

Abstract
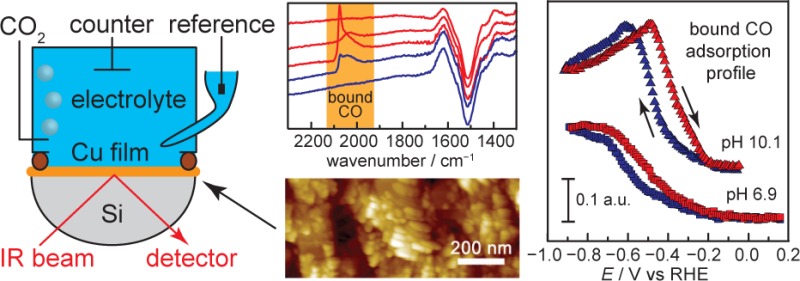
Rational design of selective CO2-to-fuels electrocatalysts requires direct knowledge of the electrode surface structure during turnover. Metallic Cu is the most versatile CO2-to-fuels catalyst, capable of generating a wide array of value-added products, including methane, ethylene, and ethanol. All of these products are postulated to form via a common surface-bound CO intermediate. Therefore, the kinetics and thermodynamics of CO adsorption to Cu play a central role in determining fuel-formation selectivity and efficiency, highlighting the need for direct observation of CO surface binding equilibria under catalytic conditions. Here, we synthesize nanostructured Cu films adhered to IR-transparent Si prisms, and we find that these Cu surfaces enhance IR absorption of bound molecules. Using these films as electrodes, we examine Cu-catalyzed CO2 reduction in situ via IR spectroelectrochemistry. We observe that Cu surfaces bind electrogenerated CO, derived from CO2, beginning at −0.60 V vs RHE with increasing surface population at more negative potentials. Adsorbed CO is in dynamic equilibrium with dissolved 13CO and exchanges rapidly under catalytic conditions. The CO adsorption profiles are pH independent, but adsorbed CO species undergo a reversible transformation on the surface in modestly alkaline electrolytes. These studies establish the potential, concentration, and pH dependencies of the CO surface population on Cu, which serve to maintain a pool of this vital intermediate primed for further reduction to higher order fuel products.
Short abstract
The dynamic nature of CO binding to Cu as a function of pH, CO concentration, and applied potential serves to establish a pool of this key intermediate primed for higher order fuel synthesis from CO2.
Introduction
The electroreduction of CO2 presents an attractive method for storing intermittent renewable electricity in energy-dense chemical bonds. Polycrystalline Cu foil surfaces are unique in mediating the conversion of CO2 to higher-order C1 and C2 fuels, including methane and ethylene, with moderate current densities (5–10 mA cm–2) and efficiencies (up to 69%).1−4 Engineering the surface structure of Cu by controlling particle morphology5,6 or by introducing defects7,8 has been shown to significantly enhance the selectivity and efficiency of CO2 and/or CO reduction to a variety of higher order products, including ethanol and acetate. In addition, for a wide array of differing Cu surface structures, it has been found that alkaline electrolytes promote the formation of C2 over C1 products.7−9 The rational design of more selective and efficient catalysts requires predictive mechanistic models that explain how Cu catalyzes the kinetically demanding multi-proton and multielectron conversion of CO2 and/or CO. Although the mechanistic proposals that have been put forward thus far differ in the assignment of the rate-limiting step for the formation of each product5,10−19 and the surface structures responsible for these rate-limiting steps,5,6,8,18,20−25 in all cases, these models invoke a common intermediate, surface-bound CO, which precedes the formation of all higher order fuels.
Despite its central role in CO2-to-fuels catalysis, the energetics and dynamics of CO adsorption to Cu under catalytic conditions have yet to be probed experimentally. As a common intermediate, the ability of the surface to form, accumulate, and transform this key species dictates the selectivity and efficiency of fuel-formation catalysis. In particular, experiments that address the following open questions are desired: (1) Does the catalyst resting state contain a high or low population of bound CO? (2) Is surface-bound CO in dynamic equilibrium with CO in solution? (3) How does the population of surface-bound CO change as a function of electrolyte pH, CO concentration, and applied potential? (4) Does bound CO proceed toward higher order products via a rate-limiting or quasi-equilibrium surface reaction? A direct probe of the surface-bound CO during CO2-to-fuels catalysis on Cu would provide insight into these key thermodynamic and kinetic questions.
The principal difficulty in tracking CO adsorption arises from the fact that CO binding to the electrode surface typically does not involve significant charge transfer and, therefore, does not give rise to a pronounced electrochemical signature. Consequently, monitoring surface-bound CO during catalysis requires robust in situ spectroscopic methods that are compatible with the extreme negative potentials required for CO2 reduction (CDR) catalysis. While both surface enhanced Raman spectroscopy (SERS)26,27 and external reflection Fourier transform infrared (FTIR)28,29 spectroscopies have been applied to observe CO on Cu surfaces, these techniques present severe limitations. SERS is known to amplify signals from hotspots on the electrode,30,31 making it difficult to extract a representative picture of the entire surface. Likewise, external reflection FTIR requires a very thin (<100 μm) electrolyte layer between the electrode and the IR-transparent prism,32 introducing large concentration gradients at the electrode surface. These gradients can convolute spectroscopic measurements, especially for gas-evolving reactions such as CDR. Conversely, surface enhanced infrared absorption spectroscopy (SEIRAS) in an attenuated total reflection (ATR) configuration has been shown to overcome these challenges.33−36 Achieving adequate surface enhancement requires a thin nanostructured film of the desired catalyst adhered to the surface of an IR transparent prism. The technique has been shown to provide a linear response in adsorbate coverage and signal intensity37 and to allow for unrestricted mass transport to the electrode surface. We have recently exploited these features to identify kinetically inert spectator CO species bound to Au during CDR catalysis.38 Unlike Au, adsorbed CO on Cu is postulated to be the common active intermediate on the pathway toward higher order fuel products, highlighting the primacy of direct observation of CO bound to Cu during CO2-to-fuels catalysis. Herein, we develop SEIRAS-active Cu electrodes on Si substrates to track the dynamics of surface-bound CO in situ under varying pH, potential, CO concentration CDR conditions.
