

Abstract
Interferon signaling plays important roles in both intestinal homeostasis and in the host response to pathogen infection. The extent to which bacterial pathogens inhibit this host pathway is an understudied area of investigation. We characterized Citrobacter rodentium strains bearing deletions in individual type III secretion system effector genes to determine whether this pathogen inhibits the host type I IFN response and which effector is responsible. The NleB effector limited host IFN-β production by inhibiting Lys63-linked ubiquitination of TNF receptor-associated factor 3 (TRAF3). Inhibition was dependent on the glycosyltransferase activity of NleB. GAPDH, a target of NleB during infection, bound to TRAF3 and was required for maximal TRAF3 ubiquitination. NleB glycosyltransferase activity inhibited GAPDH-TRAF3 binding, resulting in reduced TRAF3 ubiquitination. Collectively, our data reveal important interplay between GAPDH and TRAF3 and suggest a mechanism by which the NleB effector inhibits type I IFN signaling.
Keywords: bacterial pathogenesis, infection, innate immunity, interferon, signal transduction
Introduction
Many bacterial pathogens utilize a type III secretion system (T3SS)2 to inject virulence proteins (effectors) into host cells to subvert various host functions (1). Characterizing the identity and mechanism of bacterial effectors expressed by attaching/effacing pathogens, which include enterohemorrhagic Escherichia coli (EHEC), enteropathogenic E. coli (EPEC), and Citrobacter rodentium, has been a subject of extensive research in recent years (2). Effector subversion of host pathways regulated by the transcription factor NF-κB has been relatively well studied (3), but less attention has been given to the potential inhibition of host interferon signaling, which also functions as an important early mediator of host defense.
Virally infected cells secrete interferon to protect other cells from subsequent infection (4). Type I IFNs (e.g. IFN-α and IFN-β) bind to a common IFN-α/β receptor (5), initiating a signal transduction cascade that ultimately induces the transcription of genes with promoters containing IFN-stimulated response elements (6). Type I IFNs induce antiviral activity via double-stranded RNA-dependent protein kinase and 2′,5′-oligoadenylate synthetase to promote viral mRNA degradation and inhibit translation, respectively (7).
Type I IFN production is stimulated not only by viral infection but also by bacterial components, including LPS and flagellin (8). LPS binding to Toll-like receptor 4 (TLR4) induces a type I IFN response mediated by the TRIF-related adaptor molecule and TIR-domain-containing adapter-inducing interferon-β adaptor proteins (9). TRIF-related adaptor molecule-TIR-domain-containing adapter-inducing interferon-β signaling results in the activation of TRAF family member-associated NF-κB activator-binding kinase (TBK1) and inducible IκB kinase (IKKϵ (10)). Serine phosphorylation of interferon-regulatory factor 3 (IRF3) is mediated by TBK1/IKKϵ to induce IRF3 dimerization and nuclear translocation (11). Phosphorylated IRF3 (and IRF7) dimers then associate with AP-1, high-mobility group proteins, and NF-κB to form an IFN-β enhanceasome (12).
IFNs are important for maintaining intestinal homeostasis and for responding to pathogen infection (13). Accordingly, pathogens have also evolved strategies to interfere with host type I IFN signaling. For example, the anthrax lethal factor inhibits host IFN production by cleaving MAPKK6 (14) and by inhibiting STAT1 and ISGF3 activation (15). The deubiquitinating protease activity of Yersinia YopJ blocks TLR induction of type I IFN at the level of TAK1 activation by inhibiting TRAF3/6 ubiquitination (16). Vaccinia virus protein K7 prevents IRF3 activation by inhibiting the interaction between DEAD box protein 3 and IKKϵ (17). The Ebola virus VP35 protein blocks TBK1/IKKϵ interaction with IRF3/7 (18).
A recent study found that IFN-β is induced by EPEC infection and that the T3SS effector NleD both inhibits IFN-β induction and enhances TNF expression to promote barrier disruption (19). However, whether C. rodentium T3SS effectors may also inhibit host IFN-β induction is unclear. Here, we screened C. rodentium strains bearing deletions in individual effector genes to determine the extent to which this pathogen might inhibit the host IFN-β response. We found that the T3SS effector NleB limits host IFN-β production by inhibiting TRAF3 Lys63-linked ubiquitination.
Results
NleB Inhibits the Generation of a Type I IFN Response
To determine whether C. rodentium T3SS effectors inhibit the host type I IFN response, we monitored the survival of a recombinant vesicular stomatitis virus (VSV-G) expressing GFP (20). HeLa cells were first infected with C. rodentium strains possessing (WT) or lacking (ΔescN) a functional T3SS for 2 h or were instead treated with LPS for 30 min as a positive control. HeLa cell supernatants were then removed, filtered, and applied to Vero cells, which are highly responsive to exogenous IFN-β. After incubation for 12 h with the supernatants, the Vero cells were infected with VSV-G/GFP for 30 h. Cells were then fixed, and viral loads were quantified by measuring GFP intensity.
Transferring supernatants from cells infected with C. rodentium ΔescN inhibited VSV-G/GFP to levels similar to those induced by LPS treatment. By contrast, transferring supernatants from cells infected with WT C. rodentium had no discernible ability to inhibit VSV-GFP growth (Fig. 1A). These data suggested that a T3SS effector inhibits the production of a host factor involved in virus inhibition. We then infected HeLa cells with C. rodentium strains lacking individual T3SS effector genes and screened the cell supernatants for antiviral activity. Among the 12 individual effector deletion strains screened (Fig. 1B), ΔnleB inhibited virus replication most significantly (Figs. 1, A and B), with a minor phenotype also observed for ΔnleE.
FIGURE 1.

