

Abstract
The targeted assembly of antibody products upon antigen binding represents a novel strategy for the reconstitution of potent therapeutic activity at the site of disease, sparing healthy tissues. We demonstrate that interleukin-12, a heterodimeric pro-inflammatory cytokine consisting of the disulfide-linked p40 and p35 subunits, can be reconstituted by sequential reassembly of fusion proteins based on antibody fragments and interleukin-12 subunit mutants. Analysis of the immunostimulatory properties of interleukin-12 and its derivatives surprisingly revealed that the mutated p35 subunit partially retained the activity of the parental cytokine, whereas the p40 subunit alone was not able to stimulate T cells or natural killer cells. The concept of stepwise antibody-based reassembly of split cytokines could be useful for the development of other anticancer therapeutics with improved safety and tolerability.
Keywords: antibody, cancer, cytokine, immunotherapy, pharmacology, activity-on-demand, tumor targeting, split protein, activity reconstitution, interleukin-12
Introduction
Many pharmaceutical agents, including anticancer drugs, cause undesired toxicities to normal tissues, which prevent dose escalation to therapeutically active regimens. Paul Ehrlich's dream of a magic bullet (“Zauberkugel”), a molecule that would display toxic activity only upon binding to a target cell of interest, has to some extent been realized by the development of monoclonal antibodies directed against cell surface antigens, as these therapeutic products may preferentially redirect the cytotoxic activity of certain leukocytes (e.g. natural killer cells) to antibody-coated cells. Although certain therapeutic antibodies specific to leukemia and lymphoma antigens have demonstrated a potential to induce cancer cures in mice and humans (1–3), this pharmaceutical strategy appears to be less efficient for solid tumors and rarely leads to complete responses (4–6). To improve the therapeutic activity of anticancer monoclonal antibodies, various types of antibody derivatives have been developed, including antibody-drug conjugates, bispecific antibodies, and antibody-cytokine fusion proteins (7). However, all of these classes of “armed” antibody therapeutics lead to substantial toxicities upon systemic administration (8–10).
In principle, an ideal biopharmaceutical agent would display no toxicity when injected into the bloodstream, but should regain potent therapeutic activity upon binding to its antigen expressed on target cells. The targeted reassembly of a bioactive cytokine payload from two split cytokine constituents represents a new immunotherapeutic strategy, which could potentially accomplish this goal. The reconstitution of protein activity from two fragments has been demonstrated in vitro for a variety of proteins. For example, the enzyme ribonuclease H or green fluorescent proteins have been engineered into split proteins (11, 12), and experimental techniques that rely on protein fragment complementation have become useful tools for the study of protein interactions (13, 14). The concept of reconstituting therapeutic activity at the site of disease (“activity on demand”) has been pursued with various experimental implementations, ranging from the antigen-restricted complementation of a tri-specific antibody to the tetrazine-triggered release of drug payloads from antibody-drug conjugates (15, 16). The practical implementation of these strategies may be hindered by the fact that the reconstitution of activity at the site of disease requires relatively high local concentrations of the interaction (or reaction) partners.
Pro-inflammatory cytokines are potent activators of the immune system that have been considered for cancer therapy. Indeed, recombinant versions of interleukin-2 (IL2), interferon-α (IFNα), and tumor necrosis factor (TNF) have received marketing authorization for the treatment of certain types of malignancies (17). However, already at low doses, these biopharmaceuticals can cause systemic activation of immune cells or the endothelium, leading to cytokine release and vascular leakage. These off-target effects are clinically manifested by flu-like symptoms and hypotension, which often prevent the escalation to therapeutically active doses (18). Recombinant IL2 can be administered at doses up to 800 million IU for only 1–2 weeks and only to young cancer patients, who are in a relatively good state of health. The treatment still leads to substantial systemic side effects but also to long-term remissions in ∼10% of patients with renal cell carcinoma or with metastatic melanoma (19–21). Similarly, the recommended doses for systemic administration of IFNα and TNF are typically lower than 1 mg due to dose-limiting toxicities (18, 22).
Interleukin-12 (IL12), one of the most active cytokine products described so far, could be administered to cancer patients only at a dose of 500 ng/kg twice weekly (23). At this dose, IL12 exhibits only modest antitumor activity and may cause strong side effects primarily related to systemic IFNγ release, which have even led to fatal toxicities in a Phase II clinical trial (24). For these reasons, clinical investigations of IL12-based biopharmaceuticals have been largely abandoned until now despite promising preclinical data.
To improve the therapeutic index of cytokines for cancer therapy, tumor-homing antibody-cytokine fusion proteins (also termed “immunocytokines”) have been investigated. Studies on syngeneic mouse models of different types of solid tumors and hematological malignancies could demonstrate that immunocytokines can indeed substantially increase the therapeutic index of the corresponding cytokine payloads (25). For the most promising products, the gain in therapeutic activity could be attributed to a preferential localization at the site of disease (26–28). However, immunocytokines normally retain full cytokine activity in vitro, and cytokine-induced toxicity is mainly associated with peak concentrations of the corresponding cytokine-based pharmaceutical, which are reached shortly upon intravenous injection (8). Immunocytokines therefore often display similar side effect profiles as the parental cytokines.
