

Abstract
Protein-tyrosine phosphatase receptor type Z (PTPRZ) is predominantly expressed in the developing brain as a CS proteoglycan. PTPRZ has long (PTPRZ-A) and short type (PTPRZ-B) receptor forms by alternative splicing. The extracellular CS moiety of PTPRZ is required for high-affinity binding to inhibitory ligands, such as pleiotrophin (PTN), midkine, and interleukin-34; however, its functional significance in regulating PTPRZ activity remains obscure. We herein found that protein expression of CS-modified PTPRZ-A began earlier, peaking at approximately postnatal days 5–10 (P5–P10), and then that of PTN peaked at P10 at the developmental stage corresponding to myelination onset in the mouse brain. Ptn-deficient mice consistently showed a later onset of the expression of myelin basic protein, a major component of the myelin sheath, than wild-type mice. Upon ligand application, PTPRZ-A/B in cultured oligodendrocyte precursor cells exhibited punctate localization on the cell surface instead of diffuse distribution, causing the inactivation of PTPRZ and oligodendrocyte differentiation. The same effect was observed with the removal of CS chains with chondroitinase ABC but not polyclonal antibodies against the extracellular domain of PTPRZ. These results indicate that the negatively charged CS moiety prevents PTPRZ from spontaneously clustering and that the positively charged ligand PTN induces PTPRZ clustering, potentially by neutralizing electrostatic repulsion between CS chains. Taken altogether, these data indicate that PTN-PTPRZ-A signaling controls the timing of oligodendrocyte precursor cell differentiation in vivo, in which the CS moiety of PTPRZ receptors maintains them in a monomeric active state until its ligand binding.
Keywords: chondroitin sulfate, oligodendrocyte, phosphotyrosine signaling, pleiotrophin, proteoglycan
Introduction
Myelination is an essential feature of the vertebrate nervous system that electrically insulates axons, thereby enabling the salutatory transmission of nerve impulses. Oligodendrocyte precursor cells (OPCs)2 are the principal source of myelinating oligodendrocytes (1). Previous studies reported that protein tyrosine phosphorylation is crucially involved in the signal transduction mechanism for OPC differentiation into mature oligodendrocytes and myelin formation. FYN kinase in the Src tyrosine kinase family is the most prominent member involved in OPC differentiation and myelin formation (2). FYN activity is up-regulated during oligodendrocyte differentiation (3, 4), and Fyn-knock-out mice exhibit hypomyelination in the brain (5, 6). FYN phosphorylates p190 RhoGAP, a GTPase-activating protein (GAP) for Rho GTPase, to suppress Rho/ROCK, resulting in the maturation of oligodendrocytes and myelination (7, 8).
PTPRZ is the most abundant receptor-type protein-tyrosine phosphatase (RPTP) in OPCs (9, 10). PTPRZ dephosphorylates p190 RhoGAP, thereby acting as a counterpart of FYN (11, 12). The amounts of myelin basic protein (MBP) and myelinated axons in the brain at postnatal day 10, when myelination occurs, are significantly higher in Ptprz-deficient mice than in wild-type animals, indicating a suppressive role for PTPRZ in oligodendrocyte differentiation (11). Consistent with the earlier onset of CNS myelination, oligodendrocytes appear earlier in primary cultures from Ptprz-deficient mice than from wild-type mice (11).
Three PTPRZ isoforms are generated by alternative splicing from a single PTPRZ gene: two transmembrane isoforms, PTPRZ-A and PTPRZ-B, and one secretory isoform, PTPRZ-S (13–15). PTPRZ-A and -B protein expression levels are down-regulated by metalloproteinase or plasmin-mediated digestion under physiological conditions (16, 17). Therefore, the full-length PTPRZ-A protein is rarely observed in the adult mouse brain, and its resultant extracellular fragment (ZA-ECF) is detected at the same molecular size as PTPRZ-S (17). PTPRZ-S, also known as phosphacan, is one of the major CS proteoglycans in the extracellular matrix of the brain (18).
All three isoforms expressed in the CNS are heavily modified with CS chains (17, 19). We and others identified growth factors, such as pleiotrophin (PTN)/heparin-binding growth-associated molecule (HB-GAM) (12, 20–22), midkine (MK) (23), and IL-34 (24) as inhibitory ligands for PTPRZ. The CS moiety of PTPRZ is essential for achieving high-affinity binding sites for PTN, MK, and IL-34 (20, 23, 24). PTN has been shown to inactivate the intracellular catalytic activity of PTPRZ-B by inducing molecular oligomerization in baby hamster kidney (BHK)-21 cells (22). We recently demonstrated that PTN enhanced thyroid hormone-induced OPC differentiation in a primary culture of glial cells from wild-type mice, but not Ptprz-deficient mice, indicating that its effects are mediated by PTPRZ (12, 25).
We established the oligodendrocyte-lineage cell line, OL1, from the p53-knock-out mouse brain (12). Immature OL1 cells are neural/glial antigen 2 chondroitin sulfate (NG2) proteoglycan-positive OPC-like cells that express PTPRZ-A and PTPRZ-B as CS proteoglycans; however, their expression decreases after differentiation into oligodendrocytes, although PTPRZ-B remains weakly detectable (12). Immature OL1 cells treated with PTN show increased p190 RhoGAP phosphorylation and significantly enhanced thyroid hormone-induced differentiation to oligodendrocytes (12). Therefore, we postulated that the catalytic activity of PTPRZ maintains OPCs in an undifferentiated state, and PTN-induced PTPRZ inactivation releases this blockage of cellular differentiation (12). However, the ligand-receptor combination in PTPRZ inactivation in CNS myelination during development and the detailed regulatory mechanism of PTPRZ currently remain unclear.
We herein identified PTN as the physiological PTPRZ-A/B ligand for oligodendrocyte differentiation during CNS development and revealed the functional involvement of the CS moiety in PTPRZ inactivation by PTN through oligomerization.
Results
Inactivation of PTPRZ with Its Inhibitory Ligands Leads to OPC Differentiation in Cultures
We first examined whether inhibitory ligands for PTPRZ receptors equally promote OPC differentiation. In primary mixed glial cells from the wild-type mouse brain, OPC differentiation was augmented by the addition of MK and IL-34 as well as PTN, as described previously (12) (Fig. 1A, top pictures and left graph). In contrast, the ligand-induced enhancement of differentiation was not observed in Ptprz-deficient primary cells (Fig. 1A, bottom pictures and right graph) because they originally showed greater differentiation without the PTN stimulation (11). The three inhibitory ligands exerted the same effects on NG2-positive oligodendrocyte-lineage OL1 cells in the culture (Fig. 1B), which express the two receptor isoforms, PTPRZ-A and PTPRZ-B, as CS proteoglycans (12).
FIGURE 1.
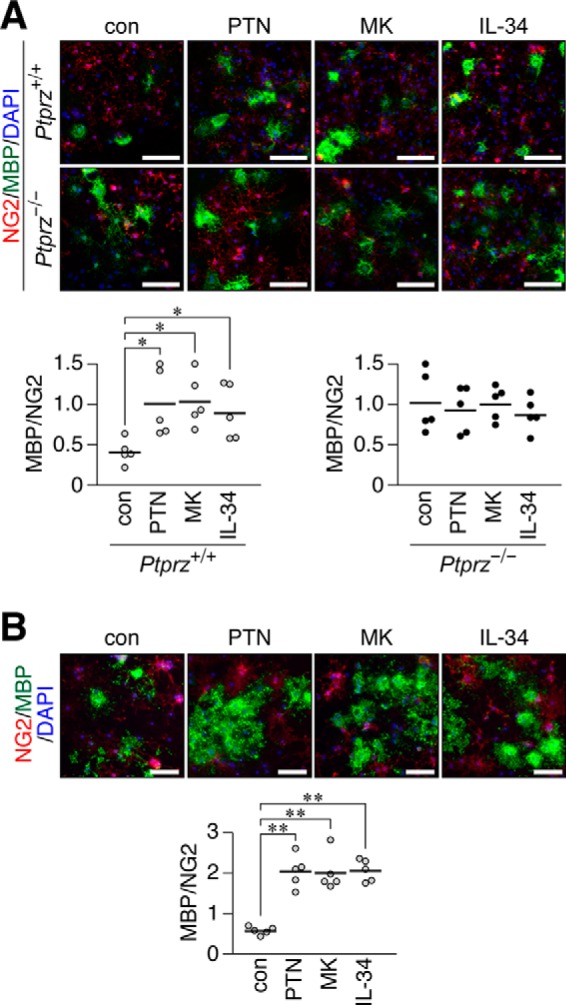
Accelerated differentiation of cultured OPCs by treatments with PTN, MK, and IL-34. A, a primary culture of mixed glial cells obtained from wild-type (Ptprz+/+) and Ptprz-deficient (Ptprz−/−) mouse brains in differentiation medium without (con) or with PTN, MK, or IL-34 (100 nm each). After 6 days, cells were fixed with formalin and stained with anti-NG2 proteoglycan (OPCs, red) and anti-MBP (oligodendrocyte, green) antibodies in conjunction with the DAPI labeling of nuclei (blue). Scale bars, 100 μm. The bottom scatter plots show the ratio of MBP-positive cells to NG2-positive cells from Ptprz+/+ mice (left) and Ptprz−/− mice (right), in which each circle corresponds to an independent cell culture (n = 5 each). *, p < 0.05, significantly different from the control group by Student's t test. B, OL1 cells cultured for 10 days in differentiation medium without (con) or with PTN, MK, or IL-34 (100 nm each). Cells were fixed and stained with anti-NG2 and anti-MBP, as in A. Scale bars, 100 μm. The scatter plot shows the ratio of MBP-positive to NG2-positive cells (n = 5 each). **, p < 0.01, significantly different from the control group by Student's t test.
