

Abstract
Activation of IKKβ is the key step in canonical activation of NF-κB signaling. Extensive work has provided insight into the mechanisms underlying IKKβ activation through the identification of context-specific regulators. However, the molecular processes responsible for its negative regulation are not completely understood. Here, we identified KLHL21, a member of the Kelch-like gene family, as a novel negative regulator of IKKβ. The expression of KLHL21 was rapidly down-regulated in macrophages upon treatment with proinflammatory stimuli. Overexpression of KLHL21 inhibited the activation of IKKβ and degradation of IκBα, whereas KLHL21 depletion via siRNA showed the opposite results. Coimmunoprecipitation assays revealed that KLHL21 specifically bound to the kinase domain of IKKβ via its Kelch domains and that this interaction was gradually attenuated upon TNFα treatment. Furthermore, KLHL21 did not disrupt the interaction between IKKβ and TAK1, TRAF2, or IκBα. Also, KLHL21 did not require its E3 ubiquitin ligase activity for IKKβ inhibition. Our findings suggest that KLHL21 may exert its inhibitory function by binding to the kinase domain and sequestering the region from potential IKKβ inducers. Taken together, our data clearly demonstrate that KLHL21 negatively regulates TNFα-activated NF-κB signaling via targeting IKKβ, providing new insight into the mechanisms underlying NF-κB regulation in cells.
Keywords: cell signaling, E3 ubiquitin ligase, NF-kappaB, protein-protein interaction, tumor necrosis factor (TNF), IKKbeta, KLHL21
Introduction
NF-κB4 is an inducible transcription factor that is critical for innate and adaptive immune responses to infection. It also plays important roles in the regulation of cell survival, differentiation, and proliferation (1). Various stimuli, including pathogen- and damage-associated molecular patterns as well as proinflammatory factors (e.g. TNFα and IL-1β), trigger specific signaling pathways that lead to the activation of NF-κB. Most of these diverse pathways converge on the activation of the IκB kinase (IKK) complex, which phosphorylates specific serine residues on the classical NF-κB inhibitors, IκBα, IκBβ, and IκBϵ. Phosphorylated IκB proteins are targeted for ubiquitination and rapid degradation via the ubiquitin-proteasome pathway, resulting in the nuclear translocation and transcriptional activity of NF-κB proteins (2, 3).
The IKK complex is composed of at least two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, NF-κB essential modulator (NEMO; also known as IKKγ). Its activation is the key event in canonical NF-κB signaling. Phosphorylation of specific serine residues within the kinase domains (KDs) of IKKα and IKKβ (Ser-177/181 for IKKβ) by upstream kinases, such as TAK1, or by autophosphorylation activates the complex (4). Several proteins, including IκBs, p105, p65, regnase-1, IRF7, Bcl-10, IRS-1, β-catenin, SNAP23, p53, TSC1/2, FOXO3a, and Aurora A, have been identified as physiological substrates of IKKβ (5, 6). Therefore, it is unsurprising that IKKβ activity is tightly regulated by multiple intracellular factors, such as HSP90, Cdc37, GβL (7), CUEDC2 (8), NLRX1 (9), NLRC5 (10), KEAP1 (11, 12), NLK (13), and RACK1 (14), under different conditions (4). IKKβ plays important roles in cell survival, cell proliferation, and cross-talk between signaling pathways. Dysregulation of IKKβ has been implicated in the pathogenesis of many human diseases, including cancers (15).
KLHL21 is a member of the Kelch-like (KLHL) gene family (16). At present, 42 KLHL genes have been identified. They encode a group of highly conserved proteins that generally possess a BTB/POZ (Bric-a-brac-Tramtrack-broad complex/poxvirus and zinc finger) domain, a BACK domain, and 5–6 Kelch motifs (16). Members of this protein family have been reported to be involved in a number of cellular and molecular processes, such as inflammatory responses, oxidative stress responses, cytokinesis, embryonic development, and lymphogenesis (11, 12, 17–19). For example, Keap1 (also known as KLHL19) was recently found to interact specifically with IKKβ. It promotes the degradation of IKKβ via autophagic (11) or 26S proteasome (12) pathways and inhibits the phosphorylation of IKKβ in response to extracellular stimuli (11, 12).
A previous study demonstrated that KLHL21 is required for successful cytokinesis by regulating translocation of the chromosomal passenger complex from chromosomes to the spindle midzone during anaphase (20). Mechanistically, KLHL21 interacts with Cul3 to mediate the ubiquitination of aurora B kinase in vitro (20). However, the functions of KLHL21 in other cellular processes remain unclear. Here, we demonstrate that KLHL21 is a novel negative regulator of IKKβ. Exogenous expression of KLHL21 suppressed TNFα-stimulated IKKβ activation, resulting in reduced NF-κB signaling, whereas depletion of KLHL21 led to enhanced IKKβ activity. This inhibitory function of KLHL21 was independent of its E3 ubiquitin ligase activity. Thus, our results reveal a novel mechanism by which IKKβ is regulated in cells.
Results
Cells Exposed to Proinflammatory Stimuli Show Down-regulated Klhl21 Expression
To identify novel genes regulated by LPS (specific agonist of TLR4 receptor) treatment, we performed a comprehensive search of the NCBI Gene Expression Omnibus (GEO) database. The expression of Klhl21 was consistently down-regulated in RAW264.7 monocytic macrophage cells upon LPS treatment (GEO accession number GDS1043) (21). Additional analyses revealed that Klhl21 expression is down-regulated in LPS-treated dendritic cells (GEO accession numbers GDS2216 and GDS883) (22, 23) and peripheral blood-derived monocytes (GEO accession number GDS2856) (24), CpG oligonucleotide-treated bone marrow-derived dendritic cells (GEO accession number GDS1315) (25), and Kaposi sarcoma-associated herpesvirus-infected primary human dermal endothelial cells (GEO accession number GDS940) (26). Taken together, these findings suggest that down-regulation of Klhl21 may be a general consequence of the inflammatory response and that KLHL21 might contribute to the regulation of related cellular processes.
We confirmed that LPS treatment induced rapid down-regulation of Klhl21 in RAW264.7 cells (Fig. 1A). KLHL21 mRNA levels were lowest by 4 h after treatment, representing <10% of the levels observed in untreated cells, before gradually increasing and reaching their peak (about 70% of the levels of untreated cells) around 24 h. However, levels cannot be completely restored even extending LPS treatment to 48 h (Fig. 1A). We observed a similar Klhl21 expression trend in LPS-treated bone marrow-derived macrophages (Fig. 1B). KLHL21 protein abundance also appeared to be temporally regulated, decreasing dramatically in RAW264.7 cells at 1 h after LPS treatment, followed by a steady increase (Fig. 1C). Further analysis revealed that Klhl21 mRNA levels were down-regulated in RAW264.7 cells exposed to Pam2CSK4 (TLR2/6 agonist), Pam3CSK4 (TLR1/2 agonist), or ODN1826 (TLR9 agonist) (Fig. 1, D and E). Treatment with TNFα also resulted in reduced expression of Klhl21 mRNA and protein (Fig. 1F). These results indicate that rapid down-regulation of Klhl21 may be a general phenomenon in response to extracellular proinflammatory stimuli in macrophages.
FIGURE 1.

Down-regulation of KLHL21 in macrophages treated with proinflammatory stimuli. The mRNA level of Klhl21 in RAW264.7 cells (A) and in bone marrow-derived macrophages (B) treated with 100 ng/ml LPS at different time points were determined by quantitative RT-PCR, respectively (n = 3). C, the protein level of KLHL21 in RAW264.7 cells stimulated with LPS was analyzed by Western blotting (n = 3). D–F, RAW264.7 cells were treated with 100 ng/ml Pam2CSK4 and Pam3CSK4 (D), 5 μm ODN1826 (E), and 20 ng/ml TNFα (F), respectively. The mRNA and protein level of KLHL21 were analyzed by using real-time PCR (n = 3) and Western blotting, respectively. IB, immunoblotting. Error bars, S.E.
