

Abstract
Aberrant Ras signaling drives numerous cancers, and drugs to inhibit this are urgently required. This compelling clinical need combined with recent innovations in drug discovery including the advent of biologic therapeutic agents, has propelled Ras back to the forefront of targeting efforts. Activated Ras has proved extremely difficult to target directly, and the focus has moved to the main downstream Ras-signaling pathways. In particular, the Ras-Raf and Ras-PI3K pathways have provided conspicuous enzyme therapeutic targets that were more accessible to conventional drug-discovery strategies. The Ras-RalGEF-Ral pathway is a more difficult challenge for traditional medicinal development, and there have, therefore, been few inhibitors reported that disrupt this axis. We have used our structure of a Ral-effector complex as a basis for the design and characterization of α-helical-stapled peptides that bind selectively to active, GTP-bound Ral proteins and that compete with downstream effector proteins. The peptides have been thoroughly characterized biophysically. Crucially, the lead peptide enters cells and is biologically active, inhibiting isoform-specific RalB-driven cellular processes. This, therefore, provides a starting point for therapeutic inhibition of the Ras-RalGEF-Ral pathway.
Keywords: autophagy, cell signaling, peptide chemical synthesis, peptides, protein-protein interaction, small GTPase, Ral, Ras, stapled peptides, therapeutics
Introduction
Ras is well established as the most frequently mutated oncogene in human cancer. This small G protein cycles between an active GTP-bound state and an inactive GDP-bound state. Molecular switching between the “on” and “off” states is positively regulated by nucleotide guanine exchange factors (GEFs)4 and negatively regulated by GTPase-activating proteins (GAPs). It is only the active, GTP-bound form of the protein that can bind to downstream effectors and facilitate signal transduction. Mutations in oncogenic Ras result in a constitutively active protein that remains fixed in the GTP-bound on-state, leading to unregulated activation of downstream pathways. These mutations are found in ∼30% of all cancers, with a higher occurrence in specific cancer types such as pancreatic (71%) and colorectal (45%) (1). This makes Ras a crucial cancer therapeutic target; nevertheless, Ras has so far evaded direct attempts at inhibition, and many Ras-driven cancers are currently deemed undruggable. Ras signaling has proven difficult to disrupt by small, drug-like molecules because its activation of downstream cascades is accomplished through protein-protein interactions, which have traditionally been avoided as drug targets due to the large, shallow surfaces involved in protein-protein interfaces (2–4). Likewise, there are no obvious clefts or small molecule binding pockets on Ras, and competitive inhibition with the nucleotide is unfeasible due to the extremely high affinity of GTP binding and its high concentration in the cellular environment (5, 6). Logical attempts to interfere with critical post-translational modifications of Ras, such as inhibition of farnesyltransferase, have also proved unsuccessful (for reviews, see Refs. 1, 6 and 7). More encouragingly, a specific Ras variant found in lung cancer, G12C, was successfully used in a recent disulfide fragment-based screen to identify small molecules that covalently modify this precise Ras mutant (8). This led to the identification of a potential allosteric site that may be targetable in this particular mutant. The problems with attacking Ras directly have driven a broader search for drug targets that will interfere with Ras signaling, with a focus on the effector proteins downstream of Ras (for reviews, see Refs. 1 and 6). After Ras activation there are several pathways that propagate the cellular signal, of which at least five have been implicated in oncogenic signaling (9). Of these, the Raf, phosphoinositide 3-kinase (PI3K), and RalGEF are the best validated (6). Both Raf and PI3K have been the object of several drug discovery initiatives in recent years, with inhibitors of both proteins currently in clinical trials (10, 11). However, the third pathway, via RalGEFs, has been less extensively studied, and no therapies exist that disrupt this signaling route. Despite this, RalA and RalB have been shown to be important drivers of cell growth, cell survival, and metastasis in many human cancers, including bladder, melanoma, colorectal, and pancreatic (12, 13).
RalGEFs act to activate RalA and RalB, themselves small GTPases of the Ras superfamily. Like Ras, the Ral proteins cycle between GTP and GDP-dependent on and off states and are involved in regulating diverse cellular processes including proliferation, motility, and maintenance of cellular architecture (13). RalA and RalB are 206 amino acid proteins that are 82% identical, with 100% identity in the nucleotide-sensitive switch regions (switch 1 and switch 2). Intriguingly both GTP-loaded proteins interact directly with the same set of downstream effectors in vitro, including RLIP76 (or RalBP1), the exocyst complex subunits Sec5 and Exo84, and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB) (12). Despite engaging the same collection of effector proteins, RalA and RalB nevertheless regulate distinct cellular functions and play different roles in tumorigenesis. This is partly attributable to the ability of the effectors themselves to promote multiple signaling pathways. For example, both Ral isoforms interact with Sec5, which can act as an integral member of the exocyst complex, controlling exocytosis (14, 15), but RalB-Sec5 can also engage and activate TBK1 and contribute to cancer cell survival (16). Interestingly, although RalA and RalB have similar affinities for the effectors that have been tested in vitro (17–19), they may actually possess different affinities in vivo (20), explaining some of their distinct cellular functions. Some differences between RalA and RalB will be due to the hypervariable C-terminal region of the proteins, which is differentially phosphorylated (21–23) and ubiquitinated (24), resulting in distinctive and specific subcellular localization for the two proteins. Differential activation and deactivation by the RalGEF and RalGAP family may also contribute to differential roles for the two Ral isoforms. However, no GEFs have been found that discriminate between the two Ral isoforms, and the one structure of a RalGEF with Ral shows that all the contacts with the GEF protein are conserved between RalA and RalB (25). Similarly, RalGAPs appear to act on both isoforms in vitro (26) and in cell lines (27).
Several studies have been reported that attempt to delineate separate cellular roles for RalA and RalB. siRNA inhibition experiments showed that knockdown of RalB in HeLa, MCF7, and SW480 cell lines resulted in apoptosis, with no effect observed in non-cancerous human cell lines, suggesting that tumor cells may become dependent on RalB survival pathways (28). Inhibition of RalA in these experiments had no effect on adherent cells but impaired anchorage-independent proliferation of cells in suspension. In contrast, Lim et al. (29) found that RalA, but not RalB, was required for oncogenic transformation of human fibroblasts and HEK-HT cells and is critical for Ras-driven tumorigenesis. Similar effects have been observed in human pancreatic cancer and colorectal cancer cell lines, and interestingly, RalB appears to be important during cell invasion and metastasis of these cancers (30, 31). The molecular basis of the divergent functions of RalA and RalB in both normal and malignant cells remains to be elucidated. It is clear, however, that both proteins play key roles in tumorigenesis and cancer progression and are, therefore, potential therapeutic targets.
The Ral proteins adopt the same overall structural fold as Ras and are, therefore, equally difficult to disrupt using small molecules. Small molecules that bind to inactive, GDP-bound forms of Ral have, however, recently been identified using in silico screens (32). Our solution structure of RalB·GMPPNP in complex with the Ral binding domain of RLIP76 (RLIP76 RBD) (33) showed novel features for a Ras family-effector complex and presented an avenue for structure-guided design of inhibitors that would target the active, GTP-bound form of the Ral proteins. The GTP-bound form is generated downstream of activated Ras, so such inhibitors would bind specifically to chronically activated Ral, as would be encountered in the disease context. The structures that are currently available reveal that most Ras and Ral effectors form intermolecular β-sheets with the small G protein or interact through loops and unstructured regions (34). In stark contrast, the RLIP76 RBD adopts a well structured coiled-coil domain consisting of two α-helices that do not significantly change conformation on Ral complex formation (33). Mimicry of these helices offers an ideal opportunity to simulate effector binding and inhibit Ral-effector interactions, stopping signaling from Ral proteins and ultimately from Ras. Biological validation of this proposition has already been reported with the observation that overexpression of the RLIP76 RBD can interfere with Ral signaling, leading to mislocalization of Ral-interacting proteins and prevention of RalA-dependent anchorage-independent growth (14, 28, 35).
In a timely confluence, the mimicry and stabilization of α-helices has been an emerging area in inhibitor design in recent years, particularly through the use of chemically “stapled” peptides. The introduction of a staple confers multiple, advantageous, drug-like qualities on the peptides; the staple stabilizes the α-helical conformation of small peptides leading to an increase in binding affinity, it improves the cell penetrating ability of the peptide, and it enhances the resistance of the peptide to protease degradation. This technique has been successfully applied to several different protein targets (36, 37), and the first stapled peptide-based therapy, a long acting growth hormone releasing hormone (GHRH) agonist, has passed Phase I clinical trials, whereas the first anti-cancer stapled peptide, targeting the reactivation of p53, has also now entered Phase I trials.5 Peptides based on a helix from the Ras exchange factor SOS have been used to target Ras (39, 40), suggesting that small G proteins are amenable to such approaches. The Ras-binding peptides designed so far are not selective for the GTP-bound form of the Ras protein, although the stapled versions bind with a high affinity to wild-type K-Ras and several oncogenic mutants (40).
Here, we report the design and characterization of peptides based on a single helix of the RLIP76 RBD that bind selectively to active, GTP-bound Ral. This work provides proof of the principle that a single helix of RLIP76 is responsible for the majority of the RLIP76 interactions with Ral and that it is possible to use this approach to obtain peptides specific to the active form of a small GTPase. We describe stapled peptides based on this helix that have cell-penetrating ability and are biologically active. These peptides provide the basis for further maturation to therapeutically useful antagonists of Ral and Ras signaling.
