabilis, and Spongionella gracilis. J. Nat. Prod. 1989;52:952–961. doi: 10.1021/np50065a007. [DOI] [Google Scholar]
- 85.Migliuolo A., Notaro G., Piccialli V., Sica D. New Tetrahydroxylated Sterols from the Marine Sponge Spongia officinalis. J. Nat. Prod. 1990;53:1414–1424. doi: 10.1021/np50072a003. [DOI] [Google Scholar]
- 86.Migliuolo A., Piccialli V., Sica D., Giordano F. New Delta-8-5-alpha,6-alpha-epoxysterols and Delta-8(14)-5-alpha,6-alpha-epoxysterols from the Marine Sponge Spongia officinalis. Steroids. 1993;58:134–140. doi: 10.1016/0039-128X(93)90050-W. [DOI] [PubMed] [Google Scholar]
- 87.Aoki S., Yoshioka Y., Miyamoto Y., Higuchi K., Setiawan A., Murakami N., Chen Z.-S., Sumizawa T., Akiyama S., Kobayashi M. Agosterol A, a Novel Polyhydroxylated Sterol Acetate Reversing Multidrug Resistance from a Marine Sponge of Spongia sp. Tetrahedron Lett. 1998;39:6303–6306. doi: 10.1016/S0040-4039(98)01336-7. [DOI] [Google Scholar]
- 88.Aoki S., Setiawan A., Yoshioka Y., Higuchi K., Fudetani R., Chen Z.-S., Sumizawa T., Akiyama S., Kobayashi M. Reversal of Muitidrug Resistance in Human Carcinoma Cell Line by Agosterols, Marine Spongean Sterols. Tetrahedron. 1999;55:13965–13972. doi: 10.1016/S0040-4020(99)00870-4. [DOI] [Google Scholar]
- 89.Chen Z.S., Aoki S., Komatsu M., Ueda K., Sumizawa T., Furukawa T., Okumura H., Ren X.Q., Belinsky M.G., Lee K., et al. Reversal of Drug Resistance Mediated by Multidrug Resistance Protein (MRP) 1 by Dual Effects of Agosterol A on MRP1 Function. Int. J. Cancer. 2001;93:107–113. doi: 10.1002/ijc.1290. [DOI] [PubMed] [Google Scholar]
- 90.Migliuolo A., Piccialli V., Sica D. Structure Elucidation and Synthesis of 3β,6α-dihydroxy-9-oxo-9,11-seco-5α-cholest-7-en-11-al, a novel 9,11-secosterol from the Sponge Spongia officinalis. Tetrahedron. 1991;47:7937–7950. doi: 10.1016/S0040-4020(01)81948-7. [DOI] [Google Scholar]
- 91.Migliuolo A., Piccialli V., Sica D. 2 New 9,11-Secosterols from the Marine Sponge Spongia officinalis. Synthesis of 9,11-seco-3-beta,6-alpha,11-Trihydroxy-5-alpha-cholest-7-en-9-one. Steroids. 1992;57:344–347. doi: 10.1016/0039-128X(92)90054-D. [DOI] [PubMed] [Google Scholar]
- 92.Adinolfi R., Migliuolo A., Piccialli V., Sica D. Isolation and Synthesis of a New 9,11-Secosterol from the Sponge Spongia officinallis. J. Nat. Prod. 1994;57:1220–1226. doi: 10.1021/np50111a005. [DOI] [Google Scholar]
- 93.Quinoa E., Kakou Y., Crews P. Fijianolides, Polyketide Heterocycles from a Marine Sponge. J. Org. Chem. 1988;53:3642–3644. doi: 10.1021/jo00250a052. [DOI] [Google Scholar]
- 94.Pettit G.R., Cichacz Z.A., Gal F., Herald C.L., Boyd M.R., Schmidt J.M., Hooperlc J.N.A. Isolation and Structure of Spongistatin 1. J. Org. Chem. 1993;58:1302–1304. doi: 10.1021/jo00058a004. [DOI] [Google Scholar]
- 95.Pettit G.R., Cichacz Z.A., Gao F., Heralds C.L., Boyd M.R. Isolation and Structure of the Remarkable Human Cancer Cell Growth Inhibitors Spongistatins 2 and 3 from an Eastern Indian Ocean Spongia sp. J. Chem. Soc. Chem. Commun. 1993;24:1166–1168. doi: 10.1039/c39930001166. [DOI] [Google Scholar]
