

Abstract
Acetyl-CoA synthase (ACS) catalyzes the reversible condensation of CO, CoA, and a methyl-cation to form acetyl-CoA at a unique Ni,Ni-[4Fe4S] cluster (the A-cluster). However, it was unknown which proteins support the assembly of the A-cluster. We analyzed the product of a gene from the cluster containing the ACS gene, cooC2 from Carboxydothermus hydrogenoformans, named AcsFCh, and showed that it acts as a maturation factor of ACS. AcsFCh and inactive ACS form a stable 2:1 complex that binds two nickel ions with higher affinity than the individual components. The nickel-bound ACS-AcsFCh complex remains inactive until MgATP is added, thereby converting inactive to active ACS. AcsFCh is a MinD-type ATPase and belongs to the CooC protein family, which can be divided into homologous subgroups. We propose that proteins of one subgroup are responsible for assembling the Ni,Ni-[4Fe4S] cluster of ACS, whereas proteins of a second subgroup mature the [Ni4Fe4S] cluster of carbon monoxide dehydrogenases.
Keywords: ATPase, biosynthesis, iron-sulfur protein, metal ion-protein interaction, metalloenzyme, nickel
Introduction
The cellular maturation of metalloenzymes, previously considered a spontaneous process in vivo, typically depends on a machinery of uptake, storage, processing, and delivery factors (1). How metalloenzymes mature has been investigated for some systems, revealing surprisingly complex maturation pathways (2–8). Enzymes containing nickel, although still relatively small in number, play critical roles in archaea, bacteria, and eukarya, through which they impact the global hydrogen (Ni,Fe-hydrogenase), nitrogen (urease), and carbon (acetyl-CoA synthase, carbon monoxide dehydrogenase, and methyl-CoM reductase) cycles (9). For most of these enzymes, we now have a good understanding of how nickel is incorporated into the active site (8, 10).
The nickel-enzymes acetyl-CoA synthase (ACS)2 and carbon monoxide dehydrogenase (CODH) are found in a variety of anaerobic microbes, including bacterial sulfate reducers, acetogens, and hydrogenogens, as well as archaeal methanogens and sulfate reducers, where they act as the prime CO2 and CO converter (11–14). ACS and CODH can be found as independent monofunctional enzymes in Carboxydothermus hydrogenoformans (15) but are typically found in other microorganisms as protein complexes: in acetogens ACS and CODH form a bifunctional (αβ)2 complex, whereas in methanogens they are part of a large (αβγδϵ)8 multienzyme complex (11, 13, 15, 16). CODHs catalyze the reversible reduction of CO2 to CO at the C-cluster, in which a single nickel ion, embedded within a 3Fe-4S scaffold with an additional iron in exo, binds and activates CO and CO2 for turnover (17–19). ACS catalyzes the reversible condensation of CO, CoA, and a methyl-cation donated by the methylated corrinoid iron-sulfur protein to form acetyl-CoA (13). ACS depends on a Ni,Ni-[4Fe4S] cluster (also called A-cluster) for activity, in which the two nickel ions have distinct coordinations: the nickel ion distal to the [4Fe4S] cluster (Nid) is coordinated by two amide nitrogen atoms and two cysteine thiolates within a Cys-X-Cys motif, holding nickel in a stable square-planar coordination environment (15, 20, 21). In contrast, the nickel ion proximal to the [4Fe4S] cluster (Nip) is weakly bound by three cysteine thiolates, is removable by 1,10-phenanthroline (22), and may be replaced by zinc and copper, thereby inactivating ACS (15, 20, 21, 23, 24). Nip likely adopts different oxidation states during turnover and is the presumed place where substrates are activated and converted (13).
Despite their ubiquity and importance in different metabolisms, little is known about the maturation of CODH and ACS, which both limits our understanding of metalloenzyme maturation and hampers their adaptation for biotechnological processes. CODH activity in vivo depends on an ATPase termed CooC (25–28). CooC belongs to the MinD-type ATPases, an enzyme family with diverse functions and a synapomorphic KGG signature in their Walker A motif (GKGGhGK(T/S)) containing a characteristic lysine residue (signature lysine) in addition to the P-loop lysine (29). Common to the MinD-type ATPases is an ATP-dependent dimerization, which is central to their function and a prerequisite to hydrolyze ATP (30). CooC binds Ni(II) through the Cys-X-Cys motifs of two monomers that combine to form a metal binding site within the dimer interface (31, 32). Additional proteins support the maturation of the CODH in Rhodospirillum rubrum (25); however, these additional proteins are not conserved in bacteria outside the Rhodospirillaceae and may be functionally replaced by non-homologous isofunctional enzymes in other organisms. Although CooC was shown to mature monofunctional CODHs (25–28), it was proposed that the homologous ATPase AcsF supports the assembly of the C-cluster in the bifunctional ACS-CODH complex in Moorella thermoacetica (33).
In contrast, how the Ni,Ni-[4Fe4S] cluster of ACS is formed in vivo is not known. The purpose of this paper is to provide insights into the role of an ATPase closely related to CooC in this process, which we named AcsFCh. We provide evidence that AcsFCh catalyzes the nickel and MgATP-dependent activation of ACS by forming a complex with apoACS that serves as a platform for nickel binding from which active ACS is generated at the expense of ATP.
Results
CooC Proteins Can Be Divided into Three Subgroups
The genome of Carboxydothermus hydrogenoformans contains three genes, annotated as cooC1, cooC2, and cooC3, which encode for proteins of the CooC family (34). The cooC1 and cooC2 genes are located in the same gene cluster as the genes encoding CODH-III and ACS (Fig. 1A). The cooC3 gene is located in proximity to the gene encoding CODH-I.
FIGURE 1.
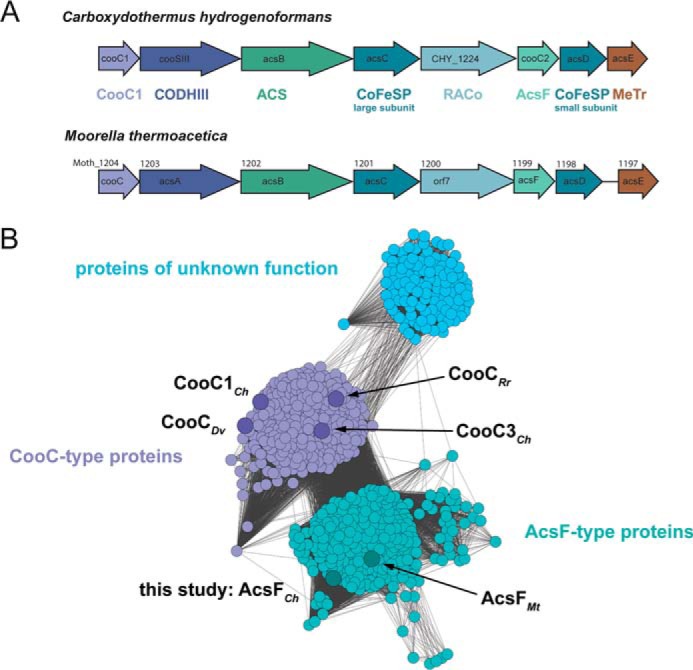
A, gene clusters containing the gene for bifunctional ACS/CODH in C. hydrogenoformans and M. thermoacetica. B, sequence similarity network of CooC proteins. The network was generated from an all-by-all distance matrix storing Kimura distances calculated from a multiple amino acid sequence alignment. Gray colored lines connect nodes (amino acid sequences) with a Kimura distance below 1.3. Nodes are arranged using the yFiles organic layout of Cytoscape 3.3.0 (59). Larger nodes represent sequences from the genomes of C. hydrogenoformans (CooC1Ch, AcsFCh, CooC3Ch), M. thermoacetica (AcsFMt), R. rubrum (CooCRr), and D. vulgaris (CooCDv). Light blue, violet, and turquoise denote subgroups of proteins of unknown function, CooC-type and AcsF-type proteins, respectively.