NleB inhibits the generation of a type I IFN response. A, HeLa cells were infected with C. rodentium strains for 2 h or treated with LPS (100 ng/ml) for 30 min. Cell supernatants were removed, filtered, and applied to Vero cells for 12 h, which were then infected with VSV-G/GFP (multiplicity of infection ∼5) for 30 h. Where indicated, Vero cells were pretreated with a type I IFN receptor blocking antibody (Ab, IFNAR2) before adding culture supernatants. B, representative images from screening multiple C. rodentium effector deletion strains. C, quantification of VSV-G/GFP intensity (arbitrary units) ± IFNAR2 antibody, normalized to DAPI intensity. *, significantly different VSV-GFP intensity compared with WT infection (one-way ANOVA, n = 3).
To test the hypothesis that this phenotype was due to the release of type I IFN from the infected HeLa cells, we used an antibody that blocks the IFN-α/β receptor. Pretreating Vero cells with this antibody blocked the ability of HeLa cell supernatants to inhibit virus replication (Fig. 1C). Thus, C. rodentium induces a host IFN-β response, and the T3SS effector NleB blocks the transduction of this response.
NleB Inhibits IFN-β and RANTES Gene Transcription
We then conducted in vitro experiments to determine the impact of NleB on IFN-β signaling pathways. To determine whether NleB inhibits transcription of the IFN-β gene, we performed quantitative RT-PCR analysis of IFNB1 transcript abundance after infection with WT, ΔescN, or ΔnleB C. rodentium. Although infection with ΔescN stimulated IFNB1 expression similar to LPS treatment, infection with the WT did not (Fig. 2A). This phenotype was attributable to NleB, as deleting nleB relieved the inhibition of IFNB1 gene transcription observed with WT C. rodentium. We obtained similar data for the chemokine gene RANTES (CCL5, Fig. 2B).
FIGURE 2.
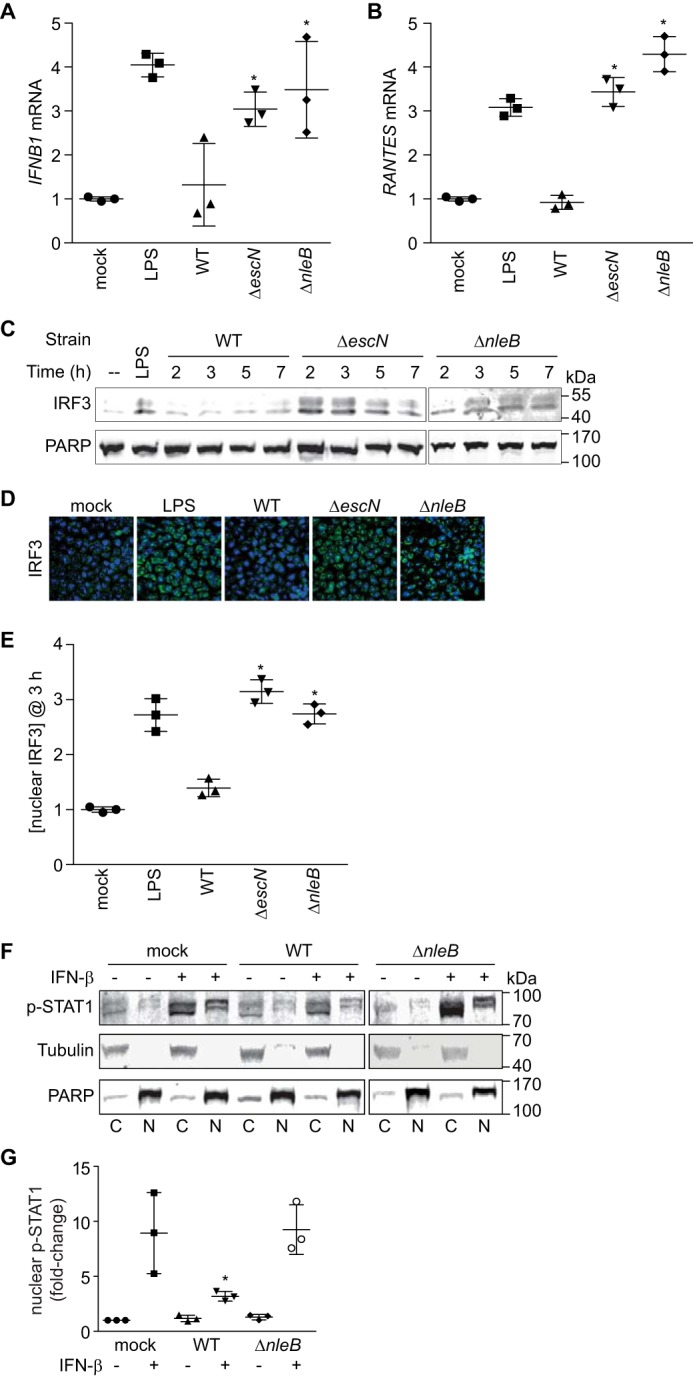
NleB inhibits IFN-β signaling. A, quantification of IFN-β mRNA abundance after LPS treatment or after 3-h C. rodentium infection. *, significantly different [ifnb1] compared with WT infection (one-way ANOVA, n = 3). B, quantification of RANTES mRNA levels. *, significantly different [rantes] compared with WT infection (one-way ANOVA, n = 3). C, IRF3 immunoblotting of nuclear extracts derived from HeLa cells infected with C. rodentium strains versus time. D, immunofluorescence microscopy analysis of IRF3 (green) and cell nuclei (blue) after LPS stimulation or C. rodentium infection. E, relative abundance of nuclear IRF3 (3 h post-infection) normalized to poly(ADP-ribose) polymerase. *, significantly different nuclear [IRF3] compared with WT infection (one-way ANOVA, n = 3). F, immunoblot analysis of cytoplasmic (C) and nuclear (N) fractions of HeLa cells infected with C. rodentium WT or ΔnleB or mock-infected with PBS in the presence or absence of IFN-β (5000 units/ml) stimulation for 30 min. Protein mobility differences between cytoplasmic and nuclear fractions are due to the differing [NaCl] in buffers used for cellular fractionation. G, quantification of the –fold change in nuclear p-STAT1 in the absence or presence of IFN-β stimulation followed by C. rodentium infection. p-STAT1 signal intensity was normalized to tubulin (cytoplasmic) and poly(ADP-ribose) polymerase (nuclear). *, significantly different nuclear [p-STAT1] (one-way ANOVA, n = 3).