We previously reported that PTPRZ-B expressed exogenously in BHK-21 cells exhibit a punctate distribution following a treatment with PTN (22); therefore, we herein examined the cell surface distribution of PTPRZ receptors (PTPRZ-A/B) in OL1 cells after the ligand treatments. Immunocytofluorescence staining revealed that PTPRZ-A/B were diffusely distributed on the cell surface in the absence of ligands; however, they exhibited a punctate appearance when exposed to PTN, MK, or IL-34 (Fig. 2A). Consistent with these results, the phosphorylation of p190 RhoGAP at Tyr-1105, which is dephosphorylated by PTPRZ (12, 26), was enhanced by the treatments with all of the ligands (Fig. 2B). These results indicated that the inhibitory ligands for PTPRZ have the common ability to promote OPC differentiation under our culture conditions.
FIGURE 2.
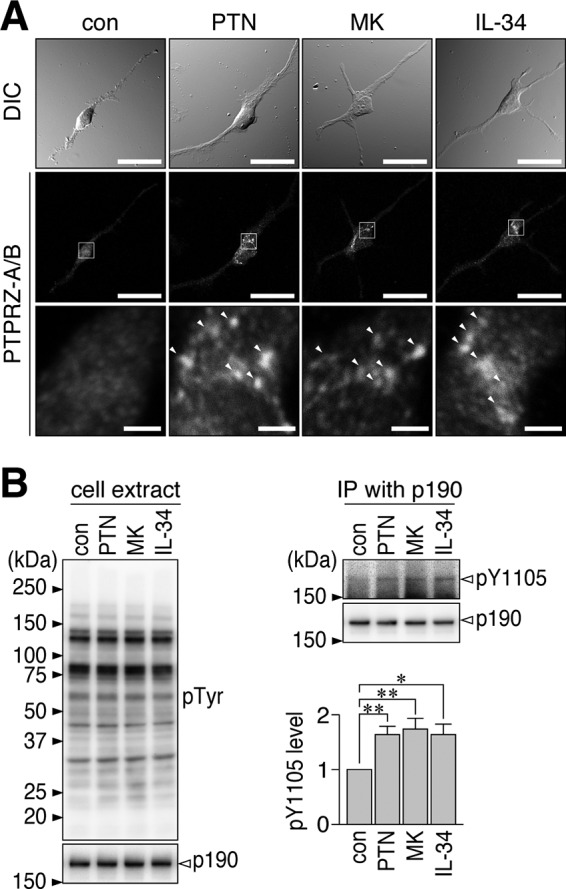
Induction of PTPRZ clustering on the surface of OL1 cells by PTN, MK, and IL-34. A, OL1 cells treated without (con) or with PTN, MK, or IL-34 (100 nm each) for 1 h. Cells were fixed with formalin, but the membrane permeabilization step was omitted to prevent antibody staining of the cytosol. Anti-PTPRZ-S, a rabbit polyclonal antibody, recognizes an extracellular epitope of PTPRZ. The bottom picture in each column is an enlarged view of the rectangular region in the middle. Arrowheads, anti-PTPRZ-S-positive puncta. Scale bars, 100 μm (top and middle pictures) and 10 μm (bottom pictures), respectively. B, the overall tyrosine phosphorylation pattern and protein expression of p190 RhoGAP in OL1 cells treated as described in A. The extracts were analyzed with anti-phosphotyrosine (PY20) and anti-p190 RhoGAP, respectively (left). The Tyr-1105 phosphorylation and protein amount of p190 RhoGAP in immunoprecipitates were detected using anti-Tyr(P)-1105 and anti-p190, respectively (top right). In the bar graph (bottom right), the phosphorylation levels were normalized to the protein amount. The graph shows the arbitrary densitometric units of Tyr-1105 phosphorylation levels relative to the average of the control group. Data are the mean ± S.E. (error bars) from four independent experiments. *, p < 0.05; **, p < 0.01, significantly different from the control group by Student's t test. DIC, differential interference contrast; IP, immunoprecipitation.
PTN Expression Is Strongly Induced at the Onset of Myelination in the Neonatal Brain
To define the PTPRZ isoform and endogenous ligand involved in CNS myelination during development, we examined the expression profile of PTPRZ isoform proteins and their ligand molecules in the mouse brain by Western blotting analyses (Fig. 3). The expression of the NG2 proteoglycan, a marker of OPCs, was detected from postnatal day 1 (P1), peaked at P10, rapidly decreased thereafter, and became almost negligible at P30. In contrast, MBP, a mature oligodendrocyte marker, appeared from P15. FYN kinase, the expression of which reportedly increases with the initiation of myelination, showed the strongest expression at P10, corresponding to the onset of myelination in mouse brain development (27).
FIGURE 3.

Developmental expression profiles of PTPRZ and related molecules in the mouse brain. A, Western blotting to analyze the expression patterns of oligodendrocyte-lineage cell markers (NG2 and MBP), FYN kinase, PTPRZ isoforms (PTPRZ-A, PTPRZ-S, and PTPRZ-B), and PTPRZ ligands (PTN, MK, and IL-34) in the whole mouse brain at P1, P5, P10, P15, P30, and P90, respectively. In analyses of NG2 and PTPRZ, brain extracts were treated with chABC before SDS-PAGE; no obvious bands of PTPRZ-A, -B, or -S were detected without the chABC treatment, as described previously (19). The graph below each blot shows the arbitrary densitometric units of the detected signal intensity normalized to the value at P1. Data are the mean ± S.E. (error bars) (n = 4 animals/group). B, Coomassie Brilliant Blue (CBB) staining was used to verify the applied protein amounts.
In the three splicing isoforms of PTPRZ, the expression of the long receptor form PTPRZ-A and secretory PTPRZ-S increased after birth, peaked at approximately P5 and P15, respectively, and then decreased toward adulthood; here, of note, the extracellular proteolytic fragment of PTPRZ-A also shows 300 kDa, corresponding to the band of PTPRZ-S (16). In contrast, the short receptor form PTPRZ-B gradually increased from P1 to P30 and maintained its expression level until P90; here it should be noted that the expression of PTPRZ-B is due to the increase of neurons and astrocytes after birth (see below). None of the bands of any of the core proteins were detected on Western blots without the removal of their CS chains by chondroitinase ABC (chABC), indicating that all PTPRZ isoforms were predominantly expressed as CS proteoglycans in the brain. Only PTN among the ligand molecules tested showed a peak at P10, corresponding to the onset of myelination, whereas MK and IL-34 were maintained at constant levels. Thus, the combination of PTN and PTPRZ-A appears to mainly contribute to the onset of myelination in vivo.
PTN Is the PTPRZ Ligand Responsible for OPC Differentiation during Brain Development
We found that MBP staining in the corpus callosum at P15 was significantly weaker in Ptn-deficient mice than in wild-type mice (Fig. 4A) but reached the same intensity levels by P90 (Fig. 4B). Immunostaining intensities on sections were consistent with the amounts of MBP proteins in whole brain extracts by Western blotting (Fig. 4, C and D). Therefore, a Ptn deficiency appeared to cause a delay in the onset of myelination. Consistent with this result, Ptprz-deficient mice exhibited the early onset of myelination (11). These findings strongly indicate that PTN-induced PTPRZ inactivation controls the timing of myelination in the postnatal brain.
FIGURE 4.