KLHL21 Inhibits Activation of the NF-κB Signaling Pathway
To investigate the potential roles of KLHL21 in the inflammatory process, we first assessed its impact on NF-κB signaling. KLHL21 was coexpressed with known inducers of NF-κB signaling in HEK293T cells carrying an NF-κB luciferase reporter. Coexpression of KLHL21 and TRAF2 (upstream regulator of the TNFR1 signaling pathway), TRAF6 (upstream regulator of the TLRs and IL-1R signaling pathways), TBK1, or the IKKβ (SS/EE) constitutively active mutant (Ser-177/181 to Glu mutations) resulted in significant inhibition of NF-κB activation (Fig. 2A). Conversely, KLHL21 had no effect on p65 (RelA)-mediated NF-κB activity (Fig. 2A). Similar findings were obtained in HeLa cells (Fig. 2B). Coexpression of KLHL21 and the IKKβ (SS/EE) mutant suppressed luciferase expression driven by the mouse NFKBIA (encoding IκBα, a NF-κB transcriptional target) promoter (Fig. 2C). Moreover, overexpression of KLHL21 inhibited TNFα-induced NF-κB-luciferase activity in HEK293T cells and IL-1β-induced NF-κB-luciferase activity in HeLa cells, respectively (Fig. 2, D and E). These results strongly imply that KLHL21 might function downstream of IKKβ and upstream of p65 to inhibit NF-κB activation.
FIGURE 2.

KLHL21 inhibits the activation of NF-κB. 293T cells (A) and HeLa cells (B) were transfected with pNFκB-TA-luc, TRAF2, TBK1, TRAF6, IKKβ SS/EE mutant, or p65, along with KLHL21, and analyzed for NF-κB-dependent luciferase activity at 24 h posttransfection (n = 3). C, 293T cells were transfected with NFKBIA-Lu (mouse NFKBIA promoter directed the expression of luciferase), IKKβ SS/EE mutant, and KLHL21 and analyzed for NFKBIA promoter-dependent luciferase activity at 24 h post-transfection (n = 3). D, 293T cells were transfected with pNFκB-TA-luc along with KLHL21. 24 h after the transfection, the cells were treated with 20 ng/ml TNFα for 2 h, and then the NF-κB-dependent luciferase activity was analyzed (n = 3). E, HeLa cells were transfected with pNFκB-TA-luc along with KLHL21. 24 h after the transfection, the cells were treated with 100 ng/ml IL-1β for 2 h, and then the NF-κB-dependent luciferase activity was analyzed (n = 3). Data are plotted as means ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ns, not significant.
In resting cells, NF-κB is retained in the cytoplasm when bound to inhibitory IκB proteins. The ubiquitin-dependent degradation of IκBs, especially IκBα, releases bound NF-κB and leads to its nuclear translocation (2). Given our observation that KLHL21 inhibits the activation of transcription factor NF-κB, we analyzed its effect on the nuclear translocation of p65/RelA in HEK293T cells upon TNFα treatment. Overexpression of KLHL21 suppressed p65/RelA nuclear translocation after 10 and 30 min of TNFα treatment, but nuclear p65/RelA reached levels similar to control cells by 60 min posttreatment (Fig. 3A). These results reveal that overexpressing KLHL21 inhibits the early phase of NF-κB/p65 nuclear translocation after TNFα treatment, thereby attenuating NF-κB activation.
FIGURE 3.

KLHL21 negatively regulates the activation of NF-κB signaling. 293T cells transfected with KLHL21-3F were treated with 20 ng/ml TNFα. Nuclear translocation of p65 (A), total IκBα protein (B), and phosphorylated IκBα (Ser-32) (E) at the indicated time points was analyzed by Western blotting. 293T cells transfected with pcDNA3 empty vector were used as a control. C, 293T cells were transfected with pooled siRNA specifically targeting human KLHL21 at the indicated concentrations. The level of KLHL21 mRNA was analyzed at 24 h after the transfection (n = 3). D and F, 293T cells were transfected with 25 nm negative control siRNA and siKLHL21 as indicated. The degradation and reaccumulation of IκBα proteins (D) and phosphorylated IκBα protein (F), respectively, in response to 20 ng/ml TNFα treatment were analyzed by Western blotting. The density ratio of p65/Lamin B (A) quantified by densitometric scanning is shown on the right (n = 3). Data from A and C are plotted as means ± S.E. (error bars). All experiments were performed at least three times. WCL, whole cell lysates; IB, immunoblotting; *, p < 0.05; **, p < 0.01.
We then examined the effects of KLHL21 on IκBα degradation. In control cells, TNFα treatment induced rapid degradation of IκBα, followed by its reaccumulation (Fig. 3B, left). In contrast, overexpression of KLHL21 suppressed the degradation and reaccumulation of IκBα (Fig. 3B, right). To corroborate these findings, we introduced chemosynthesized siRNA to knock down KLHL21 in HEK293T cells and confirmed reduced mRNA (Fig. 3C) and protein (Fig. 3D) levels. KLHL21 depletion did not affect IκBα protein in resting cells, but it enhanced the degradation of TNFα-induced IκBα (Fig. 3D).
Prior phosphorylation of both Ser-32 and Ser-36 is required for the ubiquitin-dependent degradation of IκBα protein (2). Therefore, we analyzed the consequences of KLHL21 overexpression on TNFα-induced phosphorylation of IκBα. In response to TNFα treatment, the amount of phosphorylated IκBα (Ser-32) increased substantially in control cells but was significantly suppressed in cells overexpressing KLHL21 (Fig. 3E). In contrast, knocking down KLHL21 with specific siRNAs enhanced the phosphorylation of IκBα in HEK293T cells upon TNFα treatment (Fig. 3F). Our findings that KLHL21 suppressed TNFα-induced phosphorylation and ubiquitin-dependent degradation of IκBα as well as the IKKβ (SS/EE) mutant-induced activation of NF-κB further suggest that KLHL21 might act directly on the IKK complex and inhibit its activity.
KLHL21 Directly Interacts with IKK Complex
To identify potential interacting partners of KLHL21 in the NF-κB signaling pathway, FLAG-tagged KLHL21 protein was transiently expressed in HEK293T cells. Putative interactions between KLHL21 and endogenous IKKβ, IκBα, and p65 were examined by coimmunoprecipitation assays. The interaction between KLHL21 and Cullin3 was included as a positive control (20). Only IKKβ specifically interacted with KLHL21 (Fig. 4A). An interaction between endogenous IKKβ and KLHL21 was also observed in HEK293T and RAW264.7 cells (Fig. 4B), suggesting a physiologically relevant role for this association. KLHL21 and IKKβ demonstrated a direct interaction by in vitro GST pull-down assays (Fig. 4C) and colocalized in HeLa cells (Fig. 4D). Furthermore, interaction between KLHL21 with IKKα and NEMO (other components of the IKK complex) was also detected in HEK293T cells (Fig. 4E), demonstrating that KLHL21 specifically interacts with the IKK complex in cells.
FIGURE 4.

KLHL21 protein specifically binds to IKKβ. A, 293T cells were transfected with KLHL21–3F. KLHL21-3F was immunoprecipitated with anti-FLAG beads and then analyzed by immunoblotting with the indicated antibodies. B, whole cell lysates prepared from 293T and RAW264.7 cells were immunoprecipitated with anti-KLHL21 polyclonal antibody and then analyzed by immunoblotting with the indicated antibodies. C, purified GST-KLHL21 fusion protein was incubated with whole cell lysates prepared from 293T and RAW264.7 cells, respectively. The precipitates after pull-down were analyzed by Western blotting with anti-KLHL21 antibody. D, HeLa cells were transfected with C-terminal mCherry-tagged KLHL21 (red). Cells were fixed and immunostained with anti-IKKβ antibody (green). E, 293T cells were cotransfected with vector expressing IKKα-3F, IKKβ-3F, NEMO-3F, and KLHL21-EE as indicated. Lysates were immunoprecipitated with anti-FLAG and then analyzed by immunoblot with anti-EE and anti-FLAG, respectively. F, 293T cells cotransfected with IKKβ-EE and KLHL21–3F were collected at the indicated time points after TNFα treatment. KLHL21-3F was immunoprecipitated with anti-FLAG beads and then analyzed by immunoblotting with the indicated antibodies. The density ratio of immunoprecipitated IKKβ/KLHL21 quantified by densitometric scanning is shown on the right (n = 3). All experiments were performed at least three times. IB, immunoblotting; IP, immunoprecipitation; WCL, whole cell lysates. Error bars, S.E.