An ideal immunocytokine product would display potent activity only at the site of disease, but not when the protein circulates in blood. In this study, we propose a new strategy for the development of antibody-cytokine fusions with activity on demand, based on the stepwise reassembly of cytokine subunits at the site of disease. Interleukin-12 lends itself for this purpose because of its heterodimeric architecture (29). Human IL12 exhibits structural similarity to a class I cytokine-receptor complex. The p35 subunit is homologous to IL6, whereas p40 exhibits homology to the IL6 receptor α (29). However, the functional contribution of the individual subunits to IL12 activity has so far been elusive because p35 is only secreted when co-expressed with p40 (30).
Fusion proteins based on bivalent antibody fragments of the F8 antibody, with specificity to the alternatively spliced extra-domain A (EDA)3 domain of fibronectin, can efficiently accumulate on the subendothelial extracellular matrix of the tumor neovasculature, while being rapidly cleared from circulation (31). The selective antibody uptake in different tissues or blood can be expressed as the percentage of injected dose per gram (%ID/g) at various time points. Earlier studies on mouse IL12-based antibody fusion proteins had revealed a preferential accumulation at the tumor site (32). Additionally, certain IL12-based immunocytokines, targeting splice isoforms of fibronectin, are potently active against aggressive mouse models of cancer (26, 32–34).
Within these studies, we have shown that fusion proteins, consisting of antibodies fused to mutants of the p40 and p35 subunits of mouse IL12, could selectively reassemble after product binding to the target antigen in vitro. Furthermore, these split cytokine fusion proteins were able to preferentially localize to the tumor site. Unexpectedly, we observed that antibody fusions of p35S partially retained the ability to activate CD4+ T cells, CD8+ T cells, and natural killer cells.
Results
Pharmaceutical Strategy
The hypothetical graph in Fig. 1 depicts the general pharmaceutical strategy for the antibody-based stepwise reassembly of a split cytokine at the site of disease, exemplified with heterodimeric IL12. In a first step, a bivalent p35S-based antibody fusion protein (F8-p35S-F8) is injected and allowed to selectively bind to its antigen in vivo. Once a suitable selectivity has been achieved, a second injection of a fusion protein based on the complementary p40S subunit could lead to the reconstitution of cytokine activity at the site of disease. The order and timing of the injections could be chosen on the basis of the pharmacokinetic profiles of the individual split protein fusions, to minimize reassembly in the bloodstream.
FIGURE 1.
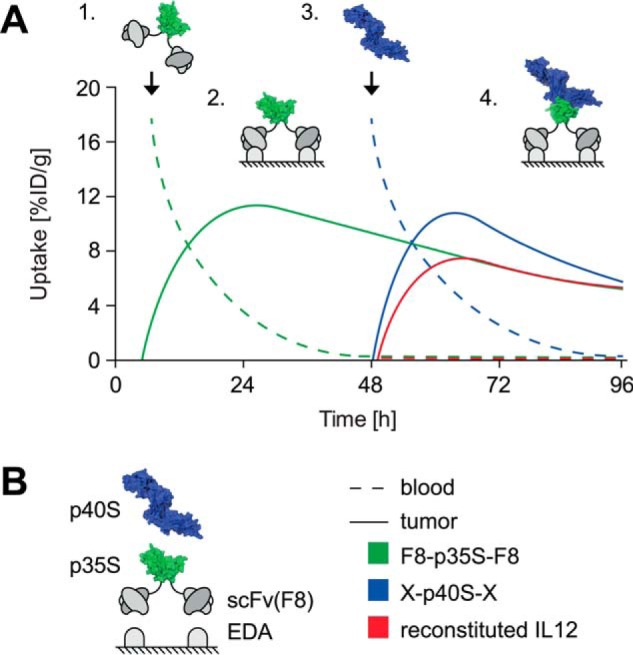
A, hypothetical graph illustrating the concept of site-specific reconstitution of non-covalent IL12 split cytokine complexes within a tumor. Step 1, administration of F8-p35S-F8 followed by clearance from blood. Step 2, F8-p35S-F8 binds to its target EDA and accumulates at the tumor neovasculature. Step 3, administration of p40S in a suitable antibody format (X = e.g. scFv(F8) or Fc). Step 4, reconstitution of active IL12 at site of disease. The time between injections (Steps 1 and 3) could be chosen to prevent systemic reassembly p35S and p40S. [%ID/g], injected dose per gram. B, explanation of schematic representation (structures based human IL12, Protein Data Bank (PDB) ID 1F45) and curve legend.
Protein Production and Characterization
The p40 subunit of IL12 is known to be secreted independently of p35 in mice (35) and patients during inflammatory conditions (36, 37) and has been produced recombinantly as mixtures of monomers and dimers (38). Homodimeric p40 is covalently linked via a disulfide bond and has the ability to antagonize IL12 by competing for IL12 receptor binding. We sought to produce monomeric p40 by mutating the cysteine at position 197 by a C197S substitution (p40S). Likewise, a C92S substitution was performed for p35 (p35S). Previous mutational studies have revealed that the intermolecular disulfide bond is not needed to form active IL12 heterodimers (29). We found that the same is also applicable to mutated murine IL12 subunits, which have an amino acid sequence homology of 60% in the case of p35 and 70% in the case of p40 when compared with their human homologs (39). Although this initial test could be performed with His-tagged IL12 mutant subunits, low expression yields prevented further analysis. The genetic fusion to single-chain variable fragments (scFv) greatly enhanced the expression of soluble homogenous monomeric p40S preparations in CHO-S cells (Fig. 2). The mass differences observed by electrospray ionization/liquid chromatography-mass spectroscopy (ESI/LC-MS) indicate the presence of different glycoforms. Earlier studies on recombinant IL12 stated that an N-terminal antibody fusion to the p40 subunit may interfere with the biological activity of the IL12 payload (32, 40). To further investigate this aspect, fusion proteins with p40S at the N- or C-terminal extremity of scFv antibody fragments (KSF-p40S and p40S-KSF) were produced and purified to homogeneity (supplemental Fig. 1).