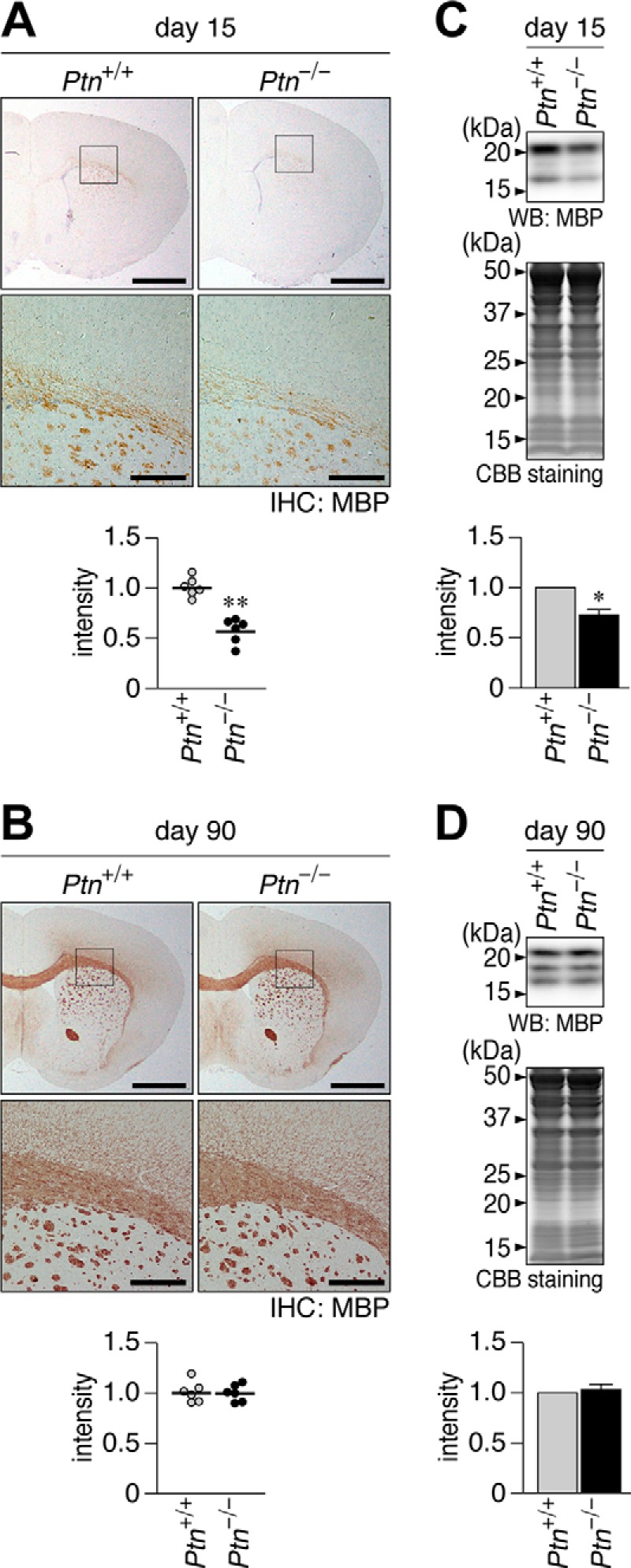
Late onset of MBP expression and myelination in the Ptn-deficient mouse brain. A and B, immunohistochemical (IHC) staining of MBP in wild-type (Ptn+/+) and Ptn-deficient (Ptn−/−) mouse brains at P15 (A) and P90 (B). The bottom pictures are enlarged views of the rectangular region in the top panels. Scale bars, 1 mm (top pictures) and 200 μm (bottom pictures), respectively. The scatter plot shows the arbitrary densitometric units of the intensity of MBP staining in the corpus callosum (n = 6 animals each), and the data were normalized to the average of the wild-type group. **, p < 0.01 by Student's t test. C and D, Western blotting analyses (WB) of MBP in the cerebral cortices of Ptn+/+ and Ptn−/− mice at P15 (C) and P90 (D). The protein amounts applied were verified by Coomassie Brilliant Blue (CBB) staining. Because the murine MBP has four isoforms with molecular masses ranging from 21.5 to 14 kDa, and their expression patterns change during myelination (51), we scanned all bands detected and quantified them as total MBP content. The graph shows the arbitrary densitometric units of total MBP relative to the averaged value for the wild type. Data are the mean ± S.E. (error bars) (n = 4 animals for P15, n = 3 animals for P90). *, p < 0.05; **, p < 0.01, significantly different from the Ptprz+/+ mouse by Student's t test.
The CS Moiety of PTPRZ Is Essential for the Inhibition of OPC Differentiation by Maintaining the Active Monomeric Form of PTPRZ
Previous studies reported that a treatment with chABC stimulated OPC differentiation in vitro (28–30); however, the target molecule of chABC on OPCs has not yet been identified. We assessed the functional significance of the CS moiety of PTPRZ by using OL1 cells, in which the PTPRZ isoforms are expressed as CS proteoglycans as in the brain (see below).
The addition of chABC to the culture medium removed almost all CS chains of PTPRZ receptors but not those of PTPRZ-S in OL1 cells (Fig. 5A). Here it is conceivable that chABC would have less access to PTPRZ-S embedded in the extracellular matrix between the cell and the culture dish. We found that the chABC treatment induced the punctate localization of PTPRZ (Fig. 5B) and increased the phosphorylation of p190 RhoGAP at Tyr-1105 (Fig. 5C), as was the case for the ligand treatments with or without chABC. The enhancement of OPC differentiation by the chABC treatment was of a magnitude similar to that caused by PTN (Fig. 5). PTN did not exert additive effects on PTPRZ clustering (Fig. 5B), PTPase activity (Fig. 5C), or OPC differentiation caused by the chABC treatment (Fig. 5D), suggesting that the removal of CS chains from cell surface proteins and the ligand stimulation of PTPRZ share a common mechanism for OPC differentiation.
FIGURE 5.
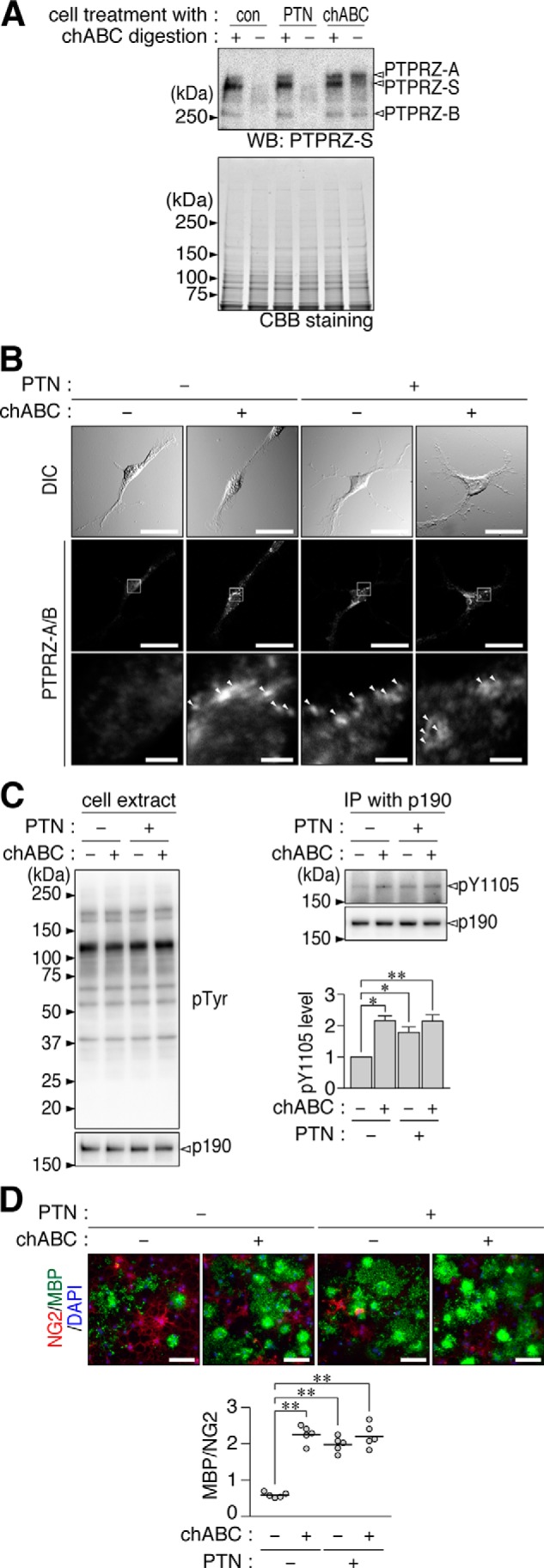
Enhanced oligodendrocyte differentiation in OL1 cells by the enzymatic removal of CS chains. A, OL1 cells treated with PTN (100 nm), chABC (6.25 microunits/ml), or vehicle (con) for 1 h. To examine CS modifications in PTPRZ proteins in cultured cells, cell extracts were treated with (+) or without (−) chABC before being applied to SDS-PAGE and subjected to Western blotting (WB) with anti-PTPRZ-S. The protein amounts applied were verified by Coomassie Brilliant Blue (CBB) staining. B, OL1 cells treated with the indicated combination of PTN (100 nm) and chABC (6.25 microunits/ml) for 1 h. After formalin fixation, cells were stained with anti-PTPRZ-S without membrane permeabilization, as in Fig. 2A. Arrowheads, punctate stains. Scale bars, 100 μm (top and middle pictures) and 10 μm (bottom pictures), respectively. C, overall tyrosine phosphorylation pattern, protein amount of p190 RhoGAP, and Tyr-1105 phosphorylation of p190 RhoGAP. OL1 cells were treated as described in B and analyzed as in Fig. 2B. The arbitrary densitometric units of Tyr-1105 phosphorylation levels normalized to the average of the control (nontreated) group are shown. Data are the mean ± S.E. (error bars) from four independent experiments. *, p < 0.05; **, p < 0.01, significantly different from the control group by Student's t test. D, OL1 cells were cultured in differentiation medium containing the indicated combinations of PTN and chABC for 10 days. Their differentiation was analyzed as in Fig. 1B. Scale bars, 100 μm. The scatter plot shows the ratio of MBP-positive cells to NG2-positive cells (n = 5 each from five independent cell cultures). **, p < 0.01, significant difference between the indicated groups using analysis of variance with Bonferroni's post hoc tests. IP, immunoprecipitation.