To define the nature of the KLHL21-IKKβ interaction under inflammatory conditions, we performed coimmunoprecipitation assays in TNFα-treated HEK293T cells and found that their interaction was gradually attenuated upon TNFα treatment, significantly reduced after 10 min (Fig. 4F). It led to the release of IKKβ, which can then be activated by its upstream inducers subsequently. It is consistent with the above findings that overexpression of KLHL21 inhibited the early phase of p65 nuclear translocation after TNF treatment. Taken together, these findings demonstrate that KLHL21 directly interacts with the IKK complex under both transient and endogenous circumstances and that TNFα-mediated immune responses may perturb this interaction to activate NF-κB signaling.
KLHL21 Binds to the KD of IKKβ via Its Kelch Domains
IKKβ contains an N-terminal KD, a ubiquitin-like domain, an elongated, α-helical scaffold/dimerization domain, and a C-terminal NEMO-binding domain (27). To identify the domain of IKKβ through which the interaction with KLHL21 occurs, we generated five deletion mutants of mouse IKKβ and examined their abilities to interact with wild-type KLHL21 (Fig. 5A). Similar to wild-type IKKβ, deletion mutants M3 (containing KD only), M4 (lacking the scaffold/dimerization domain and NEMO-binding domain), and M5 (lacking the NEMO-binding domain) were efficiently coprecipitated with KLHL21 (Fig. 5A). In contrast, deletion of the N-terminal KD (mutant M1) dramatically reduced the ability of KLHL21 to coisolate with IKKβ. The interaction between KLHL21 and IKKβ was completely abolished when both KD and ubiquitin-like domains were deleted (mutant M2) (Fig. 5A). These results indicate that the KD of IKKβ is critical for association with KLHL21.
FIGURE 5.

KLHL21 directly binds to the kinase domain of IKKβ via its Kelch domains. A, 293T cells were cotransfected with KLHL21–3F, WT, and deletion mutants of IKKβ-EE, respectively, as indicated. KLHL21–3F was immunoprecipitated with anti-FLAG beads and blotted with anti-EE and anti-FLAG. ULD, ubiquitin-like domain; SDD, scaffold/dimerization domain; NBD, NEMO-binding domain. B, 293T cells were cotransfected with IKKβ-EE and the indicated KLHL21-3F deletion constructs. KLHL21-3F was immunoprecipitated with anti-FLAG beads and blotted with anti-EE and anti-FLAG. Results are representative of three independent experiments. C, 293T cells were transfected with the indicated KLHL21 deletion constructs. KLHL21-3F was immunoprecipitated with anti-FLAG beads and blotted with anti-Cul3 and anti-FLAG. D, 293T cells were cotransfected with EE-tagged wild-type IKKβ and IKKβ E39A mutant, along with FLAG-tagged KLHL21 and KEAP1 where indicated. Lysates were immunoprecipitated with anti-FLAG and then analyzed by immunoblotting with anti-EE and anti-FLAG, respectively. IB, immunoblotting; IP, immunoprecipitation; WCL, whole cell lysates.
Next, to identify the domain of KLHL21 responsible for interaction with IKKβ, we generated four deletion mutants (Δ1, Δ2, Δ3, and Δ4) of mouse KLHL21 (Fig. 5B). Coimmunoprecipitation assays revealed that deletion mutants Δ3 and Δ4 (both containing Kelch domains) were coisolated with IKKβ. Deletion of the Kelch domains of KLHL21 (mutants Δ1 and Δ2) abolished the interaction with IKKβ. These results revealed that the C-terminal Kelch domains were required for KLHL21 to interact with IKKβ. Point mutant KLHL21M (D114A/L115A/Q117A), which is unable to bind CUL3 (Fig. 5C) (20), was also coisolated with IKKβ (Fig. 5B). This finding reveals that the BTB domain of KLHL21 is unnecessary for association with IKKβ.
KEAP1, another member of the KLHL gene family, binds to a unique (E/A)TGE motif in the KD of IKKβ via its Kelch domains and promotes the degradation of IKKβ (11, 12). To test whether KLHL21 also binds to this unique motif, Glu-39 of the mouse IKKβ motif was mutated (E39A). Consistent with previous reports, the E39A mutation dramatically reduced the interaction between IKKβ and KEAP1 (11, 12), but it had no effect on the interaction between KLHL21 and IKKβ (Fig. 5D). These results imply that KLHL21 is unlikely to compete with KEAP1 for binding to this unique motif within the KD of IKKβ in cells.
KLHL21 Negatively Regulates TNFα-induced IKKβ Activation
Activation of the IKK complex, especially via phosphorylation of both Ser-177 and Ser-181 within the KD of IKKβ, is the key step driving stimulation of NF-κB signaling. Given our observations that KLHL21 binds to the KD of IKKβ and inhibits the activation of NF-κB signaling, we hypothesized that KLHL21 may regulate the phosphorylation of IKKβ. Overexpression of IKKβ in HEK293T cells resulted in its autophosphorylation, which was dramatically suppressed when KLHL21 was coexpressed (Fig. 6A). Interestingly, the KLHL21M mutant functioned similarly to wild-type KLHL21 (Fig. 6A), demonstrating that KLHL21 inhibits the activation of IKKβ independently of its E3 ubiquitin ligase activity. Moreover, overexpression of KLHL21 significantly suppressed the phosphorylation of endogenous IKKα/β (Ser-177) induced by TNFα treatment (Fig. 6B). Conversely, siRNA-mediated depletion of KLHL21 resulted in enhanced phosphorylation of IKKα/β (Ser-177) in response to TNFα treatment without affecting total endogenous IKKβ protein levels (Fig. 6C).
FIGURE 6.

KLHL21 inhibits the activation of IKKβ. A, 293T cells were cotransfected with EE-tagged IKKβ, WT KLHL21, and KLHL21-M mutant, respectively. Phosphorylated IKKβ was analyzed by immunoblotting with anti-phospho-IKKα/β (Ser-177) (left). The density ratio of phosphorylated IKKβ/total IKKβ is shown on the right (n = 3). B and C, 293T cells were transfected with KLHL21–3F (B) and 25 nm siKLHL21 (C), respectively. Phosphorylation of IKKβ in response to TNFα treatment was analyzed by immunoblot with anti-phospho-IKKα/β (Ser-177). The mean results for densitometric scans are shown on the right (n = 3). D, 293T cells were cotransfected with siRNA 3 and mCherry-tagged WT KLHL21 (KLHL21-MC) or KLHL21M mutant (M). 24 h after the transfection, the cells were treated with 20 ng/ml TNFα for 10 min. Phosphorylated and total IKKβ and IκBα were then analyzed by Western blotting. An arrow indicates the nonspecific bands in the KLHL21 immunoblot. E, 293T cells cotransfected with HA-ubiquitin, IKKβ-3F, KLHL21-EE, and KLHL21M-EE were treated with 5 μm MG132 for 16 h. The cells were lysed by boiling in SDS-lysis buffer. The lysates were immunoprecipitated with anti-FLAG and analyzed with the indicated antibodies (long exposure: 10 min). Data are plotted as means ± S.E. (error bars). *, p < 0.05; **, p < 0.01. All experiments were performed three times. IB, immunoblotting; IP, immunoprecipitation.