The InterPro database lists 881 protein sequences as members of the CooC family (35, 36). A sequence similarity network analysis of the non-redundant sequences of the CooC proteins indicates that the sequences may be grouped into three different clusters (Fig. 1B). The two largest clusters, hereafter called “CooC-type” cluster and “AcsF-type” cluster, contain proteins typically associated with gene clusters of CODHases or ACS/CODH. The third cluster contains 116 proteins, mostly found in methanogenic archaea (Methanomicrobia, Methanococci). The CooC-type cluster harbors 298 sequences and includes all CooC proteins shown to be involved in CODH maturation, such as CooC from R. rubrum (CooCRr) (26), CooC3Ch (27), and CooC from Desulfovibrio vulgaris (CooCDv) (28). The CooC-type cluster also encompasses CooC1 from C. hydrogenoformans (CooC1Ch), which is a nickel-binding ATPase whose crystal structure has been determined (27, 31).
The AcsF-type cluster encompasses 309 protein sequences, among them AcsF from M. thermoacetica (AcsFMt). AcsFMt has been described as an ATPase of unknown function whose involvement in CODH maturation of bifunctional ACS/CODH was postulated but not shown (27, 33). The AcsF-type cluster also encompasses the gene product of cooC2 from C. hydrogenoformans, which we therefore named AcsFCh. All clusters include proteins from both bacteria and archaea. Most organisms (63%) containing a gene in the CooC-type cluster also harbor a paralogous gene of the AcsF-type cluster in their genome and only 13% of the organisms with an AcsF-type sequence have no CooC-type protein in the Interpro database.
AcsFCh Dimerizes in the Presence of ATP, Does Not Bind Nickel and Has Low ATPase Activity
Typical size exclusion chromatograms of AcsFCh showed peaks at 83.2 and 90.5 ml. The corresponding molecular masses, 57.7 and 28.8 kDa, were determined by comparison to a standard curve and agree with the predicted molecular masses of dimeric (55.4 kDa) and monomeric AcsFCh (27.7 kDa). The monomer:dimer ratio depended on the cultivation batch rather than on individual purifications. Additionally, two variants of AcsFCh were expressed and purified. The K10A-AcsFCh variant, lacking the signature lysine Lys-10, showed similar monomer:dimer ratios for as isolated protein as wild type AcsFCh, whereas the C108A-AcsFCh variant was only present as monomer.
We analyzed by size exclusion chromatography (SEC) whether monomeric AcsFCh dimerizes in the presence of MgADP and MgATP (Fig. 2A). Although the presence of MgADP had no influence on the chromatogram, we found AcsFCh in the presence of MgATP to be predominantly dimeric, as expected for a MinD-type ATPase. The monomeric fraction of the K10A-AcsFCh variant remained in the monomeric state independent of the presence of MgADP or MgATP (Fig. 2B).
FIGURE 2.

Nucleotide dependent dimerization of AcsFCh. A, the elution profile of monomeric AcsFCh without nucleotide is shown as solid line. Monomeric AcsFCh was incubated with either MgADP or MgATP and the oligomeric state was analyzed by SEC. The elution profiles of AcsFCh in the presence of MgADP or MgATP are shown as dashed and dotted lines, respectively. B, influence of nucleotide on the oligomeric state of K10A-AcsFCh. The elution profile of monomeric K10A-AcsFCh is shown as solid line and the elution profile of K10A-AcsFCh in the presence of MgADP or MgATP are shown as dashed and dotted lines, respectively. All dimerization experiments were at least performed with two independent samples.
AcsFCh shares with the other CooC proteins the CXC sequence motif. In CooC1Ch the two Cys residues of this motif are able to bind nickel, which can be followed by an increased absorption in the region 310–350 nm originating from a ligand to metal charge transfer between Cys-S− and Ni(II) (31). The UV/visible absorption spectrum of AcsFCh had a maximum at 274 nm. When we added increasing amounts of NiCl2 to AcsFCh, the UV/visible spectral features remained unchanged. Furthermore, when we titrated NiCl2 to AcsFCh in an anoxic isothermal titration calorimetry (ITC) experiment, we only observed heats of dilution. Thus, in contrast to CooC1Ch, we could not detect specific nickel binding by AcsFCh.
The amino acid sequence of AcsFCh contains the characteristic Walker A signature of the MinD-type ATPase. We determined the specific ATPase activity of AcsFCh with a coupled assay and a colorimetric assay. Determined specific activities varied with each purification batch and averaged at 7.4 nmol min−1 mg−1 at 25 °C and 25.5 nmol min−1 mg−1 at 45 °C in the coupled assay. In the colorimetric assay the average activities measured were 3.9 nmol min−1 mg−1 at 25 °C, 20.4 nmol min−1 mg−1 at 45 °C, and 56.5 nmol min−1 mg−1 at 60 °C. In contrast, we could not detect ATPase activity above background for the K10A-AcsFCh variant. There is an intrinsic risk that low ATPase activities, such as those determined for AcsFCh, could arise from minor protein impurities of high specific activities and not the protein investigated. However, all AcsFCh preparations tested had substantially higher activities than the preparation of the K10A-AcsFCh variant, which was purified using the same protocol. Furthermore, the ATPase activity increased until 60 °C, which is expected for an ATPase of a hyperthermophilic organism such as C. hydrogenoformans. We thus conclude that AcsFCh is an ATPase of low activity.
AcsFCh Forms a Complex with ApoACS
A maturing metalloenzyme typically forms a specific complex to receive metal(s) from its accessory protein(s). We therefore tested by SEC whether AcsFCh was able to form a stable complex with apoACS. When we loaded a preincubated 1:2 mixture of apoACS and AcsFCh (1:1 of apoACS and dimeric AcsFCh) on the column, the resulting chromatogram revealed a new peak at 70.2 ml, which was clearly separated from the peaks of apoACS and AcsFCh (Fig. 3A) and corresponded to a molecular mass of 199 kDa. Because apoACS and AcsFCh were incubated in a 1:2 stoichiometry, and no further peaks appeared in the chromatogram, we assume that the complex is very stable and has a 1:2 stoichiometry of apoACS and AcsFCh (141 kDa). SDS-PAGE of the peak fractions showed two bands corresponding to ACS (86.6 kDa) and AcsFCh (27.7 kDa). This confirms the presence of an ACS-AcsFCh complex (Fig. 3B). Densitometric analysis of the SDS gel indicated a 1:1.8 stoichiometry, confirming a complex of one molecule of ACS and two molecules of AcsFCh. The complex was formed regardless of whether ACS was incubated with dimeric or monomeric AcsFCh preparation.
FIGURE 3.