We previously reported that anti-PTPRZ-S, which binds to a protein epitope in the extracellular region of PTPRZ, acts as a ligand mimic for PTPRZ-B expressed in BHK-21 cells (22). However, in OL1 cells, anti-PTPRZ-S did not induce the clustering of PTPRZ receptors (Fig. 6A) and did not increase p190 RhoGAP phosphorylation (Fig. 6B). Consistent with these results, OPC differentiation was not enhanced by anti-PTPRZ-S (Fig. 6C). As expected, no additive effect was observed to those caused by the chABC treatment (Fig. 6).
FIGURE 6.
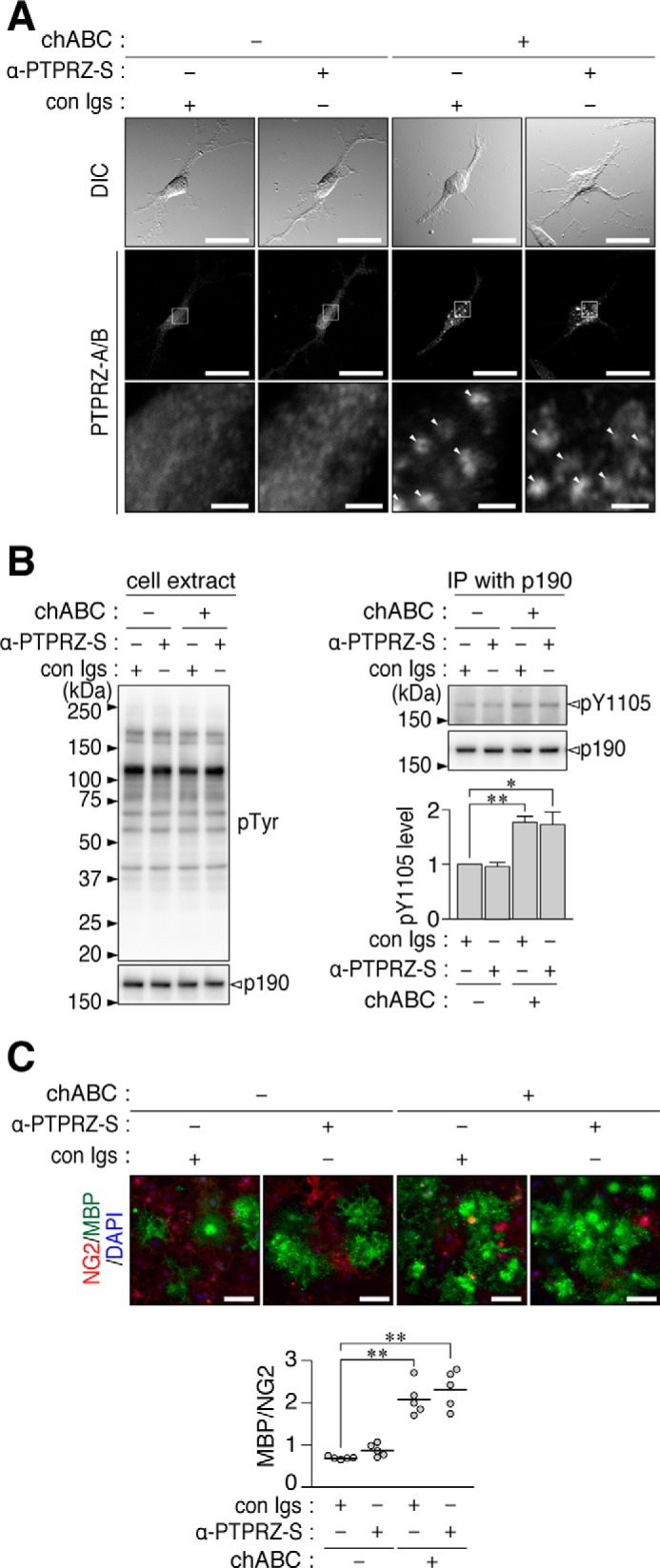
No enhancement of OL1 differentiation by the anti-PTPRZ-S treatment. A, OL1 cells incubated with the indicated combinations of chABC (6.25 microunits/ml), anti-PTPRZ-S (10 μg/ml), or non-immunized rabbit Ig fractions (con Igs; 10 μg/ml) for 1 h. After formalin fixation, cells were stained with anti-PTPRZ-S without membrane permeabilization, as in Fig. 2A. The bottom picture in each column is the enlarged view of the rectangular region in the middle. Arrowheads, punctate stains of PTPRZ receptor forms. Scale bars, 100 μm (top and middle pictures) and 10 μm (bottom pictures), respectively. B, overall tyrosine phosphorylation pattern, protein amount of p190 RhoGAP, and Tyr-1105 phosphorylation of p190 RhoGAP. OL1 cells were treated as described in A and analyzed as in Fig. 2B. The graph shows the arbitrary densitometric units of Tyr-1105 phosphorylation levels relative to the average of the control group (without both chABC and α-PTPRZ-S). Data are the mean ± S.E. (error bars) from four independent experiments. *, p < 0.05; **, p < 0.01, significantly different from the control group by Student's t test. C, OL1 cells cultured in differentiation medium containing the indicated combinations of chABC, anti-PTPRZ-S, or control Igs for 10 days. Their differentiation was analyzed as in Fig. 1B. The scatter plot shows the ratio of MBP-positive cells to NG2-positive cells (n = 5 each from five independent cell cultures). **, p < 0.01, significant difference between the indicated groups by analysis of variance with Bonferroni's post hoc tests. DIC, differential interference contrast; IP, immunoprecipitation.
We examined the reason why the ligand-like effect of anti-PTPRZ-S was not observed in OL1 cells by comparing CS modifications in PTPRZ between OL1 cells and BHK-21 cells. Western blotting indicated that PTPRZ-A and -B expressed in OL1 cells were both highly modified with CS chains (Fig. 7, A and C). In BHK-21 cells, which do not endogenously express PTPRZ proteins (22), PTPRZ-A was a fully CS-modified form when exogenously expressed; however, the degree of the CS modification in PTPRZ-B was much weaker than that in OL1 cells (Fig. 7, B and C).
FIGURE 7.
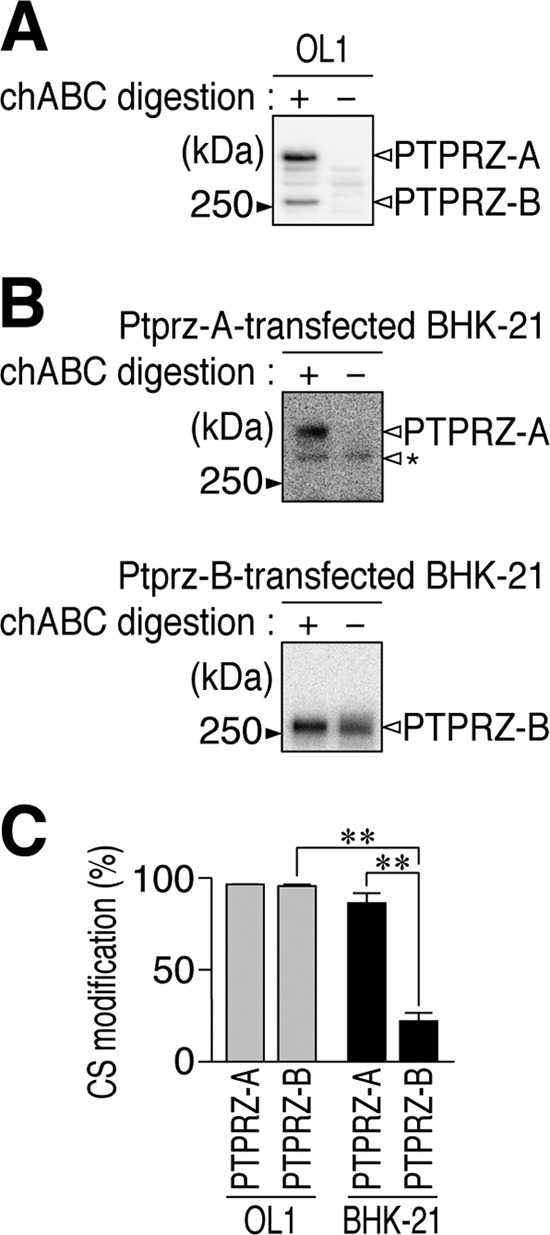
CS modification of PTPRZ-A/B receptors endogenously expressed in OL1 cells and those exogenously expressed in BHK-21 cells. A and B, Western blotting of cell extracts prepared from immature OL1 cells (A) and BHK-21 cells (B) with anti-PTPRZ-S. BHK-21 cells were transfected with an expression construct of PTPRZ-A or PTPRZ-B. Cell extracts were treated with (+) or without (−) chABC to examine the degree of their CS modification. The protein band indicated by an arrowhead with the asterisk is an immature (or abnormally processed) product of PTPRZ-A in the cell; they are typically observed as described previously (17, 19). C, graph showing the degree of CS modifications in PTPRZ-A and PTPRZ-B in the respective cells. The degrees of CS modifications were calculated as follows: 1 − (band intensity of the chABC-untreated sample/band intensity of the chABC-treated sample). Data are the mean ± S.E. (error bars) from three independent experiments. **, p < 0.01 by analysis of variance with Bonferroni's post hoc tests.