To rule out off-target effects, we performed a rescue experiment where HEK293T cells were cotransfected with siRNA specifically targeting human KLHL21 and mCherry-tagged wild-type or enzymatically inactive mutant of mouse KLHL21 construct. Consistent with above findings, knocking down KLHL21 promoted the phosphorylation of IKKβ and IκBα and the degradation of IκBα in HEK293T cells exposed to TNFα (Fig. 6D, lane 4), whereas coexpression of either WT KLHL21 or E3 ligase activity-inactive mutant KLHL21M reversed these effects (Fig. 6D, lanes 6 and 8). This result further confirmed our observation that KLHL21 negatively regulates TNFα-induced IKKβ activation, which is independent of its E3 ligase activity.
KLHL21 interacts with Cul3 and mediates the ubiquitination of aurora B kinase in vitro (20). Therefore, we next tested whether KLHL21 plays a role in the ubiquitination of IKKβ. In the presence of the proteasome inhibitor MG132, long-term exposure of the nitrocellulose membrane revealed smeared bands of polyubiquitinated IKKβ in HEK293T cells (Fig. 6E). Overexpression of either wild-type KLHL21 or the KLHL21M mutant failed to enhance the ubiquitination of IKKβ (Fig. 6E). Furthermore, exogenously expressed KLHL21 did not significantly affect endogenous levels of IKKβ protein (Fig. 6B). These results further demonstrate that KLHL21 is unlikely to target IKKβ endogenously through its E3 ubiquitin ligase function.
Given our observations that KLHL21 efficiently inhibited TBK1-, TRAF2-, and TRAF6-induced activation of NF-κB (Fig. 2A), we next tested whether KLHL21 can bind directly to TBK1, TRAF2, or TRAF6. Coimmunoprecipitation results demonstrated that KLHL21 did not bind to TBK1, TRAF2, or TRAF6 in HEK293T cells (Fig. 7A). Upon TNFα stimulation, TRAF2 rapidly recruits the IKK complex to TNFR1 in cells, which leads to TAK1-mediated phosphorylation and activation of IKKα and IKKβ, subsequently resulting in the phosphorylation and proteasomal degradation of IκBα. To examine whether KLHL21 binding to IKKβ interferes with any step of this process, we cotransfected KLHL21 and IKKβ with TRAF2, TAK1, or the undegradable IκBα mutant (S32A/S36A) in HEK293T cells. Overexpression of KLHL21 failed to disrupt the interaction between IKKβ and TRAF2, TAK1, or IκBα (Fig. 7, B–D), further suggesting that KLHL21 inhibits activation of the NF-κB signaling pathway by directly targeting IKKβ.
FIGURE 7.
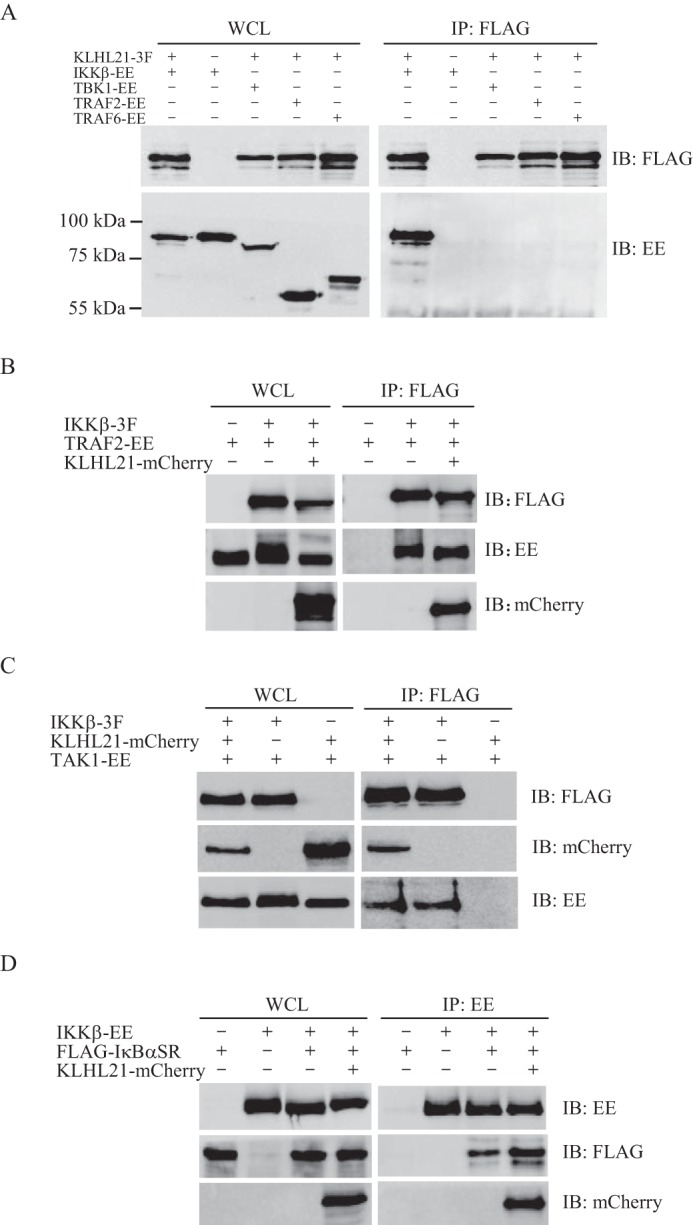
KLHL21 does not interfere with the interaction between IKKβ and TRAF2, TAK1, or IκBα. A, 293T cells were cotransfected with vector expressing IKKβ-EE, TBK1-EE, TRAF2-EE, TRAF6-EE, and KLHL21-3F as indicated. Lysates were immunoprecipitated with anti-FLAG and then analyzed by immunoblotting with anti-EE and anti-FLAG, respectively. B–D, IKKβ and KLHL21 were coexpressed with TRAF2 (B), TAK1 (C), or undegradable IκBα mutant (D), respectively. The interactions between IKKβ and TRAF2, TAK1, or IκBα were analyzed by coimmunoprecipitation. WCL, whole cell lysates; IP, immunoprecipitation; IB, immunoblotting. Error bars, S.E.
KLHL21 Differentially Modulates the Expression of NF-κB Target Genes
The above data clearly reveal that KLHL21 suppresses NF-κB signaling by directly targeting IKKβ to inhibit its activation. Next, the effects of KLHL21 on the expression of endogenous NF-κB target genes NFKBIA, DUSP1, NFKBIZ, CXCL1, TNFAIP3, and IL-8 were assessed in TNFα-treated HEK293T cells. Overexpressing KLHL21 significantly suppressed TNFα-induced transcription of the immediate early response genes, such as NFKBIA, DUSP1, and NFKBIZ (which peaked at 30 min after TNFα treatment) but, unexpectedly, enhanced the relative late phase expression of CXCL1, IL-8, and TNFAIP3 (which peaked at 2 h or even later after TNFα treatment) (Fig. 8A). Conversely, siRNA-mediated knockdown of KLHL21 resulted in marked up-regulation of NFKBIA and down-regulation of IL-8 transcript levels (2 and 4 h) but had no effect on TNFAIP3 expression (Fig. 8B). Similarly, KLHL21 depletion in LPS-treated RAW264.7 macrophages (Fig. 8C) was associated with up-regulated Nfkbia and down-regulated Tnfaip3 and Il-1b transcript levels (Fig. 8D).
FIGURE 8.