IRF3 is a transcription factor that binds IFN-β and other innate immune gene promoters (21). Because NleB inhibited IFNB1 and RANTES expression, which are both direct targets of IRF3, we tested whether IRF nuclear abundance was altered after bacterial infection. By fractionating cell lysates after bacterial infection, we observed that, although WT C. rodentium inhibited the accumulation of nuclear IRF3, infection with ΔescN and ΔnleB did not (Fig. 2C). We reached similar conclusions after using immunofluorescence microscopy and scoring the number of cells with predominantly nuclear IRF3 (Fig. 2, D and E).
NleB Reduces the Nuclear Abundance of STAT1
IFN-β stimulation results in the activation of the JAK-STAT pathway, which has a critical role in regulating the immune response to bacterial infection. After cytokine stimulation and activation of JAK, STATs, including STAT1, become phosphorylated and translocate to the nucleus, where they can activate the expression of immune response genes (22). To determine whether NleB inhibits p-STAT1 nuclear translocation, we evaluated the relative abundance p-STAT1 in HeLa cells infected with C. rodentium strains with or without IFN-β stimulation. We fractionated the cells to separate nuclear from cytoplasmic components. As expected, p-STAT1 nuclear abundance significantly increased after stimulation with IFN-β (Fig. 2, F and G). p-STAT1 nuclear abundance was reduced in samples infected with WT C. rodentium even after stimulation with IFN-β (Fig. 2, F and G). By contrast, ΔnleB C. rodentium failed to inhibit p-STAT1 nuclear translocation, indicating that NleB inhibits IFN-β-induced p-STAT1 nuclear translocation (Fig. 2, F and G).
NleB Inhibits Lys63 Ubiquitination of TRAF3
TLRs are pattern recognition receptors that sense microorganisms and are critical for the host innate immune system. A subset of liganded TLRs activates intrinsic signaling pathways to induce type I IFN production. We found that NleB also inhibited the activation of an IFN-β-dependent luciferase reporter in the presence of poly(I:C), a synthetic double-stranded RNA used for TLR3-dependent stimulation of type I IFN, in an NleB dose-dependent manner (Fig. 3A).
FIGURE 3.
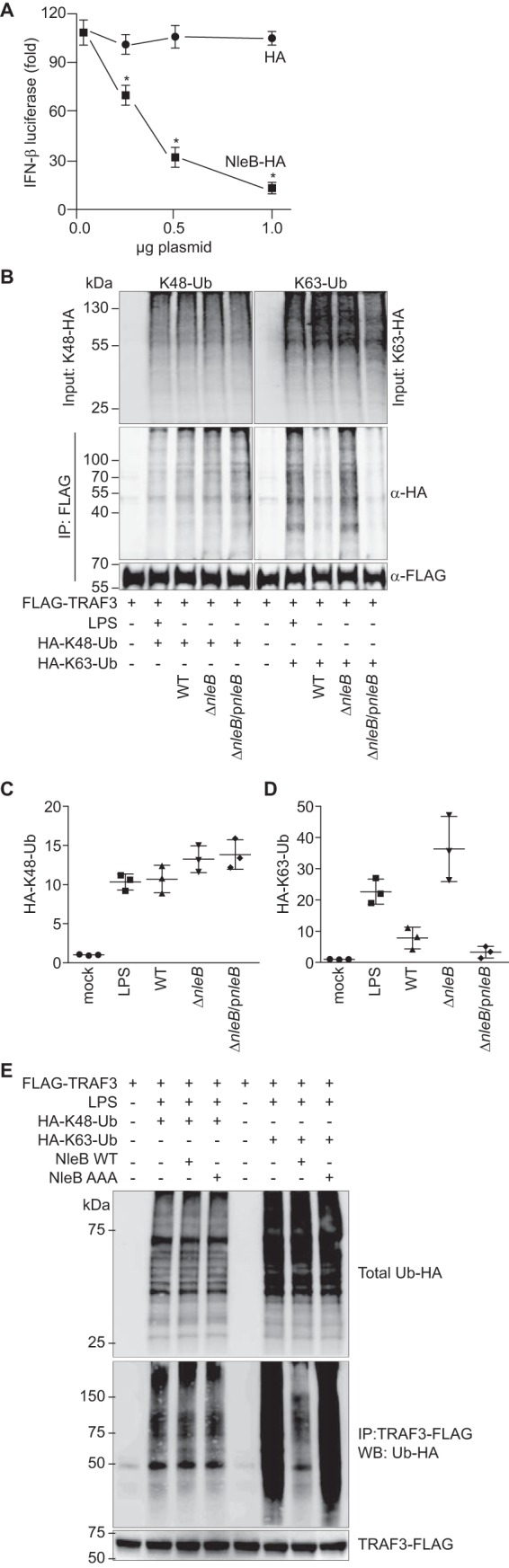
NleB inhibits Lys63-ubiquitination of TRAF3. A, cells were stimulated with 1.0 μg/ml poly(I:C) for 12 h. *, significantly different luciferase activity compared with HA control transfection (one-way ANOVA, n = 3). B, Raw 264.7 cells were transfected with TRAF3-FLAG and either treated with LPS (1.0 μg/ml, 30 min) or infected with C. rodentium strains for 3 h. Cells were lysed and immunoprecipitated using anti-FLAG antibody. Ubiquitination was assessed by immunoblotting for HA to detect ubiquitin (Ub) conjugates. C, quantification of Lys48-ubiquitin from the experiments depicted in B. D, quantification of Lys63-ubiquitin from the experiments depicted in B. *, significantly different ubiquitination levels compared with WT infection (one-way ANOVA, n = 3). E, Lys48 and Lys63 ubiquitination of TRAF3 as a function of NleB transfection. IP, immunoprecipitation; WB, Western blot.