- 96.Pettit G.R., Cichacz Z.A., Gao F., Boyd M.R., Schmidt J.M. Isolation and Structure of the Cancer Cell Growth Inhibitor Dictyostatin 1. J. Chem. Soc. Chem. Commun. 1994;9:1111–1112. doi: 10.1039/c39940001111. [DOI] [Google Scholar]
- 97.Kobayashi M., Aoki S., Sakai H., Kawazoe K., Kihara N., Sasaki T., Kitagawa I. Altohyrtin A, a Potent Anti-tumor Macrolide from the Okinawan Marine Sponge Hyrfios alturn. Tetrahedron Lett. 1993;34:2795–2798. doi: 10.1016/S0040-4039(00)73564-7. [DOI] [Google Scholar]
- 98.Fusetani N., Shinoda K., Matsunaga S. Cinachyrolide A: A Potent Cytotoxic Macrolide Possessing Two Spiro Ketals from Marine Sponge Cinachyra sp. J. Am. Chem. Soc. 1993;115:3977–3981. doi: 10.1021/ja00063a017. [DOI] [Google Scholar]
- 99.Pettit G.R., Herald C.L., Cichacz Z.A., Gao F., Boyd M.R., Christie N.D., Schmidt J.M. Antineoplastic Agents 293. the Exceptional Human Cancer Cell Growth Inhibitors Spongistatins 6 and 7. Nat. Prod. Lett. 1993;3:239–244. doi: 10.1080/10575639308043871. [DOI] [Google Scholar]
- 100.Kobayashi M., Aoki S., Gato K., Kitagawa I. Marine Natural Products. XXXVIII. Absolute stereostructures of Altohyrtins A, B, and C and 5-desacetylaltohyrtin A, a potent Cytotoxic Macrolides, from the Okinawan Marine Sponge Hyrtios altum. Chem. Pharm. Bull. 1996;44:2142–2149. doi: 10.1248/cpb.44.2142. [DOI] [Google Scholar]
- 101.Guo J., Duffy K.J., Stevens K.L., Dalko P.I., Roth R.M., Hayward M.M., Kishi Y. Total Synthesis of Altohyrtin A (Spongistatin 1): Part 1. Angew. Chem. Int. Ed. 1998;37:187–192. doi: 10.1002/(SICI)1521-3773(19980202)37:1/2<187::AID-ANIE187>3.0.CO;2-D. [DOI] [Google Scholar]
- 102.Hayward M.M., Roth R.M., Duffy K.J., Dalko P.I., Stevens K.L., Guo J., Kishi Y. Total Synthesis of Altohyrtin A (Spongistatin 1): Part 2. Angew. Chem. Int. Ed. 1998;37:192–196. doi: 10.1002/(SICI)1521-3773(19980202)37:1/2<190::AID-ANIE190>3.0.CO;2-0. [DOI] [Google Scholar]
- 103.Bai R.L., Cichacz Z.A., Herald C.L., Pettit G.R., Hamel E. Spongistatin-1, A Highly Cytotoxic, Sponge-Derived, Marine Natural Product that Inhibits Mitosis, Microtubule Assembly, and the Binding of Vinblastine to Tubulin. Mol. Pharmacol. 1993;44:757–766. [PubMed] [Google Scholar]
- 104.Luduena R.F., Roach M.C., Prasad V., Pettit G.R., Cichacz Z.A., Herald C.L. Interaction of 3 Sponge-Derived Macrocyclic Lactone Polyethers (Spongistatin-3, Halistatin-1 and Halistatin-2) With Tubulin. Drug Dev. Res. 1995;35:40–48. doi: 10.1002/ddr.430350107. [DOI] [Google Scholar]
- 105.Chen J., Forsyth C.J. Total Synthesis and Structural Assignment of Spongidepsin through a Stereodivergent Ring-Closing-Metathesis Strategy. Angew. Chem. Int. Ed. 2004;43:2148–2152. doi: 10.1002/anie.200453663. [DOI] [PubMed] [Google Scholar]
- 106.Ghosh A.K., Xu X. Assignment of Absolute Stereochemistry and Total Synthesis of (−)-Spongidepsin. Org. Lett. 2004;6:2055–2058. doi: 10.1021/ol049292p. [DOI] [PubMed] [Google Scholar]
- 107.Kalidindi R.S., Yoshida W.Y., Palermo J.A., Scheuer P.J. Pokepola ester: A Phosphat Diester from a Maui Sponge. Tetrahedron Lett. 1994;35:5579–5582. doi: 10.1016/S0040-4039(00)77251-0. [DOI] [Google Scholar]
- 108.Pettit G.R., Bond T.J., Herald D.L., Penny M., Doubek D.L., Williams M.D., Pettit R.K., Hooper J.N.A. Isolation and Structure of Spongilipid from the Republic of Singapore Marine Porifera Spongia cf. hispida. Can. J. Chem. 1997;75:920–925. doi: 10.1139/v97-110. [DOI] [Google Scholar]