Results and Discussion
SEIRAS-active and reductively stable Cu films were prepared by a two-step chemical deposition procedure.39 First, an H-terminated undoped Si substrate (Figure S1a,b) was immersed in 0.5% HF containing 750 μM CuSO4 to induce the formation of Cu seeds on the Si surface (Figure S1c,d).40 Second, Cu was grown on top of the seed layer to enhance the electrical conductivity of the Cu film. Cu overlayers (Figure S1e,f) were prepared by immersing the Cu seed layer into a bath containing disodium ethylenediaminetetraacetic acid, CuSO4, and formaldehyde as a reductant.41
The resulting films were characterized by X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), and electrochemistry. XPS spectra of the as-deposited Cu films, Figure S2, reveal peaks corresponding to Cu metal and adventitious CuOx with a ∼10% atomic surface fraction of Si, indicating the formation of a conformal Cu layer with a low fraction of exposed Si. AFM of as-deposited films reveal a faceted island morphology protruding from a conformal film with a thickness of ∼80 nm (Figures S1e,f and S3). To clean the Cu surface and reduce adventitious surface oxides, the electrode potential was cycled three times between open circuit (0.49 V vs RHE; all potentials are reported versus the reversible hydrogen electrode, RHE) and −0.10 V prior to IR measurements. Following this pretreatment, a cyclic voltammogram (CV) scan of the Cu surface, Figure 1b, reveals surface oxidation and back-reduction waves characteristic of Cu metal surfaces. Double layer capacitance measurements indicate that the Cu film has a roughness factor7 of ∼10, Figure 1b inset, which remains invariant over the course of a typical SEIRAS measurement, Figure S4. Cu films prepared by this method remain robust and conformal over multiple slow scan CVs (>1.5 h) as revealed by AFM. AFM images recorded immediately (<10 min) following a representative experimental run reveal a Si surface decorated with a continuous film of Cu consisting of interconnected ∼100 nm crystallites, Figure 1a. Together, the data indicate that this method serves to generate a nanostructured Cu electrode on IR-transparent Si substrates, which are ideality suited for ATR-SEIRAS measurements.
Figure 1.
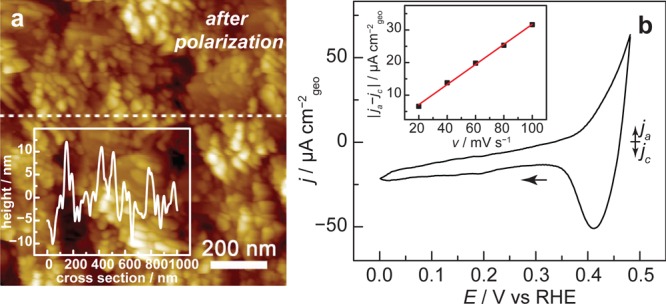
Characterization of SEIRAS-active films. (a) Atomic force microscopy image of a SEIRAS-active Cu film prepared on a Si substrate after polarization. (a, inset) Height profile along the dotted white line. (b) Cyclic voltammogram of a SEIRAS-active film recorded in 1.0 atm CO2-sparged 0.1 M NaHCO3. (b, inset) Double layer capacitance measurement of a SEIRAS-active film.
The Cu film prepared by this method effectively enhances vibrational signatures of surface adsorbates, allowing us to probe surface species during CDR catalysis in the CO2-saturated bicarbonate electrolytes commonly employed in studies of this reaction on polycrystalline Cu foils.1−4 Blank spectra were recorded at 0.25 V in CO2-saturated 0.1 M NaHCO3 electrolyte (pH 6.8), and IR traces were collected every 10 mV during a 2 mV s–1 CV shown in Figure S5. The CV displays only double layer charging current until the onset of a catalytic wave at −0.30 V, attributed to both CDR and catalytic hydrogen evolution. Upon scanning the potential to 0.03 V, we observe a pronounced bleach at 1524 cm–1, Figure 2a, attributed to (bi)carbonate anion desorption from the electrode surface.42,43 Upon scanning to more negative potentials, we observe the slow rise of a band at 1620 cm–1, Figure 2a, attributed to the δHOH bending mode of adsorbed water on the electrode surface.44,45 The rise of this feature coincides with the rise of a broad feature spanning 3500 to 3040 cm–1, Figure S6, attributed to O–H stretching modes of these adsorbed water molecules.44,45 Scanning back to more positive potentials does not lead to a significant diminishment of these features, suggesting that (bi)carbonate does not readsorb over the potential range examined. In line with this observation, subsequent traces recorded over the same potential window and blanked at the same initial potential display diminished features associated with water adsorption and (bi)carbonate desorption (Figure S7). We note that scanning to potentials negative of 0.03 V introduces a sloping of the baseline, which is most likely due to the irreversible desorption of (bi)carbonate that introduces changes in the plasmonic absorption properties of the Cu film.46 Together, the data suggest that a water adlayer decorates the Cu surface at the reducing potentials necessary for CDR.
Figure 2.
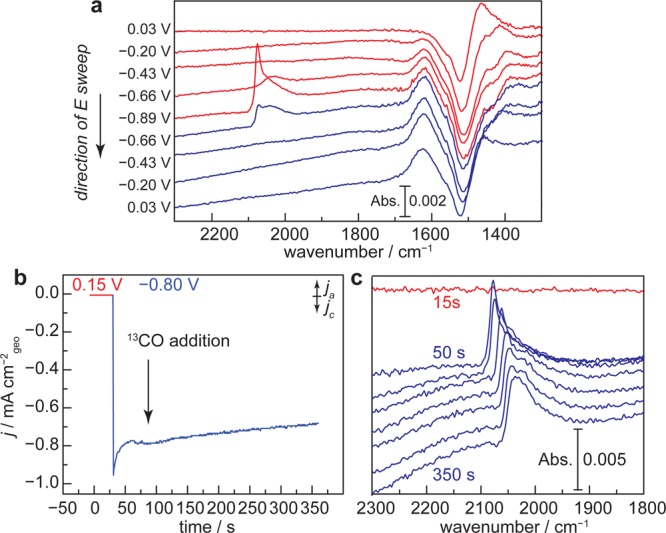
Dynamics of electrogenerated surface-adsorbed CO on Cu. SEIRA spectra (a) recorded in 1.0 atm CO2-saturated 0.1 M NaHCO3. Chronoamperometry trace (b) and corresponding SEIRA spectra (c) recorded in CO2-saturated 0.1 M NaHCO3. 13CO was introduced to the medium following 30 s of polarization at −0.80 V as denoted by the solid black arrow.