FIGURE 2.

A, short name, schematic representation (green, p35S; blue, p40S), and mass based on amino acid sequence. B, analytical SEC. C, SDS-PAGE. M, marker; NR, non-reducing conditions; R, reducing conditions. Blue and red numbers indicate apparent molecular mass in kDa. D, LC/ESI mass spectra.
In contrast to p40S, the expression and isolation of the mutated p35S subunit from eukaryotic cells presented a major challenge that has not been reported in the literature before. The biosynthesis and secretion of p35 are known to rely on the intracellular interaction with p40 and are most efficient in the presence of an excess of p40 (30). We therefore investigated whether it would be possible to express mutated p35S by transient gene expression in a stable CHO cell line previously transfected with p40. The p40 variant used for this expression strategy did not feature a purification tag and only served as a “chaperone” for the expression of p35S. Although His-tagged p35S or fusion proteins of wild-type p35 could only be isolated as aggregates, the fusion of the p35S mutant to antibody fragments greatly improved solubility and production yields. The sequential arrangement of two scFv(F8) moieties to both termini of p35S resulted in a highly stable F8-p35S-F8 fusion protein, with a suitable biochemical quality (Fig. 2) for protein reassembly experiments and in vivo studies.
To evaluate the targeting performance of purified IL12 subunit derivatives, quantitative biodistribution experiments were performed. A bivalent scFv(F8)-based fusion protein format was chosen for both IL12-derived subunit fusion proteins. The ability of F8-p35S-F8 and F8-p40S-F8 to bind with high affinity to the cognate EDA domain of fibronectin was confirmed by surface plasmon resonance (SPR) (Fig. 3A), while tumor targeting properties were evaluated by quantitative biodistribution analysis in mice bearing murine F9 teratocarcinomas (Fig. 3B). A preferential tumor uptake for F8-p35S-F8 as well as for F8-p40S-F8 was observed 24 h after intravenous administration, with average tumor-to-blood ratios of 6.7 and 8.4, respectively.
FIGURE 3.

A, experimental setup (green, p35S; blue, p40S) and corresponding EDA binding sensorgrams of bivalent F8-p35S-F8 and F8-p40S-F8 subunit fusion protein preparations tested in vivo. RU, resonance units. B, quantitative biodistribution profiles 24 h after injection of 10–12 μg of 125I-labeled protein samples into immunocompetent F9 tumor-bearing Sv129 mice (five mice per group). For F8-p40S-F8, an outlier (data point with asterisk) was excluded from the analysis because the dissected tumor was very small (10 mg) and poorly vascularized. [%ID/g], injected dose per gram. Error bars indicate ± S.D.
In Vitro Reconstitution of IL12
Different scFv antibody fragments, namely F8 (specific to EDA) and KSF (irrelevant specificity), were chosen as fusion partners for p35S and p40S, respectively, to test the in vitro reconstitution of heterodimeric IL12 in an SPR-based ligand capture experiment (Fig. 4A). In a first step, F8-p35S-F8 was immobilized by binding to the cognate antigen (an EDA-containing recombinant fragment of fibronectin) on an SPR sensor chip (Fig. 4B). A subsequent injection of the p40S-KSF or KSF-p40S fusion proteins led to the association with p35S and thus the formation of non-covalent heterodimeric IL12 moieties. The dissociation constants (KD) for the resulting heterodimers, calculated by fitting the sensorgrams of Fig. 4C, were 21.9 nm for p40S-KSF and 27.3 nm for KSF-p40S, respectively. Importantly, a trimeric scFv(KSF) fusion protein of TNF, which served as a negative control, did not exhibit detectable SPR binding, confirming that the assembly process relied on the interaction between the p35S and p40S subunits (Fig. 4B).
FIGURE 4.

A, schematic representation of the experimental setup (green, p35S; blue, p40S). B, SPR ligand capture experiment showing the p40S-to-p35S subunit interaction in a 1-to-1 binding mode. Step 1, immobilization of F8-p35S-F8 on EDA-coated chip. Step 2, injection of p40S-KSF/KSF-p40S or KSF-TNF (negative control). RU, resonance units. C, kinetic data after blank subtraction and curve fitting (black curves). Experiments were analyzed in triplicates.
Next, we assessed the biological activity of the reconstituted IL12 complexes in vitro. Using freshly isolated mouse splenocytes, equimolar mixtures of F8-p35S-F8 and KSF-p40S or p40S-KSF were able to induce IFNγ production in these cells, as shown in Fig. 5A. All fusion proteins had endotoxin levels below 0.5 endotoxin units/ml. The half-maximal effective concentrations (EC50) for the reconstituted IL12 moieties were 10–100 times lower when compared with wild-type IL12. Unexpectedly, we observed that fusion proteins containing only the p35S subunit retained the ability to induce IFNγ production, when tested as a single agent. By contrast, neither KSF-p40S nor p40S-KSF alone induced measurable IFNγ levels. To gain additional evidence for the single agent activity of F8-p35S-F8, phosphorylation of the signal transducer and activator of transcription 4 (STAT4 or pSTAT4) was assessed. Again, we compared in vitro reconstituted IL12 split cytokine mixtures with the individual mutant subunit fusion proteins. Maximal pSTAT4 levels were reached 20–25 min after stimulation with functional IL12 derivatives. Phosphorylation of STAT4 was measured in natural killer cells, as well as in activated CD4+ and CD8+ T cells (Fig. 5B). The single-agent activity of F8-p35S-F8 observed in the IFNγ release assay was confirmed in all analyzed cell populations. The data indicate that F8-p35S-F8 alone is able to bind to the murine IL12 receptor β2 (IL12Rβ2), which is required for IL12-induced STAT4 phosphorylation (41). Flow cytometry histograms are given in supplemental Fig. 2.