We found that anti-PTPRZ-S did not induce the patchy distribution of PTPRZ-A from its diffuse distribution on the cell surface in BHK-21 cells, whereas the PTN and chABC treatments induced clustering (Fig. 8A). On the other hand, the clustering of PTPRZ-B was induced in BHK-21 cells by all three treatments (Fig. 8B). The clustering of PTPRZ receptors correlated with the inhibition of their PTPase activities, leading to increases in the tyrosine phosphorylation of p190 RhoGAP (Fig. 8, C–E). Neither treatment affected p190 phosphorylation in mock transfectants. Anti-PTPRZ-S did not induce PTPRZ-A/B clustering when they were highly modified by CS, whereas PTN did. Therefore, PTN, but not anti-PTPRZ-S, likely functions to reduce CS-mediated repulsion between the PTPRZ receptor forms.
FIGURE 8.

Distinct effects of PTN, chABC, or anti-PTPRZ-S on PTPRZ clustering in BHK-21 cells. A and B, BHK-21 cells transfected with an expression plasmid for PTPRZ-A (A) and PTPRZ-B (B). Cells were treated with PTN (100 nm), chABC (6.25 microunits/ml), or anti-PTPRZ-S (10 μg/ml) for 1 h. Anti-PTPRZ-S staining of PTPRZ receptors on the cell surface was performed as in Fig. 2A. Scale bars, 100 μm (top and middle pictures) and 10 μm (bottom pictures), respectively. The bottom picture in each column is the enlarged view of the rectangular region in the middle. Arrowheads, punctate staining. C–E, BHK-21 cells transfected with FLAG-tagged p190 RhoGAP along with the PTPRZ-A (C), PTPRZ-B (D), or mock control (E) plasmid. Cells were treated as above. The overall tyrosine phosphorylation pattern and protein expression of FLAG-tagged p190 RhoGAP in cell extracts were analyzed with anti-phosphotyrosine (PY20) and anti-FLAG, respectively (left). The Tyr-1105 phosphorylation of FLAG-tagged p190 RhoGAP immunoprecipitation was assessed using anti-Tyr(P)-1105 and anti-FLAG, respectively (right). Tyr-1105 phosphorylation levels were normalized to the average of the control group. Data are the mean ± S.E. (error bars) from four independent experiments. *, p < 0.05; **, p < 0.01, significant difference between the indicated groups by analysis of variance with Bonferroni's post hoc tests. DIC, differential interference contrast; IP, immunoprecipitation.
Discussion
In the present study, we showed that inhibitory ligands for PTPRZ commonly enhance OPC differentiation in cultures by clustering PTPRZ receptors. Among these ligands, the expression level of PTN specifically and concomitantly increased at the onset of CNS myelination during brain development, together with CS-modified PTPRZ-A. In contrast to Ptprz-deficient mice, which show the early onset of myelination (11), delayed myelination was observed in Ptn-deficient mouse brains. These results indicate that the inactivation of PTPRZ-A by PTN is the molecular mechanism underlying the onset of myelination in CNS development. The enzymatic removal of the CS chains of PTPRZ also leads to the inactivation of PTPRZ through molecular clustering and induces OPC differentiation, indicating that the PTPRZ core protein originally has a clustering property when the CS modification is absent. These results provide a novel insight into the roles of PTN and the CS moiety in the regulation of PTPRZ signaling for OPC differentiation and myelination.
We previously demonstrated that PTPRZ activity maintains OPCs in an immature state by dephosphorylating p190 RhoGAP (11), which opposes the action of FYN tyrosine kinase (7, 8). FYN activity is required for OPC differentiation (27), and its expression peaks during myelination in brain development (Fig. 3). Concomitant increases in FYN and PTPRZ-A just after birth may prevent OPC differentiation until the appropriate time during brain development. On the other hand, PTPRZ-B gradually increased from P1 to P30 and maintained its expression level until P90 in the brain (Fig. 3), whereas both PTPRZ-A/B protein levels are decreased in oligodendrocyte-lineage cells after their differentiation (12). Interestingly, the expression profile of PTPRZ-B is very similar to that of glial fibrillary acidic protein, a major marker protein of astrocytes (31). It should be noted that PTPRZ is also expressed in neurons and astrocytes (32), besides oligodendrocytes, suggesting that PTPRZ-A/B play multiple roles in different cells in brain development.
The extracellular region of PTPRZ is responsible for its binding with various secretory factors (such as PTN, MK, IL-34, and basic fibroblast growth factor) (20, 23, 24, 33), extracellular matrix proteins (tenascin-C and -R) (34), and cell adhesion molecules (Nr-CAM, L1/Ng-CAM, F3/contactin, NCAM, and TAG1/axonin-1) (35). Among these molecules, PTN, MK, and IL-34 have been shown to function as inhibitory ligands against the PTPase activity of PTPRZ receptor isoforms (12, 20, 21, 23, 24). The functional significance of the binding of other molecules in PTPRZ signaling still remains unclear.
When OPCs were treated with PTN, MK, or IL-34, all enhanced cell differentiation in vitro by inducing PTPRZ clustering on the cell surface (Figs. 1 and 2). During neonatal brain development, only PTN proteins, and not MK or IL-34, exhibited an increase at the onset of myelination (Fig. 3). Ptn-deficient mice consistently exhibited delayed CNS myelination (Fig. 4). Thus, PTN is considered to be a key regulatory molecule that releases the blockage of OPC differentiation by PTPRZ inactivation at the appropriate time and location. Consistently, PTN expression has a sharp peak at P10, when CNS myelination begins. PTN is reportedly expressed by neurons, which differentiate earlier than glial cells during development (36).
PTN protein levels were lower than the detection limit in normal adult brain tissues (Fig. 3; see also Ref. 37); however, we recently found that PTN expression is activated in the adult mouse brain when demyelination is induced by feeding the copper chelator, cuprizone; PTN expression was induced in affected cortex neurons and their axon fibers in the corpus callosum (12). PTN released from damaged fibers appears to stimulate the differentiation of OPCs recruited to the demyelinated area (12).
Regarding the regulatory mechanism of PTPRZ activity, a “head-to-toe” dimerization model was recently proposed based on the x-ray crystal structure of the intracellular region of PTPRG (38), another R5 family member, which has high sequence identity to PTPRZ. Although we and others showed that ligand binding induces the inactivation of its PTPase by dimerization through the cluster formation of PTPRZ receptors on the cell surface (12, 21, 22, 39), the molecular mechanism promoting dimer and cluster formation by the ligand binding has remained unclear. In this regard, the punctate PTPRZ staining observed by confocal microscopy may also reflect other mechanisms, such as compartmentalization in lipid rafts or some other microdomains. Further experiments are thus needed to reveal the detailed relationship between the dimer (oligomer) formation and the inactivation by single-molecule fluorescence techniques.
In brain tissues, the extracellular regions of PTPRZ isoforms are highly modified with CS chains (17, 19). To the best of our knowledge, there have been no cell lines that express PTPRZ receptors in their fully modified forms with CS chains, similar to those in the brain. We recently established the immature oligodendrocyte OL1 cell line, which expresses PTPRZ receptors in the fully CS-modified form (12). As described for primary cultured OPCs (28, 30), a chABC treatment of OL1 cells also enhanced their differentiation (Fig. 5). We herein demonstrated that enzymatic CS removal was sufficient to induce PTPRZ clustering and inactivation, thereby enhancing the tyrosine phosphorylation level of p190 RhoGAP (Fig. 5). CS chains consist of repeating disaccharide units of GlcA and GalNAc with sulfate groups at various positions on sugar chains (29). Due to electrostatic repulsion between negatively charged glycosaminoglycan moieties (40), the receptor isoforms of PTPRZ may stay as monomers (Fig. 9).
FIGURE 9.

Schematic representation of the role of the CS chains in regulating PTPRZ activity. The protein domains of PTPRZ are highlighted in different colors as follows. Blue, carbonic anhydrase-like domain; black, fibronectin type III domain; brown, a serine, glycine-rich domain; pink and gray, protein-tyrosine phosphatase domains PTP-D1 and PTP-D2, respectively (52, 53). Catalytic activity is retained in membrane-proximal PTP-D1, but not in distal PTP-D2. The extracellular regions of PTPRZ-A/B are highly modified with CS chains in brain tissues and OPCs. In contrast, a lower CS modification was observed when PTPRZ-B was exogenously expressed in BHK-21 (the present study) and HEK293T cells (54) and in endogenous PTPRZ-B expressed in stomach tissue (55) and C6 glioblastoma cells (56). PTPRZ-A and PTPRZ-S contain a long serine, glycine-rich domain extracellularly that is thought to be the CS attachment region. However, this domain is largely deleted in the short receptor isoform PTPRZ-B (52), which may underlie the difference in the degree of CS modification. In the head-to-toe dimerization model (38), PTPRZ receptors function to maintain equilibrium between monomers (active PTPase) and dimers (inactive). In the present study, the high CS modification in PTPRZ was found to be essential for maintaining the monomeric active form, possibly by electrostatic repulsion, and, at the same time, provided high-affinity ligand binding sites (20). The binding of extracellular ligands, such as PTN, MK, and IL-34, appears to neutralize the negative charges of the CS chains of PTPRZ, which results in the induction of cluster formation, thereby causing the dimerization and inactivation of PTPase. This is supported by the enzymatic removal of CS chains by chABC, also inducing PTPRZ clustering and inactivation. On the other hand, the ligand-like effect of anti-PTPRZ-S, which binds to a protein epitope, is only observed in the lower CS-modified form.
