

Abstract
The endoplasmic reticulum (ER) network comprises sheets and tubules that are connected by dynamic three-way junctions. Lunapark (Lnp) localizes to and stabilizes ER three-way junctions by antagonizing the small GTPase Atlastin, but how Lnp shapes the ER network is unclear. Here, we used an affinity purification approach and mass spectrometry to identify Lnp as an interacting partner of the ER protein quality control ubiquitin ligase gp78. Accordingly, Lnp purified from mammalian cells has a ubiquitin ligase activity in vitro. Intriguingly, biochemical analyses show that this activity can be attributed not only to associated ubiquitin ligase, but also to an intrinsic ubiquitin ligase activity borne by Lnp itself. This activity is contained in the N-terminal 45 amino acids of Lnp although this segment does not share homology to any known ubiquitin ligase motifs. Despite its interaction with gp78, Lnp does not seem to have a broad function in degradation of misfolded ER proteins. On the other hand, the N-terminal ubiquitin ligase-bearing motif is required for the ER three-way junction localization of Lnp. Our study identifies a new type of ubiquitin ligase and reveals a potential link between ubiquitin and ER morphology regulation.
Keywords: endoplasmic-reticulum-associated protein degradation (ERAD), protein misfolding, ubiquitin, ubiquitin ligase, ubiquitin-conjugating enzyme (E2 enzyme), Atlastin, ER three-way junction, Lunapark/Lnp, gp78, misfolded
Introduction
In mammalian cells, the endoplasmic reticulum (ER)4 is a continuous membrane network extending from the nuclear envelop to the entire cytoplasm. It is composed of sheets and tubules. In certain specialized cell types, one form may dominate the entire volume (1). The perinuclear sheet-like ER usually provides the sites to accommodate the ribosomes, therefore is designated as rough ER (rER). rER is involved in synthesis of membrane and secretory proteins. By contrast, the peripheral tubular ER, designated as smooth ER, tends to be ribosome-free. Smooth ER is thought to regulate lipid synthesis and calcium homeostasis (2). In recent years, several membrane proteins have been reported to regulate the formation of either the sheet-like cisternae or peripheral tubules. Among them, reticulons and DP1/Yop1p shape tubular ER as well as the edge of the ER sheets (3–5). Another protein named Climp63 has been suggested to function as a spacer to control the space between ER sheets (2), whereas the GTPase Atlastin/Sey1p is involved in fusion of ER tubules (6, 7). More recently, Lnp has been identified as an ER three-way junction protein that counteracts Atlastin in shaping the dynamic polygonal ER network (8–10). Theoretical modeling suggested that reticulons and DP1/Yop1p might be membrane curvature-inducing proteins that help to generate edge lines, whereas Lnp might stabilize three-way junctions by making concave edges (11).
The ER is the subcellular organelle where secretory and membrane proteins are synthesized and assembled. To ensure that proteins exiting the ER are properly folded, cells have evolved a highly conserved protein quality control mechanism named ER-associated degradation (ERAD), which efficiently eliminates terminally misfolded or unassembled proteins (12–15). Misfolded proteins captured by chaperones in the ER lumen are retrotranslocated across the ER membrane via one or more putative retrotranslocation channels (14, 15). These channels are likely formed by large membrane protein complexes; each consists of at least one ubiquitin ligase and other assisting cofactors. Once emerging from the ER lumen, substrates are modified by these ubiquitin ligases with polyubiquitin chains and ubiquitinated substrates are then extracted from the membrane by the ATPase p97 and delivered to the 26S proteasome for degradation (16–19).
In mammalian cells, more than 20 ER-associated ubiquitin ligases have been identified (20). Among them, Hrd1 is a highly conserved ligase that mediates the degradation of a large portion of misfolded ER proteins from Saccharomyces cerevisiae to humans. It contains multiple transmembrane domains and has a ubiquitin ligase-containing RING (really interesting gene) domain facing the cytosol (21–23). In addition to Hrd1, mammalian cells also have a Hrd1-related protein named gp78. Like Hrd1, gp78 itself can mediate the ubiquitination and degradation of some misfolded ER proteins (24–28), but in some cases, gp78 assists Hrd1-mediated ERAD by maintaining the functionality of a cytosolic chaperone holdase that prevents retrotranslocated substrates from aggregation (29).
In this report, we identify Lnp as an interacting partner of gp78 in mammalian cells by affinity purification and mass spectrometry. Using a collection of biochemical assays, we uncovered a novel ubiquitin ligase activity possessed by the N-terminal domain of Lnp. Interestingly, this domain is also required for the three-way junction localization of Lnp. Although the interaction of Lnp with gp78 does not have a significant function in ER protein quality control, our findings have revealed an unexpected link between ER network formation and the ubiquitin system.
Results
Lnp Interacts with the ERAD Ubiquitin Ligase gp78
To further understand the biological function of gp78, we wished to identify its interacting proteins. We expressed FLAG-tagged gp78 in HEK293T and purified gp78 using Sepharose beads conjugated with FLAG antibodies. Protein bands uniquely present in the gp78 pulldown sample were subject to mass spectrometry analyses, which identified many previously known gp78-interacting partners such as p97, UbxD8, and BAG6. In addition, a new potential interacting protein named Lunapark (Lnp) was identified (Fig. 1A). We next confirmed the interaction of the two proteins by expressing FLAG-tagged Lnp in HEK293T cells followed by immunoblotting analysis. The result showed that overexpressed Lnp-FLAG is associated with endogenous gp78 (Fig. 1B). To detect endogenous interaction between the two proteins, cell lysates prepared from untransfected cells were incubated with protein A beads coated with affinity-purified anti-Lnp antibodies and then fractionated into a bound and unbound fraction. Immunoblotting analysis showed that this procedure effectively depleted endogenous Lnp from the lysate. Although the level of gp78 in cell lysate was only slightly reduced after Lnp depletion, a fraction of endogenous gp78 was co-precipitated with Lnp in the bound fraction (Fig. 1C), demonstrating an endogenous interaction. The interaction was specific because control IgG did not pull down any gp78, and also because HSP90, an abundant cytosolic chaperone, was not detected in the bound fraction.
FIGURE 1.