Results
A Peptide Based on RLIP76 RBD α2 Is Sufficient to Bind RalA and RalB
Our previously reported NMR solution structure of the RBD of RLIP76 bound to active, GMPPNP-loaded, RalB (33) showed that RLIP76 employs a coiled-coil motif to bind to Ral. The structure suggested that most of the key interactions with Ral are mediated by the C-terminal helix (hereafter α2) of the RLIP76 RBD coiled-coil (Fig. 1A). Indeed, 80% of the buried surface area in the complex involves helix α2, and alanine-scanning mutagenesis identified more energetic hotspots within this helix than in α1 (the N-terminal helix of the coiled-coil) (17). To determine whether helix α2 is sufficient for binding to RalB, we synthesized two separate peptides based on the contact surfaces in helix α1 and α2. Peptide 1 comprised the full helix α2 sequence, residues 423–446, to encompass all of the RalB-interacting residues in this helix. Peptide 2 incorporated residues 408–422 and comprises residues corresponding to the last two turns of helix α1 together with the short loop that connects α1 and α2. This peptide, therefore, includes all the contacts to RalB outside helix α2.
FIGURE 1.

Design and synthesis of RLIP76-based peptides. A, the structure of the RLIP76 RBD bound to RalB·GMPPNP (PDB code 2KWI) revealed key binding residues. RalB is colored blue, and the two switch regions are labeled. RLIP76 is colored as follows: the segment of the N-terminal helix that contacts RalB is colored green, the rest of the N-terminal helix is gray. The C-terminal helix is colored pink, and the side chains of residues whose mutation reduced the binding to RalB >5-fold (17) are colored yellow. The peptides synthesized are shown schematically, with the same color coding and the staple represented as a single i, i+4 olefin link. B, CD spectra of the RLIP76 RBD and the peptides generated in this study. C, sequences of the peptides used in this study and helicity were calculated from analysis of CD data of the RLIP76 RBD and the peptides. Several peptides were synthesized containing all-hydrocarbon staples of various lengths in different positions (indicated by X). Peptides 1 and SP1-SP5 were based on the sequence of the second RLIP76 RBD α-helix sequence. Peptides 2 and SP6 were based on the first α-helix. The α-helicity of the peptides assessed using CD spectroscopy confirmed that several of the stapled peptides synthesized are more helical than the unstapled version of the peptide.
Circular dichroism (CD) spectroscopy experiments performed on the complete RLIP76 RBD resulted in a characteristic α-helical spectrum with minima at 208 and 222 nm (Fig. 1B). The helical content calculated from the CD spectra was 77% (Fig. 1C), in agreement with the 78% seen in the structure of free RLIP76 RBD (Ref. 33; PDB code 2KWH). CD analysis of the two synthetic peptides revealed that Peptide 1 was 50% helical in solution, whereas Peptide 2 was only 4% helical (Fig. 1, B and C). The helical content of the residues equivalent to Peptides 1 and 2 in the context of the entire RBD is 96 and 53%, respectively. Therefore, the helices in both peptides are destabilized when they are not in the context of the coiled-coil. Peptide 1, however, clearly retains some helical propensity in isolation and presumably is in equilibrium between structured and unstructured states. Conversely, Peptide 2 shows little propensity to form any helix when it is removed from the coiled-coil, and its CD spectrum is characteristic of a random coil (Fig. 1B).
The binding of the RLIP76 RBD, Peptide 1, and Peptide 2 to RalB was investigated using isothermal titration calorimetry (ITC). The RLIP76 RBD coiled-coil bound to RalB with an affinity of 1.9 μm (Fig. 2A, Table 1) and with a similar affinity (4.6 μm) to RalA·GMPPNP (data not shown). The interaction appears to be driven by a relatively large, favorable enthalpic term that counteracts the unfavorable entropy loss that occurs when the two domains come together. Peptide 1 also bound to RalB but with a lower affinity. The Kd was around 30 μm, an order of magnitude weaker than the full coiled-coil domain of RLIP76 RBD (Fig. 2B, Table 1). In contrast, Peptide 2 showed no heat changes when titrated into RalB under the same conditions, supporting the idea that most Ral binding contacts occur through RLIP76 RBD α2 (Fig. 2C). Encouragingly, whereas ITC showed that Peptide 1 also bound to RalA, with a Kd 43 μm (Fig. 2D, Table 1), it showed no interaction with the Rho-family GTPase Rac1 (Fig. 2E), indicating that binding of this single helix is selective for Ral small G proteins.
FIGURE 2.

Peptides corresponding to helix α2 are sufficient to bind to Ral proteins. Representative data from ITC experiments are shown for titrations of the RLIP76 RBD into RalB·GMPPNP (A), Peptide 1 into RalB·GMPPNP (B), Peptide 2 into RalB·GMPPNP (C), Peptide 1 into RalA·GMPPNP (D), and Peptide 1 into Rac1·GMPPNP (E). The parameters for the fit for these individual experiments are shown in each panel. For the average parameters obtained from several experiments, see Table 1.
TABLE 1.
Summary of binding parameters obtained from ITC for RLIP76 and peptides titrated into Ral proteins
The value obtained from an orthogonal assay, FP, is included for the tightest binding peptide.
| Binding partners | Kd | ΔH | TΔS | Na |
|---|---|---|---|---|
| μm | kcal mol−1 | kcal mol−1 | ||
| RalB·GMPPNP + RLIP76 RBD (n = 2b) | 1.9 ± 0.07c | −17.5 ± 1.8 | −9.6 ± 1.8 | 0.9 |
| RalB·GMPPNP + Peptide 1 (n = 4) | 29.8 ± 7.3 | −1.6 ± 0.6 | 4.6 ± 0.7 | 1.1 |
| RalA·GMPPNP + Peptide 1 (n = 3) | 43.0 ± 10.8 | −1.8 ± 0.9 | 4.2 ± 0.9 | 1.0 |
| RalB·GMPPNP + SP1 (n = 3) | 4.7 ± 1.6 (5.6 ± 0.3d) | −0.7 ± 0.4 | 6.6 ± 0.2 | 1.0 |
| RalB·GMPPNP + SP2 (n = 2) | 10.3 ± 2.3 | −0.8 ± 0.0 | 6.0 ± 0.1 | 1.1 |
| RalB·GMPPNP + SP3 (n = 3) | 53.3 ± 17.8 | −0.7 ± 0.3 | 5.1 ± 0.5 | 0.9 |
| RalB·GMPPNP + SP4e | 50.8 | −0.4 | 5.5 | 1.0 |
| RalB·GMPPNP + SP5 (n = 3) | >24 | Not fitted | Not fitted | Not fitted |
a Stoichiometry.
b Number of experiments.
c S.D. of value obtained from multiple experiments.
d Value obtained from FP.
e Peptide behaved poorly in assays and showed low heat changes: data from a single experiment only.
The ITC data for Peptide 1 revealed that when this peptide binds to RalA and RalB, the ΔS term is positive, in contrast to the entropy loss that was observed when RLIP76 RBD and Ral proteins interact. This favorable entropy change increases the binding affinity, which would otherwise be drastically reduced by the smaller enthalpic gain when the peptides bind (Table 1).
A Peptide Based on RLIP76 RBD α2 Binds to RalB on the Same Interface as RLIP76
Given the altered thermodynamics of the interaction, it is possible that Peptide 1 binds to a different region of the Ral proteins. To ascertain whether Peptide 1 binds in a similar manner as the RLIP76 RBD, 1H,15N HSQC NMR experiments were used to map the binding surface on RalB utilized by Peptide 1. The HSQC spectra of uniformly 15N-labeled RalB·GMPPNP were recorded both alone and in the presence of increasing amounts of Peptide 1. The addition of Peptide 1 caused several RalB peaks to shift and in some cases to lose intensity (Fig. 3A), which is consistent with a mixture of fast and intermediate exchange on the NMR timescale. This is likely to be due to the switch regions of RalB undergoing conformational exchange when in the complex with the peptide. A titration was performed at a range of RalB:Peptide 1 ratios from 1:0 to 1:8, which along with the previously reported backbone assignment (41) allowed most of the cross-peak positions to be tracked. The majority of the peaks in the peptide complex were assigned, and their positions were compared with those of the peaks corresponding to the same residues in the RalB-RLIP76 RBD complex. Comparison of the HSQC spectra of free RalB·GMPPNP with RalB·GMPPNP-Peptide 1 and RalB·GMPPNP-RLIP76 RBD shows that those peaks that shifted tended to move in the same direction in both complex spectra (Fig. 3A). For example, Arg-52, which experienced a relatively large shift, moved in the Peptide 1 complex to a position very close to that of the Arg-52 cross-peak in the RLIP76 RBD complex. The peak positions in free RalB·GMPPNP and in RalB·GMPPNP-Peptide 1 were used to calculate the overall chemical shift change (Fig. 3B). The changes are concentrated in the N-terminal half of the RalB protein, which includes the two nucleotide-sensitive switch regions. The residues that experienced a significant shift change (larger than the S.D. of all changes) were mapped onto the RalB structure (Fig. 3C). These included several residues that are unlikely to directly contact Peptide 1, because they are buried in the free RalB structure (42). These shift changes, which also occurred when the RLIP76 RBD was titrated into RalB, are indicative of subtle conformational rearrangements that are often observed when small G proteins such as RalB bind to their effectors.6 The solvent accessibility of the RalB·GMPPNP residues was assessed using NACCESS (43). Residues that are shifted and are at least 50% solvent-exposed, are highlighted in Fig. 3C. All of these residues are within, or close to, the switch regions, and most of them are in direct contact with the C-terminal helix of RLIP76 RBD in the complex (Fig. 3, C and D). Two residues that shift but do not contact RLIP76 are Val-40RalB and Glu-41RalB. These are at the beginning of switch 1, whereas Asp-49RalB at the opposite end of the same switch is in direct contact. The structure and dynamics of the whole switch region changes in the RLIP76 complex, and the chemical shifts of Val-40 and Glu-41 are likely to be sensitive to this. The cross-peaks for most of switch 1 are not visible in the spectra of free RalB·GMPPNP, so it is not possible to see changes for other residues within the same loop. Overall, the correlation between cross-peak positions in the RalB·GMPPNP-Peptide 1 and RalB·GMPPNP-RLIP76 RBD spectra along with the positions of the residues that exhibit the largest chemical shift changes indicate that the contact surface for Peptide 1 on RalB is identical to that of the RLIP76 RBD helix α2.
FIGURE 3.