FIGURE 5.

A, splenocyte/IFNγ release assay comparing the biological activity of mouse IL12-derived split cytokine derivatives alone or in combination 48 h after sample addition. B, comparative pSTAT4 activation assay revealing the signaling capacity of IL12-derived split cytokine derivatives 25 min after sample addition. The sample mixtures were premixed in a molar 1-to-1 ratio 30 min before the start of the respective assay. Mean fluorescence intensities (MFI) are displayed. Experiments were analyzed in triplicates. Error bars indicate ± S.D.
Discussion
Antibody-cytokine fusion proteins represent a promising class of biopharmaceutical agents (42), whose clinical potential is limited by toxicity associated with the cytokine payload. Strategies aimed at the reconstitution of cytokine activity at the site of disease may facilitate the development of better tolerated immunomodulatory products. In this study, we have shown that the reconstitution of a heterodimeric non-covalent IL12 complex can be achieved in vitro, using mutant subunit fusion proteins. Mutation of critical cysteine residues, in the p40-p35 interface, allowed the production of split cytokine fusion proteins, which could be purified to homogeneity, eluted as a single peak in gel filtration chromatography, and retained full antigen binding activity. Notably, fusion proteins containing wild-type p35 expressed poorly and could not be produced in a non-aggregated form, whereas C92S-substituted p35S allowed the isolation of monomeric F8-p35S-F8. Both split cytokine fusion protein products were able to preferentially accumulate at the tumor site, when studied by quantitative biodistribution analysis in mice bearing F9 teratocarcinomas.
Although the reassembly of IL12 subunits was successfully demonstrated in vitro, we discovered that the p35 moiety retained a substantial portion of IL12 activity when used as a single agent. F8-p35S-F8 was the only split immunocytokine with the ability to induce IFNγ release in splenocytes. The short stimulation time (25 min) for the STAT4 phosphorylation assay indicates a direct STAT4-signaling activity of F8-p35S-F8. Our data and previous studies on mouse IL12 receptor knock-out mice (41) suggest that F8-p35S-F8 can bind to IL12Rβ2, triggering STAT4 phosphorylation. From a structural viewpoint, IL12 resembles a soluble cytokine-receptor complex (29), in which signaling events are mainly mediated by the p35 subunit. Although p40 contributes to biological activity, the p40 subunit alone does not trigger STAT4 signaling (43, 44).
For targeted activity on demand applications, the individual split cytokine fusion proteins should be less active than the reconstituted cytokine complex. Consequently, IL12 may not be ideally suited for split cytokine reassembly in vivo. However, the strategy outlined in this study could be applicable to other cytokines or bioactive proteins consisting of more than one protein domain.
A number of other cytokine-based pharmaceutical strategies, which are in principle also applicable to p35S, could be considered to achieve conditional activity at the tumor site. Inactive cytokine precursors relying on proteolytic processing at the site of disease for activation could be used as immunocytokine payloads. This strategy has been successfully implemented for monoclonal antibodies, leading to an improved therapeutic index in mice (45). Alternatively, the receptor binding sites of cytokines may be masked by inhibitory cytokine-directed antibodies, thus delaying biological activity in vivo. Masking specific cytokine epitopes has also been used to direct cytokine activity toward certain immune cell subtypes, e.g. CD8+ or regulatory T cells in the case of IL2 (46). Another recently described approach employs antibody-IL2 fusion proteins, in which the biological activity of the cytokine moiety is modulated by antibody-to-antigen binding. This “allosteric” modulation can be explained by the hinge movement of the Fab arms of the antibody upon antigen engagement and by strategic positioning of the IL2 moiety at the C-terminal end of the light chain (47).
The discovery that p35S retains partial IL12 activity may also be advantageous for IL12-based pharmacodelivery applications, considering the exceptionally high biological activity of this cytokine. Recent studies on IFNα-based immunocytokines have revealed that attenuated receptor binding affinities can result in improved target cell selectivity (48), which may reduce off-target toxicity in vivo. In the future, immunocytokines with reduced biological activities may facilitate the administration of antibody products at optimal doses for selective in vivo pharmacodelivery applications.