Lnp interacts with the ERAD ubiquitin ligase gp78. A, gp78-FLAG pulldown was performed using HEK293T cells transfected with an empty vector (control) or a gp78-FLAG construct. Proteins eluted were analyzed by SDS-PAGE by Coomassie Blue staining. B, co-immunoprecipitation confirms the interaction of Lnp with endogenous gp78. Cells transfected with an empty control vector or a Lnp-FLAG construct were lysed, and proteins immunoprecipited with FLAG beads were analyzed by immunoblotting. C, interaction of Lnp with gp78 in untransfected cells. Whole cell extracts were subject to immunoprecipitation using protein A beads preincubated with the indicated antibodies, and then fractionated into bound and unbound fractions prior to immunoblotting. D, a schematic illustration of Lnp domain structure. PR, proline rich segment; ZF, zinc finger motif. E and F, characterization of the Lnp-gp78 interaction. HEK293T cells transfected with the indicated FLAG-tagged Lnp variants were lysed in a Nonidet P-40-containing lysis buffer. The lysates were subject to immunoprecipitation by FLAG beads. G, a schematic diagram illustrating the interaction of gp78 with Lnp on the ER membrane.
Lnp has a long hydrophobic segment close to the N terminus, which is predicted to form a hairpin in the ER membrane (9). Accordingly, both the N and C termini should face the cytosol. A proline-rich segment flanking a zinc finger motif is present in the middle of the large C-terminal cytosolic region (Fig. 1D). To determine the gp78-binding site in Lnp, we made a series of truncation mutants; each bears a FLAG tag at the C terminus. We expressed these mutants and wild-type (WT) Lnp in HEK293T cells, and prepared cell lysates for immunoprecipitation by anti-FLAG beads. Compared with WT Lnp, mutants bearing just the N- or C-terminal cytosolic domains failed to interact with gp78. A mutant containing the N-terminal cytosolic domain and the transmembrane segment interacted with gp78, but more weakly than the WT protein (Fig. 1E). By contrast, a Lnp mutant missing the transmembrane segment bound gp78 similarly as WT Lnp (Fig. 1F). Together, these results suggested that the N-terminal cytosolic domains of Lnp can interact weakly with gp78 when anchored to the ER membrane, but together with the C-terminal cytosolic domain, it binds gp78 even without being properly localized to the ER membrane (Fig. 1G).
Lnp Purified from Mammalian Cells Has a Ubiquitin Ligase Activity
Because gp78 is a ubiquitin ligase, if a fraction of Lnp is associated with gp78, Lnp purified from mammalian cells should have a ubiquitin ligase activity. To test this idea, we expressed Lnp-FLAG in HEK293T cells and purified it using FLAG beads under native conditions. The purified Lnp-FLAG is relatively pure with only one major contaminant band migrating below 75 kDa as revealed by Coomassie Blue staining. A fraction of gp78 was co-purified with Lnp, but could only be detected by immunoblotting (Fig. 1B). The purified Lnp-FLAG was then incubated with purified ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), ubiquitin, and ATP. We tested UBE2G2 and UBE2D1 as the source of E2 because the former is a gp78 cognate E2 enzyme, whereas the latter is a promiscuous E2 that can assist many ubiquitin ligases to form ubiquitin chains (30). Immunoblotting analysis indeed showed that Lnp purified from mammalian cells had a ubiquitin ligase activity, but surprisingly, it was dependent on UBE2D1 but not on UBE2G2 (Fig. 2A, lane 2 versus 3).
FIGURE 2.

Lnp purified from mammalian cells has a ubiquitin ligase activity. A, Lnp-FLAG purified from HEK293T cells was visualized by Coomassie Blue staining (left panel). The purified Lnp-FLAG (+Lnp) or buffer (−Lnp) was incubated with E1, HA-ubiquitin, or ATP, in the absence (no E2), or presence of UBE2D1 or UBE2G2 for 1 h. The reaction was analyzed by immunoblotting with anti-HA antibody (right panel). B, single round ubiquitin transfer assay was done in the presence or absence of Lnp-FLAG. His-tagged UBE2D1 was charged with HA-tagged ubiquitin and then quenched to block an additional round of ubiquitin charging. The HA-ubiquitin charged E2 was then incubated with untagged ubiquitin in the absence or presence of Lnp-FLAG. Samples were analyzed by immunoblotting under non-reducing or reducing condition. The area indicated by the box under the non-reducing condition is shown with two different exposures. C, purified Lnp-FLAG was further fractionated by a Superdex 200 column to remove gp78. The resulting protein product was analyzed by Coomassie Blue staining and immunoblotting with the indicated antibodies. D, the highly purified Lnp was incubated with E1, UBE2D1, HA-ubiquitin, and ATP to assay the ubiquitin ligase activity. E, Lnp-FLAG purified from either WT or gp78 CRISPR knock-out (ko) cells was incubated with E1, UBE2D1, HA-ubiquitin, and ATP at 37 °C for 1 h. The reaction was analyzed by immunoblotting with anti-HA antibody. Where indicated, Lnp was omitted from the reaction as a negative control. Bottom panels show immunoblotting analysis of cell lysates from control and gp78 knock-out cells (Input). F, mapping of the Lnp domain required for its E3 activity. The indicated Lnp variants were expressed and purified from HEK293T cells. The purified proteins were incubated with E1, UBE2D1, HA-ubiquitin, and ATP at 37 °C for 1 h. The reaction was analyzed by immunoblotting with anti-HA antibody.
Next, we performed a single round ubiquitin turnover assay to further confirm the ability of Lnp to catalyze ubiquitin discharge from the active site of UBE2D1, a hallmark of ubiquitin ligase. To this end, we first charged His-tagged UBE2D1 with HA-tagged ubiquitin and then quenched the reaction to prevent an additional round of UBE2D1 charging. The ubiquitin-loaded UBE2D1 was then incubated with excess untagged ubiquitin as an ubiquitin acceptor. After incubation, ubiquitin-charged UBE2D1 was reduced. Concurrently, a small amount of di-ubiquitin molecules consisting of a HA-tagged ubiquitin and an untagged ubiquitin was formed (Fig. 2B, lanes 5-8). This species was not sensitive to the reducing agent β-mercaptoethanol (lanes 9–16), suggesting that the ubiquitin moieties are linked by the iso-peptide bond rather than thioester bond. Di-ubiquitin formed in the absence of Lnp is due to a low spontaneous ubiquitin transferring activity of UBE2D1. Importantly, when Lnp was present, both the decrease of the ubiquitin-UBE2D1 thioester complex and the appearance of the di-ubiquitin product were significantly accelerated, demonstrating that purified Lnp indeed has a ubiquitin ligase activity.