Electrogenerated CO, formed via reduction of CO2, is bound to the electrode surface upon reductive polarization and is labile under catalytic conditions. At −0.66 V, a broad band is observed spanning 2104 and 1970 cm–1, attributed to electrogenerated CO bound (CObound) to the Cu surface, Figure 2a. Upon polarization to more negative potentials, this CO peak takes on an asymmetric shape consisting of a sharp peak at 2075 cm–1 and a broad tail at lower stretching frequencies. The asymmetry and the large width of this peak may suggest a complex distribution of local CO binding environments on the Cu surface47,48 as well as Fano-type coupling of CO vibrations with metal-based absorptions.49−53 The range of stretching frequencies we observe encompasses the values reported in the literature for linear adsorption of CO on Cu,39,54 but we cannot rule out the possibility of other binding modes under these conditions. When the electrode was rapidly switched from 0.15 V to −0.80 V in CO2-saturated 0.1 M NaHCO3, Figure S8, saturation in the CO signal intensity was reached within ∼16 s, suggesting that the 2 mV s–1 scan rate utilized in this study was sufficient to accumulate a quasi-steady-state surface concentration of CO. Under steady-state polarization at −0.80 V, we observe that these surface-bound electrogenerated CO species can be readily exchanged for 13CO on the time scale of gas mixing into the medium (Figure 2b,c). Importantly, the CO peak shifts from 2077 to 2032 cm–1 upon isotopic substitution. This 45 cm–1 shift is in line with the 46 cm–1 shift expected using a simple harmonic oscillator model. Together, the data suggest that electrogenerated CO are bound to the electrode surface at potentials more negative than −0.66 V and that these species are labile, exchanging readily with dissolved CO.
Integrating the CO peak intensity as a function of potential reveals a gradual rise in the surface population of electrogenerated CO as the driving force for CDR catalysis increases. In 0.75 atm CO2-saturated 0.1 M NaHCO3 (pH 6.9) with 0.25 atm Ar balance, we observe that the signal corresponding to the surface population of electrogenerated CObound species rises in an exponential fashion beginning at −0.60 V and reaches a maximum at −0.80 V, Figure 3, squares. The corresponding CV, shown in Figure S9 (black), displays double layer charging current until the onset of reductive catalysis at −0.30 V, attributed to both CDR and catalytic hydrogen evolution. Control experiments in the absence of CO2 or at reduced partial pressures of CO2 would be significantly convoluted by large swings in the electrolyte pH, see below and experimental details in SI, because the pH 6.9 conditions are maintained by the water-CO2-bicarbonate equilibrium. In Figure 3, squares, the potential at which we observe the rise in CObound, −0.60 V, coincides with the onset of CO production reported for polycrystalline Cu foils,9 indicating that SEIRAS-active Cu films are catalytically active for CDR under these conditions. While a direct quantitative comparison of the selectivity and efficiency of these SEIRAS-active Cu films relative to polycrystalline foils is convoluted by partial film delamination of the Cu from the Si substrate under prolonged electrolysis, we nonetheless observe that these films are capable of producing higher order products at potentials comparable to those observed for foils (Figure S10). On the reverse potential scan, the CObound desorption profile is shifted to more positive potentials by ∼0.10 V, suggesting that CO adsorption induces restructuring of the electrode surface55 that serves to increase its own adsorption strength. As these adsorption profiles are not pH dependent, as shown below, this hysteresis cannot be explained by changes in the local pH at the electrode surface. Importantly, the observed saturation in the CObound signal at −0.80 V coincides with the potential at which CO gas production activity declines on polycrystalline Cu foils under similar conditions.9 At more negative potentials, methane and ethylene are produced in appreciable quantities.9 These experimental observations combined with the data uncovered here suggest that the potential dependence of CO adsorption, see below, on the Cu surface serves to inhibit the liberation of electrogenerated CO from the surface and, thereby, maintains a high population of CObound primed for conversion to higher order products.
Figure 3.
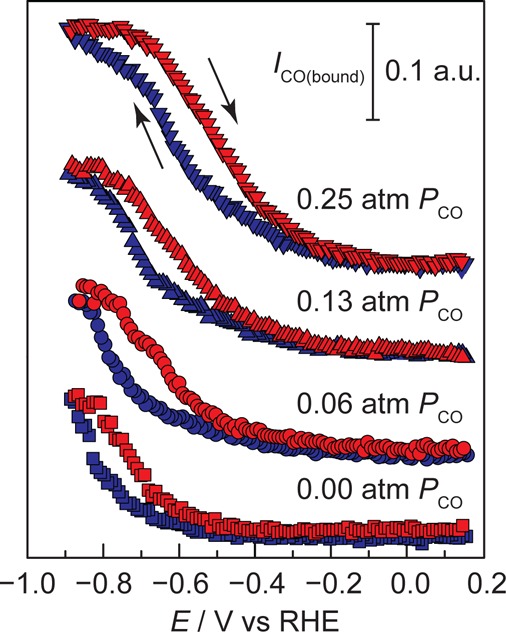
CO adsorption profiles at pH 6.9. Potential-dependence of the integrated CO band intensity for electrogenerated CO (squares), upon addition of 0.06 atm CO (circles), 0.13 atm CO (upward triangles), and 0.25 atm CO (downward triangles). Data were recorded in 0.75-atm CO2-saturated 0.1 M NaHCO3 during both negative-going (blue) and positive-going (red) potential scans.