Experimental Procedures
Cloning and Site-specific Mutagenesis
Recombinant genes of mouse IL12 (GenBankTM Accession Numbers: M86672.1 and M86671.1) subunit-antibody fusion proteins were created by PCR assembly as described before (49). Briefly, scFv fragments of the previously published human F8 and KSF antibodies were genetically fused to either the C-terminal and/or the N-terminal end of the p35 or p40 subunits of IL12 via a flexible (Gly4Ser)3 amino acid linker. The assembled fusion protein genes were cloned into a pcDNA3.1(+) vector (Thermo Scientific) using restriction endonucleases (New England Biolabs). Site-directed mutagenesis of the cysteines, normally involved in the intermolecular disulfide bond between p35 and p40, was accomplished using the method described in Ref. 50 with the help of Phusion DNA polymerase and the restriction enzyme DpnI (both from New England Biolabs). Homologous recombination and production of the mutated plasmids were achieved in chemically competent Escherichia coli TOP10 cells (Thermo Scientific). Specifically, a C92S substitution in the case of the p35 subunit and a C197S substitution for p40 (numbering based on the UniProt database) were introduced. For simplicity, the resulting split cytokine fusion proteins scFv(F8)-p35:C92S-scFv(F8), scFv(KSF)-p40:C197S, p40:C197S-scFv(KSF), and scFv(F8)-p40:C197S-scFv(F8) are herein denoted as F8-p35S-F8, KSF-p40S, p40S-KSF, and F8-p40S-F8, respectively. Likewise, the scFv(F8)-IL12-scFv(F8) fusion protein, containing the previously described single-chain mouse IL12 variant (40), is denoted as F8-IL12-F8. Recombinant wild-type mouse interleukin-12 is abbreviated as IL12.
Protein Production
IL12 antibody fusion proteins derived from mutated p40 and single-chain IL12 were produced by transient gene expression in CHO-S cells (Thermo Scientific) and purified by protein A affinity chromatography as described in Ref. 49. By contrast, the p35 subunit is not secreted in the absence of p40, and transient gene expression could therefore only be achieved in a stable cell line constitutively expressing p40 (wild-type variant without purification tag). Subsequent protein A affinity chromatography allowed complete removal of p40 and thus the isolation of F8-p35S-F8. Protein aggregates were removed by preparative size-exclusion chromatography (SEC) using an ÄKTA purifier FPLC system and a Superdex S200 column (GE Healthcare) according to the manufacturer's instructions.
Protein Characterization
Analytical SEC of purified samples (10–20 μg) was run on the same ÄKTA system and SEC column as the preparative SEC runs. Protein samples (2–4 μg) were analyzed by SDS-PAGE using the NuPAGE® Novex® Gel System (Thermo Scientific) according to the manufacturer's instructions. PageRulerTM Plus Prestained Protein Ladder (Thermo Scientific) was used as a marker. The identity of fusion proteins was confirmed by ESI/LC-MS with an ACQUITY UPLC H class system equipped with an ACQUITY BEH300 C4 column (2.1 × 50 mm, 1.7-μm particle size) sequentially coupled to a Waters Xevo G2-XS QTof ESI mass analyzer. Endotoxin levels were quantified with the PierceTM LAL Chromogenic Endotoxin Quantitation Kit (Thermo Scientific).
Binding Kinetics
Antigen binding of the F8-p35S-F8 and F8-p40S-F8 preparations tested in vivo was analyzed by SPR with a Biacore S200 using a CM5 sensor chip (GE Healthcare) initially coated with 960 resonance units of antigen (an EDA-containing recombinant fragment of fibronectin). IL12 subunit reconstitution was investigated in a ligand capture experiment on the same sensor chip using F8-p35S-F8 as capture ligand and serial dilutions of p40S-KSF or KSF-p40S fusion proteins as analytes. Trimeric scFv(KSF)-TNF (KSF-TNF) was used as a negative control. KSF-p40S was analyzed on the same sensor chip as p40S-KSF and KSF-TNF but at a later time point.
Bioactivity Assays
IL12 subunit derivatives were subjected to two different bioactivity assays. First, a splenocyte/IFNγ release assay was performed. Splenocytes were isolated from freshly dissected spleens of BL/6 mice, which served as PBS control mice in Lewis lung carcinoma therapy experiments (Animal Experimental License 27/2015, Cantonal Veterinary Authority of Zürich, Switzerland). After red blood cell lysis, the isolated cells were resuspended at a concentration of 5 × 106/ml in RPMI medium containing 10% fetal bovine serum (Thermo Scientific), 10,000 units/ml penicillin, 10 mg/ml streptomycin, 25 μg/ml amphotericin (Antibiotic-Antimycotic supplement, Thermo Scientific), and 5 mm β-mercaptoethanol (Sigma-Aldrich) before an incubation period of 2 h at 37 °C and 5% CO2. Serial dilutions of recombinant mouse IL12 (PeproTech), which served as positive control, and IL12 subunit derivatives were added followed by an incubation period of 48 h at 37 °C and 5% CO2. Cultured supernatants were analyzed by a sandwich enzyme-linked immunosorbent assay (ELISA) using the monoclonal anti-mouse IFNγ antibody R4-6A2 (1:200 dilution, eBioscience) for capture and polyclonal biotinylated anti-mouse IFNγ antibody 500-P119Bt (1:500 dilution, PeproTech) for detection.