Our observation that the ubiquitin ligase activity of Lnp depends on UBE2D1 but not on UBE2G2 suggested that the activity might not be caused by the small amount of gp78 present in the sample. Indeed, when purified Lnp was further fractionated by size exclusion chromatography to remove gp78, the highly purified Lnp still contained ubiquitin ligase activity (Fig. 2, C and D). To further exclude the involvement of gp78 in this process, we purified Lnp-FLAG from gp78 CRISPR knock-out cells and found that Lnp purified from gp78 knock-out cells contains similar activity as that from the control cells (Fig. 2E). Collectively, the findings establish Lnp as a component of a functional ubiquitin ligase complex in cells.
We next wished to identify the segment in Lnp responsible for this unexpected ubiquitin ligase activity. Lnp constructs expressing the truncated proteins shown in Fig. 1E were used to purify Lnp mutants, which were tested using the in vitro ubiquitination assay. Immunoblotting showed that the purified C-terminal cytosolic domain had absolutely no activity, whereas mutants bearing the N-terminal 45 residues (1–45 and 1–100) could synthesize ubiquitin chains in conjunction with UBE2D1 (Fig. 2F). Interestingly, a Lnp mutant bearing the TM segment in addition to the N-terminal domain appeared more active than the N-terminal cytosolic domain by itself (Fig. 2F, lane 6 versus 5), suggesting that the TM domain might bind to an unidentified ubiquitin ligase. Because Lnp(1–45) itself does not bind gp78, these results further confirm a gp78-independent ligase activity attributed to the N-terminal segment of Lnp.
Lnp Has an Intrinsic Ubiquitin Ligase Activity
To further define the ubiquitin ligase activity associated with Lnp, we wished to purify recombinant Lnp using Escherichia coli to avoid the confounding effect from contaminated mammalian proteins. Because Lnp contains a long hydrophobic segment that might be prone to aggregation, we replaced this transmembrane domain with a flexible polypeptide linker (GGS)3. This LnpΔTM mutant was expressed and purified from E. coli and tested for ligase activity by the in vitro ubiquitination assay (Fig. 3A). The results showed that when both LnpΔTM and UBE2D1 were present, ubiquitin conjugates were effectively assembled. These results suggest that Lnp itself can functionally cooperate with UBE2D1 to assemble ubiquitin chains despite lack of any known ubiquitin ligase motif.
FIGURE 3.

Recombinant Lnp purified from E. coli has a ubiquitin ligase activity. A, the indicated amount of His-LnpΔTM and UBE2D1 purified from E. coli were incubated with E1, HA-ubiquitin, and ATP at 37 °C for 1 h. The reaction was analyzed by immunoblotting with anti-HA (top panel) and anti-Lnp (bottom panel) antibodies. B, GST-Lnp(1–45) purified from E. coli was fractionated by size exclusion chromatography and compared with a molecular weight standard. Proteins in the peak fraction was also analyzed by SDS-PAGE and Coomassie Blue staining. C, increased amount of GST-Lnp(1–45) was incubated with E1, UBE2D1, HA-ubiquitin, and ATP at 37 °C for 1 h. The samples were analyzed by immunoblotting with anti-HA antibody. The graph shows the relative intensity of the ubiquitin-positive bands in the gel. Note that the truncated Lnp preferentially synthesizes ubiquitin chains containing 4–5 moieties. D, kinetic analysis of Lnp-mediated ubiquitination. The in vitro ubiquitination reaction was performed in the absence or presence of GST-Lnp(1–45). Samples taken at the indicated time points were analyzed by anti-HA immunoblotting.
Because Lnp(1–45) purified from mammalian cells was active, we also purified this segment from E. coli as a glutathione S-transferase (GST) fusion protein. We used the GST tag because it is capable of forming a dimer, which should mimic the oligomerizing effect of the C-terminal Lnp domain (31). Size exclusion chromatography confirmed that the purified GST-Lnp(1–45) protein forms a dimer (Fig. 3B). When tested by the ubiquitination assay, this N-terminal domain is sufficient to induce the formation of ubiquitin conjugates when paired with UBE2D1, although the length of the chains was shorter when compared with those formed by LnpΔTM (Fig. 3, C versus A). The amount of ubiquitin conjugates formed was dependent on both the level of GST-Lnp(1–45) and also on the incubation time (Fig. 3, C and D). All together, these results unambiguously establish that the N-terminal 45 residues of Lnp contain a ubiquitin ligase activity capable of stimulating ubiquitin chain synthesis, but also suggest that Lnp can interact with several ubiquitin ligases in cells including gp78.
Lnp Is Not an Essential ERAD Regulator
Because Lnp interacts with gp78, it might be a substrate of gp78. To test this idea, we examined whether overexpression of gp78 could induce ubiquitination of Lnp in cells. We immunoprecipitated Lnp under denaturing conditions from cells transiently expressing HA-tagged ubiquitin in the absence or presence of overexpressed gp78, and then analyzed the ubiquitination status of Lnp by immunoblotting with ubiquitin antibodies. As a positive control, we included a previously known gp78 substrate Ubl4A in the study. As shown previously, Ubl4A was ubiquitinated in cells and the level of Ubl4A ubiquitination was significantly enhanced by gp78 overexpression (Fig. 4A) (32). By contrast, no ubiquitinated Lnp was detected regardless of whether or not gp78 was co-expressed, suggesting that gp78 does not ubiquitinate Lnp.
FIGURE 4.

Lnp does not play a significant function in ERAD. A, gp78 does not ubiquitinate Lnp in cells. Endogenous Lnp and Ubl4A were immunoprecipitated (IP) under denaturing conditions from cells transiently transfected with HA-ubiquitin together with either a control or a gp78-expressing plasmid. A fraction of the whole cell extract (input) and the precipitated materials were analyzed by immunoblotting with the indicated antibodies. B, Lunapark knockdown does not significantly affect the degradation rate of TCRα-YFP. The degradation rate of the model ERAD substrate TCRα-YFP in control and Lnp knockdown cells was analyzed by a cycloheximide chase experiment using a cell line stably expressing TCRα-YFP. C, the steady state level of the ERAD substrate MHC(1–147) was examined in cells transfected with three control- and two Lnp-shRNA knockdown plasmids. Whole cell extracts prepared at 48 h post-transfection were analyzed by immunoblotting (IB). Asterisk, a nonspecific band. D, the effect of Lnp knockdown and re-expression on the degradation of MHC(1–147). The degradation of MHC(1–147) in control and Lnp knockdown cells was analyzed by a cycloheximide chase experiment using a cell line transiently expressing FLAG-tagged MHC(1–147). Where indicated, Lnp knockdown shRNA was co-transfected with a Lnp-FLAG construct.