- 109.Xu S.H., Cen Y.Z., Zeng L.M., Su J.Y. Isolation and Structural Determination of Heterocyclic Alkaloidal Compounds. Chin. J. Org. Chem. 2000;20:248–250. [Google Scholar]
- 110.Kobayashi K., Shimogawa H., Sakakura A., Teruya T., Suenaga K., Kigoshi H. Spongiacysteine, a Novel Cysteine Derivative from Marine Sponge Spongia sp. Chem. Lett. 2004;33:1262–1263. doi: 10.1246/cl.2004.1262. [DOI] [Google Scholar]
- 111.Lin C.W., Su J.Y., Zeng L.M., Wei W.X., Wei T.Y. Compounds Containing Nitrogen from Spongia zimocca Aubspecles Irregularia (Lendenfeld) Chin. J. Org. Chem. 2005;25:225–228. [Google Scholar]
- 112.Xu S.H., Yang K. Three New Ceramides from the Sponge Spongia suriganensis. Chin. J. Org. Chem. 2006;26:56–59. [Google Scholar]
- 113.Guan Z., Zeng L. A New Ceramide from a New Species of Spongia Sponge. Chem. Nat. Compd. 2010;46:287–288. doi: 10.1007/s10600-010-9590-9. [DOI] [Google Scholar]
- 114.Guan Z., Zeng L. Chemical constituents of a new species of Spongia sponge. Zhongguo Zhong Yao Za Zhi. 2010;35:1004–1008. doi: 10.4268/cjcmm20100814. [DOI] [PubMed] [Google Scholar]
- 115.Salim A.A., Rae J., Fontaine F., Conte M.M., Khalil Z., Martin S., Partonb R.G., Capon R.J. Heterofibrins: Inhibitors of Lipid Droplet Formation from a Deep-water Southern Australian Marine Sponge, Spongia (Heterofibria) sp. Org. Biomol. Chem. 2010;8:3188–3194. doi: 10.1039/c003840g. [DOI] [PubMed] [Google Scholar]
- 116.Rae J., Fontaine F., Salim A.A., Lo H.P., Capon R.J., Parton R.G., Martin S. High-Throughput Screening of Australian Marine Organism Extracts for Bioactive Molecules Affecting the Cellular Storage of Neutral Lipids. PLoS ONE. 2011;6:139. doi: 10.1371/journal.pone.0022868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carballeira N.M., Emiliano A., Morales R. Positional Distribution of Octadecadienoic Acids in Sponge Phosphatidylethanolamines. Lipids. 1994;29:523–525. doi: 10.1007/BF02578251. [DOI] [PubMed] [Google Scholar]
- 118.Junqua S., Lemonnier M., Robert L. Glycoconjugates from Spongia officinalis (Phylum Porifera). Isolation, Fractionation by Affinity-Chromatography on Lectins and Partial Characterization. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1981;69:445–453. doi: 10.1016/0305-0491(81)90334-5. [DOI] [Google Scholar]
- 119.Noyer C., Thomas O.P., Becerro M.A. Patterns of Chemical Diversity in the Mediterranean Sponge Spongia lamella. PLoS ONE. 2011;6:139. doi: 10.1371/journal.pone.0020844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Terem B., Scheuer P.J. Scalaradial Derivatives from the Nudibranch Chromodoris youngbleuthi and the Sponge Spongia oceania. Tetrahedron. 1986;42:4409–4412. doi: 10.1016/S0040-4020(01)87279-3. [DOI] [Google Scholar]
- 121.Kakou Y., Crew P. Dendrolasin and Latrunculin A from the Fijan Sponge Spongia mycofijiensis and an Associated Nudibranch Chromodoris lochi. J. Nat. Prod. 1987;50:482–484. doi: 10.1021/np50051a023. [DOI] [Google Scholar]
- 122.Crews P., Kakou Y., Quinoa E. Mycothiazole, a Polyketide Heterocycle from a Marine Sponge. J. Am. Chem. Soc. 1988;110:4365–4368. doi: 10.1021/ja00221a042. [DOI] [Google Scholar]