Peptide 1 binds to RalB in an analogous manner to RLIP76 RBD. A, section of 1H,15N HSQC NMR spectra of 15N RalB·GMPPNP alone (blue) and in the presence of excess Peptide 1 (red) or RLIP76 RBD (black). The addition of Peptide 1 caused the movement of several peaks, all of which also shifted upon the addition of RLIP76 RBD. The changes in cross-peaks when Peptide 1 is added to RalB are smaller than those observed when RLIP76 RBD is added but the peaks shift in the same direction (see the dotted lines e.g. Lys-16, Val-20, Lys-27, Tyr-51, and Arg-52). Peaks that do not shift when RLIP76 RBD is added (e.g. Asn-128) do not shift when Peptide 1 is added. B, the chemical shift changes are plotted for each residue of RalB. The secondary structural elements are shown above the graph, where β-strands are represented by arrows, and α-helices are represented by cylinders. Red lines indicate the locations of the two switch regions. The dashed blue line shows the position of the average chemical shift change (0.047), and the solid green line shows the S.D. of the chemical shift changes (0.068). C, RalB residues that shifted by >1 S.D. upon titration of Peptide 1 are highlighted on the structure of RalB·GMPPNP (blue) and show that Peptide 1 binds to RalB in the same place as RLIP76 RBD α2 (shown in pink). Residues that are shifted but that are >50% buried are shown as yellow sticks. Residues that are shifted but are solvent-exposed are shown as orange spheres and are identified with a label. D, sequence alignment of RalA/B and R-Ras in the region that interacts with RLIP76 RBD. RalA and RalB have identical sequences in this region. Residue conservation is denoted below the alignment: * = identical, : = conservative substitution. Ral residues surrounded by a gray box interact with the N-terminal helix of RLIP76 RBD (helix α1); residues colored magenta interact with the C-terminal helix of RLIP76 RBD (helix α2). The symbols above the RalA/B sequence denote residues that form hydrogen bonds or salt bridges involving backbone (+) or side-chain (*) atoms of RalB. The symbols are colored gray for RLIP76 RBD α1 interactions and magenta for α2 interactions. Red lines indicate the locations of the two switch regions.
Design and Synthesis of RLIP76-based Stapled Peptides
Having established that an RLIP76 RBD-based α2 peptide could selectively bind to Ral in an analogous manner to the RLIP76 RBD, we sought to improve its binding affinity. Chemical stapling of peptides has been shown to increase their helicity and often enhances their binding to target proteins (36). Stapled peptides were synthesized using a well established solid-phase peptide synthesis method for incorporation of all-hydrocarbon staples (44). Two unnatural amino acids containing α-methyl, α-alkenyl side chains were introduced into the peptide sequence and the staple covalently formed by ruthenium-catalyzed ring-closing metathesis (Fig. 1, A and C). Five stapled peptides were designed based on RLIP76 RBD α2 (SP1-SP5), as Peptide 1 had already been confirmed to bind Ral. One stapled peptide was also designed based on RLIP76 RBD helix α1 (SP6) to investigate whether increased secondary structure in Peptide 2 could facilitate Ral complex formation. Examination of the RalB·GMPPNP-RLIP76 RBD structure (33) together with computational and experimental alanine scanning mutagenesis data (17) revealed those RLIP76 RBD residues that were the most important for Ral binding (Fig. 1A). These included three residues in helix α1 (Leu-409, His-413, and Leu-416) and five residues in helix α2 (Leu-429, Trp-430, Arg-434, Thr-437, and Lys-440). Individual mutation of any of these residues to Ala reduced the binding affinity of RLIP76 RBD for RalA or RalB >5-fold (17). These vital binding residues were retained in the peptide sequences, and the chemical staples were positioned on the opposite face of the α-helix to allow these key residues to drive complex formation (Fig. 1, A and C). Peptides were synthesized with different staple lengths and staple positions to enable screening for optimum binding. SP1, SP2, and SP3 contained a single i, i+4 staple, that was placed in various positions along the α2 helix. SP6, based on helix α1 and the interhelix loop, only included three turns of the α-helix. This peptide also contained a single i, i+4 staple; however, only a single staple position was tested, as other positions would potentially disrupt the structure of the loop. SP4 and SP5 were also based on helix α2: SP4 contained one i, i+7 staple, and SP5 was doubly stapled, with two i, i+4 linkages (Fig. 1C). All peptides were acetylated at the N terminus and included a C-terminal amide bond. After purification, the helicity of the stapled peptides was determined by CD spectroscopy (Fig. 1, B and C). Incorporation of the staples increased the helicity of most of the RLIP76 RBD peptides compared with their unstapled counterparts: SP1, SP3, SP4, and SP5 showed extremely high helicity (80–90%), much more than the unstapled Peptide 1, and close to the ∼90% expected if the peptides were to adopt the same structure as α2. SP2 was the least helical of the α2-based stapled peptides and had the same helicity as the unstapled Peptide 1, suggesting that the staple at this position does little to stabilize the helix. Stapling of the unstructured Peptide 2 also significantly increased its helicity, although SP6 was still only 57% α-helical, less than the ∼75% expected if the peptide had the same structure as the equivalent sequence in the RLIP76 RBD.
SP1 Binds to RalB with Increased Affinity Compared with Unstapled Peptide 1
Stapled peptide binding to RalB·GMPPNP was investigated using ITC, as for the unstapled peptides (Fig. 4). All of the stapled peptides based on the RLIP76 RBD α2 sequence (SP1-SP5) were able to bind RalB, but the stapled analogue of α1, SP6, did not exhibit any heat changes despite its increased helicity. This suggests that the binding interactions made between RLIP76 RBD α1 and RalB are not strong enough to enable complex formation in the absence of the α2 helix. The α2-based peptides SP3, SP4, and SP5 bound RalB with a Kd 16–53 μm (Table 1), which is similar to the affinity of the unstapled Peptide 1 (30 μm). Compared with Peptide 1, SP1 and SP2 displayed enhanced affinity, with Kd values 4.7 μm and 10.3 μm, respectively.
FIGURE 4.

Peptide stapling can increase affinity of RalB binding. ITC data for stapled peptides titrated into RalB·GMPPNP at 298 K: SP1 (A), SP2 (B), SP3 (C), SP4 (D), SP5 (E), and SP6 (F). The parameters for the fit for the individual experiments are shown. For a summary of the parameters from several experiments, see Table 1. SP5 could not be accurately fit, although the heat changes observed indicate that the peptide does bind. A lower limit on the Kd is given, but no reliable parameters could be obtained.
The binding of the stapled peptides was characterized by similar ΔH terms (of the order of 1 kcal/mol), implying that the nature of the interfaces may be similar (Fig. 4, Table 1). The exception was SP4, which has the longer, i, i+7 staple, and was characterized by very small heat changes and non-reproducible data in the ITC experiments. This implies that SP4 binds using predominantly hydrophobic interactions and, therefore, may not utilize the same binding mode as the other peptides. Similarly, SP5, which has a double staple, bound weakly, and the data could not be accurately fitted. The stapled peptides had more favorable entropy of binding compared with the unstapled Peptide 1, and the enhanced binding of SP1 and SP2 is entirely due to the increase in ΔS.
SP1 Is Selective for Active Ral and Binds Competitively with Ral Effector Proteins
As SP1 bound to RalB with the highest affinity, this peptide was taken forward for further investigation. N-terminally fluorescein-labeled FITC-SP1 and FITC-Peptide 1 were synthesized for fluorescence polarization (FP) assays. The use of an orthogonal assay to measure peptide binding was also useful to validate the results obtained with ITC. Direct binding FP assays showed that SP1-bound RalB with a Kd value of 5.6 μm, similar to that measured by ITC (Fig. 5A). The binding of Peptide 1 measured by FP was too weak to obtain an accurate fit (Fig. 5B), which placed the Kd at ≥50 μm, in line with the value measured by ITC (Table 1). We also tested the binding of FITC-SP1 to RalA·GMPPNP and found that it bound but with a slightly lower affinity than to RalB·GMPPNP, with a Kd of 14.2 μm (Fig. 5C). This is in agreement with the lower affinities that we observed when unstapled Peptide 1 or the RLIP76 RBD was titrated into RalA·GMPPNP in ITC experiments.
FIGURE 5.

FITC-SP1 binding to Ral is specific, GTP-dependent, and competitive with Ral effectors. A, fluorescence polarization assays using FITC-labeled SP1 confirm that SP1 binds RalB (Kd 5.6 ± 0.3 μm, solid line). SP1 also bound weakly to R-Ras (Kd 30.0 ± 18.0 μm, dashed line). B, Peptide 1 bound weakly to RalB and even less tightly to R-Ras. Affinity was too low for reliable fitting. C, SP1 binds RalA·GMPPNP with similar affinity to RalB·GMPPNP (Kd 14.2 ± 8.2 μm, solid line) but did not show significant binding to RalA·GDP (dashed line). D, competition fluorescence polarization assays showed that both RLIP76 (solid line) and Sec5 (dashed line) displaced FITC-SP1 from RalB, suggesting FITC-SP1 binds to RalB on an overlapping surface. Data were fitted to give IC50 8.2 ± 2.6 μm (Sec5 RBD) and IC50 2.0 ± 1.2 μm (RLIP76 RBD). All FP experiments were performed in duplicate, and error bars show the S.D. of the duplicates. mP, millipolarization units.
To investigate the specificity of SP1 for Ral proteins, we examined its ability to bind to R-Ras, which is another small G protein in the Ras family. This is a stringent test of specificity, as R-Ras and RalB are both in the Ras family, have a sequence homology of 64%, and are conserved in the effector binding region (Fig. 3D). FITC-SP1 bound to R-Ras·GMPPNP but with an affinity of 30 μm, 5-fold weaker than RalB (Fig. 5A). The affinity of FITC-Peptide 1 for R-Ras·GMPPNP could not be determined accurately but again was weaker than that of Peptide1 for RalB·GMPPNP (Fig. 5B).