Integration of CObound as a function of exogenous CO partial pressures allows for quantitative insight into the CO adsorption profile under the conditions of CDR catalysis. Upon addition of exogenous CO at varying partial pressures while maintaining 0.75 atm of CO2 and an electrolyte pH of 6.9, we observe that the onset of CO adsorption systematically shifts to less negative potentials (Figure 3). At CO partial pressures (PCO) of 0.06, 0.13, and 0.25, CO adsorption onsets at −0.48, −0.40, and −0.30 V, respectively. At each PCO, the CObound signal rises in a sigmoidal fashion before beginning to saturate at −0.90 V, −0.80 V, and −0.73 V for PCO of 0.06, 0.13, and 0.25, respectively. Corresponding CVs shown in Figure S9 display the onset of the catalytic current at −0.30 V. The total catalytic current observed does not significantly change with increasing PCO, suggesting that the products of catalysis under these conditions do not strongly depend on PCO. Interestingly, the CO surface population rises over the span of ∼600 mV. For an adsorption process involving one electron transfer, a Langmuirian adsorption model predicts a transition from low (θ = 0.1) to high (θ = 0.9) surface coverage within the span of ∼120 mV.56 The significantly weaker potential-dependence observed here is in line with CO experiencing insignificant charge transfer upon adsorption to the surface. Indeed, the weak potential-dependence may be dominated by the potential dependence of water desorption rather than CO adsorption.57 We note that CO adsorption occurs close to the potential of zero charge of Cu58 at which the interfacial field is weakest and therefore least able to stabilize the large dipole moment of adsorbed water. Together, the data indicate that CO binds to the electrode with a weakly potential-dependent adsorption profile. The wide range of potentials over which we observe an intermediate surface concentration of CO highlights that Cu binds this intermediate neither too strongly nor too weakly under the conditions of the reaction, providing a direct experimental validation of the Sabatier principle59 in CO2-to-fuels catalysis.
The electrolyte pH is known to play a central role in dictating selectivity in CO/CO2 reduction on Cu surfaces with, in general, higher pH values favoring C2 products relative to C1 species.9,60,61 Shown in Figure 4a are CO adsorption/desorption profiles as a function of pH. For these traces, the exogenous CO partial pressure was fixed at 0.25, and the bicarbonate/carbonate concentration was fixed at 0.1 M. The electrolyte pH was adjusted from 6.9 to 10.1 by varying the partial pressure of CO2 relative to Ar. IR spectra used to construct the profiles are shown in Figure S11. As the pH is increased, the adsorption profiles shift monotonically to less negative potentials on an RHE scale. This shift is of the same magnitude as the 60 mV pH–1 shift of the RHE, and, therefore, these profiles roughly overlay when plotted on the pH-independent standard hydrogen electrode (SHE) scale, Figure 4b. These data establish that CObound accumulates in a pH-independent but potential-dependent fashion as denoted by the green region in Figure 4c. However, as shown in Figure 4c, the thermodynamic potentials for forming higher order fuels such as ethylene (red) and methane (blue), shift by 60 mV pH–1, thereby allowing for higher CO surface populations at lower effective overpotentials (η) in more alkaline electrolytes. Thus, the lack of pH dependence of the CO surface population may in part explain the general observation that CO reduction is more facile in alkaline electrolytes.7−9
Figure 4.
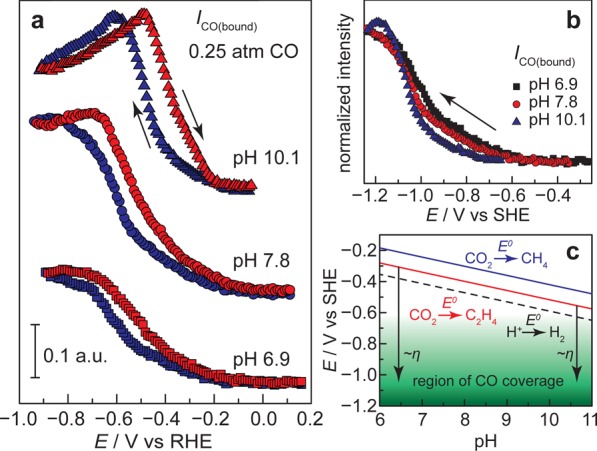
(a) CO adsorption profiles at varying pH. Integrated CO band intensities at pH 6.9 (squares), 7.8 (circles), and 10.1 (triangles) recorded in the presence of 0.25 atm of CO in 0.1 M (bi)carbonate buffer on both negative-going (blue) and positive-going (red) scans. (b) Normalized CO integrated band intensities for negative-going scans at pH 6.9 (black squares), 7.8 (red circles), and 10.1 (blue triangles) plotted vs the pH-independent SHE scale. (c) Pourbaix diagram depicting the region of observed surface-bound CO on copper (green region) and the thermodynamic potentials for hydrogen (black dashed line), methane (blue line), and ethylene (red line) production as a function of pH. Black arrows denote the approximate overpotentials (η) required to reach maximal CO surface concentrations.
Contemporary mechanistic models postulate that surface-bound CO on Cu reacts via rate-limiting chemistry to form higher order C1 and C2 products,10,13,18,62 and therefore the rise and fall in CObound population provide insight into possible chemical processes that could contribute to its further reduction on the surface. While the CObound population rises in a pH-independent fashion, see above, the saturation of the signal corresponding to the CO surface population strongly depends on the solution pH, Figure 4a. Strikingly, at pH 10.1, the CO adsorption profile does not simply rise and plateau, Figure 4a (triangles) and Figure 5. Instead, the adsorbed CO population rises, peaks, and declines as the electrode is polarized to more negative potentials. Notably, this decline is reversible, albeit with the slight hysteresis observed in all traces, indicating that this decline in CObound band intensity cannot be attributed to irreversible degradation of the film or formation of a kinetically inert surface species. Importantly, in Figure 5, inset, we observe that the rising branch of the profile (−0.08 V to −0.60 V or −0.80 V, depending on PCO) at pH 10.1 scales with PCO, in line with equilibrium adsorption, but the declining branch (−0.60 V or −0.80 V to −0.90 V depending on PCO) is largely insensitive to PCO, suggesting that this decline in CObound is not due to equilibrium desorption of CO. Instead, the data are most consistent with a reversible conversion of adsorbed CO to another surface species. Under these modestly alkaline conditions, we observe another very low intensity peak at 1801 cm–1, Figures S11d and S12. However, this peak rises and falls over a dramatically different potential (−0.10 V to −0.50 V) range than the major CObound peak.
Figure 5.