PTN and MK resemble each other and constitute a distinct growth factor family. They have positive charged regions that are involved in binding to highly sulfated CS chains (23, 41, 42). Their binding may reduce the electrostatic repulsion of, or induce conformational changes (43) to, CS chains and resultantly shift them to the dimeric or oligomeric form in the cell membrane (Fig. 9). This simple model explains why no further potentiation by PTN was observed after the removal of CS (Fig. 5) and why PTPRZ clustering was not induced in OL1 cells with the anti-PTPRZ-S antibody (Fig. 6). In BHK-21 cells, the anti-PTPRZ-S antibody enhanced the clustering of PTPRZ-B on the cell surface as well as PTN (Fig. 8; see also Ref. 22). This may be because PTPRZ-B is weakly modified by CS chains in BHK-21 cells, in contrast to OL1 cells (Fig. 7), and the antibody can make clusters of PTPRZ-B because of their weak repulsion.
We also discuss our results in relation to demyelinating diseases, such as multiple sclerosis. Most multiple sclerosis patients initially exhibit a relapsing-remitting disease course that eventually converts to a secondary progressive form of the disease with incomplete recovery (44). OPCs are present at demyelinated brain regions in multiple sclerosis patients, even at the progressive stage, but fail to differentiate at the latter stage (45). CS proteoglycans are the major components of the extracellular matrix of glial scars, which are known to be inhibitory to axonal regeneration (29, 46). Emerging findings have indicated that CS proteoglycans also accumulate in the demyelinating sites of multiple sclerosis patients (47) and are considered to be inhibitory for remyelination by impairing OPC recruitment, differentiation, and myelination (30). As one of the possible functions of CS proteoglycans deposited in lesions in multiple sclerosis, it is conceivable that they disturb the binding of PTN to PTPRZ.
Experimental Procedures
Reagents and Antibodies
Recombinant human PTN produced in yeast was described previously (48). Recombinant mouse IL-34 was purchased from R&D Systems (catalogue no. 5195-ML). Human MK was from the Peptide Institute. chABC was from Sigma-Aldrich (catalogue no. C3667). Anti-PTPRZ-S, rabbit polyclonal antibodies against the extracellular region of PTPRZ (16), and purified rabbit polyclonal antibodies against phosphorylated Tyr-1105 of p190 RhoGAP (anti-Tyr(P)-1105) (26) were described previously. The following are the specificities and sources of the commercially available antibodies used in the present study: MBP (catalogue no. sc-13914, Santa Cruz Biotechnology), NG2 proteoglycan (catalogue no. AB5320, Millipore), FYN (catalogue no. P2992, Sigma-Aldrich), RPTPβ (a monoclonal antibody against the intracellular domain of PTPRZ receptors, catalogue no. 610179, BD Biosciences), PTN (catalogue no. ab79411, Abcam), MK (catalogue no. sc-20715, Santa Cruz Biotechnology), IL-34 (catalogue no. PRS4781, Sigma-Aldrich), p190 RhoGAP (catalogue no. 610150, BD Biosciences; catalogue no. 12164, Cell Signaling Technology), phosphotyrosine (clone PY20; catalogue no. ab16389, Abcam), and anti-FLAG (catalogue no. F7425, Sigma-Aldrich).
Ethics Statement and Experimental Animals
All animal experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee of the National Institutes of Natural Sciences, Japan (approval numbers 14A151, 15A096, 16A145, and 16A148). All surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize suffering. Ptprz-deficient mice (32) and Ptn-deficient mice (49) were back-crossed with the inbred C57BL/6 strain for over 10 generations.
Primary Mixed Glial Culture
A primary mixed glial cell culture was performed as described previously (11). Cortex tissues obtained from mouse brains on P1 were dissociated with papain (Worthington), and cells (2.0 × 104 cells) were cultured on a poly-l-ornithine-coated 35-mm dish with a differentiation medium containing DMEM mixed 1:1 with Ham's F-12 (DMEM/F-12; Life Technologies, Inc.), 1× GlutaMAX (Life Technologies), 1× N2 supplement (Life Technologies), 10 μg/ml of the AA homodimeric form of platelet-derived growth factor (PDGF-AA; Wako Pure Chemical), 0.5% FBS (Nichirei Biosciences), 100 μg/ml BSA (Sigma-Aldrich), 10 nm biotin (Sigma-Aldrich), and 30 ng/ml thyronine/thyroxine (Sigma-Aldrich) in a humidified incubator at 37 °C with 5% CO2. On the sixth day of culture, cells were fixed and stained with anti-NG2 (a specific marker for OPCs) and anti-MBP (a marker for matured oligodendrocytes). Differentiation from OPC to oligodendrocyte was expressed as the ratio of MBP-positive cells to NG2-positive cells.
Cell Culture and DNA Transfection
The preparation of mouse oligodendrocyte-lineage OL1 cells was described previously (12). In the differentiation assay, OL1 cells (2.0 × 104 cells) were cultured on a poly-l-ornithine-coated 35-mm plastic dish in knock-out DMEM/F-12 (Life Technologies) supplemented with 1× GlutaMAX, 1× StemPro neural supplement (Life Technologies), 10 μg/ml PDGF-AA, 10 nm biotin, and 30 ng/ml thyronine/thyroxine. On the 10th day of culture, differentiation from OPC to oligodendrocyte was assessed as above.
Hamster kidney BHK-21 cells that have been maintained in our laboratory were used. BHK-21 cells were cultured in DMEM (Life Technologies) supplemented with 10% FBS. The mammalian expression plasmids, pZeo-PTPζ-A (for full-length rat PTPRZ-A) (16), pZeoPTPζ (for PTPRZ-B) (21), and pFLAG-p190 (for FLAG-tagged p190 RhoGAP) (50) were described previously. DNA transfection was carried out using Lipofectamine 2000 reagent (Thermo Fisher Scientific) according to the manufacturer's directions.
Ligand, Antibody, or chABC Treatments of Cultured Cells
Stock solutions of PTN, MK, IL-34, and anti-PTPRZ-S were made as 100 μg/ml with PBS (4.3 mm Na2HPO4, 1.4 mm KH2PO4, 137 mm NaCl, and 2.7 mm KCl at pH 7.4) containing 100 μg/ml BSA. A lyophilized powder of chABC was reconstituted to 25 units/ml according the manufacturer's instructions. They were stored at −30 °C until use. Before cell treatments, these stocks were diluted in the cell culture medium to the desired concentrations and immediately applied to the cells by replacing the old medium.
Immunocytofluorescence and Immunohistochemistry Staining
Cells were fixed with 4% paraformaldehyde in PBS for 30 min. Mouse brains were removed and immediately immersed in 4% paraformaldehyde in PBS at 4 °C overnight and then subjected to paraffin embedding. Fixed cells or deparaffinized sections (3 μm) were permeabilized and blocked with 4% nonfat dry milk and 0.1% Triton X-100 in TBS (10 mm Tris-HCl, pH 7.4, 150 mm NaCl) for 30 min and then incubated with the respective primary antibodies at 4 °C overnight. To detect PTPRZ, fixed cells were microwaved in 10 mm citrate buffer (pH 6.0) at 98 °C for 5 min as an antigen retrieval step. Bound primary antibodies on tissue sections were visualized with HRP-conjugated secondary antibodies along with the Dako liquid diaminobenzidine chromogen system. In immunocytofluorescence staining, Alexa Fluor-conjugated secondary antibodies (Life Technologies) or DyLight amine-reactive dyes (DyLight 488 NHS Ester, Thermo Fisher Scientific) were used according to the standard procedure. Digital photomicrographs of individual specimens were taken with the Eclipse microscope Ci-L with a DS-Fi2 CCD camera (Nikon), Biozero BZ-800 (Keyence), or LSM 700 confocal microscope (Zeiss).
Extraction of Tissues and Cells and chABC Digestion of the Extracts
For Western blotting and immunoprecipitation experiments, mouse brains or cultured cells were extracted with 1% Nonidet P-40 in TBS containing 1 mm vanadate, 10 mm NaF, and protease inhibitors (EDTA-free Complete, Roche Applied Science). When extracts were subjected to chABC digestion, tissues or cells were extracted with 1% Nonidet P-40 in TBS containing protease inhibitors (Complete, Roche Applied Science), and 10-μl aliquots were mixed with an equal volume of 0.2 m Tris-HCl, 60 mm sodium acetate, 10 mm EDTA, pH 7.5, containing 250 microunits of chABC or not (control), for 1 h at 37 °C.