Differential ubiquitination of TNF receptor (TNFR)-associated factors can selectively activate or suppress the expression of different groups of cytokines (23). For example, TRAF3 Lys63 ubiquitination typically activates IFN responses, whereas TRAF3 Lys48 ubiquitination mediated by TRAF6 typically induces pro-inflammatory cytokine expression (24). Lys48-ubiquitin linkages typically result in proteasome-mediated protein degradation, whereas Lys63-ubiquitin linkages are generally associated with regulating protein subcellular localization and activation (24).
We then hypothesized that NleB might inhibit TRAF3 ubiquitination. We transfected RAW264.7 cells with plasmids expressing TRAF3-FLAG and with ubiquitin-HA mutants that contain lysine only at a single position (e.g. Lys48 or Lys63). After 36 h, cells were treated with LPS (100 ng/ml, 30 min) to induce TRAF signaling. TRAF3-FLAG was immunoprecipitated, and these samples were immunoblotted for HA to detect ubiquitin conjugates. As expected, LPS treatment induced significant Lys48- and Lys63-linked TRAF3 ubiquitination (Fig. 3B). Quantification of these data is shown in Fig. 3, C and D.
Performing these studies with cells that were subsequently infected with C. rodentium strains possessing or lacking NleB revealed the selective impact of NleB on Lys63-linked TRAF3 ubiquitination. Although Lys48-linked TRAF3 ubiquitination did not differ among infection conditions, Lys63-linked TRAF3 ubiquitination was significantly inhibited in cells infected with either WT or ΔnleB/pnleB C. rodentium in contrast to its ubiquitination after infection with ΔnleB C. rodentium (Fig. 3, B–D). We corroborated these data by transfecting NleB in the absence of bacterial infection. Transfecting NleB into RAW264.7 cells inhibited Lys63-linked but not Lys48-linked TRAF3 ubiquitination (Fig. 3E). Overall, these data suggest that NleB selectively inhibits Lys63-linked TRAF3 ubiquitination.
NleB Targets GAPDH to Regulate Lys63 Ubiquitination of TRAF3
We showed previously that NleB functions as a translocated glycosyltransferase enzyme that covalently modifies host proteins with GlcNAc to subvert their normal functions (25). Specifically, we found that NleB disrupts TRAF2 signaling, leading to inhibition of the pro-inflammatory NF-κB pathway (25). We also revealed that the glycolysis enzyme GAPDH functions as a co-activator of TRAF2 activity. The modification of GAPDH with GlcNAc by NleB prevents GAPDH from binding to and activating TRAF2, leading to a reduced NF-κB response to infection (25).
To determine the extent to which the phenotypes we observed with TRAF3 were also dependent upon GAPDH, we first determined whether GAPDH interacts with TRAF3. By transfecting TRAF3-FLAG, we observed that GAPDH immunoprecipitated with TRAF3, similar to its ability to immunoprecipitate with TRAF2 (Fig. 4A). Co-incubation of GAPDH with NleB almost abolished the binding of GAPDH to TRAF3, at low concentrations of TRAF3, as shown in pulldown assays and in ELISAs (Fig. 4, B and C), similar to our previous observations with GAPDH binding to TRAF2 (25). Although a ∼20% complex formation in the presence of NleB was achieved, this is contrast to ∼100% complex formation in the absence of NleB (Fig. 4C).
FIGURE 4.