We then considered the possibility that the interaction of gp78 with Lnp might reflect a functional collaboration of these proteins in ERAD. To see whether Lnp is involved in ERAD, we first used the classical membrane ERAD substrate TCRα as a model. We examined its steady state level and degradation rate under Lnp depletion conditions. Intriguingly, the steady state level of TCRα was moderately increased in Lnp knockdown cells when compared with control cells. A translational shutdown assay revealed a small increase in the half-life of TCRα when Lnp was knocked down (Fig. 4B). Moreover, when we examined the steady state level of a luminal soluble ERAD substrate, the truncated MHC class I heavy chain (MHC(1–147)) by immunoblotting, we found that knockdown of Lnp also increased the MHC(1–147) protein level (Fig. 4C). However, when we co-transfected FLAG-tagged Lnp together with Lnp shRNA, even though the exogenous Lnp was expressed at a level similar to endogenous Lnp in control knockdown cells, the degradation rate of MHC(1–147) was not restored to that of control cells (Fig. 4D). We therefore conclude that Lnp is not an essential ERAD regulator. The mild ERAD phenotype observed in Lnp knockdown cells might be due to subtle changes in ER morphology as a result of Lnp depletion, which indirectly impacts ERAD (see “Discussion”).
Because Lnp was previously shown to regulate the shape and dynamics of the ER network, we also considered the possibility that the interaction of gp78 with Lnp may implicate gp78 in the ER shaping process. However, confocal microscopy analyses showed no distinction in ER morphology between control and gp78 knock-out HeLa cells (Data not shown), suggesting that gp78 does not play a significant role in regulating the ER network or three-way junction morphology.
The N-terminal Domain of Lnp Is Required for Three-way Junction Localization
Because Lnp was known to localize to ER three-way junctions, we wished to map the domain in Lnp that is required for this localization. We used FLAG antibodies to stain COS7 cells that transiently expressed either full-length WT Lnp or several Lnp truncation mutants bearing the FLAG tag. We found that in cells expressing low levels of WT Lnp, it was localized to punctae that are consistent with ER three-way junctions, but when expressed at high levels, Lnp transformed ER network into tangled fiber-like structures (Fig. 5, A and B, upper panels). When a fragment consisting of the N-terminal 45 amino acids was expressed, it is distributed throughout the cell, but a small fraction appeared to associate with the ER (Fig. 5A). Interestingly, when a Lnp variant lacking the N-terminal 39 amino acids was expressed, the protein was localized to the ER, but it was not concentrated in three-way junctions as the staining pattern appeared continuous rather than puncta-like (Fig. 5, A and B, bottom panels). In addition, even expressed at high levels, this mutant failed to transform the ER network structure into fiber-like structures.
FIGURE 5.

The N-terminal domain of Lnp is necessary for its localization to the three-way junction. A, COS7 cells transiently transfected with the indicated FLAG-tagged Lnp constructs together with the ER marker Derlin1-GFP were fixed and stained with anti-FLAG antibody. Scale bars, 5 μm. Two examples of cells expressing wild-type Lnp are shown, representing two distinct ER morphologies seen in these cells. B, a close-up view of cells expressing either Lnp-FLAG or Lnp-FLAG(40–427) is shown in A. Scale bars, 3 μm. C, mCherry-tagged Lnp was transiently expressed together with GFP-Atlastin3 in COS7 cells. Images taken at different time points show strong co-localization of Lnp with Atlastin3. Scale bars, 10 μm.
To better define the mechanism of Lnp localization, we imaged cells expressing mCherry-tagged Lnp together with GFP-Atlastin3 in live cells by two-color confocal microscopy. Atlastin3 is a GTPase that has been shown to localize to ER three-way junctions (8–10). As anticipated, the time course recording of the fluorescence signals showed that Lnp co-localizes with Atlastin3, confirming the ER three-way junction localization of Lnp (Fig. 5C). Next, we imaged cells expressing mCherry-tagged Lnp variants together with the ER marker mCitrine-ER in live cells. The results further confirmed that WT Lnp forms punctae of irregular shape, and it is mostly localized to ER three-way junctions, whereas the mutant lacking the first 39 amino acids is uniformly distributed along the ER network (Fig. 6A). By contrast, Lnp(1–45), a fragment lacking any TM domain, is mostly localized to the cytosol with a fraction bound to the ER. Intriguingly, when a Lnp fragment lacking the C-terminal cytosolic domain was expressed, it is entirely localized to vesicles that are associated with the ER network (Fig. 6A). These results suggest that the Lnp C-terminal domain is required for ER retention of Lnp, whereas the N-terminal domain promotes the three-way junction localization and also contains an ER shaping activity.
FIGURE 6.