Attempts were made to perform chemical shift mapping experiments with SP1 and 15N-labeled RalB·GMPPNP, analogous to those shown in Fig. 3. It was, however, not possible to find conditions in which both SP1 and RalB were stable at sufficiently high concentrations for NMR. As the peptides were designed based on helices from a Ral effector and the NMR chemical shift mapping had suggested that Peptide 1 bound close to the switch regions, we reasoned that the peptide binding should be nucleotide-dependent. Nucleotide-dependent binding of SP1 to Ral would also imply that SP1 binds to Ral in the same regions as Peptide 1. When RalA·GDP was titrated into FITC-SP1 (Fig. 5C) no significant binding was observed, indicating that SP1 binding to Ral is dependent upon the GTP-bound conformation of the switch regions. Therefore, as is the case for GTPase effector proteins, SP1 preferentially binds active, GTP-bound Ral over the inactive, GDP-bound state.
To determine whether effector proteins could compete with SP1 for binding to RalB, a competition FP assay was performed. Increasing amounts of the RLIP76 RBD were titrated into a preformed complex of RalB·GMPPNP-FITC-SP1. The resulting decrease in FP signal (Fig. 5D) indicates that FITC-SP1 and the RLIP76 RBD bind competitively to RalB·GMPPNP. The RLIP76 RBD was able to fully displace FITC-SP1 with an IC50 of 2.0 μm, which is comparable with the Kd for RalB·GMPPNP-RLIP76 RBD measured by ITC. This indicates that the peptide and RLIP76 RBD use the same interface to bind RalB·GMPPNP. We were interested in exploring whether other Ral effectors could also compete with SP1 for RalB·GMPPNP binding. The Sec5 RBD binds to Ral proteins with a higher affinity than RLIP76; both RalA and RalB bind Sec5 RBD with a Kd of ∼100 nm (42). When the Sec5 RBD was titrated into the RalB·GMPPNP-FITC-SP1 complex, the FP signal was reduced. However, Sec5 did not cause complete loss of the FP signal even at high concentrations and resulted in an IC50 of 8.2 μm, i.e. much higher than its Kd. This indicates that although there is some overlap in the binding sites of SP1 and Sec5 RBD, even at high Sec5 concentrations some binding remains between FITC-SP1 and RalB·GMPPNP. This can be explained by the observation that the RLIP76 RBD binds to both switch regions of RalB (33), whereas Sec5 RBD only contacts switch 1 (18). Therefore, the Sec5 RBD binding to switch 1 is unable to displace SP1 completely, which can continue to contact switch 2 even in the presence of Sec5. Taken together, the evidence indicates that FITC-SP1 binds in an analogous manner to α2 of the RLIP76 RBD, which contacts both switch regions and cannot be fully displaced by the Sec5 RBD.
FITC-SP1 Can Enter Mammalian Cells
For stapled peptides to have experimental or therapeutic value they must be able to access their intracellular targets. To assess the cell penetrating properties of RLIP76 RBD-based peptides, we monitored the cellular uptake of FITC-Peptide 1 and FITC-SP1 using confocal microscopy. HEK293T cells were treated with 10 μm FITC-Peptide 1, FITC-SP1, or FITC alone overnight at 37 °C and fixed with formaldehyde before imaging (Fig. 6, A and B). Consistent with previous studies on stapled peptides (36, 37, 45, 46), robust penetration of SP1 into cells was observed. In contrast, uptake of FITC alone or the unconstrained FITC-Peptide 1 was not detectable in any cells. These data also indicate that the stapled peptide is stable for at least 16 h. Single-cell images indicate that FITC-SP1 localizes throughout the cell, entering the nucleus as well as diffusing through the cytosol (Fig. 6B). Encouragingly, the cells treated with FITC-SP1 were observed to be in a similar state of health and confluency as the control cells, suggesting that the peptide is not inherently toxic.
FIGURE 6.

FITC-SP1 can enter mammalian cells and inhibits the interaction between RalB and Sec5. Shown are wide-field (A) and single-cell images (B) of HEK293T cells treated with 10 μm FITC-Peptide 1, 10 μm FITC-SP1, or 10 μm FITC control overnight. Chemical stapling enables FITC-SP1 to penetrate the cell membrane and localize in both the cytosol and nucleus. C, SP1 disrupted RalB-Sec5 interaction in cells. HEK293T cells were treated for 24 h with SP1 and SP6 as indicated. The numbers above the Western blot represent the relative intensities of the Sec5 band, normalized to the no-peptide control. Endogenous RalB was immunoprecipitated (IP) with mouse anti-RalB antibody. Immunoblotting was conducted with primary antibodies against RalB and Sec5. * marks the band specific to Sec5. Ab, mouse anti-RalB antibody alone. WCL, whole cell lysate.
Once we had established cellular uptake of SP1, we wanted to confirm that the inhibition of Ral-effector interactions that we had already demonstrated in vitro for the stapled peptide were maintained in a cellular context. To judge this, we examined the ability of SP1 to inhibit co-immunoprecipitation of RalB with an effector protein. We incubated HEK293T cells in the presence or absence of SP1, immunoprecipitated endogenous RalB, and immunoblotted for the presence of Sec5. SP6, the stapled helical peptide based on helix α1, which showed no heat changes in ITC experiments (Fig. 4F), was used as a control. We used Sec5 to investigate whether SP1 acts as a pan-RalB inhibitor in cells with similar activity to that which we had already observed in vitro. Endogenous Sec5 was observed to co-immunoprecipitate with RalB in the absence of peptide (Fig. 6C), but the interaction was almost completely inhibited by SP1 24 h after treatment. Interestingly, SP6 also appeared to attenuate the RalB-Sec5 interaction, although not as strongly as SP1. This suggests that even though it showed no heat changes in the ITC experiments, SP6 exhibits some low affinity binding to RalB. We can, therefore, conclude that SP1 enters cells, and once it is intracellular it retains the ability to inhibit RalB interactions with its downstream effector proteins.
SP1 Can Inhibit RalB-driven Autophagosome Assembly in Cells
We next sought to examine whether SP1 retained the biological activity of the RLIP76 RBD and could interfere with Ral signaling in cells. Previous studies have demonstrated the involvement of RalB in nutrient deprivation-induced autophagosome assembly through its interaction with the downstream effector Exo84 (35). RalB is localized to nascent autophagosomes and is activated in response to nutrient deprivation. In its active conformation, RalB·GTP binds directly to its effector Exo84, inducing the assembly of catalytically active ULK1 and Beclin1-VPS34 complexes on the exocyst, which drive isolation membrane formation and maturation. Thus, RalB signaling is necessary and sufficient to engage autophagy. In contrast, RalA has no involvement in the control of autophagy, so monitoring the effects of SP1 on autophagy specifically reveals the effects of its inhibition of RalB. Autophagosome assembly can be monitored through the loss of the marker LC3, which is first recruited to autophagosomes before its degradation in autophagolysosomes (Fig. 7, A and B). Exogenous expression of the RLIP76 RBD inhibits RalB-Exo84 complex formation and prevents autophagosome formation and maturation (35). To investigate whether SP1 had the same effect, HeLa cells stably expressing GFP-LC3 were treated with the cell-permeable SP1 for 24 h and assayed for autophagosomal flux with quantitative microtiter plate-based assays. SP1 but not SP6 exposure resulted in dose-dependent inhibition of GFP-LC3 turnover that was not reversed by nutrient starvation, indicating robust inhibition of autophagosomal maturation at 20 μm SP1 (Fig. 7C). These observations imply that SP1 is able to penetrate cells and achieve a sufficiently high intracellular concentration to bind to RalB and inhibit its biological activity. To confirm that the effects observed were due to the interaction between SP1 and RalB, the localization of TFEB was evaluated. TFEB is a direct mTORC1 substrate that is sequestered in the cytosol under nutrient replete conditions (47, 48). Nutrient starvation results in mTORC1 inactivation and consequent translocation of TFEB to the nucleus where it induces adaptive gene expression programs. TFEB localization was unimpaired in SP1-treated cells (Fig. 7D), demonstrating that the SP1-dependent inhibition of autophagosome maturation is unlikely to be through non-selective disruption of cellular responses to nutrient deprivation.
FIGURE 7.

SP1 can prevent RalB-driven autophagosome assembly in HeLa cells. Shown are RalB-Exo84-dependent autophagy (A) and epifluorescent images (B) of GFP-LC3 in HeLa cells stably expressing GFP-LC3. Diffuse and punctate GFP-LC3 fluorescence (left panel) is quickly quenched upon recruitment to maturing autophagolysosomes (right panel). C, quantitation of total GFP-LC3 fluorescence/cell detected under the indicated treatment conditions. Values represent population-based fluorescence-intensity ratios of GFP-LC3:Hoechst stain after 24 h of pretreatment with peptides in GFP-LC3 HeLa cells. Error bars indicate S.D. from the mean, n = 4. D, subcellular distribution of GFP-TFEB fusion protein was monitored under the indicated conditions in HeLa cells stably expressing GFP-TFEB. Cells were treated with 20 μm SP1 and SP6. Images are representative of multiple fields of view.
Discussion
The Ras signaling pathway has been proven to be a key driver of numerous cancers and is, therefore, a crucial anti-cancer drug discovery target. The difficulty in obtaining drug-like compounds that inhibit Ras itself has led to broader targeting of downstream Ras pathways, including the kinases Raf and PI3K. These efforts have been hampered by unexpected discoveries; the first class of Raf inhibitors turned out to activate the pathway in Ras-driven cancers (10), whereas MEK and PI3K inhibitors are also less efficacious due to the presence of feedback loops and poor tolerance (11). The third main Ras-driven signaling pathway activates the small GTPases RalA and RalB through the RalGEFs. Evidence indicates that this pathway is equally important in conveying signals from oncogenic Ras to drive tumorigenesis and is, therefore, a valuable therapeutic target (1). This signaling route has not been as extensively studied as the Raf and PI3K systems and does not exhibit as many conventionally exploitable targets. In fact until very recently there have not been any chemical inhibitors reported that act on this branch of the Ras cascade. Recently, a potential allosteric site was identified on RalA and used as the basis for a virtual small molecule screen. This generated hits that inhibit RalA and RalB by binding to the small G proteins in their GDP-bound form, locking them in their inactive conformation. These molecules have biological activity in cell lines and are also efficacious in mouse xenografts driven by K-Ras/Ral, demonstrating the utility of Ral inhibition for therapeutic gain and representing an exciting step forward in cancer therapeutics (32). Here, we have used our structure of the RalB-RLIP76 RBD complex to design and synthesize all-hydrocarbon-stapled peptides that bind to both RalA and RalB in the active, GTP-bound conformation and compete with their effector proteins.