Immunoprecipitation and Western Blotting
After precleaning the extracts with Protein G-Sepharose (GE Healthcare), the extracted samples were subjected to immunoprecipitation with a combination of anti-p190 RhoGAP and Protein G-Sepharose beads or with anti-FLAG M2 magnetic beads (catalogue no. M8823, Sigma-Aldrich), and the immunocomplexes were then separated by SDS-PAGE, followed by semidry electroblotting onto a PVDF membrane. After blocking with 4% nonfat dry milk and 0.1% Triton X-100 in TBS, the membranes were incubated overnight with the respective antibodies. The binding of these antibodies was detected with Luminata Forte Western HRP substrate (Millipore). To detect tyrosine phosphorylated proteins, 1% BSA and 0.1% Triton X-100 in TBS was used as a blocking solution, and antibodies (PY20 or anti-Tyr(P)-1105) were diluted with this blocking solution.
Image and Statistical Analyses
Quantitative image analyses were performed using ImageJ software (National Institutes of Health) or Adobe Photoshop CS6 software (Adobe Systems). Statistical analyses were performed using IBM SPSS Statistics version 20 software.
Author Contributions
K. K., A. F., and M. N. designed the research; K. K., A. F., R. S., and N. T. performed the research; K. K., A. F., and M. N. analyzed the data; and K. K., A. F., and M. N. wrote the paper.
Acknowledgments
We thank Dr. Hisako Muramatsu (Nagoya University Graduate School of Medicine) for providing Ptn-deficient mice. We thank Yoshiko Isoshima and Norie Nakanishi for technical assistance and Akiko Kodama for secretarial assistance. Immunofluorescence photomicrograph images were acquired at the Spectrography and Bioimaging Facility, NIBB Core Research Facilities.
This work was supported in part by Grants-in-Aid for Young Scientists (B) (Japan Society for the Promotion of Science (JSPS) KAKENHI Grant JP26830050 (to K. K.)) and for Scientific Research on Innovative Areas (Grant JP26110722 (to A. F.)) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT). The authors declare that they have no conflicts of interest with the contents of this article.
- OPC
- oligodendrocyte precursor cell
- PTPRZ
- protein-tyrosine phosphatase receptor type Z
- CS
- chondroitin sulfate
- PTN
- pleiotrophin
- GAP
- GTPase-activating protein
- RPTP
- receptor-type protein tyrosine phosphatase
- MBP
- myelin basic protein
- HB-GAM
- heparin-binding growth-associated molecule
- MK
- midkine
- NG2
- neural/glial antigen 2 chondroitin sulfate
- chABC
- chondroitinase ABC
- P
- postnatal day
- BHK
- baby hamster kidney.
References
- 1. Buffo A., and Rossi F. (2013) Origin, lineage and function of cerebellar glia. Prog. Neurobiol. 109, 42–63 [DOI] [PubMed] [Google Scholar]
- 2. Krämer-Albers E. M., and White R. (2011) From axon-glial signalling to myelination: the integrating role of oligodendroglial Fyn kinase. Cell Mol. Life Sci. 68, 2003–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu Z., Ku L., Chen Y., and Feng Y. (2005) Developmental abnormalities of myelin basic protein expression in fyn knock-out brain reveal a role of Fyn in posttranscriptional regulation. J. Biol. Chem. 280, 389–395 [DOI] [PubMed] [Google Scholar]
- 4. White R., Gonsior C., Krämer-Albers E. M., Stöhr N., Hüttelmaier S., and Trotter J. (2008) Activation of oligodendroglial Fyn kinase enhances translation of mRNAs transported in hnRNP A2-dependent RNA granules. J. Cell Biol. 181, 579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Umemori H., Kadowaki Y., Hirosawa K., Yoshida Y., Hironaka K., Okano H., and Yamamoto T. (1999) Stimulation of myelin basic protein gene transcription by Fyn tyrosine kinase for myelination. J. Neurosci. 19, 1393–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seiwa C., Sugiyama I., Yagi T., Iguchi T., and Asou H. (2000) Fyn tyrosine kinase participates in the compact myelin sheath formation in the central nervous system. Neurosci. Res. 37, 21–31 [DOI] [PubMed] [Google Scholar]
- 7. Wolf R. M., Wilkes J. J., Chao M. V., and Resh M. D. (2001) Tyrosine phosphorylation of p190 RhoGAP by Fyn regulates oligodendrocyte differentiation. J. Neurobiol. 49, 62–78 [DOI] [PubMed] [Google Scholar]
- 8. Liang X., Draghi N. A., and Resh M. D. (2004) Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J. Neurosci. 24, 7140–7149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sim F. J., Lang J. K., Waldau B., Roy N. S., Schwartz T. E., Pilcher W. H., Chandross K. J., Natesan S., Merrill J. E., and Goldmanm S. A. (2006) Complementary patterns of gene expression by human oligodendrocyte progenitors and their environment predict determinants of progenitor maintenance and differentiation. Ann. Neurol. 59, 763–779 [DOI] [PubMed] [Google Scholar]
- 10. Ranjan M., and Hudson L. D. (1996) Regulation of tyrosine phosphorylation and protein tyrosine phosphatases during oligodendrocyte differentiation. Mol. Cell Neurosci. 7, 404–418 [DOI] [PubMed] [Google Scholar]
- 11. Kuboyama K., Fujikawa A., Masumura M., Suzuki R., Matsumoto M., and Noda M. (2012) Protein tyrosine phosphatase receptor type z negatively regulates oligodendrocyte differentiation and myelination. PLoS One 7, e48797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuboyama K., Fujikawa A., Suzuki R., and Noda M. (2015) Inactivation of protein tyrosine phosphatase receptor type Z by pleiotrophin promotes remyelination through activation of differentiation of oligodendrocyte precursor cells. J. Neurosci. 35, 12162–12171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maeda N., Hamanaka H., Shintani T., Nishiwaki T., and Noda M. (1994) Multiple receptor-like protein tyrosine phosphatases in the form of chondroitin sulfate proteoglycan. FEBS Lett. 354, 67–70 [DOI] [PubMed] [Google Scholar]
- 14. Faissner A., Clement A., Lochter A., Streit A., Mandl C., and Schachner M. (1994) Isolation of a neural chondroitin sulfate proteoglycan with neurite outgrowth promoting properties. J. Cell Biol. 126, 783–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maurel P., Rauch U., Flad M., Margolis R. K., and Margolis R. U. (1994) Phosphacan, a chondroitin sulfate proteoglycan of brain that interacts with neurons and neural cell-adhesion molecules, is an extracellular variant of a receptor-type protein tyrosine phosphatase. Proc. Natl. Acad. Sci. U.S.A. 91, 2512–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chow J. P., Fujikawa A., Shimizu H., and Noda M. (2008) Plasmin-mediated processing of protein-tyrosine phosphatase receptor type Z in the mouse brain. Neurosci. Lett. 442, 208–212 [DOI] [PubMed] [Google Scholar]
- 17. Chow J. P., Fujikawa A., Shimizu H., Suzuki R., and Noda M. (2008) Metalloproteinase- and γ-secretase-mediated cleavage of protein-tyrosine phosphatase receptor type Z. J. Biol. Chem. 283, 30879–30889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maeda N., Hamanaka H., Oohira A., and Noda M. (1995) Purification, characterization and developmental expression of a brain-specific chondroitin sulfate proteoglycan, 6B4 proteoglycan/phosphacan. Neuroscience 67, 23–35 [DOI] [PubMed] [Google Scholar]
- 19. Nishiwaki T., Maeda N., and Noda M. (1998) Characterization and developmental regulation of proteoglycan-type protein tyrosine phosphatase ζ/RPTPβ isoforms. J. Biochem. 123, 458–467 [DOI] [PubMed] [Google Scholar]
- 20. Maeda N., Nishiwaki T., Shintani T., Hamanaka H., and Noda M. (1996) 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTPβ, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM). J. Biol. Chem. 271, 21446–21452 [DOI] [PubMed] [Google Scholar]
- 21. Kawachi H., Fujikawa A., Maeda N., and Noda M. (2001) Identification of GIT1/Cat-1 as a substrate molecule of protein tyrosine phosphatase ζ/β by the yeast substrate-trapping system. Proc. Natl. Acad. Sci. U.S.A. 98, 6593–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukada M., Fujikawa A., Chow J. P., Ikematsu S., Sakuma S., and Noda M. (2006) Protein tyrosine phosphatase receptor type Z is inactivated by ligand-induced oligomerization. FEBS Lett. 580, 4051–4056 [DOI] [PubMed] [Google Scholar]
- 23. Maeda N., Ichihara-Tanaka K., Kimura T., Kadomatsu K., Muramatsu T., and Noda M. (1999) A receptor-like protein-tyrosine phosphatase PTPζ/RPTPβ binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPζ. J. Biol. Chem. 274, 12474–12479 [DOI] [PubMed] [Google Scholar]