KLHL21 regulates the induced expression of NF-κB target genes. 293T cells were transiently transfected with KLHL21–3F (A) and 25 nm siKLHL21 (B), respectively. The expression of Nfkbia, Dusp1, Nfkbiz, Cxcl1, Il-8, and Tnfaip3 induced by 20 ng/ml TNFα was determined by real-time PCR. C, 50 nm siKLHL21 was used to knock down the expression of Klhl21 in RAW264.7 cells. 24 h post-transfection, mRNA and protein level of KLHL21 was analyzed by quantitative RT-PCR (n = 3) and Western blotting, respectively. D, RAW264.7 cells were transfected with a 50 nm siRNA pool targeting mouse Klhl21. The expression of Nfkbia, Tnfaip3, and Il-1b induced by LPS were analyzed by using quantitative RT-PCR. E, 293T cells transfected with KLHL21-3F were treated with 20 ng/ml TNFα. Nuclear translocation of p65 at the indicated time points was analyzed by Western blotting. 293T cells transfected with pcDNA3 empty vector were used as a control. Data are plotted as means ± S.E. (error bars). *, p < 0.05; **, p < 0.01. All experiments were performed three times. IB, immunoblotting.
To clarify the discrepancy that overexpressing KLHL21 inhibited the activation of IKKβ yet enhanced the expression of some relative late phase NF-κB target genes, such as CXCL1, IL-8, and TNFAIP3, we then analyzed the nuclear duration of p65 in HEK293T cells upon TNFα treatment. By 2 and 4 h after TNFα treatment, nuclear levels of p65 were dramatically reduced in control cells compared with KLHL21-overexpressing cells (Fig. 8E). These findings suggest that KLHL21 overexpression prolongs the duration of p65 in the nucleus, which may partly account for the up-regulation of IL-8 and TNFAIP3 by 4 h after TNFα treatment.
Discussion
Here, we demonstrate that KLHL21 specifically binds to and negatively regulates the activation of IKKβ in resting cells. The interaction between KLHL21 and IKKβ is gradually attenuated in cells upon TNFα treatment. This leads to the release of IKKβ, which can then be activated, suggesting that IKKβ activity is finely tuned by KLHL21 under inflammatory conditions. The expression of KLHL21 is also dynamically regulated in macrophages treated with diverse proinflammatory stimuli, highlighting its potentially important function in the appropriate activation of NF-κB signaling.
KLHL21 was previously reported to interact with Cul3 and to mediate the ubiquitination of aurora B in vitro (20). However, exogenous expression of KLHL21 had no effect on the ubiquitination states of endogenous IKKβ or its general translation. The KLHL21M mutant, which is unable to interact with Cul3, functioned in a similar manner to wild-type KLHL21 by suppressing the autophosphorylation of IKKβ. These findings demonstrate that KLHL21 negatively regulates IKKβ independently of its E3 ubiquitin ligase activity. It is not uncommon for an E3 ubiquitin ligase to carry out alternative cellular functions independent of its traditional enzymatic activity (8, 28, 29). For example, the E3 ubiquitin ligase CUEDC2 inhibits the activity of IKK by recruiting phosphatase PP1 to the IKK complex (8).
Furthermore, KLHL21 did not interfere with the interaction between IKKβ and TRAF2, TAK1, or IκBα. KLHL21 probably inhibits IKKβ by masking the KD residues to be phosphorylated. Given that KLHL21 also suppressed the activity of the IKKβ SS/EE mutant, it is plausible that it may also sterically block access to the activated KD from IKKβ substrates. However, we cannot exclude the possibility that KLHL21 may negatively regulate IKKβ by recruiting other unidentified proteins. Future studies are necessary to clarify this hypothesis.
KEAP1 was recently demonstrated to bind specifically to a unique (E/A)TGE motif in the KD of IKKβ through its Kelch domains (11, 12). KEAP1 promotes the degradation of IKKβ via the autophagic (11) or 26S proteasome (12) pathways and inhibits the phosphorylation of IKKβ in response to extracellular stimuli. Consistent with previous reports, mutation of the second glutamate to alanine (E39A) in this motif dramatically suppressed the interaction between IKKβ and KEAP1 (11, 12) but failed to disrupt the IKKβ-KLHL21 interaction. These findings imply that KLHL21 may bind to other motifs within the KD of IKKβ and probably does not compete with KEAP1 for binding to IKKβ endogenously. This possibility further confirms that the interaction between KLHL21 and IKKβ is specific.
IKKβ-mediated phosphorylation and degradation of IκBα lead to the nuclear translocation of NF-κB and the induced expression of its target genes, including NFKBIA (encoding IκBα protein). The IκBα protein then translocates to the nucleus and terminates the activity of NF-κB, thereby generating a negative feedback loop for NF-κB signaling and acting as an internal timer for the first phase of NF-κB activity. Reduced IκBα expression results in longer retention of NF-κB in the nucleus and enhanced expression of its late phase target genes (30, 31). In this study, overexpression of KLHL21 in TNFα-stimulated HEK293T cells resulted in the inhibition of IKKβ activation, leading to reduced phosphorylation and subsequent ubiquitin-dependent degradation of IκBα. However, the interaction between KLHL21 and IKKβ was dynamically attenuated upon TNFα treatment, which led to the release of IKKβ and its delayed activation. Thus, overexpression of KLHL21 is only sufficient to inhibit TNFα-induced nuclear translocation of p65 in the early phase, thus leading to reduced expression of IκBα (an immediate early response gene). It results in prolonged nuclear retention of NF-κB, which potentially accounts, at least in part, for the enhanced expression of TNFAIP3, CXCL1, and IL-8 in the relatively late phase (at 2 and 4 h post-TNFα treatment). But we also cannot exclude the possibility that KLHL21 may differentially regulate the expression of those NF-κB target genes through targeting other unidentified components in NF-κB signaling pathway. More detailed studies are needed to elucidate its mechanism.
Attenuated interactions between IKKβ and its regulators in response to extracellular stimuli have also been reported in many other cellular contexts (8, 9, 13). However, the differential regulation of the expression of NF-κB target genes appears to be unique to KLHL21. This information extends our understanding of NF-κB signaling regulation. Differential modulation of NF-κB target gene expression by KLHL21 may also reflect a unique function of IKKβ in NF-κB signaling. IKKβ deficiency was recently shown to affect NF-κB target gene expression selectively (32). CCL2, TNFAIP3, and TNF demonstrated normal or slightly reduced expression levels, whereas CCL5, CXCL3, and CXCL10 had markedly reduced expression levels in IKKβ-deficient human primary fibroblasts in response to TNFα treatment (32).
Interestingly, KLHL21 expression was reported in the NCBI GEO database to be up-regulated in synovial macrophages from patients with rheumatoid arthritis (RA), a chronic inflammatory joint disease (GEO accession number GDS3192) (33). TNFα levels are elevated in synovial fluid from RA patients in proportion to cartilage destruction (34). As a master proinflammatory cytokine, TNFα induces the expression of other cytokines, such as IL-8, IL-1β, and IL-6, all of which are also up-regulated in the synovium of RA patients (34). Based on our finding that overexpression of KLHL21 leads to the up-regulation of IL-8 and TNFAIP3 in response to prolonged TNFα treatment, KLHL21 up-regulation in synovial macrophages from RA patients may also play a role in the induction of IL-8, IL-1β, and other proinflammatory cytokines. Further detailed studies are needed to decipher the potential relationship between KLHL21 and RA development.
In conclusion, we have identified KLHL21 as a novel negative regulator of IKKβ. KLHL21 specifically binds to the KD of IKKβ through its Kelch domains and inhibits IKKβ activation. The rapid down-regulation of KLHL21 in macrophages treated with diverse proinflammatory stimuli and the dynamically attenuated interaction between IKKβ and KLHL21 after TNFα stimulation suggest that IKKβ activation is tightly controlled by KLHL21 under physiological conditions (Fig. 9).
FIGURE 9.