Complex formation of apoACS with AcsFCh. A, complex formation monitored by SEC. The apoACS-AcsFCh sample was prepared by incubating 13 nmol of apoACS with 13 nmol of dimeric AcsFCh for 10 min. For comparison, apoACS and dimeric AcsFCh were also loaded individually on the column. The elution profile of the apoACS-AcsFCh incubation mixture is shown as dotted line and the elution profiles of the individual proteins apoACS and AcsFCh are shown as dashed and solid lines, respectively. Gray arrows mark the elution volumes of the molecular weight standards. The apoACS-AcsFCh complex elutes at 70.2 ml, whereas apoACS and AcsFCh elute at 76.0 and 83.2 ml, respectively. Complex formation of apoACS with AcsFCh was observed in more than five independent samples. B, SDS-PAGE analysis of SEC experiments. From each chromatography run, the main peak fractions were loaded on a gel (apoACS-AcsFCh complex, 67 to 76 ml; apoACS, 72.5 to 80 ml; AcsFCh, 80.5 to 85.5 ml). Lanes 1, 5, 9, and 12 show a marker with the indicated molecular masses. C, CooC1Ch, AcsFCh, or CooC3Ch were individually incubated with apoACS and complex formation was analyzed by SEC. The elution profiles of the incubation mixtures of apoACS with CooC1Ch, AcsFCh, and CooC3Ch are shown as solid, dashed, and dotted lines, respectively. Complex formation experiments of apoACS with CooC1Ch and CooC3Ch were carried out with two independent samples. D, complex formation of the AcsFCh variants K10A-AcsFCh and C108A-AcsFCh with apoACS. Either monomeric K10A-AcsFCh or monomeric C108A-AcsFCh was incubated with apoACS and complex formation was followed by SEC. The elution profiles of apoACS incubated with K10A-AcsFCh and C108A-AcsFCh are shown as dashed and dotted lines, respectively. Complex formation experiments of apoACS with K10A-AcsFCh and C108A-AcsFCh were carried out with two independent samples.
CooC1Ch and CooC3Ch belong to the CooC-type proteins (Fig. 1) and share 28.6 and 28.3% amino acid sequence identity with AcsFCh, respectively. Despite the similarity, apoACS did not form a stable complex with CooC1Ch or CooC3Ch, indicating that its interaction with AcsFCh is specific (Fig. 3C).
Furthermore, we determined the ability of the AcsFCh variants K10A-AcsFCh and C108A-AcsFCh to form a complex with apoACS (Fig. 3D). Although the K10A-AcsFCh variant formed a complex with apoACS, we did not observe a complex between apoACS and the C108A-AcsFCh, indicating that Cys-108 is involved in complex formation.
ApoACS Can Be Activated by AcsFCh, NiCl2, and MgATP
The UV/visible spectrum of ACS had the characteristic absorption shoulder of a [4Fe4S]2+ cluster at 420 nm. Metal analysis by inductively coupled plasma optic emission spectrometry (ICP-OES) confirmed the presence of iron in as isolated ACS, whereas nickel could not be detected (Table 1). In agreement with the lack of nickel, the enzyme is inactive. Therefore, the as isolated ACS is designated as apoACS.
TABLE 1.
Results from ICP-OES analysis
Values show the number of metal ions per molecule of ACS. The metal content measurements were replicated three times for all ACS samples and twice for the ACS-AcsFCh sample.
| Nickel | Zinc | Iron | Nickel per 4 Fe | |
|---|---|---|---|---|
| ApoACS | NDa | 1.33 ± 0.18 | 4.83 ± 0.11 | 0 |
| ND | 0.73 ± 0.11 | 3.25 ± 0.16 | 0 | |
| ACS after ITC | 0.54 ± 0.14 | ND | 4.96 ± 0.22 | 0.43 |
| ACS-AcsFCh after ITC | 2.26 ± 0.05 | ND | 4.88 ± 0.09 | 1.85 |
a ND, not detected.
The lack of nickel and catalytic activity in apoACS served as a starting point to investigate the maturation of ACS. When we added stoichiometric amounts of nickel, MgATP, and AcsFCh to ACS, it reached within 2 h a specific activity of 70 nmol min−1 mg−1 (Fig. 4A). In contrast, when we omitted AcsFCh from the mixture, the activity of ACS increased only marginally. Activation of ACS by AcsFCh strictly depended on MgATP addition. Thus, AcsFCh catalyzes the Ni2+ and MgATP-dependent maturation of ACS. The process is catalytic as also substoichiometric amounts of AcsFCh (1:20 AcsFCh:apoACS) sufficed to activate apoACS.
FIGURE 4.

Activation of apoACS. A, time dependent activation of apoACS in the presence of AcsFCh, MgATP, and nickel. 20 μm apoACS were incubated with 40 μm AcsFCh, 10 mm MgATP, and 40 μm NiCl2 at 60 °C. Aliquots were taken from the activation assay at the indicated time points and the specific activity was measured at 25 °C by following the conversion of Mecob(III)inamid to cob(I)inamid by UV/visual spectroscopy. Assay conditions included a sample of the activation assay, Ti(III)citrate, Mecob(III)inamid, CO, and CoA in 0.1 m HEPES, pH 7.2. Each data point was measured in triplicate and error bars represent the standard deviation. Four control experiments were performed, in which AcsFCh, MgATP, NiCl2, or apoACS was omitted from the activation assay. Data points for the control experiments were measured once. A monoexponential function was fitted to the data points giving an observed rate constant of 1.47 h−1 for the activation. B, activation of apoACS after 60 (dark) and 180 min (light) in the presence of AcsFCh, K10A-AcsFCh, C108A-AcsFCh, CooC1Ch, or CooC3Ch, which was measured once. The inset shows a magnification of the data points for K10A-AcsFCh, C108A-AcsFCh, CooC1Ch, and CooC3Ch.
In a next step, we investigated how the amino acid exchanges K10A and C108A influence the capability of AcsFCh to activate apoACS. Neither the ATPase-deficient variant K10A-AcsFCh, nor the C108A-AcsFCh variant was able to activate apoACS (Fig. 4B). This indicates that the ATPase activity of AcsFCh, as well as the presence of the CXC motif, are essential for the activation process.
AcsFCh is a specific maturation factor of ACS, as neither CooC1Ch nor CooC3Ch can replace AcsFCh in the activation assay (Fig. 4B). Even though the data may indicate a slight increase of ACS activity over time (Fig. 4B, inset), the determined activities of ACS are within the error of the activity assay. They remained within the range detected in the absence of AcsFCh and are more than 37 times lower than when incubated with AcsFCh. Thus only AcsFCh, not CooC1Ch or CooC3Ch, acts as a specific maturation factor of ACS.
The ACS-AcsFCh Complex Binds Nickel Stoichiometrically and Can Be Subsequently Activated by MgATP
We determined whether there is a difference between the nickel binding properties of apoACS and the apoACS-AcsFCh complex. Therefore, we titrated the proteins with nickel using ITC and analyzed the metal contents of these samples by ICP-OES after removal of excess nickel. When we titrated apoACS with NiCl2, we observed substoichiometric nickel binding; the ICP-OES metal analysis detected only 0.43 nickel atoms per 4 Fe atoms in nickel-titrated apoACS (Table 1). The apoACS-AcsFCh complex, by contrast, binds nickel stoichiometrically; the ICP-OES analysis detected 1.85 nickel atoms. Thus, whereas apoACS alone cannot bind Ni2+ stoichiometrically, the apoACS-AcsFCh complex is able to bind approximately two Ni2+ ions (Table 1).
As the apoACS-AcsFCh complex has the full complement of metal ions after titration with nickel, we determined its specific activity. Despite the presence of two nickel ions, the nickel-bound apoACS-AcsFCh complex lacked ACS activity. But when we removed excess Ni2+ and incubated the nickel-bound apoACS-AcsFCh complex with 10 mm MgATP (1 h, 60 °C), it gained an activity of 67 nmol min−1 mg−1, the same specific ACS activity as observed when all components were present in our activation assay. Thus, MgATP is needed to convert the active site in the ACS-AcsFCh complex from an inactive nickel-bound to an active nickel-bound state.