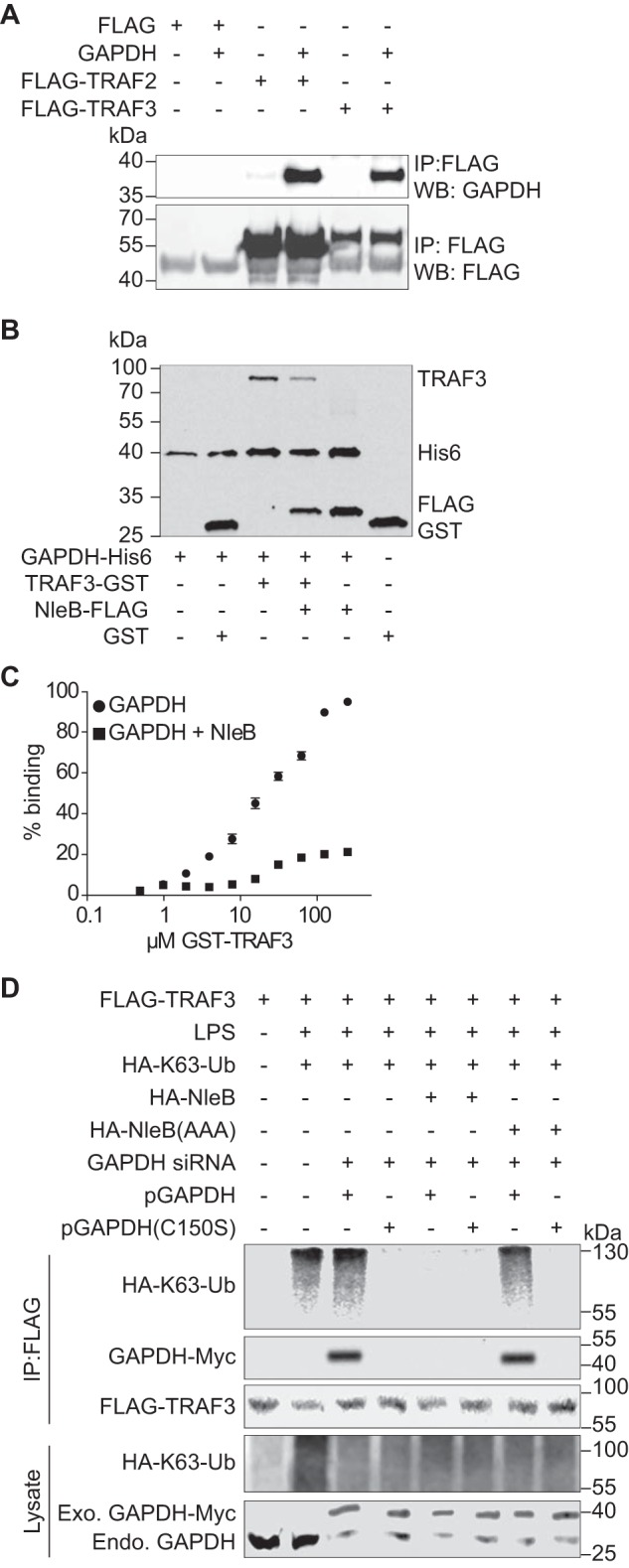
Role of GAPDH in TRAF3 ubiquitination. A, HEK293T cells were co-transfected with Myc-GAPDH and FLAG-TRAF2 or FLAG-TRAF3. After 48 h, cell lysates were immunoprecipitated (IP) with anti-FLAG antibody (Sigma), followed by immunoblotting with anti-GAPDH or anti-FLAG antibody. WB, Western blot. B, His-GAPDH immobilized to nickel-nitrilotriacetic acid-agarose beads were incubated with GST, GST-TRAF3, FLAG-NleB, or GST-TRAF3 + FLAG-NleB. Protein interactions were analyzed using immunoblotting. C, Immulon-2 plates were coated with 1 μg of His-GAPDH and overlaid with GST-TRAF3 in the presence or absence of FLAG-NleB. Protein binding was detected using anti-GST antibody and 1-Step Ultra TMB-ELISA solution. Absorbance at 450 nm was measured. D, HEK-Blue hTLR4 cells were transfected with the indicated plasmids and GAPDH siRNAs. After 72 h, cells were treated with 200 ng/ml LPS for 40 min. Cell lysates were immunoprecipitated using FLAG antibody and immunoblotted for FLAG-TRAF3, HA-K63-ubiquitin, and GAPDH-Myc.
Then we investigated whether the GAPDH-TRAF3 interaction is important for TRAF3 activity in response to LPS. We transfected GAPDH siRNA targeting the GAPDH 3′ UTR and complemented these knockdown cells with two GAPDH-Myc plasmids, either WT or a C150S GAPDH mutant (which does not interact with TRAFs (25)). After LPS treatment, Lys63-Ub levels on TRAF3 were significantly elevated, as expected (Fig. 4D). Complementing GAPDH knockdown cells with WT, but not with C150S GAPDH, supported TRAF3 ubiquitination. Co-transfecting WT NleB, but not the glycosyltransferase-deficient mutant NleB(AAA) (25), significantly reduced Lys63-linked TRAF3 ubiquitination and inhibited the association of GAPDH with TRAF3 (Fig. 4D).
Discussion
NleB is a translocated glycosyltransferase enzyme that was originally studied for its ability to suppress NF-κB activation (26, 27). We found previously that NleB disrupts TRAF2 signaling by modifying GAPDH with GlcNAc and preventing GAPDH from binding to and activating TRAF2 (25). The EPEC NleB1 effector modifies arginine residues of host proteins (N-GlcNAcylation) rather than the more typical targeting of serine/threonine residues (O-GlcNAcylation (28, 29)). In particular, it was found that EPEC NleB1 glycosylates the death domains of TRADD, FADD, RIPK1, and TNFR1, blocking the assembly of the TNFR1 complex. This N-GlcNAcylation disrupts TNF signaling in EPEC-infected cells, thus impacting NF-κB activation, apoptosis, and necroptosis (28, 29). A recent paper defined numerous amino acids that are important to the N-GlcNAcylation of FADD by EPEC NleB1 and also used the C. rodentium infection model to show the role of these amino acids in virulence (30). To date, the studies that have evaluated the arginine-GlcNAcylation of substrates by NleB orthologs have only described a single addition site for each protein. This modification occurs on Arg117 in FADD (29) and on Arg235 in TRADD (28). Labeling of an arginine residue by NleB1 was also detected for TNFR1 and RIPK1 (28). It is conceivable that some NleB orthologs could label other substrates, including GAPDH, on single or multiple arginine residues, a topic we are investigating in ongoing studies.
Here we observed an additional role of C. rodentium NleB affecting type I IFN signaling by inhibiting Lys63-linked TRAF3 ubiquitination. EPEC suppresses type I IFN production using the T3SS effector NleD (19), which we did not observe in our screening of C. rodentium mutants. We used the VSV-G/GFP system as the readout for type I IFN production, in which VSV replication is very sensitive to type I IFN secreted from bacterially infected cells. By contrast, Long et al. (19) measured IFN-β mRNA levels in Caco2 cells 3 h after EPEC infection. Because the expression of innate immunity genes is dynamic, mRNA levels assayed at a single time point might not fully reflect type I IFN production during bacterial infection. Additionally, the activities of the EHEC, EPEC, and C. rodentium NleB proteins may be functionally distinct (25, 28, 29).