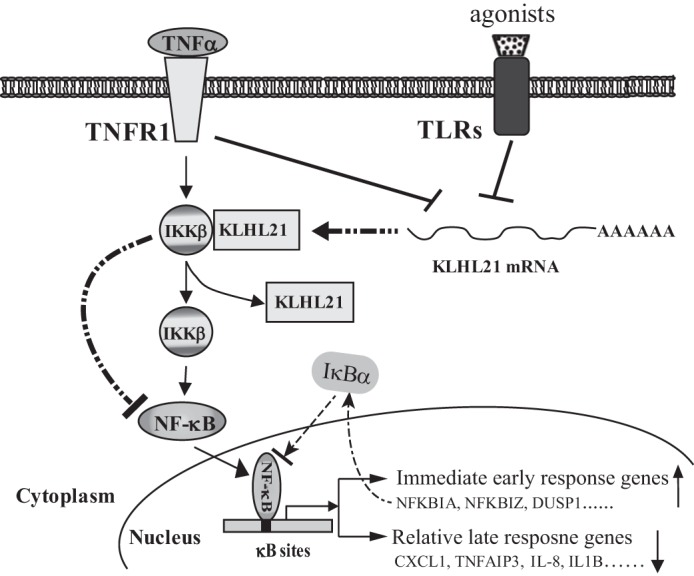
Proposed model illustrating how KLHL21 modulates the NF-κB signaling pathway via targeting IKKβ. In resting cells, KLHL21 protein specifically binds to IKKβ and inhibits its activation, which leads to the suppression of NF-κB signaling. Their interaction is gradually attenuated in response to TNFα treatment, which leads to the release of IKKβ and the activation of the NF-κB signaling pathway. This results in the up-regulation of those immediate early response genes, including NFKBIA (encoding IκBα). Up-regulation of IκBα leads to the termination of NF-κB signaling and down-regulation of its relative late phase target genes, such as CXCL1, IL-8, and IL-1B. Furthermore, the expression of KLHL21 is rapidly down-regulated in macrophages upon extracellular proinflammatory stimuli treatment, which also releases its inhibitory effects on IKKβ activation.
Experimental Procedures
Materials
Rabbit monoclonal antibodies against IKKβ (catalog no. 1601-1, lot YD012503), Lamin B (catalog no. 6581-1, lot YJ022918S), Cul3 (catalog no. 2489-1, lot YF052714), and phospho-IκBα (S32) (catalog no. 2798-1, lot YG071308C) were purchased from Epitomics (Burlingame, CA). Rabbit anti-KLHL21 (catalog no. 16952-1-AP, lot 10013204), anti-β-actin (catalog no. 20536-1-AP, lot 10001772), and anti-β-tubulin (catalog no. 10068-1-AP, lot 1001352) polyclonal antibodies were from Proteintech Biotechnology (Wuhan, China). Mouse anti-IκBα (L35A5) (catalog no. 4814, lot 10) and anti-ubiquitin (P4D1) (catalog no. 3936, lot 13), rabbit anti-phospho-IKKα (Ser-176)/IKKβ (Ser-177) (C84E11) (catalog no. 2078, lot 13), and anti-NF-κB p65 (D14E12) (catalog no. 8242, lot 4) antibodies were obtained from Cell Signaling Technology (Beverly, MA). Polyclonal anti-KLHL21 (catalog no. AB111604, lot GR173841-1) was obtained from Abcam (Cambridge, MA). Mouse anti-mCherry (catalog no. E022110, lot MY2301) was obtained from Earthox (Beijing, China). Monoclonal anti-FLAG M2 (catalog no. F3165, lot SLBH1191V) was from Sigma-Aldrich. Rabbit anti-EE (Glu-Glu) antibody (catalog no. A190-111A, lot A190-111A-3) was from Bethyl Laboratories (Montgomery, TX). The specificity of all antibodies was confirmed by Western blotting.
Protein A/G-agarose beads were purchased from Sangon Biotech (Shanghai, China). TRIzol reagent, polyclonal goat anti-rabbit Alexa-488 antibody, and Lipofectamine RNAiMAX were obtained from Life Technologies, Inc. LPS (from Escherichia coli 0111:B4) and phosphatase inhibitor mixture were obtained from Sigma-Aldrich. The Dual-Luciferase Reporter Assay system was purchased from Promega (Madison, WI). FastStart Universal SYBR Green Master (Rox) and EDTA-free complete protease inhibitors were purchased from Roche Diagnostics GmbH (Mannheim, Germany). SuperSignal chemiluminescence reagents and NE-PER nuclear and cytoplasmic extraction reagents were obtained from Pierce. ReverTra Ace® quantitative RT-PCR kit was purchased from Toyobo Life Science (Osaka, Japan). Pam3CSK4 and pam2CSK4 were obtained from Invivogen (San Diego, CA). Recombinant human and mouse TNFα, IL-1β, and M-CSF were purchased from PeproTech (Rocky Hill, NJ).
Cell Culture and Stimulation
RAW264.7 cells and human embryonic kidney (HEK) 293T cell lines were cultured in DMEM (Life Technologies) supplemented with 10% fetal bovine serum in a humidified atmosphere of 5% CO2 at 37 °C. Bone marrow-derived macrophages were prepared as adherent cultures using procedures described previously (35). In selected experiments, cells were treated with 100 ng/ml LPS, 100 ng/ml pam3CSK4, and 100 ng/ml pam2CSK4, 5 μm ODN1826, or 20 ng/ml TNFα for NF-κB activation respectively.
cDNAs and Site-directed Mutagenesis
Total RNA was extracted from RAW264.7 cells and reverse transcribed into cDNA. It was used to amplify the cDNAs of mouse Klhl21, Traf2, Traf6, Tbk1, Ikkβ, p65 (RelA), and Keap1. These cDNAs were subcloned into pcDNA3 expression plasmids with appropriate 3′ tag 3×FLAG (3F), Glu-Glu (EE), or mCherry (MC), respectively, as specified. Point mutations were introduced in the expression plasmids using the QuikChange site-directed mutagenesis protocol (Stratagene) and verified by DNA sequencing.
Luciferase Assay
Luciferase reporter plasmid pNFκB-TA-luc, which contains 6×κB binding sites and a minimal TA promoter, was obtained from Beyotime Biotechnology (Jiangsu, China). HEK293T cells were transfected with 0.2 μg of pNFκB-TA-luc plus 0.02 μg of Renilla reporter pRL-TK as an internal control and various amounts of Klhl21, Traf2, Traf6, Tbk1, Ikkβ (SS/EE), and p65 (RelA) expression vectors, respectively. 24 h after the transfection, the cells were harvested and subjected to the Dual-Luciferase Reporter Assay system according to the manufacturer's recommendations.
Quantitative Real-time PCR Analysis
Total RNA was extracted by using TRIzol reagent according to the manufacturer's instruction. RNA was reverse transcribed by using the ReverTra Ace quantitative RT-PCR kit. For detection and quantification, PCRs were performed using the FastStart Universal SYBR Green Master (Rox) (Roche Diagnostics) and the 7500 real-time PCR system (Applied Biosystems). The primers for quantitative PCR are listed in Table 1.
TABLE 1.