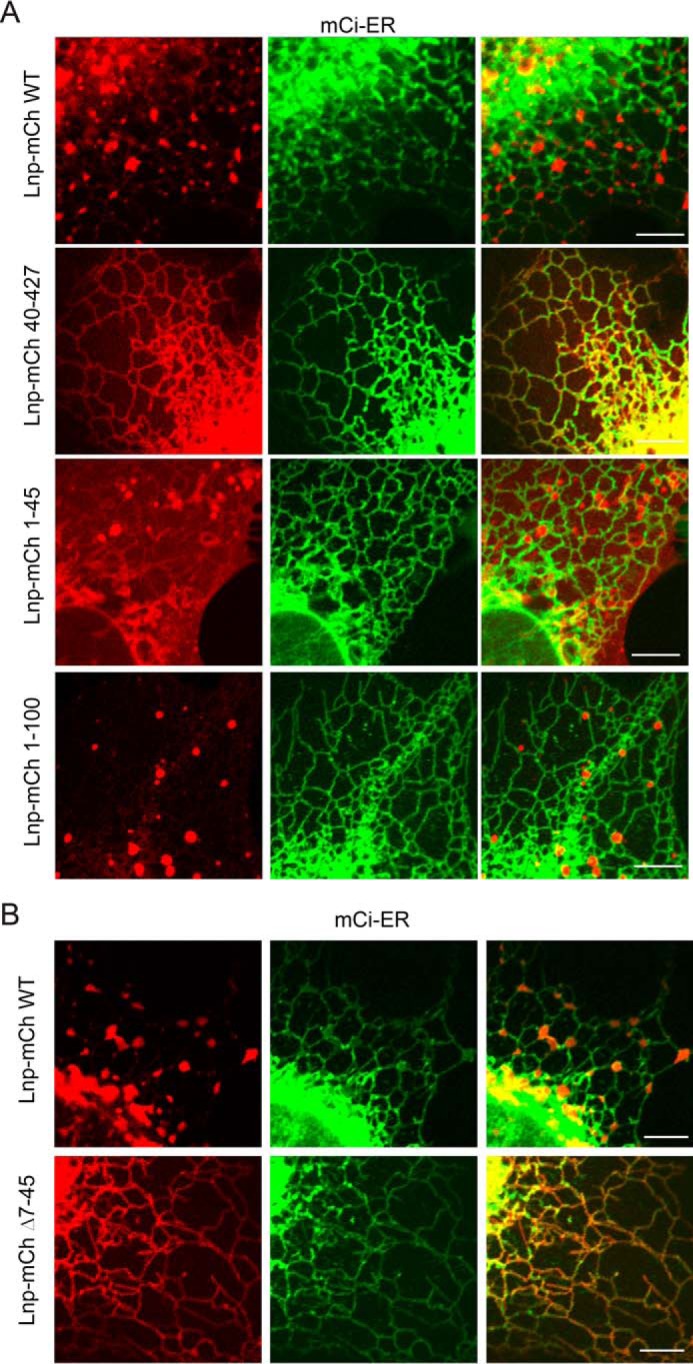
Live cell imaging analysis of mCherry-tagged Lnp subcellular localization. A, the indicated Lnp variants fused with mCherry were transiently expressed together with mCitrine (mCi)-ER in COS7 cells. 24 h post-transfection, cells were directly imaged by a confocal microscope. Scale bars, 2 μm. B, as in A, except that the indicated Lnp variants were analyzed. Scale bars, 2 μm.
Because it was recently reported that Lnp is myristoylated at the N terminus and this modification regulates its localization and ER-shaping activity (33), the effect of the deletion of the N-terminal 39 amino acids might be simply due to a deficiency in myristoylation. To exclude this possibility, we generated another truncated Lnp mutant construct, which encodes a mutant Lnp protein lacking residues 7–45 but retaining the N-terminal myristoylation site. Intriguingly, this truncated mutant also failed to concentrate at the ER three-way junctions and its overexpression did not lead to any abnormal ER morphology (Fig. 6B). Together, these data suggested that the Lnp N-terminal segment that has a ubiquitin ligase activity is also required for ER shaping activity of the Lnp and for its proper localization to ER three-way junctions.
Discussion
Lnp is an ER membrane protein localized to the three-way junctions of the ER network and it regulates tubular ER formation together with reticulons, DP1/Yop1p, and Atlastin (9–11). The Lnp homologous protein Lnp1p in budding yeast acts in conjunction with Rtn1p to antagonize Sey1p, the yeast homologue of Atlastin (9). Recently, it was reported that the localization of Lnp to the ER three-way junctions plays a role in stabilizing the polygonal network of the dynamic ER structure (8). Here, we identify Lnp as an interacting protein for the ER-anchored ubiquitin ligase gp78, an enzyme with a well established function in ER-associated protein quality control. The finding suggests a possible link between ER morphology and protein quality control. Importantly, we uncover an unexpected ubiquitin ligase activity that is associated with the N-terminal domain of Lnp, and thus linking the ubiquitin pathway to ER morphology regulation.
The known ubiquitin ligases can be categorized into three major classes depending on whether a RING finger, a U-box, or a HECT-domain is present (34–36). RING finger and U-box ubiquitin ligases usually contain cysteine residues that serve a scaffolding function. These domains can transiently associate with a cognate ubiquitin-conjugating enzyme to promote the transfer of ubiquitin to substrate. By contrast, a HECT domain ubiquitin ligase contains an active site cysteine that receives ubiquitin from a conjugating enzyme (37). Ubiquitin is then transferred from the E3 active site to a substrate. In this regard, it is surprising that Lnp has a ubiquitin ligase activity even though it does not contain any of these known ubiquitin ligase domains. We have also compared the sequence Lnp with the ubiquitin ligases from bacteria (38), but did not find any homology. In fact, Lnp does not even have any cysteine residues in the N-terminal domain. Thus, it appears that the N-terminal segment of Lnp may contain a new ubiquitin ligase motif.
It is noteworthy that ubiquitin conjugates are formed more efficiently by full-length Lnp purified from mammalian cells than by recombinant LnpΔTM from E. coli as ubiquitin conjugates formed by the former contain more high molecular weight species. Although it is possible that Lnp might fold better in mammalian cells than in E. coli, our data are more consistent with the presence of an assisting cofactor or an associated ubiquitin ligase in samples purified from mammalian cells. The fact that Lnp(1–100) has a stronger ligase activity than Lnp(1–45) suggests that this factor probably binds Lnp through its TM domain. Thus, in addition to gp78, which binds the cytosol-exposed Lnp domains, Lnp may also bind another ubiquitin ligase using its TM domain. Thus, our data establish Lnp as a component of ubiquitin ligase complex with several ligase activities contributing to the overall ubiquitination activity. Because Lnp has been implicated in regulating the formation of tubular ER, we tested whether any previously identified Lnp-interacting proteins could be subject to ubiquitination by Lnp. However, none of the proteins tested including reticulon4, DP1/Yop1p, or Atlastin could be detected in ubiquitinated form in a significant amount (data not shown). Further studies are required to identify endogenous substrates of the Lnp ligase complex, which would shed insights on the physiological function of this unusual enzyme activity.