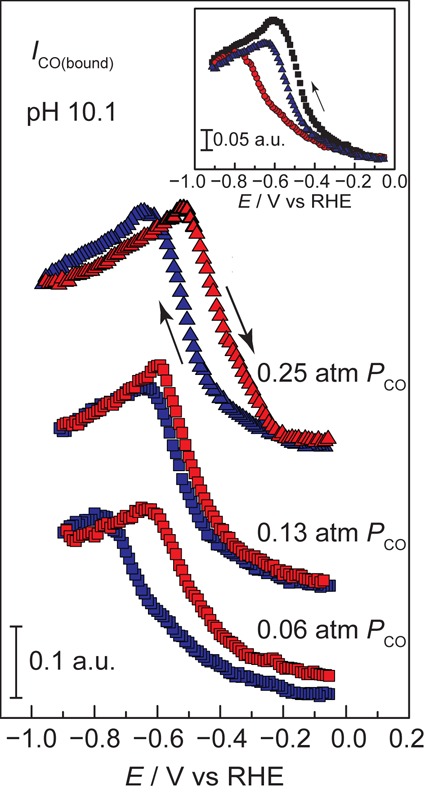
CO adsorption profiles recorded at pH 10.1. Potential-dependence of the integrated CO band intensity upon exposure to 0.06 atm CO, 0.13 atm CO, and 0.25 atm CO. Data were recorded in Ar-saturated 0.1 M (bi)carbonate during both negative-going (blue) and positive-going (red) potential scans. Inset: Overlay of CO integrated band intensities of negative-going scans at 0.06 atm CO (red), 0.13 atm CO (blue), and 0.25 atm CO (black).
To estimate the spectral signatures of putative intermediates downstream of CO, we calculated the stretching frequencies of a surface-bound CO dimer that retains two carbon–metal bonds, OCCO, and a surface-bound formyl with a metal–carbon linkage, CHO (Figure S13). Using computed structures reported previously,19 vibrational frequencies for these species were calculated on Cu (100) slabs using periodic Kohn–Sham density functional theory (see SI for details). While the calculations were performed with the absence of both explicit solvent and temperature, we find that the computed values for CO bound to low index facets of Cu appear between 2032 and 2049 cm−1 (Table S1). These values lie within the 2104 to 1970 cm−1 range observed experimentally, suggesting that the computational methods employed permit a reasonable estimation of the experimental vibrational frequencies observed in situ. Surface-bound OCCO, formed via the reductive coupling of two surface bound CO species or the combination of a surface-bound CO species with COg, has been postulated to be a key intermediate in the formation of C–C bonds on copper surfaces.10,14,15,19,63 We compute that C–O vectors of the surface-bound OCCO species contribute to symmetric and asymmetric stretches at 1510 and 1541 cm–1, respectively (Table S1 and Figure S13a). Both of these stretches lie in the region of (bi)carbonate desorption in the IR spectra, Figure S11d. Thus, the observation of an OCCO species could be obscured by the concomitant electrolyte desorption observed under these conditions. Conversely, a commonly proposed one-electron, one-proton, reduced intermediate from CO, surface-bound formyl, CHO,10−12,62,63 was computed to possess a C–O stretching frequency of 1741 cm–1 (Figure S13b). While this region of the spectrum is unobscured by other peaks (Figure S11d), we cannot conclusively rule out the formation of CHO because this species may possess lower C–O oscillator strengths64 and/or large vibrational vector components parallel to the surface, which would be canceled out by the surface selection rule.33−36 Together, our spectroscopic and computational data do not allow us to rule out the possibility that CO is transformed to CHO, OCCO, or other surface species in modestly alkaline media, but the spectroscopic observation of a reversible CO conversion motivates further investigations.
While we only observe surface conversion of CObound in quasi-equilibrium at pH 10.1, this chemistry may also be at play at the neutral pH conditions typical of most CDR studies. In particular, for weakly buffered media or highly porous electrodes, the local pH at the Cu surface is expected to be in excess of 9.65 Notwithstanding, the observation of reversible conversion of adsorbed CO runs counter to many contemporary mechanistic schemes,10,13,18,62 which posit that CO reacts via rate-limiting chemistry to form higher order C1 and C2 products.
Concluding Remarks
Herein, we establish a synthesis of nanostructured Cu films on IR transparent Si substrates, and we use these films as electrodes for surface enhanced infrared absorption spectroscopy (SEIRAS) measurements. Using SEIRAS, we probe the variation in the aggregate CO surface population as a function of the key parameters (potential, CO concentration, pH) relevant to CO2 reduction catalysis. We find that electrogenerated CO species bind to Cu surfaces at potentials more negative than −0.60 V vs RHE. The surface population rises in a weakly potential-dependent fashion upon polarization to more negative values and reaches a potential-invariant maximum at −0.80 V vs RHE. This potential is in line with the observed decline in CO gas production and the rise in higher order product formation, highlighting that the potential dependence of CO adsorption serves to inhibit its liberation from the Cu electrode and maintain a large pool of adsorbed CO available for further reduction. Furthermore, electrogenerated bound CO remains in quasi-equilibrium with dissolved CO, exhibiting facile exchange with 13CO. These observations provide the first experimental evidence for the Sabatier Principle in CO2-to-fuels catalysis: Cu’s intermediate affinity for CO underlies its unique ability to reduce CO2 beyond CO to higher order products. Additionally, we show that CO adsorption is pH-independent, providing an explanation for the general phenomenon that CO reduction occurs more readily in alkaline electrolytes. Whereas at intermediate pH, the CO adsorption profile reaches a maximum and plateaus off, in more alkaline environments, surface-bound CO appears to undergo a reversible transformation on the Cu surface, challenging the prevailing mechanistic models that postulate that product formation occurs via a rate-limiting transformation of adsorbed CO. Together, our data suggest that the relative rates of higher order production formation on Cu are dictated by the population of active sites capable of CO conversion as well as the pool of available surface-bound CO, which is highly dependent on the pH, CO concentration, and potential. This work, therefore, provides a rich picture of the dynamic nature of CO binding to Cu, which serves to establish the surface population landscape that underpins CO2-to-fuels conversion.
Acknowledgments
We acknowledge Tomohiro Fukushima for facilitating studies conducted at HU. A.W. and Y.S. gratefully acknowledge the MIT International Science and Technology Initiatives and the Hayashi Seed Grant for Travel Funds to HU. This research was supported by the Air Force Office of Scientific Research under AFOSR Award No. FA9550-15-1-0135 and by the MIT Department of Chemistry through junior faculty funds for Y.S. Computational investigations made use of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (NSF) (grant ACI-1053575). Work at HU was partially supported by the New Energy and Industrial Technology Development Organization (NEDO). A.W. is supported by a Graduate Research Fellowship from the National Science Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00155.