List of primers used in the quantitative PCR analysis
| Gene name | Primers | Sequences (5′–3′) |
|---|---|---|
| Mouse β-actin | Forward | ACGGCCAGGTCATCACTATTG |
| Reverse | AGAGGTCTTTACGGATGTCAACGT | |
| Mouse Klhl21 | Forward | CTCCCACCGTGGTCCTTTG |
| Reverse | GATCTTGTCCCATTCGTCTTTAGTG | |
| Mouse Nfkbia | Forward | GAAGAGAAGCCGCTGACCAT |
| Reverse | CAGAAGTGCCTCAGCAATTCC | |
| Mouse Tnfaip3 | Forward | AGCTCGTGGCTCTGAAAACC |
| Reverse | TTCCTCAGGACCAGGTCAGTATC | |
| Mouse Il-1b | Forward | TGACAGTGATGAGAATGACCTGTTC |
| Reverse | TGCTGCTGCGAGATTTGAAG | |
| Human β-actin | Forward | CGCCAACACAGTGCTGTCT |
| Reverse | TCAGGAGGAGCAATGATCTTG | |
| Human KLHL21 | Forward | CCCAAGACTGCGACTCTAAACG |
| Reverse | GATTCATGGACGGGATCTTGTC | |
| Human NFKBIA | Forward | AAGTGATCCGCCAGGTGAAG |
| Reverse | AGCAATTTCTGGCTGGTTGGT | |
| Human TNFAIP3 | Forward | GAATGCTACAGATACCCCATTGTTCT |
| Reverse | GTGGAACAGCTCGGATTTCAG | |
| Human IL-8 | Forward | CTTTCCACCCCAAATTTATCAAAG |
| Reverse | GGGTCCAGACAGAGCTCTCTTC | |
| Human DUSP1 | Forward | GCAGTACCCCACTCTACGATCAG |
| Reverse | GACGTTGATCAAGGCAGTGATG | |
| Human NFKBIZ | Forward | ACCTCTGCATGTGTGTGCTGAG |
| Reverse | ACTGATTACTTCCCACTGCTCCCTT | |
| Human CXCL1 | Forward | CAATCCTGCATCCCCCATAG |
| Reverse | ACAGCCACCAGTGAGCTTCCT |
Coimmunoprecipitation, GST Pull-down, and Immunoblotting
Cells transfected with various plasmids as specified were incubated for 24 h before analysis. Cell lysate preparation, immunoprecipitation, and immunoblotting analysis were done as described previously (36). Nuclear proteins were isolated using NE-PER nuclear and cytoplasmic extraction reagents according to the manufacturer's instructions (Pierce). For detection of ubiquitinated proteins in vivo, transfected cells were treated with 5 μm MG132 for 16 h before collection. Then the cells were rapidly lysed by boiling in a buffer containing 2% SDS, 150 mm NaCl, 10 mm Tris-HCl, and 1 mm DTT. This will inactivate cellular ubiquitin hydrolases and therefore preserve ubiquitin-protein conjugates present in cells before lysis. For immunoprecipitation, these lysates were diluted 5-fold in buffer lacking SDS and incubated with anti-FLAG antibody. Immunoprecipitated proteins were analyzed by immunoblotting with anti-FLAG and anti-ubiquitin antibodies. The blots were developed using Supersignal chemiluminescence reagents, and images were captured with an Eastman Kodak Co. Image Station 1000.
pGEX4T plasmid containing the C-terminal BACK and Kelch domains of mouse KLHL21 (amino acids 134–597) was introduced into E. coli BL21 (DE3) strain, and expression of the GST fusion protein was induced by the addition of 0.1 m isopropyl β-d-1-thiogalactopyranoside at 25 °C for 4 h. Whole cell lysates of HEK293T and RAW264.7 were incubated with GST-KLHL21 protein purified with glutathione-Sepharose 4B for 4 h at 4 °C. The beads were washed four times with cell lysis buffer, and the bound proteins were analyzed by Western blotting with anti-IKKβ antibody.
RNA Interference and Rescue
Chemosynthesized siRNAs CGTCCATGAATCAGGTACA (siRNA 1), GCATCTTCCGCCAGTTCAT (siRNA 2), and GCTGGGCAATGACATCTAC (siRNA 3) targeting human KLHL21; siRNAs GGTACGACAACACCTTTGA (siRNA 4) and GCTGAACAGTGGTAGCAAT (siRNA 5) targeting mouse KLHL21; and TTCTCCGAACGTGTCACGT (negative control siRNA) were all obtained from GenePharma (Shanghai, China). To achieve efficient knockdown and limit off-target effects, a pool of siRNA oligonucleotides was used by mixing the different siRNAs at equal concentration. The siRNAs were transfected at the specified concentrations by Lipofectamine RNAiMax. For siRNA rescue experiments, HEK293T cells were cotransfected with chemosynthesized siRNA 3, which does not target mouse KLHL21 mRNA, and mCherry-tagged wild type or enzymatically inactive mutant (KLHL21M) of mouse KLHL21 construct by using jetPRIME (PolyPlus) according to the manufacturer's protocol. At 24 h post-transfection, the cells were treated with TNFα or LPS and harvested to analyze the expression of KLHL21 and IκBα protein by Western blotting or the mRNA level of KLHL21, NFKBIA, TNFAIP3, and IL-8 by real-time PCR as specified.
Fluorescent Confocal Microscopy
HeLa cells were grown on poly-l-lysine-coated glass coverslips and transfected C-terminal mCherry-tagged KLHL21 by using jetPRIME (PolyPlus) according to the manufacturer's protocol. At 24 h post-transfection, the cells were fixed with 4% formaldehyde for 15 min, followed by blocking with 5% normal goat serum for 1 h. Anti-IKKβ antibody was used for detecting endogenous IKKβ. Goat anti-rabbit Alexa Fluor 488 was used as secondary antibody. Confocal microscopy images were obtained using Zeiss LSM 700 confocal microscope (Carl Zeiss) with the excitation source at 488 and 587 nm for Alexa Fluor 488 and mCherry, respectively. The acquired images were processed in ZEN 2012.
Statistical Analysis
Unless otherwise indicated, experiments were performed in triplicate and repeated at least three times. The figures show the results from one representative experiment. The intensity of the protein bands was determined by densitometry using ImageJ. All data are presented as means ± S.E. Data sets were tested using two-tailed Student's t test. Statistical analyses were carried out using Origin version 8 software (OriginLab). Differences were considered statistically significant for values of p < 0.05.
Author Contributions
Z.-Z. M., J. L., and Y. J. conceived and designed the experiments. Z.-Z. M., X.-Y. C., S.-W. H., N. W., X.-L. O., J. W., H.-H. L., and J. L. performed the experiments. Z.-Z. M., J. L., and Y. J. analyzed the data. Z.-Z. M., J. L., and Y. J. wrote the manuscript.
Acknowledgments
We thank Dr. Jianghuai Wang (University College Cork) and Dr. Timothy R. Billiar (University of Pittsburgh) for critical reading and editing of the manuscript.
This work was supported by National Natural Science Foundation of China Grants 81272149 (to J. L.) and 81030055 and 81372030 (to Y. J.); Science and Technology Innovation Projects of the Department of Education of Guangdong Province Grant 2013KJCX0040 (to Z. Z. M.); and Project Supported by Guangdong Natural Science Foundation Grant 2015A030313294 (to Z. Z. M.). The authors declare that they have no conflicts of interest with the contents of this article.
- NF-κB
- nuclear factor κ-light chain enhancer of activated B cells
- IKK
- IκB kinase
- EE tag
- Glu-Glu tag
- NEMO
- NF-κB essential modulator
- KD
- kinase domain
- RA
- rheumatoid arthritis.