The signaling capacity of IL12 derivatives was analyzed by assessing the phosphorylation state of STAT4 by flow cytometry. Splenocytes were isolated according to the procedure outlined above. IL12-induced pSTAT4 activation in natural killer cells could be directly assessed without any other stimuli than the IL12-derived mutant fusion proteins. By contrast pSTAT4 signaling in CD4+ and CD8+ T cells required pre-activation of splenocytes in culture flasks pre-coated with a mix of anti-mouse CD3 (clone 145-2C11) and CD28 (clone 37.51) antibodies (1 μg/ml each, eBioscience). Sufficient pre-activation in supplemented RPMI medium was achieved after an incubation period of 48 h at 37 °C and 5% CO2. The splenocytes (1.5 × 106 cells/well in 100 μl) were then stimulated with serial dilutions of IL12 derivatives. F8-IL12-F8 featuring single chain IL12 (34, 40) as bioactive payload was used as a positive control. After 25 min, the cells were fixed by the addition of 1.5% p-formaldehyde (Santa Cruz Biotechnology) and permeabilized with ice-cold methanol (Sigma-Aldrich). Cell type-specific surface marker staining and intracellular pSTAT4 staining were performed using the following monoclonal anti-mouse antibody-fluorophore conjugates: anti-CD8b-APC-eFluor780 (1:200 dilution, eBioscience) and anti-CD4-FITC (1:200 dilution, eBioscience) together with anti-CD3ϵ-eFluor710 (1:300 dilution, eBioscience) were used to discriminate CD8+ and CD4+ T cells from other cells. Natural killer cells were identified within the CD3ϵ-negative population with anti-NK1.1-FITC (1:200 dilution, BD Biosciences), and intracellular pSTAT4 in all assayed cell populations was revealed using a monoclonal anti-pSTAT4-Alexa Fluor 647 antibody conjugate (1:50 dilution, BD Biosciences). Nonspecific mouse Fc receptor binding was prevented by the addition of anti-CD16/CD32 monoclonal antibodies (1:1000 dilution, Mouse BD Fc BlockTM, BD Biosciences). The stained cell populations were analyzed on a BD FACSCanto (BD Biosciences) device using the corresponding FACSDiva software. Data analysis was performed with FlowJo (FlowJo LLC). The specificity of used monoclonal anti-pSTAT4-Alexa Fluor 647 antibody conjugate has been confirmed with a fluorescence minus one control.
Animal Experiments
Quantitative biodistribution studies were carried out as described before (49) except that Pierce Iodination Tubes (iodogen method) were used for the chemical activation of 125I. F9 teratocarcinoma cells (2 × 107 cells) were subcutaneously injected into the flank of female Sv129 mice (Charles River Laboratories, age: 12 weeks) 5 days before the experiment. Mice were randomized (five mice per group) and grouped to have similar tumor size distribution at the day of injection. The study was non-blinded, and no experimental data were excluded. The experiments were performed under the Animal Experimental License 27/2015 granted to Prof. Dario Neri by the Cantonal Veterinary Authority of Zurich, Switzerland.
Author Contributions
D. N. and D. V. conceived and coordinated the study, designed experiments and wrote the paper. B. W., D. K., and D. V. performed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This research was supported by the ETH Zürich, the Swiss National Science Foundation (Project 310030B_163479/1), the Commission for Technology and Innovation (Project 17072.1), and the European Research Council (Project 670603). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. 1 and 2.
- EDA
- extra-domain A
- ESI
- electrospray ionization
- SEC
- size-exclusion chromatography
- pSTAT4
- phosphorylated signal transducer and activator of transcription 4
- scFv
- single-chain variable fragments.
References
- 1. Coiffier B., Lepage E., Briere J., Herbrecht R., Tilly H., Bouabdallah R., Morel P., Van Den Neste E., Salles G., Gaulard P., Reyes F., Lederlin P., and Gisselbrecht C. (2002) CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 235–242 [DOI] [PubMed] [Google Scholar]
- 2. Zhang Z., Zhang M., Goldman C. K., Ravetch J. V., and Waldmann T. A. (2003) Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD52 monoclonal antibody, Campath-1H. Cancer Res. 63, 6453–6457 [PubMed] [Google Scholar]
- 3. Gallamini A., Zaja F., Patti C., Billio A., Specchia M. R., Tucci A., Levis A., Manna A., Secondo V., Rigacci L., Pinto A., Iannitto E., Zoli V., Torchio P., Pileri S., and Tarella C. (2007) Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood 110, 2316–2323 [DOI] [PubMed] [Google Scholar]
- 4. Pietras R. J., Pegram M. D., Finn R. S., Maneval D. A., and Slamon D. J. (1998) Remission of human breast cancer xenografts on therapy with humanized monoclonal antibody to HER-2 receptor and DNA-reactive drugs. Oncogene 17, 2235–2249 [DOI] [PubMed] [Google Scholar]
- 5. Tassev D. V., and Cheung N. K. (2009) Monoclonal antibody therapies for solid tumors. Expert Opin. Biol. Ther. 9, 341–353 [DOI] [PubMed] [Google Scholar]
- 6. Weiner L. M., Surana R., and Wang S. (2010) Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 10, 317–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hess C., Venetz D., and Neri D. (2014) Emerging classes of armed antibody therapeutics against cancer. Medchemcomm 5, 408–431 [Google Scholar]