Full experimental details, AFM characterization of copper films, XPS characterization of copper films, double layer capacitance measurements of copper films during sequential spectroscopic measurements, cyclic voltammograms accompanying all spectroscopic measurements, spectra shown over larger spectral range, time-dependent spectra at a single potential, and vibrational frequency calculations of surface-bound intermediates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hori Y.; Kikuchi K.; Suzuki S. Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1985, 14, 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Hori Y.; Kikuchi K.; Murata A.; Suzuki S. Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide at Copper Electrode in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1986, 15, 897–898. 10.1246/cl.1986.897. [DOI] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R.; Suzuki S. Enhanced Formation of Ethylene and Alcohols at Ambient Temperature and Pressure in Electrochemical Reduction of Carbon Dioxide at a Copper Electrode. J. Chem. Soc., Chem. Commun. 1988, 17–19. 10.1039/c39880000017. [DOI] [Google Scholar]
- Noda H.; Ikeda S.; Oda Y.; Ito K. Potential Dependencies of the Products on Electrochemical Reduction of Carbon Dioxide at a Copper Electrode. Chem. Lett. 1989, 18, 289–292. 10.1246/cl.1989.289. [DOI] [Google Scholar]
- Manthiram K.; Beberwyck B. J.; Alivisatos A. P. Enhanced Electrochemical Methanation of Carbon Dioxide with a Dispersible Nanoscale Copper Catalyst. J. Am. Chem. Soc. 2014, 136, 13319–13325. 10.1021/ja5065284. [DOI] [PubMed] [Google Scholar]
- Loiudice A.; Lobaccaro P.; Kamali E. A.; Thao T.; Huang B. H.; Ager J. W.; Buonsanti R. Tailoring Copper Nanocrystals towards C2 Products in Electrochemical CO2 Reduction. Angew. Chem., Int. Ed. 2016, 55, 5789–5792. 10.1002/anie.201601582. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Ciston J.; Kanan M. W. Electroreduction of Carbon Monoxide to Liquid Fuel on Oxide-Derived Nanocrystalline Copper. Nature 2014, 508, 504–507. 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Feng X.; Jiang K.; Fan S.; Kanan M. W. A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci. 2016, 2, 169–174. 10.1021/acscentsci.6b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R. Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution. J. Chem. Soc., Faraday Trans. 1 1989, 85, 2309–2326. 10.1039/f19898502309. [DOI] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Peterson A. A.; Abild-Pedersen F.; Studt F.; Rossmeisl J.; Norskov J. K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. 10.1039/c0ee00071j. [DOI] [Google Scholar]
- Nie X.; Esopi M. R.; Janik M. J.; Asthagiri A. Selectivity of CO2 Reduction on Copper Electrodes: The Role of the Kinetics of Elementary Steps. Angew. Chem., Int. Ed. 2013, 52, 2459–2462. 10.1002/anie.201208320. [DOI] [PubMed] [Google Scholar]
- Schouten K. J. P.; Kwon Y.; van der Ham C. J. M.; Qin Z.; Koper M. T. M. A New Mechanism for the Selectivity to C1 and C2 Species in the Electrochemical Reduction of Carbon Dioxide on Copper Electrodes. Chem. Sci. 2011, 2, 1902–1909. 10.1039/c1sc00277e. [DOI] [Google Scholar]
- Calle-Vallejo F.; Koper M. T. M. Theoretical Considerations on the Electroreduction of CO to C2 Species on Cu(100) Electrodes. Angew. Chem., Int. Ed. 2013, 52, 7282–7285. 10.1002/anie.201301470. [DOI] [PubMed] [Google Scholar]
- Montoya J. H.; Shi C.; Chan K.; Nørskov J. K. Theoretical Insights into a CO Dimerization Mechanism in CO2 Electroreduction. J. Phys. Chem. Lett. 2015, 6, 2032–2037. 10.1021/acs.jpclett.5b00722. [DOI] [PubMed] [Google Scholar]
- Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. 10.1039/c2ee21234j. [DOI] [Google Scholar]
- Hori Y.Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas C., White R., Gamboa-Aldeco M., Eds.; Springer: New York, 2008; Vol. 42, pp 89–189. [Google Scholar]
- Gattrell M.; Gupta N.; Co A. A Review of the Aqueous Electrochemical Reduction of CO2 to Hydrocarbons at Copper. J. Electroanal. Chem. 2006, 594, 1–19. 10.1016/j.jelechem.2006.05.013. [DOI] [Google Scholar]
- Goodpaster J. D.; Bell A. T.; Head-Gordon M. Identification of Possible Pathways for C–C Bond Formation during Electrochemical Reduction of CO2: New Theoretical Insights from an Improved Electrochemical Model. J. Phys. Chem. Lett. 2016, 7, 1471–1477. 10.1021/acs.jpclett.6b00358. [DOI] [PubMed] [Google Scholar]
- Verdaguer-Casadevall A.; Li C. W.; Johansson T. P.; Scott S. B.; McKeoen J. T.; Kumar M.; Stephens I. E. L.; Kanan M. W.; Chorkendorff I. Probing the Active Surface Sites for CO Reduction on Oxide- Derived Copper Electrocatalysts. J. Am. Chem. Soc. 2015, 137, 9808–9811. 10.1021/jacs.5b06227. [DOI] [PubMed] [Google Scholar]
- Schouten K. J. P.; Qin Z.; Pérez Gallent E.; Koper M. T. M. Two Pathways for the Formation of Ethylene in CO Reduction on Single-Crystal Copper Electrodes. J. Am. Chem. Soc. 2012, 134, 9864–9867. 10.1021/ja302668n. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Takahashi I.; Koga O.; Hoshi N. Electrochemical Reduction of Carbon Dioxide at Various Series of Copper Single Crystal Electrodes. J. Mol. Catal. A: Chem. 2003, 199, 39–47. 10.1016/S1381-1169(03)00016-5. [DOI] [Google Scholar]