TRAF3, a ubiquitin ligase, together with TRAF6, are recruited by the mitochondrion adaptor mitochondrial antiviral-signaling protein to activate the non-canonical IKK-related kinase TBK1 with various adaptor proteins, including TRAF family member-associated NF-κB activator, NAP1, and NF-κB essential modulator. The TBK1 complex induces the phosphorylation and dimerization of IRF3, which is required for IRF3 nuclear translocation. IRF3 is a master transcriptional regulator of type I IFN gene expression. TLR4 activation induces Lys63-linked TRAF3 ubiquitination, which is required for IRF3 activation.
GAPDH is a stress sensor involved in many biological processes, including immune signaling and apoptosis (31). We showed that the interaction between GAPDH and TRAF2 is enhanced by TNF and is required for maximal NF-κB activation after TNF stimulation (25). Here we found that GAPDH also interacts with TRAF3 and enhances TRAF3 Lys63-linked ubiquitination. Given that TRAF family members are widely involved in mammalian immune signaling pathways, our data suggest that GAPDH may have an unappreciated role in regulating the immune system. There are six distinct TRAF proteins in mammals, each containing a ring finger domain (except TRAF1), a zinc finger motif, an amino-terminal TRAF domain, and a carboxy-terminal TRAF domain (TRAF-C). TRAF-C is highly conserved among all TRAFs and is responsible for the association of TRAFs with other proteins. It will be interesting to investigate whether GAPDH interacts with other TRAFs under different conditions.
NleB inhibited the TRAF3 and GAPDH interaction by modifying GAPDH with GlcNAc. Other studies have shown that EPEC NleB1 glycosylates the death domains of TRADD, FADD, and other death domain-containing proteins (28, 29). Although these results are able to explain NleB function in the TNF/NF-κB signaling pathway, they fail to explain the function of NleB in the TLR4/type I IFN pathway we report here. The role of GAPDH in T cell activation has been described recently (32). GAPDH functions as a “switcher” of T cell activation by binding to AU-rich elements within the 3′ UTR of IFN-γ mRNA to suppress the translation of IFN-γ (32). Therefore, selective targeting of GAPDH might provide an interesting framework with which to understand the multiple effects of NleB on immune signaling pathways. Overall, our data suggest that C. rodentium NleB affects both NF-κB and interferon signaling.
Experimental Procedures
VSV-GFP assay
HeLa cells (ATCC) were grown in 6-well plates and infected with C. rodentium strains for 3 h. After infection, the cell supernatants were collected and filtered (0.25 μm). Vero cells (ATCC) were grown in 24-well plates on coverslips and treated with 500 μl of cell-free HeLa supernatants for 12 h and then infected with VSV-G/GFP (multiplicity of infection = 5) for 30 h. Vero cells were fixed with 3.7% paraformaldehyde before processing for immunofluorescence microscopy. Where indicated, Vero cells were treated with IFNAR2 antibody (1.0 μg/ml) before adding HeLa supernatants.
IRF3 Nuclear Translocation
HeLa cells were grown in 6-well plates and infected with C. rodentium strains when 70% confluent for 2–7 h. LPS (100 ng/ml, 1 h) was used as a positive control to induce IRF nuclear translocation. Nuclear extraction was performed as described previously (33), and nuclear proteins were resolved using SDS-PAGE and immunoblotted for IRF3 (Cell Signaling Technology).
STAT1 Assays
Overnight cultures of C. rodentium were diluted 1:10 in DMEM and grown for 3 h without shaking to an A600 of 0.9–1.0. HeLa cells were treated with IFN-β (5000 units/ml, PBL Assay Science) for 30 min to promote STAT1 phosphorylation and nuclear translocation (34). Cells were then inoculated with 100 μl of C. rodentium cultures for 3 h. Cells were subsequently washed with PBS, and cell fractionation was conducted as described (35). The concentration of p-STAT1 in the nuclear and cytoplasmic subcellular fractions was normalized to poly(ADP-ribose) polymerase and tubulin abundance, respectively.
Immunoprecipitation and Immunoblotting
Raw 264.7 cells (ATCC) were transfected, treated, and infected as indicated. Cells were then harvested into PBS, pooled, and centrifuged at 16,200 × g for 5 min. Supernatants were removed, and cells pellets were lysed in 20 mm Tris-HCl (pH 8.0), 2 mm EDTA, 137 mm NaCl, 1% (v/v) Nonidet P-40, and 10% (v/v) glycerol. Samples were incubated on ice for 30 min, and cell lysates were collected by centrifugation at 7800 × g for 10 min at 4 °C. Protein G Dynabeads (Invitrogen) were used with appropriate antibodies for immunoprecipitation. Western blots were imaged using an Odyssey infrared imaging system (LI-COR).
Luciferase Assays
HEK 293T cells (ATCC) were co-transfected at a ratio of 10:1 (1.0 μg of total DNA) with a firefly luciferase construct driven by a consensus IFN-β promoter together with a Renilla luciferase plasmid and with NleB-HA or an HA control plasmid. After 24 h, cells were stimulated with 1.0 μg/ml poly(I:C) for 12 h. Cells were then lysed with passive lysis buffer, and lysates were analyzed by using the Dual-Luciferase kit (Promega) with firefly fluorescence units normalized to Renilla fluorescence units. Luciferase assays were performed in triplicate with at least three independently transfected cell populations.