Interestingly, our study shows that one of the Lnp-interacting ubiquitin ligases is gp78, an ER-associated ubiquitin ligase that mediates the degradation of misfolded ER proteins. This suggests a potential functional link between the two processes. gp78 is known to interact with several ubiquitin ligases such as Hrd1, TRIM25, and RMA1 (29, 39–41). It was reported that under certain conditions, the collaboration between gp78 and these ligases in which gp78 serves as a ubiquitin chain elongating factor is required for efficient ubiquitin chain assembly (39, 41). This model may also be applicable to the gp78-Lnp partnership. It is conceivable that the two ligases may function together to regulate a certain aspect of ER network construction by mediating ubiquitination of an unknown factor. Although our study does not reveal an apparent abnormality in ER morphology in gp78 CRISPR knock-out cells, it is noteworthy that the ER morphology change observed in Lnp knockdown mammalian cells is subtle (8). Thus, more sensitive assays are necessary to reveal the function of the Lnp-gp78 complex in ER morphology regulation.
Given the three-way junction localization of Lnp, the interaction of Lnp with gp78 implies that a fraction of gp78 is also localized to the ER three-way junction. Due to the high membrane curvature, the ER three-way junctions do not offer a suitable environment for every membrane protein, yet its continuation with the rest of the ER would imply that most ER membrane proteins would have access to this ER domain. In this context, we propose that the interplay between gp78 and Lnp might play a role in degradation of certain ER proteins mis-localized to the ER three-way junctions, which maintains the unique proteome of this specialized ER domain. This model, if correct, would not be revealed by testing the degradation of the conventional misfolded ERAD substrates because their degradation occurs throughout the ER. Instead, a careful comparison of the protein composition of the ER three-way junction between control and gp78- or Lnp-deficient cells may help to clarify the function of the gp78-Lnp interaction.
Lnp is one of a small handful of ER proteins known to be concentrated at the ER three-way junctions (9). Another three-way junction protein is Atlastin, which appears to antagonize Lnp to this ER domain, but the precise mechanism by which either Lnp or Atlastin is enriched in the ER three-way junctions is unclear (9). It was shown that N-terminal myristoylation of Lnp is also required for this localization (33), but myristoylation by itself is not sufficient because the LnpΔ7–45 mutant bearing the myristoylation sequence is not concentrated at the ER three-way junctions. Interestingly, when expressed at high levels, WT Lnp, but not the LnpΔ7–45 mutant, transformed ER into tangled fiber-like structures. Thus, it seems that the ubiquitin ligase activity, the ER three-way junction localization, and ER shaping activity are to some extent coupled by the Lnp N-terminal domain. A thorough characterization of the Lnp interactome as well as its substrates should provide important clues on how the Lnp ubiquitin ligase complex functions at this unusual intramembrane domain.
Experimental Procedures
Cell Lines, Plasmids, and Antibodies
The HEK293T, HeLa, and COS7 cell lines were obtained from ATCC. Plasmid expressing FLAG-tagged gp78 was described previously (42). Plasmid expressing FLAG-tagged Lnp was purchased from Origene (Rockville, MD). The Lnp-truncated variants were constructed by cloning corresponding coding DNA fragments as indicated in Fig. 1 into the SgfI and MluI sites in pCMV6-ENTRY, the same vector as the purchased FLAG-tagged full-length Lnp. mCherry-tagged Lnp and the truncated variants were generated by inserting the Lnp or truncation mutant DNA fragments as indicated in Fig. 1 into XhoI and EcoRI sites in the mCherry2-N1 vector. The construct for expression of His-tagged UBE2G2 was described previously (43). The construct for expression of His-tagged UBE2D1 was kindly provided by Dr. Cynthia Wolberger (John Hopkins University, Baltimore, MD) (44). The construct for expression of GFP-Atlastin3 was kindly provided by Dr. Gia Voeltz (University of Colorado Boulder) (45). His-LnpΔTM was generated by cloning the Lnp coding sequence, with 45–99 amino acids replaced by a linker (GGS)3, into NcoI and XhoI sites of pET32a, with a tobacco etch virus site following the NcoI site. GST-Lnp(1–45) amino acids was made by cloning the Lnp(1–45) amino acid into the SalI and NotI sites of pET42b(+) vector (Novagen/Merck, Germany). For Lnp knockdown experiments, the target sequences are as follows: Lnp shRNA number 1: GGAAGTGTGCTTTCATCAGACAACCAGTT; Lnp shRNA number 2: TGAGCCGCCATCTGCTGGAGCAGCTGTAA. The knockdown constructs were purchased from Origene (Rockville, MD). Lipofectamine2000 (Invitrogen) was used for DNA plasmid transfection.
Lnp and gp78 CRISPR knock-out cells were generated using the CRISPR/Cas9 technology (46). The vector pX330 (47) for inserting the guide sequence was purchased from Addgene. Pair of oligos of Lnp or gp78 were: forward Lnp primer: 5′-CACCGTGGATTATTTTCTCGATGG, reverse Lnp primer: 5′-AAACCCATCGAGAAAATAATCCAC; forward gp78 primer: 5′-CACCGCCCAGCCTCCGCACCTACA; reverse gp78 primer: 5′-AAACTGTAGGTGCGGAGGCTGGGC.
These primers were annealed and the resulting double strand DNA fragment was inserted into pX330 at the BbsI sites. The construct was then transfected into HeLa cells following the standard protocol. 48 h post-transfection, 50% of the cells were used to prepare genomic DNA for SURVOR assay to validate the cleavage of the target DNA. The remaining cells were cloned by infinite dilution. Immunoblotting was used to validate the positive knock-out clones.
The antibody to Lnp was raised against recombinant GST-Lnp(358–427) amino acids. Antibody to gp78 and Ubl4A were described previously (42). Other antibodies used are FLAG (M2) (Sigma), HSP90 (Santa Cruz Biotechnology), HA (Sigma), Tubulin (Sigma), GFP (Invitrogen), and calreticulin (Thermo/Pierce). HRP-linked (Jackson ImmunoResearch Laboratories) or fluorescence-labeled secondary antibodies (Rockland immunochemicals, Limerick, PA) were used for immunoblotting detection. GST-E1 and ubiquitin were purchased from Boston Biochem.