- Tang W.; Peterson A. A.; Varela A. S.; Jovanov Z. P.; Bech L.; Durand W. J.; Dahl S.; Norskov J. K.; Chorkendorff I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Electroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. 10.1039/C1CP22700A. [DOI] [PubMed] [Google Scholar]
- Kas R.; Kortlever R.; Milbrat A.; Koper M. T. M.; Mul G.; Baltrusaitis J. Electrochemical CO2 Reduction on Cu2O-Derived Copper Nanoparticles: Controlling the Catalytic Selectivity of Hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. 10.1039/c4cp01520g. [DOI] [PubMed] [Google Scholar]
- Reske R.; Mistry H.; Behafarid F.; Roldan Cuenya B.; Strasser P. Particle Size Effects in the Catalytic Electroreduction of CO2 on Cu Nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. 10.1021/ja500328k. [DOI] [PubMed] [Google Scholar]
- Oda I.; Ogasawara H.; Ito M. Carbon Monoxide Adsorption on Copper and Silver Electrodes during Carbon Dioxide Electroreduction Studied by Infrared Reflection Absorption Spectroscopy and Surface-Enhanced Raman Spectroscopy. Langmuir 1996, 12, 1094–1097. 10.1021/la950167j. [DOI] [Google Scholar]
- Batista E. A.; Temperini M. L. A. Spectroscopic Evidences of the Presence of Hydrogenated Species on the Surface of Copper during CO2 Electroreduction at Low Cathodic Potentials. J. Electroanal. Chem. 2009, 629, 158–163. 10.1016/j.jelechem.2009.02.010. [DOI] [Google Scholar]
- Hori Y.; Koga O.; Watanabe Y.; Matsuo T. FTIR Measurements of Charge Displacement Adsorption of CO on Poly- and Single Crystal (100) of Cu Electrodes. Electrochim. Acta 1998, 44, 1389–1395. 10.1016/S0013-4686(98)00261-8. [DOI] [Google Scholar]
- Hori Y.; Koga O.; Yamazaki H.; Matsuo T. Infrared Spectroscopy of Adsorbed CO and Intermediate Species in Electrochemical Reduction of CO2 to Hydrocarbons on a Cu Electrode. Electrochim. Acta 1995, 40, 2617–2622. 10.1016/0013-4686(95)00239-B. [DOI] [Google Scholar]
- Surface Enhanced Raman Scattering; Chang R. K., Furtak T. E., Eds.; Plenum Press: New York, 1982. [Google Scholar]
- Moskovits M. Surface-Enhanced Spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826. 10.1103/RevModPhys.57.783. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Spectroelectrochemistry and Other Coupled Characterization Methods. In Electrochemical Methods; John Wiley & Sons, Inc.: New York, 2001; p 700. [Google Scholar]
- Osawa M. Dynamic Processes in Electrochemical Reactions Studied by Surface-Enhanced Infrared Absorption Spectroscopy(SEIRAS). Bull. Chem. Soc. Jpn. 1997, 70, 2861–2880. 10.1246/bcsj.70.2861. [DOI] [Google Scholar]
- Osawa M.Surface Enhanced Infrared Absorption Spectroscopy. In Handbook of Vibrational Spectroscopy; Chalmers J. M., Griffiths P. R., Eds.; Wiley-VCH: Chichester, UK, 2002; pp 785–800. [Google Scholar]
- Osawa M.In-Situ Surface-Enhanced Infrared Spectroscopy of the Electrode/Solution Interface. In Diffraction and Spectroscopic Methods in Electrochemistry (Advances in Electrochemical Science and Engineering); Alkire R. C., Kolb D. M., Lipkowski J., Ross P. N., Eds.; Wiley-VCH: New York, 2006; pp 269–314, Vol. 9. [Google Scholar]
- Osawa M.Surface-Enhanced Infrared Absorption. In Near-Field Optics and Surface Plasmon Polaritons. Topics in Applied Physics; Kawata S., Ed.; Springer: Berlin, 2001; Vol. 81, pp 163–187. [Google Scholar]
- Samjeské G.; Komatsu K.; Osawa M. Dynamics of CO Oxidation on a Polycrystalline Platinum Electrode: A Time-Resolved Infrared Study. J. Phys. Chem. C 2009, 113, 10222–10228. 10.1021/jp900582c. [DOI] [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited Proton Transfer Enhances Au-Catalyzed CO2-to-Fuels Selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-F.; Yan Y.-G.; Huo S.-J.; Cai W.-B.; Xu Q.-J.; Osawa M. Seeded Growth Fabrication of Cu-on-Si Electrodes for in Situ ATR-SEIRAS Applications. Electrochim. Acta 2007, 52, 5950–5957. 10.1016/j.electacta.2007.03.042. [DOI] [Google Scholar]
- Ye S.; Ichihara T.; Uosaki K. Spectroscopic Studies on Electroless Deposition of Copper on a Hydrogen-Terminated Si(111) Surface in Fluoride Solutions. J. Electrochem. Soc. 2001, 148, C421–C426. 10.1149/1.1370964. [DOI] [Google Scholar]
- Shimizu S. The Present and Future Trends in Electroless Copper Plating. Hyomen Gijutsu 2007, 58, 81–86. 10.4139/sfj.58.81. [DOI] [Google Scholar]
- Berná A.; Rodes A.; Feliu J. M.; Illas F.; Gil A.; Clotet A.; Ricart J. M. Structural and Spectroelectrochemical Study of Carbonate and Bicarbonate Adsorbed on Pt(111) and Pd/Pt(111) Electrodes. J. Phys. Chem. B 2004, 108, 17928–17939. 10.1021/jp048082r. [DOI] [Google Scholar]
- Arihara K.; Kitamura F.; Ohsaka T.; Tokuda K. Characterization of the Adsorption State of Carbonate Ions at the Au(111) Electrode Surface Using in Situ IRAS. J. Electroanal. Chem. 2001, 510, 128–135. 10.1016/S0022-0728(01)00498-3. [DOI] [Google Scholar]