We used ITC to determine the affinities and thermodynamics of binding of the peptides. Interestingly, we obtained a Kd of 1.9 μm for the RalB·GMPPNP-RLIP76 RBD complex by ITC, 10-fold higher than the Kd of 184 nm that we had previously determined by scintillation proximity assay (33). There are several likely reasons for these differences. First, different techniques used to determine affinities often yield different Kd values. This has been shown for other Ral-effector complexes; for example RalA·GMPPNP binding to Sec5 RBD had a Kd of 137 nm measured by ITC (18) but a Kd of 10 nm measured by surface plasmon resonance (19). We also found that Cdc42·GMPPNP binding to PAK gave lower affinities with ITC (Kd 150 nm) than the scintillation proximity assay (Kd 10 nm) (49). The difference in the RalB-RLIP76 RBD affinity may also be affected by the use of the nucleotide used in the different experiments. In the ITC experiments, RalB was bound to the nucleotide analogue GMPPNP, whereas a scintillation proximity assay utilized 3H-labeled GTP. All the peptides synthesized in this study were examined for binding by ITC, so their relative values can be compared. Furthermore, the affinities measured using FP for the peptides were similar to those obtained by ITC, allowing comparison between the affinities measured with these techniques.
We have shown that a peptide based on a single helix from RLIP76 (Peptide 1) is sufficient to bind Ral, albeit with an ∼14-fold weaker affinity than the full coiled-coil domain that comprises the RBD. NMR mapping indicates that Peptide 1 interacts with RalB at the same binding interface as RLIP76 RBD, so the reduction in affinity is due to the smaller peptide making fewer contacts with Ral rather than to a different mode of binding. ITC was used to measure the affinities, but it also yielded useful information about the thermodynamics of the interactions (Table 1). The interaction between RalB·GMPPNP and the full RLIP76 RBD is driven solely by a large negative ΔH, as the entropy change is in fact unfavorable. An analysis of the interactions between the two proteins suggests that the interface comprises a mixture of hydrogen bonds/salt bridges and hydrophobic interactions, which involve both α-helices of the RLIP76 RBD. Presumably the hydrophobic interactions between the two proteins are not sufficiently numerous to offset the entropy loss on association. In contrast, when the peptides bind, the enthalpy term is reduced 10-fold or more (although it is still negative), but the entropy term is now positive (Table 1). Some of the change in enthalpy compared with the full RBD binding is likely to be due to the loss of the three hydrogen bonds involving helix α1 predicted from the structure: His-413(Nϵ)–Tyr-82(OH), Leu-416(CO)–Asn-81(Hδ), Gln-417(Hϵ)–Asp-74(CO), which are not present in the peptide complexes. The switch in the entropic term from negative in the full RBD to positive in the peptides is probably due to the larger number of exposed hydrophobic side chains in the peptides, which lowers the entropy of the free peptides. The stapled peptides bind more tightly than the unstapled versions because the entropic term is even more favorable, and apart from SP4 (which gave poor, non-reproducible data), ΔS is proportional to the affinity (Table 1). Peptide stapling reduces the entropic cost of the peptide folding upon binding by forcing the helix to be pre-formed. The subtle differences between the stapled peptides may be due to slight conformational differences when the staple position is moved. These could change the extent of burial of side chains in the interface.
The helicity of the stapled peptides shows little obvious correlation with their affinities. The helical content of SP1, SP3, SP4, and SP5 is almost equivalent, whereas SP2, which has the second tightest binding, is no more helical than Peptide 1 (Fig. 1C). The helical content estimated by CD is, however, the average in solution. It is reasonable to assume that Peptide 1 exists in an equilibrium between helical and unstructured, and that, as it is 50% helical, the two populations are approximately equal. Binding to RalB then pushes the equilibrium to the helical state. In SP2, on the other hand, the stapling ensures that the center of the peptide is helical, so that at least part of the peptide is helical all of the time. This shorter helix would then act as a nucleation site for the formation of the remainder of the helix on binding. For peptides such as SP3, which exhibits high helicity but low affinity, we can speculate that the staple position has locked the side chains into a position where they can no longer make favorable interactions with RalB. It is noteworthy that SP3 and SP4, neither of which binds tightly, both have their staple positioned behind the Trp within the peptide. Mutation of Trp-430 in the context of the RLIP76 RBD knocks out binding completely (17), and the structure shows that this residue makes extensive interactions with RalB (33). It is likely that the rigidity imposed by stapling across the back surface of this Trp restricts at least some of these interactions.
The most potent RLIP76 RBD α2-based stapled peptide (SP1) bound to RalB with a 5-fold increase in affinity over the unstapled peptide. Previously published studies on stapled peptides have reported affinity increases between 2- and 100-fold, depending on the protein target (36, 37). The 5-fold increase noted here, therefore, is rather a modest effect. However, in this case the native unstapled peptide is already significantly helical (unlike many other peptides) and has a reasonable affinity compared with that of the RLIP76 RBD. The final affinity of the best stapled peptide is only ∼2.5-fold weaker than the complete progenitor domain, the RLIP76 RBD, and as such, SP1 provides an excellent starting point for further optimization. Interestingly, the small molecule inhibitors identified for Ral also have a Kd of ∼5–8 μm and are still effective inhibitors in both cell culture and animal models (32). These low μm Kd values reflect the difficulty in designing chemical inhibitors for Ral proteins and highlight the importance of our peptide.
Constrained peptides have already been designed to bind to small G proteins of the Ras superfamily. Ras itself has been targeted with stabilized α-helical peptides that were based on a helix from the exchange factor SOS. Peptides stabilized by the hydrogen bond surrogate method (39) bound to Ras·GDP with a Kd of 160 μm, 10-fold weaker than the catalytic domain of SOS. On the other hand, hydrocarbon-stapled peptides based on the same SOS helix bound to K-Ras with an affinity of around 0.1 μm but were not selective for the GTP-bound form, as expected for a peptide based on an exchange factor (40). Hydrocarbon-stapled peptides based on Rab-binding proteins, including effectors and exchange factors, have been screened against several members of the Rab small G protein family. Several of these bound preferentially to nucleotide-free Rabs, but a Kd of 22 μm was obtained for the most potent stapled peptide with GMPPNP-bound Rab8a, which was also selective for this Rab isoform (50).
We exploited the helical nature of an effector protein in this work to specifically target the GTP-bound form of the Ral proteins on the basis that this is the form that is downstream of oncogenic Ras and so is active in a disease scenario. We have shown that this is a viable approach, as the highest affinity peptide was indeed specific for the active Ral proteins and showed no interaction with the GDP-bound forms. Can this approach be applied to other small G proteins? For Ras itself it is difficult to envisage, as all the known effectors utilize an intermolecular β-sheet to bind the G protein (for review, see Ref. 34). Apart from Ras, the largest structural class of effectors for small G proteins is actually that which bind using a helical pair. We recently classified these into six subclasses, based on their helix orientations, which cover interactions between effectors and members of the Ras, Rho, Arf, and Rab families (34). In several cases one of the helices dominates, making the majority of the interactions with the nucleotide-sensitive switch regions, and therefore the relevant small G proteins, open to this approach to inhibitor design. Representative examples from five of the six classes, where one helix makes the most interactions, are RalB-RLIP76, Arf1-GGA, Rac1-PRK1, Arl1-Arfaptin1, and Arf6-JIP4. Stabilized helical peptides based on effectors may, therefore, be useful starting points for design of molecules that bind (and inhibit) members of the Arf and Rho families as well as Rab and Ral.
The stapled peptides that we have designed bind to the nucleotide-sensitive switch regions of Ral. These regions, as well as being responsible for effector binding, are the sites of interaction with the GEFs and GAPs, the regulators of the G proteins. We tested whether SP1 bound competitively with Ral effectors and found that it could be fully displaced by RLIP76 and only partially displaced by Sec5. This indicates that SP1 binds to both switch 1 and switch 2. The recent structure of Ral with a GEF protein (Rlf) shows that switch 2 is involved in the GEF interaction and is also important for Ral versus Ras selectivity (25). There are currently no structures of Rals in complex with cognate GAPs, but the RalGAP proteins have the closest homology with RapGAPs, and a Rap1b·Rap1GAP complex shows that both switch regions of the Rap protein are involved in the interaction (51). As our peptides do not bind to the GDP-bound form of Ral, they should not compete with GEF proteins for binding, but they are likely to bind competitively with the GAP proteins. This implies that they would not prevent Ral proteins being activated (i.e. becoming GTP-bound) in the cell, but as the peptides would be competitive with GAP binding, they could prevent deactivation. Nevertheless, we have demonstrated here the important overall effect of SP1 in cells, which is to prevent Ral signaling by competing with effector binding. Furthermore, GTP-specific peptides would be particularly useful in cells that have reduced RalGAP activity, for example invasive bladder cancer cell lines (27).