Protein Expression and Purification
The purification of Lnp-FLAG, Lnp(1–45aa)-FLAG, Lnp(1–100aa)-FLAG, and Lnp(99–427aa)-FLAG were all purified using the FLAG affinity chromatography procedure described before (42). His-tagged LnpΔTM and GST-Lnp(1–45), and His-tagged UBE2D1 were purified from E. coli according to a previously described method (48). Protein eluted from glutathione beads (GE Healthcare) or nickel-nitrilotriacetic acid beads (Qiagen) were fractionated on a Superdex 200 HR (10/30) column in a buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm potassium chloride, 2 mm magnesium chloride, 2 mm DTT, and 5% glycerol.
Immunoprecipitation, Pulldown, and Immunoblotting
Cells were lysed in the DeoxyBIGCHAP lysis buffer with 30 mm Tris-HCl, pH 7.4, 37.5 mm potassium acetate, 4 mm magnesium acetate, 1% DeoxyBIGCHAP, and a protease inhibitor mixture. Whole cell extract was used for the experiments. For immunoprecipitation, the whole cell extract was incubated with FLAG-agarose beads (Sigma) or protein A-Sepharose CL-4B (GE Healthcare) bound with antibodies against specific proteins. For denatured immunoprecipitation, cells were first lysed in a buffer containing 1% SDS and 5 mm DTT. The lysates were heated at 65 °C for 15 min and then diluted 10-fold by the Nonidet P-40 lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm sodium chloride, 2 mm magnesium chloride, 0.5% Nonidet P-40, and a protease inhibitor mixture). The samples were subject to centrifugation at 20,000 × g for 10 min and the supernatant fractions were used for immunoprecipitation by the indicated antibodies. Immunoblotting was performed according to the standard protocol.
Immunofluorescence Microscopy
To detect the subcellular localization of protein by fluorescence labeling, cells were seeded on coverglass and transiently transfected. Cells were then fixed with phosphate-buffered saline containing 4% paraformaldehyde for 20 min at room temperature. For immunostaining experiments, fixed cells were permeabilized in PBS containing 0.1% Nonidet P-40 and 5% fetal bovine serum, and stained with antibodies in the same buffer according to a standard protocol. Images were acquired with a Zeiss LSM780 confocal microscope. For live cell imaging, cells were seeded at 1.5 × 105/well in a 35-mm u-dish coated with fibronectin (ibidi GmbH, Germany). Cells were immediately transfected with 1 μg of mCherry-tagged Lnp plasmids and 1 μg of mCitrine-ER plasmid (a gift from Michael Davidson (Addgene plasmid number 56557)) using Lipofectamine 2000. 24 h post-transfection, cells were incubated with phenol red-free minimum Eagle's medium at 37 °C and imaged by a Zeiss LSM780 confocal microscope.
In Vitro Ubiqutination Assay and Single Round Ubiquitin Transfer Assay
Ubiquitination experiments were described before (48). Briefly, E1 (60 nm), UBE2D1 (2 μm), Lnp-FLAG (500 nm), or GST-Lnp (1 μm), or as indicated in the figures, were incubated with HA-tagged ubiquitin (30 μm) at 37 °C for 1 h, or as indicated in the figures, in the ubiquitination reaction buffer containing 25 mm Tris-HCl, pH 7.4, 2 mm magnesium/ATP, and 0.1 mm DTT. For the single round ubiquitin transfer assay, two reactions were performed. First, UBE2D1 (2 μm) was incubated with E1 (60 nm), HA-tagged ubiquitin (1 μm) at 30 °C in the ubiquitination reaction buffer as described above. The reaction was treated with 50 mm EDTA and 10 mm N-ethylmaleimide for 15 min at 25 °C to prevent a further round of charging. The reaction was then incubated (chase) with untagged ubiquitin (200 μm) at 37 °C in the presence of either buffer control or Lnp-FLAG. The reaction was stopped by Laemmli buffer and analyzed by immunoblotting with anti-HA antibodies. For reducing conditions, samples were treated with 500 mm β-mercaptoethanol before SDS-polyacrylamide gel electrophoresis analyses.
Miscellaneous Biochemical Assay
Cycloheximide chase experiments were performed by incubating the cells in DMEM containing 50 μg/ml of cycloheximide at 37 °C. An equal number of cells was taken at 0, 1, 2, and 3 h for immunoblotting analysis.
Author Contributions
Y. Y. and Y. L. designed the study and wrote the paper. T. Z. designed and constructed plasmids for Lnp expression and performed the imaging experiments. Y. L., Y. Z., and H. H. purified Lnp proteins and characterize the enzyme activity. All authors analyzed the results and approved the content of the manuscript.
Acknowledgments
We thank Cynthia Wolberger (John Hopkins University), Tom Rapoport (Harvard Medical School), Craig Blackstone (NIH), and Gia Voeltz (University of Colorado Boulder) for plasmids and reagents, Jeff Reece at the NIDDK imaging core for assistance with live cell imaging, and the Harvard Taplin Mass Spectrometry Core for assistance in protein identification.
This work was supported, in whole or in part, by National Institutes of Health Intramural Research Program NIDDK Grant 1ZIADK033008-06 and National Natural Science Foundation of China Grant 31570781. The authors declare no conflict of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ER
- endoplasmic reticulum
- rER
- rough ER
- ERAD
- ER-associated degradation
- Lnp
- Lunapark
- TM
- transmembrane.