- 24. Nandi S., Cioce M., Yeung Y. G., Nieves E., Tesfa L., Lin H., Hsu A. W., Halenbeck R., Cheng H. Y., Gokhan S., Mehler M. F., and Stanley E. R. (2013) Receptor-type protein-tyrosine phosphatase ζ is a functional receptor for interleukin-34. J. Biol. Chem. 288, 21972–21986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujikawa A., and Noda M. (2016) Role of pleiotrophin-protein tyrosine phosphatase receptor type Z signaling in myelination. Neural Regen. Res. 11, 549–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tamura H., Fukada M., Fujikawa A., and Noda M. (2006) Protein tyrosine phosphatase receptor type Z is involved in hippocampus-dependent memory formation through dephosphorylation at Y1105 on p190 RhoGAP. Neurosci. Lett. 399, 33–38 [DOI] [PubMed] [Google Scholar]
- 27. Krämer E. M., Klein C., Koch T., Boytinck M., and Trotter J. (1999) Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J. Biol. Chem. 274, 29042–29049 [DOI] [PubMed] [Google Scholar]
- 28. Karus M., Ulc A., Ehrlich M., Czopka T., Hennen E., Fischer J., Mizhorova M., Qamar N., Brüstle O., and Faissner A. (2016) Regulation of oligodendrocyte precursor maintenance by chondroitin sulphate glycosaminoglycans. Glia 64, 270–286 [DOI] [PubMed] [Google Scholar]
- 29. Dyck S. M., and Karimi-Abdolrezaee S. (2015) Chondroitin sulfate proteoglycans: key modulators in the developing and pathologic central nervous system. Exp. Neurol. 269, 169–187 [DOI] [PubMed] [Google Scholar]
- 30. Lau L. W., Keough M. B., Haylock-Jacobs S., Cua R., Döring A., Sloka S., Stirling D. P., Rivest S., and Yong V. W. (2012) Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann. Neurol. 72, 419–432 [DOI] [PubMed] [Google Scholar]
- 31. Fox I. J., Paucar A. A., Nakano I., Mottahedeh J., Dougherty J. D., and Kornblum H. I. (2004) Developmental expression of glial fibrillary acidic protein mRNA in mouse forebrain germinal zones: implications for stem cell biology. Brain Res. Dev. Brain Res. 153, 121–125 [DOI] [PubMed] [Google Scholar]
- 32. Shintani T., Watanabe E., Maeda N., and Noda M. (1998) Neurons as well as astrocytes express proteoglycan-type protein tyrosine phosphatase ζ/RPTPβ: analysis of mice in which the PTPζ/RPTPβ gene was replaced with the LacZ gene. Neurosci. Lett. 247, 135–138 [DOI] [PubMed] [Google Scholar]
- 33. Milev P., Monnerie H., Popp S., Margolis R. K., and Margolis R. U. (1998) The core protein of the chondroitin sulfate proteoglycan phosphacan is a high-affinity ligand of fibroblast growth factor-2 and potentiates its mitogenic activity. J. Biol. Chem. 273, 21439–21442 [DOI] [PubMed] [Google Scholar]
- 34. Peles E., Schlessinger J., and Grumet M. (1998) Multi-ligand interactions with receptor-like protein tyrosine phosphatase β: implications for intercellular signaling. Trends Biochem. Sci. 23, 121–124 [DOI] [PubMed] [Google Scholar]
- 35. Xu C., Zhu S., Wu M., Han W., and Yu Y. (2014) Functional receptors and intracellular signal pathways of midkine (MK) and pleiotrophin (PTN). Biol. Pharm. Bull. 37, 511–520 [DOI] [PubMed] [Google Scholar]
- 36. Wanaka A., Carroll S. L., and Milbrandt J. (1993) Developmentally regulated expression of pleiotrophin, a novel heparin binding growth factor, in the nervous system of the rat. Brain Res. Dev. Brain Res. 72, 133–144 [DOI] [PubMed] [Google Scholar]
- 37. Vanderwinden J. M., Mailleux P., Schiffmann S. N., and Vanderhaeghen J. J. (1992) Cellular distribution of the new growth factor pleiotrophin (HB-GAM) mRNA in developing and adult rat tissues. Anat. Embryol. 186, 387–406 [DOI] [PubMed] [Google Scholar]
- 38. Barr A. J., Ugochukwu E., Lee W. H., King O. N., Filippakopoulos P., Alfano I., Savitsky P., Burgess-Brown N. A., Müller S., and Knapp S. (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136, 352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meng K., Rodriguez-Peña A., Dimitrov T., Chen W., Yamin M., Noda M., and Deuel T. F. (2000) Pleiotrophin signals increased tyrosine phosphorylation of β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ. Proc. Natl. Acad. Sci. U.S.A. 97, 2603–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sattelle B. M., Shakeri J., Cliff M. J., and Almond A. (2015) Proteoglycans and their heterogeneous glycosaminoglycans at the atomic scale. Biomacromolecules 16, 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iwasaki W., Nagata K., Hatanaka H., Inui T., Kimura T., Muramatsu T., Yoshida K., Tasumi M., and Inagaki F. (1997) Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 16, 6936–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ryan E., Shen D., and Wang X. (2016) Structural studies reveal an important role for the pleiotrophin C-terminus in mediating interactions with chondroitin sulfate. FEBS J. 283, 1488–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solera C., Macchione G., Maza S., Kayser M. M., Corzana F., de Paz J. L., and Nieto P. M. (2016) Chondroitin sulfate tetrasaccharides: synthesis, three-dimensional structure and interaction with midkine. Chemistry 22, 2356–2369 [DOI] [PubMed] [Google Scholar]
- 44. Frohman E. M., Racke M. K., and Raine C. S. (2006) Multiple sclerosis: the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955 [DOI] [PubMed] [Google Scholar]
- 45. Göttle P., and Küry P. (2015) Intracellular protein shuttling: a mechanism relevant for myelin repair in multiple sclerosis? Int. J. Mol. Sci. 16, 15057–15085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Viapiano M. S., and Matthews R. T. (2006) From barriers to bridges: chondroitin sulfate proteoglycans in neuropathology. Trends Mol. Med. 12, 488–496 [DOI] [PubMed] [Google Scholar]
- 47. Lau L. W., Cua R., Keough M. B., Haylock-Jacobs S., and Yong V. W. (2013) Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat. Rev. Neurosci. 14, 722–729 [DOI] [PubMed] [Google Scholar]
- 48. Murasugi A., Kido I., Kumai H., and Asami Y. (2003) Efficient production of recombinant human pleiotrophin in yeast, Pichia pastoris. Biosci. Biotechnol. Biochem. 67, 2288–2290 [DOI] [PubMed] [Google Scholar]
- 49. Muramatsu H., Zou P., Kurosawa N., Ichihara-Tanaka K., Maruyama K., Inoh K., Sakai T., Chen L., Sato M., and Muramatsu T. (2006) Female infertility in mice deficient in midkine and pleiotrophin, which form a distinct family of growth factors. Genes Cells 11, 1405–1417 [DOI] [PubMed] [Google Scholar]
- 50. Fukada M., Kawachi H., Fujikawa A., and Noda M. (2005) Yeast substrate-trapping system for isolating substrates of protein tyrosine phosphatases: isolation of substrates for protein tyrosine phosphatase receptor type z. Methods 35, 54–63 [DOI] [PubMed] [Google Scholar]
- 51. Boggs J. M. (2006) Myelin basic protein: a multifunctional protein. Cell Mol. Life Sci. 63, 1945–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krueger N. X., and Saito H. (1992) A human transmembrane protein-tyrosine-phosphatase, PTP ζ, is expressed in brain and has an N-terminal receptor domain homologous to carbonic anhydrases. Proc. Natl. Acad. Sci. U.S.A. 89, 7417–7421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fujikawa A., Chow J. P. H., Shimizu H., Fukada M., Suzuki R., and Noda M. (2007) Tyrosine phosphorylation of ErbB4 is enhanced by PSD95 and repressed by protein tyrosine phosphatase receptor type Z. J. Biochem. 142, 343–350 [DOI] [PubMed] [Google Scholar]
- 54. Fujikawa A., Fukada M., Makioka Y., Suzuki R., Chow J. P., Matsumoto M., and Noda M. (2011) Consensus substrate sequence for protein-tyrosine phosphatase receptor type Z. J. Biol. Chem. 286, 37137–37146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fujikawa A., Shirasaka D., Yamamoto S., Ota H., Yahiro K., Fukada M., Shintani T., Wada A., Aoyama N., Hirayama T., Fukamachi H., and Noda M. (2003) Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 33, 375–381 [DOI] [PubMed] [Google Scholar]
- 56. Fujikawa A., Nagahira A., Sugawara H., Ishii K., Imajo S., Matsumoto M., Kuboyama K., Suzuki R., Tanga N., Noda M., Uchiyama S., Tomoo T., Ogata A., Masumura M., and Noda M. (2016) Small-molecule inhibition of PTPRZ reduces tumor growth in a rat model of glioblastoma. Sci. Rep. 6, 20473. [DOI] [PMC free article] [PubMed] [Google Scholar]