- Osawa M.; Tsushima M.; Mogami H.; Samjeské G.; Yamakata A. Structure of Water at the Electrified Platinum–Water Interface: A Study by Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. C 2008, 112, 4248–4256. 10.1021/jp710386g. [DOI] [Google Scholar]
- Ataka K.; Yotsuyanagi T.; Osawa M. Potential-Dependent Reorientation of Water Molecules at an Electrode/Electrolyte Interface Studied by Surface-Enhanced Infrared Absorption Spectroscopy. J. Phys. Chem. 1996, 100, 10664–10672. 10.1021/jp953636z. [DOI] [Google Scholar]
- Osawa M.; Ikeda M. Surface-Enhanced Infrared Absorption of P-Nitrobenzoic Acid Deposited on Silver Island Films: Contributions of Electromagnetic and Chemical Mechanisms. J. Phys. Chem. 1991, 95, 9914–9919. 10.1021/j100177a056. [DOI] [Google Scholar]
- Truong C. M.; Rodriguez J.; Goodman D. W. CO Adsorption Isotherms on Cu(100) at Elevated Pressures and Temperatures Using Infrared Reflection Absorption Spectroscopy. Surf. Sci. 1992, 271, L385–L391. 10.1016/0039-6028(92)90896-E. [DOI] [Google Scholar]
- Hoffmann F. Infrared Reflection-Absorption Spectroscopy of Adsorbed Molecules. Surf. Sci. Rep. 1983, 3, 107–192. 10.1016/0167-5729(83)90001-8. [DOI] [Google Scholar]
- Wu C.-X.; Lin H.; Chen Y.-J.; Li W.-X.; Sun S.-G. Abnormal IR Effects of Pt Nanostructured Surfaces upon CO Chemisorption due to Interaction and Electron-Hole Damping. J. Chem. Phys. 2004, 121, 1553–1556. 10.1063/1.1763135. [DOI] [PubMed] [Google Scholar]
- Su Z.-F.; Sun S.-G.; Wu C.-X.; Cai Z.-P. Study of Anomalous Infrared Properties of Nanomaterials through Effective Medium Theory. J. Chem. Phys. 2008, 129, 044707. 10.1063/1.2953441. [DOI] [PubMed] [Google Scholar]
- Krauth O.; Fahsold G.; Magg N.; Pucci A. Anomalous Infrared Transmission of Adsorbates on Ultrathin Metal Films: Fano Effect near the Percolation Threshold. J. Chem. Phys. 2000, 113, 6330–6333. 10.1063/1.1309133. [DOI] [Google Scholar]
- Wang H. C.; Sun S. G.; Yan J. W.; Yang H. Z.; Zhou Z. Y. In Situ STM Studies of Electrochemical Growth of Nanostructured Ni Films and Their Anomalous IR Properties. J. Phys. Chem. B 2005, 109, 4309–4316. 10.1021/jp046313o. [DOI] [PubMed] [Google Scholar]
- Priebe A.; Sinther M.; Fahsold G.; Pucci A. The Correlation between Film Thickness and Adsorbate Line Shape in Surface Enhanced Infrared Absorption. J. Chem. Phys. 2003, 119, 4887–4890. 10.1063/1.1597232. [DOI] [Google Scholar]
- Pritchard J.; Catterick T.; Gupta R. K. Infrared Spectroscopy of Chemisorbed Carbon Monoxide on Copper. Surf. Sci. 1975, 53, 1–20. 10.1016/0039-6028(75)90113-2. [DOI] [Google Scholar]
- Kim Y.-G.; Baricuatro J. H.; Javier A.; Gregoire J. M.; Soriaga M. P. The Evolution of the Polycrystalline Copper Surface, First to Cu(111) and Then to Cu(100), at a Fixed CO2RR Potential: A Study by Operando EC-STM. Langmuir 2014, 30, 15053–15056. 10.1021/la504445g. [DOI] [PubMed] [Google Scholar]
- Gileadi E.Intermediates in Electrode Reactions. In Physical Electrochemistry, Fundamentals, Techniques and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2011; pp 151–164. [Google Scholar]
- Bockris J. O.; Devanathan M. A. V.; Muller K. On the Structure of Charged Interfaces. Proc. R. Soc. London, Ser. A 1963, 274, 55–79. 10.1098/rspa.1963.0114. [DOI] [Google Scholar]
- Łukomska A.; Sobkowski J. Potential of Zero Charge of Monocrystalline Copper Electrodes in Perchlorate Solutions. J. Electroanal. Chem. 2004, 567, 95–102. 10.1016/j.jelechem.2003.11.063. [DOI] [Google Scholar]
- Sabatier P. Hydrogénations et Déshydrogénations Par Catalyse. Ber. Dtsch. Chem. Ges. 1911, 44, 1984–2001. 10.1002/cber.19110440303. [DOI] [Google Scholar]
- Hori Y.; Takahashi R.; Yoshinami Y.; Murata A. Electrochemical Reduction of CO at a Copper Electrode. J. Phys. Chem. B 1997, 101, 7075–7081. 10.1021/jp970284i. [DOI] [Google Scholar]
- Varela A. S.; Kroschel M.; Reier T.; Strasser P. Controlling the Selectivity of CO2 Electroreduction on Copper: The Effect of the Electrolyte Concentration and the Importance of the Local pH. Catal. Today 2016, 260, 8–13. 10.1016/j.cattod.2015.06.009. [DOI] [Google Scholar]
- Peterson A. A.; Nørskov J. K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. 10.1021/jz201461p. [DOI] [Google Scholar]
- Schouten K. J. P.; Kwon Y.; van der Ham C. J. M.; Qin Z.; Koper M. T. M. A New Mechanism for the Selectivity to C1 and C2 Species in the Electrochemical Reduction of Carbon Dioxide on Copper Electrodes. Chem. Sci. 2011, 2, 1902–1909. 10.1039/c1sc00277e. [DOI] [Google Scholar]
- Serrano-Andres L.; Forsberg N.; Malmqvist P.-Å. Vibronic Structure in Triatomic Molecules: The Hydrocarbon Flame Bands of the Formyl Radical (HCO). A Theoretical Study. J. Chem. Phys. 1998, 108, 7202–7216. 10.1063/1.476138. [DOI] [Google Scholar]
- Gupta N.; Gattrell M.; MacDougall B. Calculation for the Cathode Surface Concentrations in the Electrochemical Reduction of CO2 in KHCO3 Solutions. J. Appl. Electrochem. 2006, 36, 161–172. 10.1007/s10800-005-9058-y. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.