Discussion
Role of AcsFCh in Ni,Ni-[4Fe4S] Cluster Formation
Our analysis allows first mechanistic conclusions about how AcsFCh supports the assembly of the Ni,Ni-[4Fe4S] cluster of ACS (Fig. 5). In the first step, two AcsFCh monomers or one AcsFCh dimer form a complex with apoACS. Complex formation creates at least one additional high-affinity nickel binding site in the complex, as only the apoACS-AcsFCh complex binds the full complement of nickel when presented with low physiological concentrations: we detected no affinity for AcsFCh to bind nickel; apoACS binds less than one nickel ion when titrated with nickel; however, the apoACS-AcsFCh complex binds two nickel ions under the same conditions. Thus, complex formation likely induces conformational changes, creating new binding sites for nickel, in apoACS, AcsFCh, or at the interface between both.
FIGURE 5.

Preliminary model of ACS activation by AcsFCh. ApoACS (1), which contains the [4Fe4S] cluster, but no nickel, can form a complex with either one dimer or two monomers of AcsFCh. The apoACS-AcsFCh complex (2) is then able to bind two nickel ions. The nickel ions may bind either sequentially (3) or simultaneously. However, neither the details of the nickel binding mechanism nor the location of the nickel binding site are known. ATP hydrolysis by AcsFCh converts inactive (4) to active ACS (5).
When the Cys-X-Cys motif in AcsFCh is altered, the complex does not form, suggesting that it is part of the ACS-AcsFCh interface. It is tempting to assume that the presence of strictly conserved Cys-X-Cys motifs in both AcsFCh and ACS is not purely coincidental (37), but serves to have binding sites with similar affinity for Ni2+ in both proteins.
Once nickel is bound to the apoACS-AcsFCh complex, MgATP is required to activate ACS. Although MgATP binding and its hydrolysis are key to the activation, their impact on the bound nickel ions remains to be defined. Furthermore, it should be noted that the observed rate constant of apoACS activation of 1.47 h−1 is substantially lower than expected from the determined rate of ATP hydrolysis by AcsFCh alone.
Two Classes of CooC Proteins Are Responsible for the Maturation of ACS and CODH
Despite the sequence similarity between AcsF and the paralogous CooC1 and CooC3 proteins of C. hydrogenoformans, it is obvious that the CooC proteins, as defined by InterPro, can be grouped in at least three classes (Fig. 1). We suggest that enzymes clustered in the CooC-type group may act in the maturation of the [Ni4Fe4S] cluster of CODHs, whereas those that belong to the AcsF-type group, such as AcsFCh and AcsFMt, catalyze the MgATP and nickel-dependent maturation of the Ni,Ni-[4Fe4S] cluster of ACS. A possible division of selected CooC proteins in two classes has also been proposed based on a phylogenetic tree (27).
CODH and ACS differ not only in their function and overall fold, but their active sites differ in (i) composition: CODH has 1 nickel ion per 4 iron ions, whereas ACS has 2 nickel ions; (ii) architecture: in CODH nickel is integrated into an FeS scaffold, whereas in ACS the 2 nickel ions are bound next to a [4Fe4S] cluster; and (iii) accessibility: in CODH the Ni,Fe-cluster is connected to the surface via small channels, whereas the Ni,Fe-cluster of ACS is surface exposed in at least one conformation of the protein (21). It is surprising that despite all these differences the maturases involved in nickel- and MgATP-dependent activation of the enzymes belong to the same ATPase family, have remarkably similar amino acid sequences, and likely evolved from a common ancestor.
Comparison with Homologous NTPases
AcsFCh is homologous and functionally related to other NTP-hydrolyzing maturation factors, which act, for example, in the assembly of ureases (UreG), Ni,Fe-hydrogenases (HypB), nitrogenases (NifH), and cytosolic Fe/S proteins (Cfd1/Nbp35). UreG and HypB are homologous GTPases that are essential for the maturation of ureases and hydrogenases, respectively. Both are like AcsFCh SIMIBI NTPases, but belong to the G3E family and carry the signature motif (ESGG) and the guanine specificity loop (NKTD) characteristic for this family (29, 38). In contrast to AcsFCh, they act together with other maturation factors to transfer a metal to a target protein: UreG forms a complex with the additional factors UreD (UreH), UreE and UreF to facilitate apo-urease maturation (8, 39, 40), whereas HypB is believed to process and deliver Ni2+ in cooperation with HypA and SlyD to the large subunit of Ni,Fe-hydrogenase (7, 10, 41, 42). In contrast, AcsFCh is able to mature apo-ACS in vitro without additional proteins required.
The CooC/AcsF proteins are not the only MinD-type ATPases involved in metalloenzyme maturation: they are joined by Cfd1/Nbp35 and NifH. Cfd1/Nbp35 is a heterodimeric ATPase essential for cytosolic iron-sulfur cluster maturation (43), whereas NifH is involved in molybdenum-homocitrate transfer to the maturing Mo,Fe-cluster on NifEN (44). Mutagenesis studies indicate that Cys residues in the dimer interface of Cfd1/Nbp35 are involved in coordinating the [4Fe4S] cluster to be transferred (45). Although NifH and Cfd1/Nbp35 are part of a larger maturation machinery, they seem capable, just like AcsFCh, of directly transferring a metal or metal cluster to a maturing metalloenzyme. It is thus tempting to assume that the prototypical conformational changes in the dimer interface of MinD-type ATPases upon ATP hydrolysis are used by Cfd1/Nbp35, NifH, and AcsFCh to transfer one or more metal ions from one protein to another.
ApoACS can also be activated by incubating it for several hours with an excess of nickel (46–48). However, in common with many other metalloenzymes, under physiological conditions the maturation of apoACS requires an accessory enzyme, which can mature apoACS without the need for high nickel concentrations and long incubation times. In contrast to other metalloenzymes, the maturation of the A-cluster may require only one specific maturation factor, which gives us the unique opportunity to study the assembly of a complex metal cluster in a well defined system in vitro.
Experimental Procedures
Bioinformatic Methods
881 sequences were extracted from InterPro entry IPRO14433 (35, 36). The sequence of CooC from D. vulgaris (CooCDv) was added, because it was missing in the InterPro entry. After removal of redundant (identical) sequences 723 sequences remained. These sequences were aligned using MAFFT (49) and a distance matrix was calculated from the multiple sequence alignment using protdist/PHYLIP (50) using the Kimura distance as a criterion for similarity. The distance cutoff used to define an interaction in the sequence similarity plot was 1.3. Cytoscape 3.3.0 was used for clustering and displaying the sequence similarity network using the yFiles organic layout provided in Cytoscape (51).
Chemicals
All chemicals were at least of analytical grade and purchased from Sigma, AppliChem, or Roth. Restriction enzymes, DNA polymerases, and T4 DNA ligase were purchased from New England Biolabs, Fermentas (Thermo Scientific), or Agilent Genomics. N2, N2/H2 (95/5%), and CO were purchased from Air Liquide. Chromatography columns were purchased from GE Healthcare or IBA. Bottles for metal-free buffers were rinsed with dilute HCl and then washed with MilliQ/H2O. Chelex 100 resin (Bio-Rad) was added to all metal-free buffers (5 to 10 g liter−1). All anaerobic solutions were prepared at a Schlenk line in a bottle equipped with a butyl rubber septum by at least four cycles of evacuating and purging with N2. CO-saturated buffer was prepared by bubbling CO through the buffer for at least 10 min. Ti(III)citrate was prepared as previously described (52). Methylcobinamide was synthesized from methylcobalamin using the method from Zou et al. (53) except that it was purified with a SepPak C18 reversed phase column (Waters).