References
- 1. Baltimore D. (2011) NF-κB is 25. Nat. Immunol. 12, 683–685 [DOI] [PubMed] [Google Scholar]
- 2. Hayden M. S., and Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 3. Smale S. T. (2011) Hierarchies of NF-κB target-gene regulation. Nat. Immunol. 12, 689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu F., Xia Y., Parker A. S., and Verma I. M. (2012) IKK biology. Immunol. Rev. 246, 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schröfelbauer B., Polley S., Behar M., Ghosh G., and Hoffmann A. (2012) NEMO ensures signaling specificity of the pleiotropic IKKβ by directing its kinase activity toward IκBα. Mol. Cell 47, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang F., Tang E., Guan K., Wang C. Y. (2003) IKKβ plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 170, 5630–5635 [DOI] [PubMed] [Google Scholar]
- 7. Kim Y. L., Kim J. E., Shin K. J., Lee S., Ahn C., Chung J., Kim D. H., Seong J. Y., and Hwang J. I. (2008) GβL regulates TNFα-induced NF-κB signaling by directly inhibiting the activation of IκB kinase. Cell. Signal. 20, 2127–2133 [DOI] [PubMed] [Google Scholar]
- 8. Li H. Y., Liu H., Wang C. H., Zhang J. Y., Man J. H., Gao Y. F., Zhang P. J., Li W. H., Zhao J., Pan X., Zhou T., Gong W. L., Li A. L., and Zhang X. M. (2008) Deactivation of the kinase IKK by CUEDC2 through recruitment of the phosphatase PP1. Nat. Immunol. 9, 533–541 [DOI] [PubMed] [Google Scholar]
- 9. Xia X., Cui J., Wang H. Y., Zhu L., Matsueda S., Wang Q., Yang X., Hong J., Songyang Z., Chen Z. J., and Wang R. F. (2011) NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 34, 843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cui J., Zhu L., Xia X., Wang H. Y., Legras X., Hong J., Ji J., Shen P., Zheng S., Chen Z. J., and Wang R. F. (2010) NLRC5 negatively regulates the NF-κB and type I interferon signaling pathways. Cell 141, 483–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim J. E., You D. J., Lee C., Ahn C., Seong J. Y., and Hwang J. I. (2010) Suppression of NF-κB signaling by KEAP1 regulation of IKKβ activity through autophagic degradation and inhibition of phosphorylation. Cell. Signal. 22, 1645–1654 [DOI] [PubMed] [Google Scholar]
- 12. Lee D. F., Kuo H. P., Liu M., Chou C. K., Xia W., Du Y., Shen J., Chen C. T., Huo L., Hsu M. C., Li C. W., Ding Q., Liao T. L., Lai C. C., Lin A. C., et al. (2009) KEAP1 E3 ligase-mediated downregulation of NF-κB signaling by targeting IKKβ, Mol. Cell. 36, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li S. Z., Zhang H. H., Liang J. B., Song Y., Jin B. X., Xing N. N., Fan G. C., Du R. L., and Zhang X. D. (2014) Nemo-like kinase (NLK) negatively regulates NF-κB activity through disrupting the interaction of TAK1 with IKKβ. Biochim. Biophys. Acta 1843, 1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yao F., Long L. Y., Deng Y. Z., Feng Y. Y., Ying G. Y., Bao W. D., Li G., Guan D. X., Zhu Y. Q., Li J. J., and Xie D. (2014) RACK1 modulates NF-κB activation by interfering with the interaction between TRAF2 and the IKK complex. Cell Res. 24, 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bradford J. W., and Baldwin A. S. (2014) IKK/nuclear factor-κB and oncogenesis: roles in tumor-initiating cells and in the tumor microenvironment. Adv. Cancer Res. 121, 125–145 [DOI] [PubMed] [Google Scholar]
- 16. Dhanoa B. S., Cogliati T., Satish A. G., Bruford E. A., and Friedman J. S. (2013) Update on the Kelch-like (KLHL) gene family, Hum. Genomics 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Angers S., Thorpe C. J., Biechele T. L., Goldenberg S. J., Zheng N., MacCoss M. J., Moon R. T. (2006) The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-β-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol. 8, 348–357 [DOI] [PubMed] [Google Scholar]
- 18. Sumara I., Quadroni M., Frei C., Olma M. H., Sumara G., Ricci R., Peter M. (2007) A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev. Cell. 12, 887–900 [DOI] [PubMed] [Google Scholar]
- 19. Kroll J., Shi X., Caprioli A., Liu H. H., Waskow C., Lin K. M., Miyazaki T., Rodewald H. R., and Sato T. N. (2005) The BTB-kelch protein KLHL6 is involved in B-lymphocyte antigen receptor signaling and germinal center formation. Mol. Cell. Biol. 25, 8531–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maerki S., Olma M. H., Staubli T., Steigemann P., Gerlich D. W., Quadroni M., Sumara I., and Peter M. (2009) The Cul3-KLHL21 E3 ubiquitin ligase targets aurora B to midzone microtubules in anaphase and is required for cytokinesis. J. Cell Biol. 187, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murray R. Z., Wylie F. G., Khromykh T., Hume D. A., and Stow J. L. (2005) Syntaxin 6 and Vti1b form a novel SNARE complex, which is up-regulated in activated macrophages to facilitate exocytosis of tumor necrosis factor-α. J. Biol. Chem. 280, 10478–10483 [DOI] [PubMed] [Google Scholar]
- 22. Macagno A., Molteni M., Rinaldi A., Bertoni F., Lanzavecchia A., Rossetti C., and Sallusto F. (2006) A cyanobacterial LPS antagonist prevents endotoxin shock and blocks sustained TLR4 stimulation required for cytokine expression. J. Exp. Med. 203, 1481–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kane C. M., Cervi L., Sun J., McKee A. S., Masek K. S., Shapira S., Hunter C. A., Pearce E. J. (2004) Helminth antigens modulate TLR-initiated dendritic cell activation. J. Immunol. 173, 7454–7461 [DOI] [PubMed] [Google Scholar]
- 24. Stolovitzky G. A., Kundaje A., Held G. A., Duggar K. H., Haudenschild C. D., Zhou D., Vasicek T. J., Smith K. D., Aderem A., and Roach J. C. (2005) Statistical analysis of MPSS measurements: application to the study of LPS-activated macrophage gene expression. Proc. Natl. Acad. Sci. U.S.A. 102, 1402–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stetson D. B., and Medzhitov R. (2006) Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 [DOI] [PubMed] [Google Scholar]
- 26. Hong Y. K., Foreman K., Shin J. W., Hirakawa S., Curry C. L., Sage D. R., Libermann T., Dezube B. J., Fingeroth J. D., Detmar M. (2004) Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 36, 683–685 [DOI] [PubMed] [Google Scholar]
- 27. Xu G., Lo Y. C., Li Q., Napolitano G., Wu X., Jiang X., Dreano M., Karin M., and Wu H. (2011) Crystal structure of inhibitor of κB kinase β. Nature 472, 325–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Djiane A., Shimizu H., Wilkin M., Mazleyrat S., Jennings M. D., Avis J., Bray S., and Baron M. (2011) Su(dx) E3 ubiquitin ligase-dependent and -independent functions of polychaetoid, the Drosophila ZO-1 homologue. J. Cell Biol. 192, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu G., Wang X., Saunders D. N., Henderson M., Russell A. J., Herring B. P., and Zhou J. (2010) Modulation of myocardin function by the ubiquitin E3 ligase UBR5. J. Biol. Chem. 285, 11800–11809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Werner S. L., Kearns J. D., Zadorozhnaya V., Lynch C., O'Dea E., Boldin M. P., Ma A., Baltimore D., and Hoffmann A. (2008) Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20, Genes Dev. 22, 2093–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klement J. F., Rice N. R., Car B. D., Abbondanzo S. J., Powers G. D., Bhatt P. H., Chen C. H., Rosen C. A., and Stewart C. L. (1996) IκBα deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol. Cell. Biol. 16, 2341–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pannicke U., Baumann B., Fuchs S., Henneke P., Rensing-Ehl A., Rizzi M., Janda A., Hese K., Schlesier M., Holzmann K., Borte S., Laux C., Rump E. M., Rosenberg A., Zelinski T., et al. (2013) Deficiency of innate and acquired immunity caused by an IKBKB mutation. N. Engl. J. Med. 369, 2504–2514 [DOI] [PubMed] [Google Scholar]
- 33. Yarilina A., Park-Min K. H., Antoniv T., Hu X., Ivashkiv L. B. (2008) TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 9, 378–387 [DOI] [PubMed] [Google Scholar]
- 34. Siebert S., Tsoukas A., Robertson J., McInnes I. (2015) Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacol Rev. 67, 280–309 [DOI] [PubMed] [Google Scholar]
- 35. Liu J., Buckley J. M., Redmond H. P., and Wang J. H. (2010) ST2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J. Immunol. 184, 5802–5808 [DOI] [PubMed] [Google Scholar]
- 36. Mei Z. Z., Xia R., Beech D. J., Jiang L. H. (2006) Intracellular coiled-coil domain engaged in subunit interaction and assembly of melastatin-related transient receptor potential channel 2. J. Biol. Chem. 281, 38748–38756 [DOI] [PMC free article] [PubMed] [Google Scholar]