Interestingly, we found that RalA·GMPPNP binding to both Peptide 1 and SP1 was of a lower affinity than RalB·GMPPNP binding to the same peptides. The peptides were designed based on the structure of RalB·GMPPNP with RLIP76 RBD. Although the RalA·GMPPNP and RalB·GMPPNP sequences are identical in the regions that bind to RLIP76, mutagenesis of the RLIP76 RBD has revealed some isoform differences (17). Of interest is the observation that mutation of Leu-412 in helix α1 to Ala reduces RalA binding 4-fold but does not affect RalB binding. This implies that helix α1 may contribute more to RalA interactions than to RalB binding and raises the exciting prospect that it might be possible to design Ral-isoform-specific inhibitors. We also found that the related G protein R-Ras was able to bind to SP1, albeit with a reduced affinity. The binding of R-Ras is not surprising when considering the sequence identity between these two members of the Ras family. Of the 14 residues that contact helix α2 in the structure (Fig. 3D and Ref. 33) 11 are identical or represent conservative substitutions in R-Ras. Only Ala-48, Arg-52, and Ser-85 are not conserved in R-Ras. Ala-48RalB backbone forms a hydrogen bond with the side chain of Gln-433RLIP76; the replacement of Ala by Glu in R-Ras should not prevent this directly, although it may alter the conformation of switch 1. Arg-52RalB forms a hydrogen bond with the backbone of the RLIP76 coiled coil and makes contacts with RLIP76 residues Arg-434, Thr-437, and Ala-438. Replacement of Arg-52 by the smaller side chain of Thr in R-Ras would lead to a rearrangement of these interactions. Finally, Ser-85RalB forms a hydrogen bond with Glu-427RLIP76. This would not be able to form when R-Ras binds to RLIP76 because it has an Ala at this position. Therefore, the selectivity of SP1 for Ral proteins over R-Ras probably lies in just two residues in Ral, Arg-52 and Ser-85. These residues also both experience chemical shift changes when SP1 binds and hence contact SP1 directly.
Importantly, SP1 was able to enter mammalian cells, confirming its potential for use as a chemical probe and as a starting point for designing therapeutic peptides. The ability of hydrocarbon staples to enhance cell penetration is perhaps their most important attribute. Several classes of cell penetration enhancement sequences have been identified and successfully employed (52). Enhancement by chemical stapling appears to be due to an increase in the overall hydrophobicity of the peptide as a result of the chemical clamp and is sequence-independent. Cell penetration is likely to be further enhanced by the overall positive charge carried by SP1, as previous studies have suggested that positive charge is often favorable for uptake of constrained peptides into cells (36). A systematic study of >200 peptides showed that charges of +1 to +7 are optimal for cellular uptake; as SP1 has a charge of +2, it fits the profile of a peptide that readily enters cells (53). The same study showed that the mechanism of cell uptake of stapled peptides likely involves ATP-dependent endocytosis but is not dependent on caveolin or clathrin. Rather, it may be dependent on anionic cell-surface proteoglycans, explaining the necessity for positive charge.
Our cellular uptake assays indicated that SP1 has a cellular lifetime of at least 16 h (Fig. 6). Furthermore, the activity assays were performed 24 h after peptide treatment, demonstrating that the peptide was effective for a minimum of 24 h. Similar activity assays using Peptide 1 revealed some activity in cells after 12 h, but this had fallen back to the control levels by 24 h, presumably due to degradation of the unstapled and, therefore, unprotected peptide (data not shown). This is borne out by the lack of signal for FITC-Peptide 1 in cell penetration assays after 16 h.
Once in cells, SP1 shows clear biological activity. We chose to use inhibition of nutrient starvation-induced autophagocytosis as a cellular readout for peptide activity. RalB, but not RalA, is required for autophagosome biogenesis and is sufficient to activate autophagy. Thus, autophagosome biogenesis is an isoform-specific Ral controlled pathway that gives a clean readout for inhibition of RalB activity. Our peptides were designed using the RalB-RLIP76 RBD structure and indeed have a slightly higher affinity for RalB over RalA. In this assay, clear inhibition by SP1 was achieved at low micromolar concentrations. We also demonstrated the ability of SP1 to inhibit the cellular interaction between RalB and its immediate downstream effector, Sec5, by inhibition of co-immunoprecipitation. We expected that SP1 should be able to interfere with multiple RalB effector interactions due to the overlap of all known binding sites. RalB activates autophagy via its effector protein, Exo84. The mechanism of action of RalB in autophagy is known to be through initiation of vesicle nucleation by assembly of the ULK1-Beclin1-VPS34 complex directly on Exo84. Thus inhibition of autophagy suggests that the Exo84 interaction is also inhibited by SP1 in cells. Taken together, the data demonstrate that SP1 inhibits RalB-specific pathways via its engineered mode of action by acting as a pan RalB-effector complex inhibitor.
The role of autophagy in cancer has been the subject of much investigation recently. In some contexts autophagy may suppress tumorigenesis, but in the majority of situations autophagy is thought to promote tumorigenesis (54). Autophagy is found to be up-regulated in Ras-driven cancers, and although this seems to be context dependent, autophagy inhibitors may well have utility in defined disease scenarios (55). Thus, the action of our stapled peptides in a cellular pathway important to Ras-driven cancers indicates their utility as both a cellular probe and as a potential therapeutic starting point.
In summary, we have exploited the α-helical nature of the RBD of the Ral effector RLIP76 and rationally designed stabilized α-helical peptides that bind to Ral proteins and inhibit their interactions with downstream effectors. The constrained peptide displaying the highest affinity for RalB in vitro was also capable of entering cells and inhibiting RalB-specific cellular functions. Thus this approach represents a potential strategy to interfere with Ras and Ral signaling as well as providing a chemical tool to investigate Ral function in vivo. SP1 binds to Ral and competes with effector proteins, suggesting that it would be able to switch off Ral signaling completely and, therefore, contribute to the down-regulation of oncogenic Ras. Therapeutics against Ras-driven cancers are desperately required; this peptide provides an excellent starting point for development of such a drug that could be used alone or in conjunction with Raf or PI3K inhibitors in a multipronged attack on aberrant Ras signaling. These data also provide proof of principle that small G proteins in their active conformation can be targeted using this strategy, which has ramifications for many other small G protein-controlled pathways and, therefore, diseases. Work is ongoing in our laboratories to improve the affinity of the peptide and further explore in vivo effects.
Experimental Procedures
Peptide Synthesis and Characterization
All peptides were synthesized using Fmoc-based solid-phase peptide synthesis (as previously published; Ref. 44) on Rink Amide (MBHA) resin (30 μmol scale). Resin was swollen using 1 ml of N-methyl-2-pyrrolidone (NMP) for 10 min, then Fmoc-deprotection was carried out using 2 × 1.5 ml of piperidine (25% v/v) in NMP for 10 min followed by a pair of consecutive dichloromethane and NMP washes. Standard amino acid couplings were performed with 800 μl of activated amino acid solution consisting of Fmoc-Xaa-OH (375 μl, 0.4 m, 5 eq), PyClock (375 μl, 0.4 m, 5 eq), and N,N-diisopropylethylamine (52 μl, 10 eq). Each residue was double-coupled at room temperature for 2 × 1 h. Unnatural amino acid solutions contained Fmoc-protected amino acid (300 μl, 0.4 m, 4 eq), PyClock (300 μl, 0.4 m, 4 eq), N,N-diisopropylethylamine (42 μl, 8 equivalents) and were single-coupled for 2 h. Peptides containing unnatural amino acids were chemically stapled by reaction with 2 × 1 ml of Grubbs' first generation catalyst (6 mm) in 1,2-dichloroethane at room temperature for 2 h. After the final Fmoc deprotection, peptide N termini were acetylated with 2 ml of acetylation solution (4% acetic anhydride, 16% N,N-diisopropylethylamine in N-methyl-2-pyrrolidone) for 1 h or labeled with FITC with 500 μl of FITC solution (82 mg FITC (7 eq), 73 μl of N,N-diisopropylethylamine (14 eq) in dimethylformamide) overnight. Peptide resins were extensively washed with dichloromethane, methanol, and diethyl ether and dried in a vacuum desiccator overnight before deprotection using 95% trifluoroacetic acid, 2.5% triisopropylsilane, 2.5% water (1.5 ml) for 2.5 h. Peptides were purified by HPLC and analyzed using LCMS and MALDI. The concentrations of stock solutions were checked by amino acid analysis. See supplemental Table S1 for characterization data for all peptides synthesized. 6-Carboxyfluorescein peptides were purchased from Eurogentec.
Circular Dichroism Spectroscopy
CD spectra were recorded at 1-nm intervals between 190 and 260 nm using a Chirascan CD spectrometer with a 1-mm path length quartz cuvette. Three scans were recorded for each peptide, the data were averaged, and buffer background was subtracted. Peptides were measured at 10 μm in 20 mm phosphate buffer, pH 7.5, at 298 K. The helical content of each peptide was determined using CDSSTR, Set 7, and DichroWeb (56).
Protein Expression
RalB (residues 1–185, Q72L), RalA (1–184, Q72L), and RLIP76 RBD (residues 393–446, C411S) were expressed and purified as described previously (33, 41). Cultures used to express 15N-labeled RalB were grown in MOPS minimal media supplemented with 15NH4Cl, as described previously. All protein tags were cleaved before use, as previously described (33). Exchange of nucleotide for GMPPNP was performed as described previously (49). R-Ras (residues 23–201, Q89L) was cloned into pET16b (Novagen) and transformed into BL21(DE3) Escherichia coli. Its expression was induced by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h, and the protein was purified using an iminodiacetic acid-Sepharose column (GE Healthcare) and gel filtration (S75, GE Healthcare).
Isothermal Titration Calorimetry
ITC data were collected using a MicroCal iTC200 calorimeter at 298 K in 50 mm Tris-HCl, pH 7.5, 1 mm MgCl2. Peptide (1–2 mm) was titrated into protein (70 μm) in 19 × 2-μl additions with 120-s spacing between injections. Control experiments were performed by titrating peptides (1–2 mm) into buffer. Data were fitted using MicroCal Origin 7.0 software using a single-site binding model (n = 1). All data shown are representative of at least two independent experiments.
Fluorescence Polarization
Fluorescence polarization experiments were measured on a BMG Labtech Pherastar fluorimeter with excitation 485 nm and emission 520 nm at 298 K. Solutions were made up in black, flat-bottomed 384-well plates with 30-μl total volume per well. Ral proteins were serially diluted (doubling dilutions) in 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 1 mm MgCl2 (maximum final concentration 50 μm) and added to the plate followed by FITC peptide (final concentration, 10 nm). Plates were spun at 1000 × g for 1 min and then read both immediately and after 30 min of incubation at 298 K. Data were fitted to a single-site binding model using non-linear regression analysis in GraphPad Prism 6.0 to obtain Kd values and their S.E.