Cloning, Mutagenesis, and Expression
The cooC2 gene was amplified by PCR from genomic DNA of C. hydrogenoformans Z-2901 using Pfu DNA polymerase with primer C2_fw with an NdeI restriction site and C2_rv with an Eco31I restriction site (primer sequences are listed in Table 2), which produces a BamHI compatible ligation site. The PCR product was digested with Eco31I and NdeI and then ligated into an NdeI/BamHI-digested pET11a vector (Novagen). The ligation product was named pET11a_AcsF. From this plasmid, the cooC2 gene was cloned into a modified pET28a vector with an N-terminal streptag and a tobacco etch virus (TEV) protease cleavage site, named pET28aTEVstrep, via polymerase incomplete primer extension (PIPE) cloning (54, 55). For PIPE cloning the vector PCR contained 0.1 ng of template, 1 μm of each primer (PIPE_vec_fw and PIPE_vec_rv; Table 2), and 25 μl of Phusion High Fidelity PCR master mix in a 50-μl reaction volume. The DNA was amplified using the Mastercycler pro PCR machine (Eppendorf) with the following protocol: 2 min initial denaturation at 98 °C, 35 cycles of 98 °C for 10 s, 65 °C for 10 s, and 72 °C for 2 min, followed by immediate cooling to 4 °C. The PCR product was purified and aliquots were stored at −20 °C. Using appropriate primers and templates, the set up for the insert PCR was the same as for the vector PCR. The reaction mixture was treated as follows: 2 min initial denaturation at 98 °C, 35 cycles of 10 s at 98 °C, 10 s at 62 °C, and 12 s at 72 °C, followed by immediate cooling to 4 °C. After purification of the insert, 1 μl of vector was annealed with 1 μl of insert for 1 min on ice and then directly transformed into Escherichia coli DH5α. The plasmid was named pET28a_AcsFstrep. K10A-AcsFCh and C108A-AcsFCh variants were prepared using the QuikChange method following standard procedures. The cooC3 gene was amplified by PCR from genomic DNA of C. hydrogenoformans Z-2901 using Herculase II DNA fusion polymerase with primer C3_fw with an NdeI restriction site and C3_rv with a HindIII restriction site (Table 2). The PCR product was digested with the respective restriction enzymes and ligated into the original pET28a vector and the modified pET28aTEVstrep vector. The latter plasmid was named pET28a_C3strep. The acsB gene was cloned into the pET28aTEVstrep vector from genomic DNA of C. hydrogenoformans Z-2901 (10 ng) using PIPE cloning as described above with primers PIPE_ACS_fw and PIPE_ACS_rv (Table 2). The plasmid was named pET28a_ACSstrep. The pET28a_ACSstrep plasmid was also digested with NdeI and BamHI and the digestion product was ligated into a pET28a_twinstrep vector, which has a twin-streptag and a TEV protease cleavage site. The plasmid was named pET28a_ACStwinstrep. All plasmids were transformed into E. coli DH5α and verified by nucleotide sequencing (Eurofins Genomics). For expression, all plasmids were transformed into E. coli BL21(DE3). Cells containing the pET28a_AcsFstrep or pET28a_C3strep plasmid were grown aerobically in LB medium containing 50 μg ml−1 of kanamycin at 37 °C. After the optical density at 600 nm (A600) reached 0.7 ± 0.1, the culture was induced with 0.2 mm isopropyl β-d-thiogalactopyranoside and the temperature was decreased to 28 °C. Cells were harvested 16 to 24 h after induction and washed with 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 2 mm tris(2-carboxyethyl)phosphine, and 2 mm EDTA. Cell pellets were frozen in liquid N2 and stored at −80 °C. Cells with the pET28a_ACSstrep or pET28a_ACStwinstrep plasmid were cultivated in modified TB medium containing 50 μg ml−1 of kanamycin in 5-liter bottles in a water bath at 37 °C. The culture was stirred aerobically and after 1 h the medium was supplemented with 0.1 mm FeSO4 and 0.5 mm cysteine. When the A600 reached 0.6 ± 0.1 cultures were induced with 0.5 mm isopropyl β-d-thiogalactopyranoside and transitioned to anaerobic growth by closing the bottles with a butyl rubber septum. After 24 h cells were quickly harvested aerobically, frozen in liquid N2, and stored at −80 °C.
TABLE 2.
PCR primers used for cloning
Restriction sites are underlined.
| Name | Sequence |
|---|---|
| C2_fw | 5′-GGA ATT CCA TAT GGC CTT TAA AAT TGC GGT TG-3′ |
| C2_rv | 5′-CAT GGT CTC GGA TCC TCA GAT AAT ACC CGC ATT TG-3′ |
| C3_fw | 5′-GGA ATT CCA TAT GCA GGT GTC TG-3′ |
| C3_rv | 5′-CCC AAG CTT CTA ACA ATT CTG CC-3′ |
| PIPE_vec_fw | 5′-GGA TCC GAA TTC GAG CTC CGT CG-3′ |
| PIPE_vec_rv | 5′-CAT ATG GCC CTG GAA ATA CAA GTT TTC GGT CG-3′ |
| PIPE_C2_fw | 5′-CTT GTA TTT CCA GGG CCA TAT GGC CTT TAA AAT TGC GGT TGC AG-3′ |
| PIPE_C2_rv | 5′-CGA CGG AGC TCG AAT TCG GAT CCC TAG ATA ATA CCC GCA TTT TGT AAA ATT TTC-3′ |
| PIPE_ACS_fw | 5′-CTT GTA TTT CCA GGG CCA TAT GAG CGA AGT TAT TAA TTT TGA TCA AAT TTT TG-3′ |
| PIPE_ACS_rv | 5′-CGA CGG AGC TCG AAT TCG GAT CCT TAG AGT AGT GGC TCC ATG GAA AGA GCT GG-3′ |
Purification
All purifications were carried out at room temperature in an anaerobic glove box (model B; COY Laboratory Products) using metal-free buffers. CooC1Ch was prepared as previously described (31). For AcsFCh, 5 to 10 g of frozen cells were resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 2 mm tris(2-carboxyethyl)phosphine) with a small amount of avidin and lysozyme. The cell suspension was sonicated (Bandelin Sonoplus 2200) three times for 5 min each (5 × cycle, 50% amplitude) in a rosette cooling cell on ice. It was then centrifuged in a polycarbonate bottle with cap assembly at 142,400 × g for 1 h at 12 °C. The supernatant was loaded on a StrepTactin-Sepharose High Performance column (10–30 ml) equilibrated in buffer A. After washing the column with 3 column volumes of buffer A, AcsFCh was eluted with a linear gradient of 0–2.5 mm desthiobiotin in the same buffer. The streptag of AcsFCh was cleaved by incubation with a strep-tagged TEV protease for ∼16 h. The protein solution was applied to a PD-10 desalting column and was then reloaded on the StrepTactin column equilibrated with buffer A. The flow-through was collected, concentrated, and loaded on a Superdex 200 Hiload 16/60 column equilibrated in buffer B (50 mm Tris-HCl, pH 8.0, 100 mm NaCl). Fractions corresponding to monomeric and dimeric AcsFCh were pooled, concentrated, frozen in liquid N2 (in glass vials equipped with a PTFE/silicone septum), and stored at −80 °C. For K10A-AcsFCh, C108A-AcsFCh, and CooC3Ch, the purification procedure was carried out as described above. ACS was prepared following the same basic strategy except that buffers did not contain tris(2-carboxyethyl)phosphine and the streptag or the twin-streptag was not cleaved. Cells containing the pET28a_ACSstrep or pET28a_ACStwinstrep plasmid were lysed, centrifuged, and captured on the streptactin column as described above. The protein was then eluted with desthiobiotin, concentrated, and directly loaded on the Superdex 200 size exclusion column. The fractions corresponding to monomeric ACS were collected, concentrated, frozen in liquid N2 (in glass vials equipped with a PTFE/silicone septum), and stored at −80 °C.