RT-PCR
cDNA was prepared from 1 μg of RNA by using the Superscript First Strand System (Invitrogen) with oligo(dT) primer. Real-time PCR was performed in triplicate using SYBR Green PCR Master Mix (Ambion) in a Fast 7500 sequence detection system (Applied Biosystems). Relative transcription levels were calculated by using the ΔΔCt method.
Ubiquitination Assays
Anti-FLAG M2 affinity resin was washed twice with cold TBS buffer (50 mm Tris HCl (pH 7.4) and 150 mm NaCl) and centrifuged at 8000 × g for 30 s at 4 °C. HEK-Blue hTLR4 cells (InvivoGen) were transfected and treated with LPS. Cells were harvested and washed with cold PBS. Cell pellets were lysed in lysis buffer (50 mm Tris HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100) on ice for 30 min and then mixed with anti-FLAG M2 affinity resin and rotated at 4 °C overnight. Resins were centrifuged at 8000 × g for 30 s at 4 °C and then washed three times with cold TBS buffer. Eluates were immunoblotted with FLAG-TRAF3, GAPDH-Myc, and HA-Lys63-ubiquitin.
ELISAs
Immulon-2 96-well plates (Dynatech) were coated with 1.0 μg of GAPDH and incubated at 37 °C for 1 h. Plates were washed with 0.05% PBS-Tween and blocked in 5% (w/v) nonfat milk in PBS-Tween. After washing, the plates were overlaid with different amounts of GST-TRAF3 or with GST-TRAF3 that had been labeled with GlcNAc by NleB. After 1-h incubation at 37 °C, primary and secondary antibodies were added. Plates were developed with 1-Step Ultra TMB-ELISA solution (Thermo Scientific) and then quenched with 2 m H2SO4. Absorbance at 450 nm was measured.
Pulldown Assays
GAPDH was immobilized on nickel-nitrilotriacetic acid-agarose beads (Qiagen) and then incubated with 1.0 μg of purified GST or GST-TRAF3 in the presence or absence of FLAG-NleB for 4 h at 4 °C. Beads were centrifuged and washed three times with 20 mm HEPES (pH 7.9), 150 mm KCl, 0.2 mm EDTA, 0.1% Nonidet P-40, 10% glycerol, and 1.0 mm DTT. Samples were analyzed using immunoblotting.
Statistical Analyses
Immunoblotting, luciferase, and RT-PCR assays were analyzed statistically using one-way ANOVA and Dunn's multiple comparisons test. Data shown are the mean ± S.E. of at least three replicates. p < 0.05 was considered significant.
Author Contributions
P. R. H. conceived and coordinated the study and wrote the paper. X. G., T. H. P., L. A. F., K. C., M. P. H., and G. S. designed, performed, and analyzed the experiments. C. R. and R. H. G. assisted with analysis and interpretation of data. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by NIAID, National Institutes of Health Grant AI093913. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- T3SS
- type III secretion system
- EHEC
- enterohemorrhagic Escherichia coli
- EPEC
- enteropathogenic Escherichia coli
- TLR
- Toll-like receptor
- VSV
- vesicular stomatitis virus
- TNFR
- TNF receptor
- TRAF
- TNF receptor-associated factor
- ANOVA
- analysis of variance.
References
- 1. Cornelis G. R. (2010) The type III secretion injectisome, a complex nanomachine for intracellular 'toxin' delivery. Biol. Chem. 391, 745–751 [DOI] [PubMed] [Google Scholar]
- 2. Sanchez-Villamil J., and Navarro-Garcia F. (2015) Role of virulence factors on host inflammatory response induced by diarrheagenic Escherichia coli pathotypes. Future Microbiol. 10, 1009–1033 [DOI] [PubMed] [Google Scholar]
- 3. Rahman M. M., and McFadden G. (2011) Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 9, 291–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Isaacs A., and Lindenmann J. (1957) Virus interference: I: the interferon. Proc. R Soc. Lond. B Biol. Sci. 147, 258–267 [PubMed] [Google Scholar]
- 5. Mogensen K. E., Lewerenz M., Reboul J., Lutfalla G., and Uzé G. (1999) The type I interferon receptor: structure, function, and evolution of a family business. J. Interferon Cytokine Res. 19, 1069–1098 [DOI] [PubMed] [Google Scholar]
- 6. Levy D. E., Marié I., and Prakash A. (2003) Ringing the interferon alarm: differential regulation of gene expression at the interface between innate and adaptive immunity. Curr. Opin. Immunol. 15, 52–58 [DOI] [PubMed] [Google Scholar]
- 7. van den Broek M. F., Müller U., Huang S., Zinkernagel R. M., and Aguet M. (1995) Immune defence in mice lacking type I and/or type II interferon receptors. Immunol. Rev. 148, 5–18 [DOI] [PubMed] [Google Scholar]
- 8. Belardelli F. (1995) Role of interferons and other cytokines in the regulation of the immune response. APMIS 103, 161–179 [DOI] [PubMed] [Google Scholar]
- 9. Karaghiosoff M., Steinborn R., Kovarik P., Kriegshäuser G., Baccarini M., Donabauer B., Reichart U., Kolbe T., Bogdan C., Leanderson T., Levy D., Decker T., and Müller M. (2003) Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 4, 471–477 [DOI] [PubMed] [Google Scholar]
- 10. Fitzgerald K. A., McWhirter S. M., Faia K. L., Rowe D. C., Latz E., Golenbock D. T., Coyle A. J., Liao S. M., and Maniatis T. (2003) IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 [DOI] [PubMed] [Google Scholar]
- 11. Sharma S., tenOever B. R., Grandvaux N., Zhou G. P., Lin R., and Hiscott J. (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151 [DOI] [PubMed] [Google Scholar]