- 8. Eigentler T. K., Weide B., de Braud F., Spitaleri G., Romanini A., Pflugfelder A., González-Iglesias R., Tasciotti A., Giovannoni L., Schwager K., Lovato V., Kaspar M., Trachsel E., Menssen H. D., Neri D., and Garbe C. (2011) A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin. Cancer Res. 17, 7732–7742 [DOI] [PubMed] [Google Scholar]
- 9. Verma S., Miles D., Gianni L., Krop I. E., Welslau M., Baselga J., Pegram M., Oh D. Y., Diéras V., Guardino E., Fang L., Lu M. W., Olsen S., Blackwell K., and EMILIA Study Group (2012) Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 367, 1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Topp M. S., Gökbuget N., Zugmaier G., Klappers P., Stelljes M., Neumann S., Viardot A., Marks R., Diedrich H., Faul C., Reichle A., Horst H. A., Brüggemann M., Wessiepe D., Holland C., et al. (2014) Phase II trial of the anti-CD19 bispecific T cell-engager blinatumomab shows hematologic and molecular remissions in patients with relapsed or refractory B-precursor acute lymphoblastic leukemia. J. Clin. Oncol. 32, 4134–4140 [DOI] [PubMed] [Google Scholar]
- 11. Hostomsky Z., Hostomska Z., Hudson G. O., Moomaw E. W., and Nodes B. R. (1991) Reconstitution in vitro of RNase H activity by using purified N-terminal and C-terminal domains of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 88, 1148–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cabantous S., Terwilliger T. C., and Waldo G. S. (2005) Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102–107 [DOI] [PubMed] [Google Scholar]
- 13. Remy I., and Michnick S. W. (2007) Application of protein-fragment complementation assays in cell biology. BioTechniques 42, 137–141 [DOI] [PubMed] [Google Scholar]
- 14. Morell M., Ventura S., and Avilés F. X. (2009) Protein complementation assays: approaches for the in vivo analysis of protein interactions. FEBS Lett. 583, 1684–1691 [DOI] [PubMed] [Google Scholar]
- 15. Banaszek A. (2013) Dual Antigen-restricted Complementation of a Two-part Trispecific Antibody for Targeted Immunotherapy of Blood Cancer. Ph. D thesis, Julius-Maximilians-Universität of Würzburg [Google Scholar]
- 16. Versteegen R. M., Rossin R., ten Hoeve W., Janssen H. M., and Robillard M. S. (2013) Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem. Int. Ed. Engl. 52, 14112–14116 [DOI] [PubMed] [Google Scholar]
- 17. Walsh G. (2014) Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 32, 992–1000 [DOI] [PubMed] [Google Scholar]
- 18. Motzer R. J., Hutson T. E., Tomczak P., Michaelson M. D., Bukowski R. M., Rixe O., Oudard S., Negrier S., Szczylik C., Kim S. T., Chen I., Bycott P. W., Baum C. M., and Figlin R. A. (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124 [DOI] [PubMed] [Google Scholar]
- 19. Donohue J. H., and Rosenberg S. A. (1983) The fate of interleukin-2 after in vivo administration. J. Immunol. 130, 2203–2208 [PubMed] [Google Scholar]
- 20. Boyman O., and Sprent J. (2012) The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 12, 180–190 [DOI] [PubMed] [Google Scholar]
- 21. Rosenberg S. A. (2014) IL-2: the first effective immunotherapy for human cancer. J. Immunol. 192, 5451–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vadhan-Raj S., Keating M., LeMaistre A., Hittelman W. N., McCredie K., Trujillo J. M., Broxmeyer H. E., Henney C., and Gutterman J. U. (1987) Effects of recombinant human granulocyte-macrophage colony-stimulating factor in patients with myelodysplastic syndromes. N. Engl. J. Med. 317, 1545–1552 [DOI] [PubMed] [Google Scholar]
- 23. Gollob J. A., Mier J. W., Veenstra K., McDermott D. F., Clancy D., Clancy M., and Atkins M. B. (2000) Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-γ induction is associated with clinical response. Clin. Cancer Res. 6, 1678–1692 [PubMed] [Google Scholar]
- 24. Leonard J. P., Sherman M. L., Fisher G. L., Buchanan L. J., Larsen G., Atkins M. B., Sosman J. A., Dutcher J. P., Vogelzang N. J., and Ryan J. L. (1997) Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-γ production. Blood 90, 2541–2548 [PubMed] [Google Scholar]
- 25. Pasche N., and Neri D. (2012) Immunocytokines: a novel class of potent armed antibodies. Drug Discov. Today 17, 583–590 [DOI] [PubMed] [Google Scholar]
- 26. Halin C., Rondini S., Nilsson F., Berndt A., Kosmehl H., Zardi L., and Neri D. (2002) Enhancement of the antitumor activity of interleukin-12 by targeted delivery to neovasculature. Nat. Biotechnol. 20, 264–269 [DOI] [PubMed] [Google Scholar]
- 27. Carnemolla B., Borsi L., Balza E., Castellani P., Meazza R., Berndt A., Ferrini S., Kosmehl H., Neri D., and Zardi L. (2002) Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood 99, 1659–1665 [DOI] [PubMed] [Google Scholar]
- 28. Borsi L., Balza E., Carnemolla B., Sassi F., Castellani P., Berndt A., Kosmehl H., Biro A., Siri A., Orecchia P., Grassi J., Neri D., and Zardi L. (2003) Selective targeted delivery of TNFα to tumor blood vessels. Blood 102, 4384–4392 [DOI] [PubMed] [Google Scholar]