Analysis of Dimerization of AcsFCh and K10A-AcsFCh in the Presence of Nucleotide
The dimerization behavior of AcsFCh and K10A-AcsFCh in the presence of nucleotide was analyzed by size exclusion chromatography. Monomeric AcsFCh was incubated in 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 2 mm tris(2-carboxyethyl)phosphine, 0.25 mm MgCl2, and 0.25 mm ADP or ATP for 10 min. The incubation mixture was then loaded on a Superdex 200 HiLoad 16/60 column equilibrated with the incubation buffer containing the corresponding nucleotide. The flow rate was 1 ml min−1.
UV/Visible Spectroscopy
UV/visible spectra were obtained inside the glove box, using an Agilent 8453 photodiode array spectrophotometer with a Peltier temperature controller at 25 °C. Black walled quartz cuvettes with 1-cm path length were used to record spectra.
Nickel Binding Assay
20 μm AcsFCh in 50 mm Tris-HCl, pH 8.0, 100 mm NaCl were titrated with NiCl2 to yield concentrations of 1, 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, and 40 μm Ni2+. After each addition one spectrum was recorded. Titrations and UV/visible spectroscopy were performed inside the glove box.
Measurements of ATPase Activity
To measure the ATPase activity of AcsFCh a coupled assay using pyruvate kinase and lactate dehydrogenase was performed (56). In this assay the conversion of one molecule of NADH to NAD+ by lactate dehydrogenase equals the consumption of one molecule of ATP by AcsFCh. The rate of ATP hydrolysis was determined by measuring the decrease of absorption at 340 nm for 10 min and calculating the reaction rates using the extinction coefficient of NADH ϵ340 nm = 6220 m−1 cm−1. The assay solution was prepared in 50 mm HEPES, pH 7.2, containing 150 μm KCl, 10 mm MgCl2, 2.3 mm phosphoenolpyruvate, 140 μm NADH, 9 units of lactate dehydrogenase, 6 units of pyruvate kinase, and 1 mm MgATP. The assay was started by addition of 5 μm AcsFCh. This assay was performed at two different temperatures, at 25 or 45 °C. The velocities obtained by the coupled assay were confirmed with a modified malachite green ammonium molybdate assay (57). This assay was performed at 25, 45, and 60 °C. In this assay the amount of phosphate released by the hydrolysis of ATP is detected by following the increase of absorption at 630 nm, which is due to the formation of a malachite green phosphomolybdate complex. The rate calculation was based on a standard curve, which was prepared with KH2PO4 in the range from 1 to 9 μm. The assay solution contained 5 μm AcsFCh, 10 mm MgCl2, and 1 mm MgATP in 0.1 mm HEPES, pH 7.2. Aliquots were taken at different time points, incubated with the malachite green ammonium molybdate mixture and 34% (w/v) sodium citrate for 15 min, and then the absorption was measured at 630 nm. Control experiments in the absence of AcsFCh were performed to assess the rate of autohydrolysis of ATP at different temperatures.
Analysis of Complex Formation Behavior between ACS and AcsF and CooC Proteins
ApoACS and AcsFCh were incubated for 10 min inside the glove box and loaded on a Superdex 200 HiLoad 16/60 column equilibrated in 50 mm Tris-HCl, pH 8.0, 100 mm NaCl. The flow rate was 1 ml min−1. Elution profiles were recorded by following the absorption at 280 nm. As a control apoACS and AcsFCh were also loaded individually on the column. From these three chromatography runs, the fractions were analyzed by SDS-polyacrylamide gel electrophoresis. The stoichiometry of the complex was analyzed with the software GelAnalyzer 2010a. Complex formation of apoACS with K10A-AcsFCh, C108A-AcsFCh, CooC1Ch, and CooC3Ch was also analyzed by size exclusion chromatography as described for wild type AcsFCh. A calibration of the Superdex HiLoad 16/60 column was performed with the following molecular mass standards: aprotinin (6.5 kDa), RNase A (13.7 kDa), DNase I (31 kDa), ovalbumin (44 kDa), conalbumin (75 kDa), lactate dehydrogenase (140 kDa), aldolase (161 kDa), and catalase (250 kDa). The decadic logarithm of the molecular weight was plotted against the elution volume and the calibration curve was determined by linear regression.
Reconstitution of ApoACS
All incubation and reaction mixtures were set up inside an anaerobic glove box (model B; COY Laboratory Products). ApoACS was reconstituted by incubating 20 μm apoACS with 40 μm NiCl2, 40 μm AcsFCh, 10 mm MgCl2, and 10 mm MgATP in 0.1 m HEPES, pH 7.2, at 60 °C. Four control experiments were set up; in each case, one component of the incubation mixture was missing. Reconstitution experiments were also performed with K10A-AcsFCh, C108A-AcsFCh, CooC1Ch, and CooC3Ch instead of AcsFCh. Aliquots were taken at different time points and the rate of acetyl-CoA formation was analyzed by UV/visible spectroscopy.
Measurements of Acetyl-CoA Formation Activity
Acetyl-CoA synthesis activity of ACS from CO, methylcobinamide, and CoA was determined by following the conversion of methylcob(III)inamide (MeCbi) to cob(I)inamide (Cbi). The change of absorption was measured at 387 and 462 nm. The reaction rates were calculated using the differences of the extinction coefficients, Δϵ387 nm(MeCbi-Cbi) = −17,410 m−1 cm−1 and Δϵ465 nm(MeCbi-Cbi) = 7,840 m−1 cm−1, which were calculated from spectra of Robertson et al. (58). The reaction was set up inside the glove box as follows: 0.1 m HEPES, pH 7.2, 50 μm MeCbi, and 300 μm Ti(III)citrate were placed in a screw cap cuvette; the ACS incubation mixture (see ACS reconstitution) was then added to yield a final concentration of 2 μm ACS. After closing the cap, CO-saturated buffer (10% of the final volume) was injected with a syringe and the reaction was started by adding 200 μm CoA. All measurements were performed in darkness.
Isothermal Titration Calorimetry
ITC experiments were performed inside a glove box (LABstar, MBRAUN) with a MicroCal VP-ITC system (GE Healthcare) at 25 °C in 50 mm Tris, 100 mm NaCl. Three different experiments were performed: (a) 15 μm AcsFCh were titrated with 300 μm NiCl2, (b) 24.7 μm apoACS were titrated with 750 μm NiCl2, and (c) 24.4 μm ACS-AcsFCh complex were titrated with 750 μm NiCl2. The concentrations of apoACS and ACS-AcsFCh complex were normalized to the amount of iron, which was determined after ITC measurements by ICP-OES. The enzyme was present in the reaction cell (1.4 ml) and NiCl2 was in the syringe. The initial injection was 3 μl (lasting 6 s) and the 47 subsequent injections were 6 μl (lasting 12 s). There was an interval of 300 s between injections. The solution in the reaction cell was stirred at 307 rpm.
Removal of Excess Metal from ITC Samples
When the ITC experiment was finished, samples b and c were used for further experiments, i.e. activity measurements and metal analysis. The protein samples were concentrated to ∼300 μl and the excess metal was removed using a PD MiniTrap-G25 column. Only colored fractions were collected. To ensure that all excess metal was removed a buffer sample was prepared for metal analysis: 130 μm NiCl2 were added to the buffer and the sample was treated exactly the same as the protein samples.