- 123.Guella G., Mancini I., Chiasera G., Pietra F. Rogiolol Acetate: A Novel β-chamigrene-type Sesquiterpene Isolated from a Marine Sponge. Helv. Chim. Acta. 1990;73:1612–1620. doi: 10.1002/hlca.19900730606. [DOI] [Google Scholar]
- 124.Guella G., Pietra F. Rogiolenyne-A, Rogiolenyne-B, and Rogiolenyne-C—The first Branched Marine C-15 Acetogenins. Isolation from the Red Seaweed Laurencia microcladia or the Sponge Spongia-zimocca of Il-Rogiolo. Helv. Chim. Acta. 1991;74:47–54. doi: 10.1002/hlca.19910740107. [DOI] [Google Scholar]
- 125.Guella G., Mancini I., Pietra F. C-15 Acetogenins and Terpenes of the Dictyoceratid Sponge Spongia-zimocca of Il-Rogiolo: A Case of Seaweed-Metabolite Transfer to, and Elaboration within, a Sponge? Comp. Biochem. Phys. B Comp. Biochem. 1992;103:1019–1023. doi: 10.1016/0305-0491(92)90232-G. [DOI] [Google Scholar]
- 126.Brown J.W., Kesler C.T., Neary J.T., Fishman L.M. Effects of Marine Sponge Extracts on Mitogen-Activated Protein Kinase (MAPK/ERK1,2) Activity in SW-13 Human Adrenal Carcinoma Cells. Toxicon. 2001;39:1835–1839. doi: 10.1016/S0041-0101(01)00138-6. [DOI] [PubMed] [Google Scholar]
- 127.Brown J.W., Cappell S., Perez-Stable C., Fishman L.M. Extracts from Two Marine Sponges Lower Cyclin B1 Levels, Cause a G2/M Cell Cycle Block and Trigger Apoptosis in SW-13 Human Adrenal Carcinoma Cells. Toxicon. 2004;43:841–846. doi: 10.1016/j.toxicon.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 128.Bartolotta S.A., Scuteri M.A., Hick A.S., Palermo J., Brasco M.F.R., Hajdu E., Mothes B., Lerner C., Campos M., Carballo M.A. Evaluation of Genotoxic Biomarkers in Extracts of Marine Sponges from Argentinean South Sea. J. Exp. Mar. Biol. Ecol. 2009;369:144–147. doi: 10.1016/j.jembe.2008.11.008. [DOI] [Google Scholar]
- 129.Devi P., Wahidulla S., Kamat T., D’Souza L. Screening Marine Organisms for Antimicrobial Activity against Clinical Pathogens. Indian J. Mar. Sci. 2011;40:338–346. [Google Scholar]
- 130.Dellai A., Mansour H.B., Clary-Laroche A., Deghrigue M., Bouraoui A. Anticonvulsant and Analgesic Activities of Crude Extract and Its Fractions of the Defensive Secretion from the Mediterranean Sponge, Spongia officinalis. Cancer Cell Int. 2012;12:15. doi: 10.1186/1475-2867-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dellai A., Deghrigue M., Laroche-Clary A., Masour H.B., Chouchane N., Robert J., Bouraoui A. Anti-inflammatory and Antiproliferative Activities of Crude Extract and Its Fractions of the Defensive Secretion From the Mediterranean Sponge, Spongia officinalis. Drug Dev. Res. 2010;71:412–418. doi: 10.1002/ddr.20392. [DOI] [Google Scholar]
- 132.Dellai A., Deghrigue M., Laroche-Clary A., Masour H.B., Chouchane N., Robert J., Bouraoui A. Evaluation of Antiproliferative and Anti-inflammatory Activities of Methanol Extract and Its Fractions from the Mediterranean Sponge. Cancer Cell Int. 2012;12:18. doi: 10.1186/1475-2867-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lakshmi V., Ghosal S. Antiamoebic Activity of Marine Sponge Spongia officinalis var. ceylonensis Dendy. Bangladesh Pharm. J. 2014;17:38–42. [Google Scholar]