- 29. Yoon C., Johnston S. C., Tang J., Stahl M., Tobin J. F., and Somers W. S. (2000) Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J. 19, 3530–3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jalah R., Rosati M., Ganneru B., Pilkington G. R., Valentin A., Kulkarni V., Bergamaschi C., Chowdhury B., Zhang G. M., Beach R. K., Alicea C., Broderick K. E., Sardesai N. Y., Pavlakis G. N., and Felber B. K. (2013) The p40 subunit of interleukin (IL)-12 promotes stabilization and export of the p35 subunit: implications for improved IL-12 cytokine production. J. Biol. Chem. 288, 6763–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Villa A., Trachsel E., Kaspar M., Schliemann C., Sommavilla R., Rybak J. N., Rösli C., Borsi L., and Neri D. (2008) A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int. J. Cancer 122, 2405–2413 [DOI] [PubMed] [Google Scholar]
- 32. Gafner V., Trachsel E., and Neri D. (2006) An engineered antibody-interleukin-12 fusion protein with enhanced tumor vascular targeting properties. Int. J. Cancer 119, 2205–2212 [DOI] [PubMed] [Google Scholar]
- 33. Sommavilla R., Pasche N., Trachsel E., Giovannoni L., Roesli C., Villa A., Neri D., and Kaspar M. (2010) Expression, engineering and characterization of the tumor-targeting heterodimeric immunocytokine F8-IL12. Protein Eng. Des. Sel. 23, 653–661 [DOI] [PubMed] [Google Scholar]
- 34. Pasche N., Wulhfard S., Pretto F., Carugati E., and Neri D. (2012) The antibody-based delivery of interleukin-12 to the tumor neovasculature eradicates murine models of cancer in combination with paclitaxel. Clin. Cancer Res. 18, 4092–4103 [DOI] [PubMed] [Google Scholar]
- 35. Heinzel F. P., Hujer A. M., Ahmed F. N., and Rerko R. M. (1997) In vivo production and function of IL-12 p40 homodimers. J. Immunol. 158, 4381–4388 [PubMed] [Google Scholar]
- 36. Lauwerys B. R., Van Snick J., and Houssiau F. A. (2002) Serum IL-12 in systemic lupus erythematosus: absence of p70 heterodimers but presence of p40 monomers correlating with disease activity. Lupus 11, 384–387 [DOI] [PubMed] [Google Scholar]
- 37. Shigehara K., Shijubo N., Ohmichi M., Kamiguchi K., Takahashi R., Morita-Ichimura S., Ohchi T., Tatsuno T., Hiraga Y., Abe S., and Sato N. (2003) Increased circulating interleukin-12 (IL-12) p40 in pulmonary sarcoidosis. Clin. Exp. Immunol. 132, 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gillessen S., Carvajal D., Ling P., Podlaski F. J., Stremlo D. L., Familletti P. C., Gubler U., Presky D. H., Stern A. S., and Gately M. K. (1995) Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur. J. Immunol. 25, 200–206 [DOI] [PubMed] [Google Scholar]
- 39. Schoenhaut D. S., Chua A. O., Wolitzky A. G., Quinn P. M., Dwyer C. M., McComas W., Familletti P. C., Gately M. K., and Gubler U. (1992) Cloning and expression of murine IL-12. J. Immunol. 148, 3433–3440 [PubMed] [Google Scholar]
- 40. Lieschke G. J., Rao P. K., Gately M. K., and Mulligan R. C. (1997) Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat. Biotechnol. 15, 35–40 [DOI] [PubMed] [Google Scholar]
- 41. Wu C., Wang X., Gadina M., O'Shea J. J., Presky D. H., and Magram J. (2000) IL-12 receptor β2 (IL-12R β2)-deficient mice are defective in IL-12-mediated signaling despite the presence of high affinity IL-12 binding sites. J. Immunol. 165, 6221–6228 [DOI] [PubMed] [Google Scholar]
- 42. Neri D., and Sondel P. M. (2016) Immunocytokines for cancer treatment: past, present and future. Curr. Opin. Immunol. 40, 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gubler U., Chua A. O., Schoenhaut D. S., Dwyer C. M., McComas W., Motyka R., Nabavi N., Wolitzky A. G., Quinn P. M., Familletti P. C., et al. (1991) Coexpression of two distinct genes is required to generate secreted bioactive cytotoxic lymphocyte maturation factor. Proc. Natl. Acad. Sci. U.S.A. 88, 4143–4147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ling P., Gately M. K., Gubler U., Stern A. S., Lin P., Hollfelder K., Su C., Pan Y. C., and Hakimi J. (1995) Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J. Immunol. 154, 116–127 [PubMed] [Google Scholar]
- 45. Desnoyers L. R., Vasiljeva O., Richardson J. H., Yang A., Menendez E. E., Liang T. W., Wong C., Bessette P. H., Kamath K., Moore S. J., Sagert J. G., Hostetter D. R., Han F., Gee J., Flandez J., et al. (2013) Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 5, 207ra144. [DOI] [PubMed] [Google Scholar]
- 46. Boyman O., Kovar M., Rubinstein M. P., Surh C. D., and Sprent J. (2006) Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 311, 1924–1927 [DOI] [PubMed] [Google Scholar]
- 47. Gillies S. D. (2013) A new platform for constructing antibody-cytokine fusion proteins (immunocytokines) with improved biological properties and adaptable cytokine activity. Protein Eng. Des. Sel. 26, 561–569 [DOI] [PubMed] [Google Scholar]
- 48. Garcin G., Paul F., Staufenbiel M., Bordat Y., Van der Heyden J., Wilmes S., Cartron G., Apparailly F., De Koker S., Piehler J., Tavernier J., and Uzé G. (2014) High efficiency cell-specific targeting of cytokine activity. Nat. Commun. 5, 3016. [DOI] [PubMed] [Google Scholar]
- 49. Venetz D., Hess C., Lin C. W., Aebi M., and Neri D. (2015) Glycosylation profiles determine extravasation and disease-targeting properties of armed antibodies. Proc. Natl. Acad. Sci. U.S.A. 112, 2000–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li J., Li C., Xiao W., Yuan D., Wan G., and Ma L. (2008) Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli. Anal. Biochem. 373, 389–391 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.