Metal Analysis
Metal contents were analyzed using a PerkinElmer Optima 2100 DV ICP-OES spectrometer. Samples included two different purifications of apoACS and ITC, samples b and c, which were treated as described above. 500-μl protein samples were wet-washed overnight in a 1:1 mixture with 65% nitric acid (Suprapur, Merck, Darmstadt, Germany) at 100 °C. The samples were diluted with 4 ml of H2O prior to their injection into the ICP-OES. The multielement standard solution XVI (Merck) was used as a reference.
Author Contributions
C. M. G., S. G., J. H. J., and H. D. designed research; C. M. G. and S. G. performed research; C. M. G., J. H. J., and H. D. analyzed and interpreted data; and C. M. G. and H. D. wrote the paper.
Acknowledgments
We thank the group of Silke Leimkühler, especially Jasmin Kurtzke, for performing the metal analysis. We thank members of our group for helpful discussions.
The work was supported by Deutsche Forschungsgemeinschaft Project Grant DO 785/6-1. The authors declare that they have no conflicts of interest with the contents of this article.
- ACS
- acetyl-CoA synthase
- CODH
- carbon monoxide dehydrogenase
- SEC
- size exclusion chromatography
- ITC
- isothermal titration calorimetry
- ICP-OES
- inductively coupled plasma optic emission spectrometry
- TEV
- tobacco etch virus
- PIPE
- polymerase incomplete primer extension.
References
- 1. Waldron K. J., and Robinson N. J. (2009) How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 7, 25–35 [DOI] [PubMed] [Google Scholar]
- 2. Ribbe M. W., Hu Y., Hodgson K. O., and Hedman B. (2014) Biosynthesis of nitrogenase metalloclusters. Chem. Rev. 114, 4063–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu Y., and Ribbe M. W. (2013) Nitrogenase assembly. Biochim. Biophys. Acta 1827, 1112–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frazzon J., and Dean D. R. (2002) Biosynthesis of the nitrogenase iron-molybdenum-cofactor from Azotobacter vinelandii. Met. Ions Biol. Syst. 39, 163–186 [PubMed] [Google Scholar]
- 5. Netz D. J., Mascarenhas J., Stehling O., Pierik A. J., and Lill R. (2014) Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol. 24, 303–312 [DOI] [PubMed] [Google Scholar]
- 6. Zamble D. B. (2015) Nickel in biology. Metallomics 7, 588–589 [DOI] [PubMed] [Google Scholar]
- 7. Watanabe S., Sasaki D., Tominaga T., and Miki K. (2012) Structural basis of [NiFe] hydrogenase maturation by Hyp proteins. Biol. Chem. 393, 1089–1100 [DOI] [PubMed] [Google Scholar]
- 8. Farrugia M. A., Macomber L., and Hausinger R. P. (2013) Biosynthesis of the urease metallocenter. J. Biol. Chem. 288, 13178–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boer J. L., Mulrooney S. B., and Hausinger R. P. (2014) Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 544, 142–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leach M. R., and Zamble D. B. (2007) Metallocenter assembly of the hydrogenase enzymes. Curr. Opin. Chem. Biol. 11, 159–165 [DOI] [PubMed] [Google Scholar]
- 11. Appel A. M., Bercaw J. E., Bocarsly A. B., Dobbek H., DuBois D. L., Dupuis M., Ferry J. G., Fujita E., Hille R., Kenis P. J., Kerfeld C. A., Morris R. H., Peden C. H., Portis A. R., et al. (2013) Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bender G., Pierce E., Hill J. A., Darty J. E., and Ragsdale S. W. (2011) Metal centers in the anaerobic microbial metabolism of CO and CO2. Metallomics 3, 797–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Can M., Armstrong F. A., and Ragsdale S. W. (2014) Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase. Chem. Rev. 114, 4149–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thauer R. K. (1998) Biochemistry of methanogenesis: a tribute to Marjory Stephenson. 1998 Marjory Stephenson Prize Lecture. Microbiology 144, 2377–2406 [DOI] [PubMed] [Google Scholar]
- 15. Svetlitchnyi V., Dobbek H., Meyer-Klaucke W., Meins T., Thiele B., Römer P., Huber R., and Meyer O. (2004) A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc. Natl. Acad. Sci. U.S.A. 101, 446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeoung J. H., Fesseler J., Goetzl S., and Dobbek H. (2014) Carbon monoxide. Toxic gas and fuel for anaerobes and aerobes: carbon monoxide dehydrogenases. Met. Ions Life Sci. 14, 37–69 [DOI] [PubMed] [Google Scholar]
- 17. Drennan C. L., Heo J., Sintchak M. D., Schreiter E., and Ludden P. W. (2001) Life on carbon monoxide: x-ray structure of Rhodospirillum rubrum Ni-Fe-S carbon monoxide dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 98, 11973–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dobbek H., Svetlitchnyi V., Gremer L., Huber R., and Meyer O. (2001) Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science 293, 1281–1285 [DOI] [PubMed] [Google Scholar]
- 19. Jeoung J. H., and Dobbek H. (2007) Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 318, 1461–1464 [DOI] [PubMed] [Google Scholar]
- 20. Doukov T. I., Iverson T. M., Seravalli J., Ragsdale S. W., and Drennan C. L. (2002) A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science 298, 567–572 [DOI] [PubMed] [Google Scholar]
- 21. Darnault C., Volbeda A., Kim E. J., Legrand P., Vernède X., Lindahl P. A., and Fontecilla-Camps J. C. (2003) Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat. Struct. Biol. 10, 271–279 [DOI] [PubMed] [Google Scholar]
- 22. Shin W., and Lindahl P. A. (1992) Function and CO binding properties of the NiFe complex in carbon monoxide dehydrogenase from Clostridium thermoaceticum. Biochemistry 31, 12870–12875 [DOI] [PubMed] [Google Scholar]
- 23. Gencic S., and Grahame D. A. (2003) Nickel in subunit β of the acetyl-CoA decarbonylase/synthase multienzyme complex in methanogens: catalytic properties and evidence for a binuclear Ni-nickel site. J. Biol. Chem. 278, 6101–6110 [DOI] [PubMed] [Google Scholar]
- 24. Seravalli J., Xiao Y., Gu W., Cramer S. P., Antholine W. E., Krymov V., Gerfen G. J., and Ragsdale S. W. (2004) Evidence that NiNi acetyl-CoA synthase is active and that the CuNi enzyme is not. Biochemistry 43, 3944–3955 [DOI] [PubMed] [Google Scholar]
- 25. Kerby R. L., Ludden P. W., and Roberts G. P. (1997) In vivo nickel insertion into the carbon monoxide dehydrogenase of Rhodospirillum rubrum: molecular and physiological characterization of cooCTJ. J. Bacteriol. 179, 2259–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jeon W. B., Cheng J., and Ludden P. W. (2001) Purification and characterization of membrane-associated CooC protein and its functional role in the insertion of nickel into carbon monoxide dehydrogenase from Rhodospirillum rubrum. J. Biol. Chem. 276, 38602–38609 [DOI] [PubMed] [Google Scholar]
- 27. Inoue T., Takao K., Fukuyama Y., Yoshida T., and Sako Y. (2014) Over-expression of carbon monoxide dehydrogenase-I with an accessory protein co-expression: a key enzyme for carbon dioxide reduction. Biosci. Biotechnol. Biochem. 78, 582–587 [DOI] [PubMed] [Google Scholar]
- 28. Hadj-Saïd J., Pandelia M. E., Léger C., Fourmond V., and Dementin S. (2015) The carbon monoxide dehydrogenase from Desulfovibrio vulgaris. Biochim. Biophys. Acta 1847, 1574–1583 [DOI] [PubMed] [Google Scholar]