Competition fluorescence polarization assays were run as above using 10 nm FITC-Peptide 5 and 1 μm RalB·GMPPNP in the presence of doubling dilutions of Sec5 or RLIP76 Ral binding domains (maximum final concentration 30 μm). Data were fitted to a sigmoidal dose-response curve using non-linear regression analysis in GraphPad Prism 6.0 to obtain Kd values and their S.E.
1H,15N HSQC NMR Spectroscopy
Experiments were recorded on a Bruker DRX500 at 298 K using 200 μm 15N-labeled RalB·GMPPNP in 50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm MgCl2, 10% D2O. For the titration experiments, one, two, five, and eight eq of peptide were added to the protein solution, and the spectra were recorded after each peptide addition. NMR data were processed using the AZARA package7 and analyzed using CCPN ANALYSIS (38). Overall chemical shift perturbations, δ, were calculated using the equation,
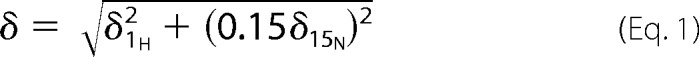 |
where δ1H and δ15N are the chemical shift changes for the 1H and 15N dimensions, respectively.
Cellular Uptake of Peptides
5 × 105 HEK293T cells were seeded into 35-mm glass-bottom culture dishes and incubated in 1 ml of DMEM supplemented with 10% FBS. After 24 h, FITC alone, FITC-labeled unstapled Peptide 1, or FITC-labeled stapled peptide SP1 were added to a final concentration of 10 μm. After 16 h the cells were fixed in 4% w/v formaldehyde. Images were acquired using a Fluoview 300 Laser scanning confocal microscope with a PLAPON 60×1.42 PLan Apo oil objective and a 60-μm confocal aperture. Cells were excited at 488 nm, and emitted light was detected at 510–570 nm. All images were recorded using the same instrument settings.
Co-immunoprecipitation Assays
5 × 106 HEK293T cells were seeded 48 h before the end point into 10-cm dishes (3 per condition) in DMEM supplemented with 10% FBS. The medium was aspirated, and fresh DMEM (with 10% FBS) with and without 6-carboxyfluorescein (FAM)-SP1 or -SP6 (at 100 μm each) was added 24 h before the end point. The medium was then replaced with 1× EBSS (Earle's balanced salt solution) 90 min before the end point. At the end point, cells were lysed in lysis buffer (20 mm Tris-HCl, pH 7.4, 137 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 10% glycerol, 10 mm MgCl2, 2 mm EGTA, 1 mm PMSF, 50 mm NaF, 1 mm NaVO4, and 80 mm β-glycerophosphate plus EDTA-free protease inhibitor mixture (Roche Applied Science 04693159001)). After 15 min lysis at 4 °C, lysates were cleared at 20,000 × g for 20 min at 4 °C. Cell lysates were diluted with lysis buffer to prepare 120 μl of whole cell lysate (4 μg/μl) and 1400 μl of immunoprecipitation lysate (6 μg/μl) for each condition. Endogenous RalB was immunoprecipitated by the addition of 30 μl of mouse anti-RalB antibody (a kind gift from Larry Feig, Tufts University) to immunoprecipitation lysates at 4 °C for 4 h. Protein A/G-agarose beads (Santa Cruz Biotechnology Inc., sc-2003) were added for 1 h at 4 °C to precipitate antibody-antigen complexes. Precipitated complexes were washed 3 times with lysis buffer for 5 min at 4 °C. The samples were then separated via SDS-PAGE and transferred to PVDF membranes (Immobilon-P). Membranes were probed by immunoblotting with the following primary antibodies: rabbit anti-RalB (Cell Signaling Technology cs-3523, lot 1) and mouse anti-Sec5 (a kind gift from Charles Yeaman, University of Iowa).
GFP-LC3 Fluorescence Quantification and Imaging
72 h before the end point, 8 × 103 HeLa cells stably expressing GPF-LC3 were plated per well of glass-bottomed 96-well plates. Cells were plated in 100 ml of DMEM supplemented with 10% FBS and grown for 48 h. 24 h before the end point, the medium was changed with 100 ml of fresh DMEM (with 10% FBS). DMSO and peptides were added 24 h before the end point at the final indicated concentrations (final concentration of DMSO per well was 1%). 4 h before the end point, cells were washed twice with PBS (with Ca2+ and Mg2+) and then fed with either 100 ml of 1× (EBSS) or serum-free DMEM as indicated. At the end point, EBSS-treated cells were washed once, and serum-free DMEM-treated cells were washed twice with 1× PBS (with Ca2+ and Mg2+). Cells were then fixed in 4% paraformaldehyde in 1× PBS (with Ca2+ and Mg2+) for 10 min. Cells were then washed twice as before and then stained with 0.01% Hoechst in 1× PBS (with Ca2+ and Mg2+) for 20 min. Total fluorescence intensity for GFP and Hoechst was measured using the PheraStar FS plate-reader. For epifluorescence imaging, HeLa cells stably expressing GPF-LC3 were grown in 384-well plates in DMEM (with 10% FBS) for 48 h. The medium was not changed for DMEM-treated cells, and EBSS-treated cells were starved for 2 h before live-cell imaging at 20× via the (BD Biosciences) Pathway 855 High-Content Bioimager.
GFP-TFEB Activation
5 × 103 HeLa cells stably expressing GFP-TFEB (48) were plated in 500 μl of DMEM (with 10% FBS) per well of 8-Chamber Lab-Tek® II Chambered #1.5 German Coverglass slides and grown overnight. 24 h before imaging, the medium was changed with 265 μl of fresh DMEM (with 10% FBS) and DMSO or peptides at the final concentrations indicated (final concentration of DMSO per well was 1%). 24 h after peptide addition, live-cell images were acquired with an Andor Spinning Disc Confocal Microscope (oil immersion and 60× objective) under normal cell culture conditions (37 °C, 5% CO2). Initial images of fed-state cells were taken 24 h after peptide addition. The medium in each well was then changed with 265 μl of 1 × EBSS, and subsequent images were taken at the times indicated.
Author Contributions
J. C. T. synthesized all of the peptides and performed all of the biophysical and structural analyses. H. R. M. performed the NMR experiments and data processing. N. S. C. and D. O. undertook the cell penetration experiments. J. M. C. and C. W. performed the co-immunoprecipitation and autophagy assays. M. A. W. oversaw all cell biology. C. A. oversaw all the chemistry. D. O. and H. R. M. directed all aspects of the overall project. J. C. T., H. R. M., and D. O. wrote the manuscript. All co-authors participated in discussions and in editing the final manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Nick Pugh and Dr. Stephanie Jung for assistance with confocal microscopy, Dr. Arooj Shafiq for providing RalA, and Dr. Karthik Rajasekar for R-Ras protein. We are also grateful to Dr. Chiara Valenzano and Dr Katherine Stott for helpful discussions.
This work was supported in part by a Cambridge Cancer Centre Pump Priming award (to C. A., D. O., and H. R. M.), a BBSRC (Biotechnology and Biological Sciences Research Council) studentship (to N. S. C.), National Institutes of Health Grant CA71443, and Welch Foundation Grant I-1414 (to M. A. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table 1.
Aileron Therapeutics (2013) Aileron therapeutics initiates phase 1 cancer study of ALRN-6924 in advanced hematologic and solid malignancies with wild type p53. This trial successfully completed stapled peptide Phase 1 clinical trials (February 12, 2015).
D. Owen and H. R. Mott, unpublished observations.
W. Boucher, unpublished data.
- GEF
- guanine nucleotide exchange factor
- GAP
- GTPase activating protein
- RBD
- Ral binding domain
- FP
- fluorescence polarization
- ITC
- isothermal titration calorimetry
- HSQC
- heteronuclear single quantum correlation
- EBSS
- Earle's balanced salt solution
- LC3
- microtubule-associated protein 1A/1B-light chain 3 (LC3)
- ULK1
- Unc-51 like autophagy activating kinase 1
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- GMPPNP
- guanosine 5′-[β,γ-imido] triphosphate
- TFEB
- transcription factor EB.