- 12. Caillaud A., Hovanessian A. G., Levy D. E., and Marié I. J. (2005) Regulatory serine residues mediate phosphorylation-dependent and phosphorylation-independent activation of interferon regulatory factor 7. J. Biol. Chem. 280, 17671–17677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Katakura K., Lee J., Rachmilewitz D., Li G., Eckmann L., and Raz E. (2005) Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Invest. 115, 695–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dang O., Navarro L., Anderson K., and David M. (2004) Cutting edge: anthrax lethal toxin inhibits activation of IFN-regulatory factor 3 by lipopolysaccharide. J. Immunol. 172, 747–751 [DOI] [PubMed] [Google Scholar]
- 15. Gold J. A., Hoshino Y., Hoshino S., Jones M. B., Nolan A., and Weiden M. D. (2004) Exogenous γ and α/β interferon rescues human macrophages from cell death induced by Bacillus anthracis. Infect. Immun. 72, 1291–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sweet C. R., Conlon J., Golenbock D. T., Goguen J., and Silverman N. (2007) YopJ targets TRAF proteins to inhibit TLR-mediated NF-κB, MAPK and IRF3 signal transduction. Cell. Microbiol. 9, 2700–2715 [DOI] [PubMed] [Google Scholar]
- 17. Schröder M., Baran M., and Bowie A. G. (2008) Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKϵ-mediated IRF activation. EMBO J. 27, 2147–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prins K. C., Cárdenas W. B., and Basler C. F. (2009) Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKϵ and TBK-1. J. Virol. 83, 3069–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Long T. M., Nisa S., Donnenberg M. S., and Hassel B. A. (2014) Enteropathogenic Escherichia coli inhibits type I interferon- and RNase L-mediated host defense to disrupt intestinal epithelial cell barrier function. Infect. Immun. 82, 2802–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nusinzon I., and Horvath C. M. (2006) Positive and negative regulation of the innate antiviral response and β interferon gene expression by deacetylation. Mol. Cell Biol. 26, 3106–3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hiscott J., Pitha P., Genin P., Nguyen H., Heylbroeck C., Mamane Y., Algarte M., and Lin R. (1999) Triggering the interferon response: the role of IRF-3 transcription factor. J. Interferon Cytokine Res. 19, 1–13 [DOI] [PubMed] [Google Scholar]
- 22. Shuai K., and Liu B. (2003) Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 [DOI] [PubMed] [Google Scholar]
- 23. Tseng P. H., Matsuzawa A., Zhang W., Mino T., Vignali D. A., and Karin M. (2010) Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 11, 70–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tao M., Scacheri P. C., Marinis J. M., Harhaj E. W., Matesic L. E., and Abbott D. W. (2009) ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr. Biol. 19, 1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao X., Wang X., Pham T. H., Feuerbacher L. A., Lubos M. L., Huang M., Olsen R., Mushegian A., Slawson C., and Hardwidge P. R. (2013) NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-κB activation. Cell Host Microbe 13, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nadler C., Baruch K., Kobi S., Mills E., Haviv G., Farago M., Alkalay I., Bartfeld S., Meyer T. F., Ben-Neriah Y., and Rosenshine I. (2010) The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog. 6, e1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newton H. J., Pearson J. S., Badea L., Kelly M., Lucas M., Holloway G., Wagstaff K. M., Dunstone M. A., Sloan J., Whisstock J. C., Kaper J. B., Robins-Browne R. M., Jans D. A., Frankel G., Phillips A. D., et al. (2010) The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog. 6, e1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S., Zhang L., Yao Q., Li L., Dong N., Rong J., Gao W., Ding X., Sun L., Chen X., Chen S., and Shao F. (2013) Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501, 242–246 [DOI] [PubMed] [Google Scholar]
- 29. Pearson J. S., Giogha C., Ong S. Y., Kennedy C. L., Kelly M., Robinson K. S., Lung T. W., Mansell A., Riedmaier P., Oates C. V., Zaid A., Mühlen S., Crepin V. F., Marches O., Ang C.-S., et al. (2013) A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature 501, 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong Fok Lung T., Giogha C., Creuzburg K., Ong S. Y., Pollock G. L., Zhang Y., Fung K. Y., Pearson J. S., and Hartland E. L. (2016) Mutagenesis and functional analysis of the bacterial arginine glycosyltransferase effector NleB1 from enteropathogenic Escherichia coli. Infect. Immun. 84, 1346–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., and Sawa A. (2005) S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 32. Chang C. H., Curtis J. D., Maggi L. B. Jr., Faubert B., Villarino A. V., O'Sullivan D., Huang S. C., van der Windt G. J., Blagih J., Qiu J., Weber J. D., Pearce E. J., Jones R. G., and Pearce E. L. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gao X., Wan F., Mateo K., Callegari E., Wang D., Deng W., Puente J., Li F., Chaussee M. S., Finlay B. B., Lenardo M. J., and Hardwidge P. R. (2009) Bacterial effector binding to ribosomal protein s3 subverts NF-κB function. PLoS Pathog. 5, e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rani M. R., Croze E., Wei T., Shrock J., Josyula A., Kalvakolanu D. V., and Ransohoff R. M. (2010) STAT-phosphorylation-independent induction of interferon regulatory factor-9 by interferon-β. J. Interferon Cytokine Res. 30, 163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang X., Gao X., and Hardwidge P. R. (2012) Heat-labile enterotoxin-induced activation of NF-κB and MAPK pathways in intestinal epithelial cells impacts enterotoxigenic Escherichia coli (ETEC) adherence. Cell. Microbiol. 14, 1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]