References
- 1. Shibata Y., Voeltz G. K., and Rapoport T. A. (2006) Rough sheets and smooth tubules. Cell 126, 435–439 [DOI] [PubMed] [Google Scholar]
- 2. Shibata Y., Shemesh T., Prinz W. A., Palazzo A. F., Kozlov M. M., and Rapoport T. A. (2010) Mechanisms determining the morphology of the peripheral ER. Cell 143, 774–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., and Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 [DOI] [PubMed] [Google Scholar]
- 4. Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M. M., Rapoport T. A., and Prinz W. A. (2008) Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 319, 1247–1250 [DOI] [PubMed] [Google Scholar]
- 5. Shibata Y., Voss C., Rist J. M., Hu J., Rapoport T. A., Prinz W. A., and Voeltz G. K. (2008) The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 283, 18892–18904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hu J., Shibata Y., Zhu P. P., Voss C., Rismanchi N., Prinz W. A., Rapoport T. A., and Blackstone C. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Orso G., Pendin D., Liu S., Tosetto J., Moss T. J., Faust J. E., Micaroni M., Egorova A., Martinuzzi A., McNew J. A., and Daga A. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983 [DOI] [PubMed] [Google Scholar]
- 8. Chen S., Desai T., McNew J. A., Gerard P., Novick P. J., and Ferro-Novick S. (2015) Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 112, 418–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen S., Novick P., and Ferro-Novick S. (2012) ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell. Biol. 14, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen S., Novick P., and Ferro-Novick S. (2013) ER structure and function. Curr. Opin. Cell. Biol. 25, 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shemesh T., Klemm R. W., Romano F. B., Wang S., Vaughan J., Zhuang X., Tukachinsky H., Kozlov M. M., and Rapoport T. A. (2014) A model for the generation and interconversion of ER morphologies. Proc. Natl. Acad. Sci. U.S.A. 111, E5243–E5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vembar S. S., and Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith M. H., Ploegh H. L., and Weissman J. S. (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruggiano A., Foresti O., and Carvalho P. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ye Y., Meyer H. H., and Rapoport T. A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 17. Bays N. W., and Hampton R. Y. (2002) Cdc48-Ufd1-Npl4: stuck in the middle with Ub. Curr. Biol. 12, R366–371 [DOI] [PubMed] [Google Scholar]
- 18. Jarosch E., Taxis C., Volkwein C., Bordallo J., Finley D., Wolf D. H., and Sommer T. (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol. 4, 134–139 [DOI] [PubMed] [Google Scholar]
- 19. Rabinovich E., Kerem A., Fröhlich K. U., Diamant N., and Bar-Nun S. (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 22, 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neutzner A., Neutzner M., Benischke A. S., Ryu S. W., Frank S., Youle R. J., and Karbowski M. (2011) A systematic search for endoplasmic reticulum (ER) membrane-associated RING finger proteins identifies Nixin/ZNRF4 as a regulator of calnexin stability and ER homeostasis. J. Biol. Chem. 286, 8633–8643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., and Hampton R. Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 22. Gardner R. G., Swarbrick G. M., Bays N. W., Cronin S. R., Wilhovsky S., Seelig L., Kim C., and Hampton R. Y. (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling: transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 151, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bordallo J., Plemper R. K., Finger A., and Wolf D. H. (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9, 209–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang S., Ferrone M., Yang C., Jensen J. P., Tiwari S., and Weissman A. M. (2001) The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 98, 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen Y., Ballar P., and Fang S. (2006) Ubiquitin ligase gp78 increases solubility and facilitates degradation of the Z variant of α1-antitrypsin. Biochem. Biophys. Res. Commun. 349, 1285–1293 [DOI] [PubMed] [Google Scholar]
- 26. Lee J. N., Song B., DeBose-Boyd R. A., and Ye J. (2006) Sterol-regulated degradation of Insig-1 mediated by the membrane-bound ubiquitin ligase gp78. J. Biol. Chem. 281, 39308–39315 [DOI] [PubMed] [Google Scholar]
- 27. Cao J., Wang J., Qi W., Miao H. H., Wang J., Ge L., DeBose-Boyd R. A., Tang J. J., Li B. L., and Song B. L. (2007) Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab. 6, 115–128 [DOI] [PubMed] [Google Scholar]
- 28. Jo Y., Sguigna P. V., and DeBose-Boyd R. A. (2011) Membrane-associated ubiquitin ligase complex containing gp78 mediates sterol-accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 286, 15022–15031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang T., Xu Y., Liu Y., and Ye Y. (2015) gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell 26, 4438–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye Y., and Rape M. (2009) Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Casey A. K., Chen S., Novick P., Ferro-Novick S., and Wente S. R. (2015) Nuclear pore complex integrity requires Lnp1, a regulator of cortical endoplasmic reticulum. Mol. Biol. Cell 26, 2833–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y., Soetandyo N., Lee J. G., Liu L., Xu Y., Clemons W. M. Jr., and Ye Y. (2014) USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. Elife 3, e01369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moriya K., Nagatoshi K., Noriyasu Y., Okamura T., Takamitsu E., Suzuki T., and Utsumi T. (2013) Protein N-myristoylation plays a critical role in the endoplasmic reticulum morphological change induced by overexpression of protein Lunapark, an integral membrane protein of the endoplasmic reticulum. PLoS ONE 8, e78235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deshaies R. J., and Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 35. Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 36. Bedford L., Lowe J., Dick L. R., Mayer R. J., and Brownell J. E. (2011) Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 10, 29–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scheffner M., Nuber U., and Huibregtse J. M. (1995) Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 373, 81–83 [DOI] [PubMed] [Google Scholar]
- 38. Maculins T., Fiskin E., Bhogaraju S., and Dikic I. (2016) Bacteria-host relationship: ubiquitin ligases as weapons of invasion. Cell Res. 26, 499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morito D., Hirao K., Oda Y., Hosokawa N., Tokunaga F., Cyr D. M., Tanaka K., Iwai K., and Nagata K. (2008) Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRDeltaF508. Mol. Biol. Cell 19, 1328–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ballar P., Ors A. U., Yang H., and Fang S. (2010) Differential regulation of CFTRΔF508 degradation by ubiquitin ligases gp78 and Hrd1. Int. J. Biochem. Cell Biol. 42, 167–173 [DOI] [PubMed] [Google Scholar]
- 41. Wang Y., Ha S. W., Zhang T., Kho D. H., Raz A., and Xie Y. (2014) Polyubiquitylation of AMF requires cooperation between the gp78 and TRIM25 ubiquitin ligases. Oncotarget 5, 2044–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Q., Liu Y., Soetandyo N., Baek K., Hegde R., and Ye Y. (2011) A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble States for proteasome degradation. Mol. Cell 42, 758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li W., Tu D., Brunger A. T., and Ye Y. (2007) A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature 446, 333–337 [DOI] [PubMed] [Google Scholar]
- 44. Wiener R., DiBello A. T., Lombardi P. M., Guzzo C. M., Zhang X., Matunis M. J., and Wolberger C. (2013) E2 ubiquitin-conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat. Struct. Mol. Biol. 20, 1033–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. English A. R., and Voeltz G. K. (2013) Rab10 GTPase regulates ER dynamics and morphology. Nat. Cell Biol. 15, 169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ran F. A., Hsu P. D., Lin C. Y., Gootenberg J. S., Konermann S., Trevino A. E., Scott D. A., Inoue A., Matoba S., Zhang Y., and Zhang F. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ye Y., Meyer H. H., and Rapoport T. A. (2003) Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]