References
- 1. Stephen A. G., Esposito D., Bagni R. K., and McCormick F. (2014) Dragging Ras back in the ring. Cancer Cell 25, 272–281 [DOI] [PubMed] [Google Scholar]
- 2. Jones S., and Thornton J. M. (1996) Principles of protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 93, 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arkin M. R., and Wells J. A. (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3, 301–317 [DOI] [PubMed] [Google Scholar]
- 4. Arkin M. R., Tang Y., and Wells J. A. (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem. Biol. 21, 1102–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gysin S., Salt M., Young A., and McCormick F. (2011) Therapeutic strategies for targeting Ras proteins. Genes Cancer 2, 359–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., and Der C. J. (2014) Drugging the undruggable RAS: mission possible? Nat. Rev. Drug Discov. 13, 828–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fiordalisi J. J., Johnson R. L. 2nd, Weinbaum C. A., Sakabe K., Chen Z., Casey P. J., and Cox A. D. (2003) High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J. Biol. Chem. 278, 41718–41727 [DOI] [PubMed] [Google Scholar]
- 8. Ostrem J. M., Peters U., Sos M. L., Wells J. A., and Shokat K. M. (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baines A. T., Xu D., and Der C. J. (2011) Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem. 3, 1787–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holderfield M., Deuker M. M., McCormick F., and McMahon M. (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fruman D. A., and Rommel C. (2014) PI3K and cancer: lessons, challenges, and opportunities. Nat. Rev. Drug Discov. 13, 140–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bodemann B. O., and White M. A. (2008) Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer 8, 133–140 [DOI] [PubMed] [Google Scholar]
- 13. Neel N. F., Martin T. D., Stratford J. K., Zand T. P., Reiner D. J., and Der C. J. (2011) The RalGEF-Ral effector signaling network: the road less traveled for anti-Ras drug discovery. Genes Cancer 2, 275–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moskalenko S., Henry D. O., Rosse C., Mirey G., Camonis J. H., and White M. A. (2002) The exocyst is a Ral effector complex. Nat. Cell Biol. 4, 66–72 [DOI] [PubMed] [Google Scholar]
- 15. Sugihara K., Asano S., Tanaka K., Iwamatsu A., Okawa K., and Ohta Y. (2002) The exocyst complex binds the small GTPase RaIA to mediate filopodia formation. Nat. Cell Biol. 4, 73–78 [DOI] [PubMed] [Google Scholar]
- 16. Chien Y., Kim S., Bumeister R., Loo Y. M., Kwon S. W., Johnson C. L., Balakireva M. G., Romeo Y., Kopelovich L., Gale M. Jr., Yeaman C., Camonis J. H., Zhao Y., and White M. A. (2006) RaIB GTPase-mediated activation of the IκB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127, 157–170 [DOI] [PubMed] [Google Scholar]
- 17. Campbell L. J., Peppa M., Crabtree M. D., Shafiq A., McGough N. F., Mott H. R., and Owen D. (2015) Thermodynamic mapping of effector protein interfaces with RalA and RalB. Biochemistry 54, 1380–1389 [DOI] [PubMed] [Google Scholar]
- 18. Fukai S., Matern H. T., Jagath J. R., Scheller R. H., and Brunger A. T. (2003) Structural basis of the interaction between RalA and Sec5, a subunit of the sec6/8 complex. EMBO J. 22, 3267–3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin R., Junutula J. R., Matern H. T., Ervin K. E., Scheller R. H., and Brunger A. T. (2005) Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J. 24, 2064–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shipitsin M., and Feig L. A. (2004) RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol. Cell. Biol. 24, 5746–5756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lim K.-H., Brady D. C., Kashatus D. F., Ancrile B. B., Der C. J., Cox A. D., and Counter C. M. (2010) Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol. Cell. Biol. 30, 508–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang H., Owens C., Chandra N., Conaway M. R., Brautigan D. L., and Theodorescu D. (2010) Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 70, 8760–8769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin T. D., Mitin N., Cox A. D., Yeh J. J., and Der C. J. (2012) Phosphorylation by protein kinase Cα regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J. Biol. Chem. 287, 14827–14836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neyraud V., Aushev V. N., Hatzoglou A., Meunier B., Cascone I., and Camonis J. (2012) RalA and RalB proteins are ubiquitinated GTPases, and ubiquitinated RalA increases lipid raft exposure at the plasma membrane. J. Biol. Chem. 287, 29397–29405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Popovic M., Schouten A., Rensen-de Leeuw M., and Rehmann H. (2016) The structure of the guanine nucleotide exchange Factor Rlf in complex with the small G-protein Ral identifies conformational intermediates of the exchange reaction and the basis for the selectivity. J. Struct. Biol. 193, 106–114 [DOI] [PubMed] [Google Scholar]
- 26. Shirakawa R., Fukai S., Kawato M., Higashi T., Kondo H., Ikeda T., Nakayama E., Okawa K., Nureki O., Kimura T., Kita T., and Horiuchi H. (2009) Tuberous sclerosis tumor suppressor complex-like complexes act as GTPase-activating proteins for Ral GTPases. J. Biol. Chem. 284, 21580–21588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saito R., Shirakawa R., Nishiyama H., Kobayashi T., Kawato M., Kanno T., Nishizawa K., Matsui Y., Ohbayashi T., Horiguchi M., Nakamura T., Ikeda T., Yamane K., Nakayama E., Nakamura E., Toda Y., Kimura T., Kita T., Ogawa O., and Horiuchi H. (2013) Downregulation of Ral GTPase-activating protein promotes tumor invasion and metastasis of bladder cancer. Oncogene 32, 894–902 [DOI] [PubMed] [Google Scholar]
- 28. Chien Y., and White M. A. (2003) RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Reports 4, 800–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim K. H., Baines A. T., Fiordalisi J. J., Shipitsin M., Feig L. A., Cox A. D., Der C. J., and Counter C. M. (2005) Activation of RaIA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 7, 533–545 [DOI] [PubMed] [Google Scholar]
- 30. Lim K.-H., O'Hayer K., Adam S. J., Kendall S. D., Campbell P. M., Der C. J., and Counter C. M. (2006) Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 16, 2385–2394 [DOI] [PubMed] [Google Scholar]
- 31. Martin T. D., and Der C. J. (2012) Differential involvement of RalA and RalB in colorectal cancer. Small GTPases 3, 126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yan C., Liu D., Li L., Wempe M. F., Guin S., Khanna M., Meier J., Hoffman B., Owens C., Wysoczynski C. L., Nitz M. D., Knabe W. E., Ahmed M., Brautigan D. L., Paschal B. M., Schwartz M. A., Jones D. N., Ross D., Meroueh S. O., and Theodorescu D. (2014) Discovery and characterization of small molecules that target the GTPase Ral. Nature 515, 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fenwick R. B., Campbell L. J., Rajasekar K., Prasannan S., Nietlispach D., Camonis J., Owen D., and Mott H. R. (2010) The RaIB-RLIP76 complex reveals a novel mode of RaI-effector interaction. Structure 18, 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mott H. R., and Owen D. (2015) Structures of Ras superfamily effector complexes: what have we learnt in two decades? Crit. Rev. Biochem. Mol. Biol. 50, 85–133 [DOI] [PubMed] [Google Scholar]
- 35. Bodemann B. O., Orvedahl A., Cheng T., Ram R. R., Ou Y.-H., Formstecher E., Maiti M., Hazelett C. C., Wauson E. M., Balakireva M., Camonis J. H., Yeaman C., Levine B., and White M. A. (2011) RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walensky L. D., and Bird G. H. (2014) Hydrocarbon-stapled peptides: principles, practice, and progress. J. Med. Chem. 57, 6275–6288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wittrup K. D., and Verdine G. L. (2012) Protein engineering for therapeutics, Part A. Preface. Methods Enzymol. 502, xiii–xiv [DOI] [PubMed] [Google Scholar]
- 38. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 39. Patgiri A., Yadav K. K., Arora P. S., and Bar-Sagi D. (2011) An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 7, 585–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leshchiner E. S., Parkhitko A., Bird G. H., Luccarelli J., Bellairs J. A., Escudero S., Opoku-Nsiah K., Godes M., Perrimon N., and Walensky L. D. (2015) Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl. Acad. Sci. U.S.A. 112, 1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Prasannan S., Fenwick R. B., Campbell L. J., Evetts K. A., Nietlispach D., Owen D., and Mott H. R. (2007) 1H, 13C, and 15N resonance assignments for the small G protein RalB in its active conformation. Biomol. NMR Assign. 1, 147–149 [DOI] [PubMed] [Google Scholar]
- 42. Fenwick R. B., Prasannan S., Campbell L. J., Nietlispach D., Evetts K. A., Camonis J., Mott H. R., and Owen D. (2009) Solution structure and dynamics of the small GTPase RalB in its active conformation: significance for effector protein binding. Biochemistry 48, 2192–2206 [DOI] [PubMed] [Google Scholar]
- 43. Hubbard S. J., and Thornton J. M. (1993) NACCESS. Department of Biochemistry and Molecular Biology, University College London, London [Google Scholar]
- 44. Kim Y.-W., Grossmann T. N., and Verdine G. L. (2011) Synthesis of all-hydrocarbon stapled α-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 6, 761–771 [DOI] [PubMed] [Google Scholar]
- 45. Moellering R. E., Cornejo M., Davis T. N., Del Bianco C., Aster J. C., Blacklow S. C., Kung A. L., Gilliland D. G., Verdine G. L., and Bradner J. E. (2009) Direct inhibition of the NOTCH transcription factor complex. Nature 462, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grossmann T. N., Yeh J. T., Bowman B. R., Chu Q., Moellering R. E., and Verdine G. L. (2012) Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proc. Natl. Acad. Sci. U.S.A. 109, 17942–17947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peña-Llopis S., Vega-Rubin-de-Celis S., Schwartz J. C., Wolff N. C., Tran T. A., Zou L., Xie X.-J., Corey D. R., and Brugarolas J. (2011) Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30, 3242–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roczniak-Ferguson A., Petit C. S., Froehlich F., Qian S., Ky J., Angarola B., Walther T. C., and Ferguson S. M. (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5, ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson G., Owen D., Chalk P. A., and Lowe P. N. (1998) Delineation of the Cdc42/Rac-binding domain of p21-activated kinase. Biochemistry 37, 7885–7891 [DOI] [PubMed] [Google Scholar]
- 50. Spiegel J., Cromm P. M., Itzen A., Goody R. S., Grossmann T. N., and Waldmann H. (2014) Direct targeting of Rab-GTPase-effector interactions. Angew. Chem. Int. Ed. Engl. 53, 2498–2503 [DOI] [PubMed] [Google Scholar]
- 51. Scrima A., Thomas C., Deaconescu D., and Wittinghofer A. (2008) The Rap-RapGAP complex: GTP hydrolysis without catalytic glutamine and arginine residues. EMBO J. 27, 1145–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kauffman W. B., Fuselier T., He J., and Wimley W. C. (2015) Mechanism matters: a taxonomy of cell penetrating peptides. Trends Biochem. Sci. 40, 749–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chu Q., Moellering R. E., Hilinski G. J., Kim Y.-W., Grossmann T. N., Yeh J. T. H., and Verdine G. L. (2015) Towards understanding cell penetration by stapled peptides. Med. Chem. Commun. 6, 111–119 [Google Scholar]
- 54. Yang Z. J., Chee C. E., Huang S., and Sinicrope F. A. (2011) The role of autophagy in cancer: therapeutic implications. Mol. Cancer Ther. 10, 1533–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. White E. (2015) The role for autophagy in cancer. J. Clin. Invest. 125, 42–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Whitmore L., and Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.