- 29. Leipe D. D., Wolf Y. I., Koonin E. V., and Aravind L. (2002) Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 317, 41–72 [DOI] [PubMed] [Google Scholar]
- 30. Bange G., and Sinning I. (2013) SIMIBI twins in protein targeting and localization. Nat. Struct. Mol. Biol. 20, 776–780 [DOI] [PubMed] [Google Scholar]
- 31. Jeoung J. H., Giese T., Grünwald M., and Dobbek H. (2009) CooC1 from Carboxydothermus hydrogenoformans is a nickel-binding ATPase. Biochemistry 48, 11505–11513 [DOI] [PubMed] [Google Scholar]
- 32. Jeoung J. H., Giese T., Grünwald M., and Dobbek H. (2010) Crystal structure of the ATP-dependent maturation factor of Ni,Fe-containing carbon monoxide dehydrogenases. J. Mol. Biol. 396, 1165–1179 [DOI] [PubMed] [Google Scholar]
- 33. Loke H. K., and Lindahl P. A. (2003) Identification and preliminary characterization of AcsF, a putative Ni-insertase used in the biosynthesis of acetyl-CoA synthase from Clostridium thermoaceticum. J. Inorg. Biochem. 93, 33–40 [DOI] [PubMed] [Google Scholar]
- 34. Wu M., Ren Q., Durkin A. S., Daugherty S. C., Brinkac L. M., Dodson R. J., Madupu R., Sullivan S. A., Kolonay J. F., Haft D. H., Nelson W. C., Tallon L. J., Jones K. M., Ulrich L. E., Gonzalez J. M., Zhulin I. B., Robb F. T., and Eisen J. A. (2005) Life in hot carbon monoxide: the complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 1, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Apweiler R., Attwood T. K., Bairoch A., Bateman A., Birney E., Biswas M., Bucher P., Cerutti L., Corpet F., Croning M. D., Durbin R., Falquet L., Fleischmann W., Gouzy J., Hermjakob H., et al. (2001) The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29, 37–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mitchell A., Chang H. Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., Sangrador-Vegas A., Scheremetjew M., Rato C., Yong S. Y., Bateman A., et al. (2015) The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43, D213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Denny J. A., and Darensbourg M. Y. (2015) Metallodithiolates as ligands in coordination, bioinorganic, and organometallic chemistry. Chem. Rev. 115, 5248–5273 [DOI] [PubMed] [Google Scholar]
- 38. Boer J. L., Quiroz-Valenzuela S., Anderson K. L., and Hausinger R. P. (2010) Mutagenesis of Klebsiella aerogenes UreG to probe nickel binding and interactions with other urease-related proteins. Biochemistry 49, 5859–5869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soriano A., and Hausinger R. P. (1999) GTP-dependent activation of urease apoprotein in complex with the UreD, UreF, and UreG accessory proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 11140–11144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fong Y. H., Wong H. C., Yuen M. H., Lau P. H., Chen Y. W., and Wong K. B. (2013) Structure of UreG/UreF/UreH complex reveals how urease accessory proteins facilitate maturation of Helicobacter pylori urease. PLos Biol. 11, e1001678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaluarachchi H., Chan Chung K. C., and Zamble D. B. (2010) Microbial nickel proteins. Nat. Prod. Rep. 27, 681–694 [DOI] [PubMed] [Google Scholar]
- 42. Chung K. C., and Zamble D. B. (2011) The Escherichia coli metal-binding chaperone SlyD interacts with the large subunit of [NiFe]-hydrogenase 3. FEBS Lett. 585, 291–294 [DOI] [PubMed] [Google Scholar]
- 43. Netz D. J., Pierik A. J., Stümpfig M., Mühlenhoff U., and Lill R. (2007) The Cfd1-Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nat. Chem. Biol. 3, 278–286 [DOI] [PubMed] [Google Scholar]
- 44. Hu Y., Corbett M. C., Fay A. W., Webber J. A., Hodgson K. O., Hedman B., and Ribbe M. W. (2006) Nitrogenase Fe protein: a molybdate/homocitrate insertase. Proc. Natl. Acad. Sci. U.S.A. 103, 17125–17130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Netz D. J., Pierik A. J., Stümpfig M., Bill E., Sharma A. K., Pallesen L. J., Walden W. E., and Lill R. (2012) A bridging [4Fe-4S] cluster and nucleotide binding are essential for function of the Cfd1-Nbp35 complex as a scaffold in iron-sulfur protein maturation. J. Biol. Chem. 287, 12365–12378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loke H. K., Tan X., and Lindahl P. A. (2002) Genetic construction of truncated and chimeric metalloproteins derived from the α subunit of acetyl-CoA synthase from Clostridium thermoaceticum. J. Am. Chem. Soc. 124, 8667–8672 [DOI] [PubMed] [Google Scholar]
- 47. George S. J., Seravalli J., and Ragsdale S. W. (2005) EPR and infrared spectroscopic evidence that a kinetically competent paramagnetic intermediate is formed when acetyl-coenzyme A synthase reacts with CO. J. Am. Chem. Soc. 127, 13500–13501 [DOI] [PubMed] [Google Scholar]
- 48. Gencic S., Duin E. C., and Grahame D. A. (2010) Tight coupling of partial reactions in the acetyl-CoA decarbonylase/synthase (ACDS) multienzyme complex from Methanosarcina thermophila: acetyl C-C bond fragmentation at the a cluster promoted by protein conformational changes. J. Biol. Chem. 285, 15450–15463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Katoh K., and Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Felsenstein J. (1989) PHYLIP: phylogeny inference package (version 3.2). Cladistics 5, 164–166 [Google Scholar]
- 51. Demchak B., Hull T., Reich M., Liefeld T., Smoot M., Ideker T., and Mesirov J. P. (2014) Cytoscape: the network visualization tool for GenomeSpace workflows. F1000Res 3, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zehnder A. J., and Wuhrmann K. (1976) Titanium(III) citrate as a nontoxic oxidation-reduction buffering system for the culture of obligate anaerobes. Science 194, 1165–1166 [DOI] [PubMed] [Google Scholar]
- 53. Zou X., Evans D. R., and Brown K. L. (1995) Efficient and convenient method for axial nucleotide removal from vitamin B12 and its derivatives. Inorg. Chem. 34, 1634–1635 [Google Scholar]
- 54. Klock H. E., and Lesley S. A. (2009) The polymerase incomplete primer extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 498, 91–103 [DOI] [PubMed] [Google Scholar]
- 55. Klock H. E., Koesema E. J., Knuth M. W., and Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins Struct. Funct. Genet. 71, 982–994 [DOI] [PubMed] [Google Scholar]
- 56. Lindsley J. E. (2001) Use of a real-time, coupled assay to measure the ATPase activity of DNA topoisomerase II. Methods Mol. Biol. 95, 57–64 [DOI] [PubMed] [Google Scholar]
- 57. Lanzetta P. A., Alvarez L. J., Reinach P. S., and Candia O. A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95–97 [DOI] [PubMed] [Google Scholar]
- 58. Robertson W. D., Bovell A. M., and Warncke K. (2013) Cobinamide production of hydrogen in a homogeneous aqueous photochemical system, and assembly and photoreduction in a (βα)8 protein. J. Biol. Inorg. Chem. 18, 701–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Su G., Morris J. H., Demchak B., and Bader G. D. (2014) Biological network exploration with cytoscape 3. Curr. Protoc. Bioinformatics 47, 8.13.1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]