

Abstract
An important contributor to brain ischemia is known to be extracellular acidosis, which activates acid-sensing ion channels (ASICs), a family of proton-gated sodium channels. Lines of evidence suggest that targeting ASICs may lead to novel therapeutic strategies for stroke. Investigations of the role of ASICs in ischemic brain injury have naturally focused on the role of extracellular pH in ASIC activation. By contrast, intracellular pH (pHi) has received little attention. This is a significant gap in our understanding because the ASIC response to extracellular pH is modulated by pHi, and activation of ASICs by extracellular protons is paradoxically enhanced by intracellular alkalosis. Our previous studies show that acidosis-induced cell injury in in vitro models is attenuated by intracellular acidification. However, whether pHi affects ischemic brain injury in vivo is completely unknown. Furthermore, whereas ASICs in native neurons are composed of different subunits characterized by distinct electrophysiological/pharmacological properties, the subunit-dependent modulation of ASIC activity by pHi has not been investigated. Using a combination of in vitro and in vivo ischemic brain injury models, electrophysiological, biochemical, and molecular biological approaches, we show that the intracellular alkalizing agent quinine potentiates, whereas the intracellular acidifying agent propionate inhibits, oxygen-glucose deprivation-induced cell injury in vitro and brain ischemia-induced infarct volume in vivo. Moreover, we find that the potentiation of ASICs by quinine depends on the presence of the ASIC1a, ASIC2a subunits, but not ASIC1b, ASIC3 subunits. Furthermore, we have determined the amino acids in ASIC1a that are involved in the modulation of ASICs by pHi.
Keywords: acid sensing ion channel (ASIC), brain, electrophysiology, ischemia, patch clamp
Introduction
A common feature of brain ischemia is acidosis (1–5), which plays a critical role in the ensuing brain injury. However, the mechanisms of acidosis-induced injury remain poorly understood. Acidosis activates acid-sensing ion channels (ASICs),4 a family of proton-gated cation channels that belong to the degenerin and epithelial Na+ channel superfamily of ion channels (6). These channels are expressed in neurons throughout the peripheral and central nervous systems (6–9). In sensory neurons, ASICs play important roles in nociception (9–11), mechanosensation (12, 13), and taste transduction (14), whereas in the central nervous system (CNS), ASICs are involved in processes such as synaptic plasticity, learning, and memory (15–17). The specific ASIC important in acidosis-induced injury is the ASIC1a subtype (5, 18). This subtype has a higher sensitivity to protons and is both sodium- and calcium-permeable. Neuronal injury induced by ASIC activation is independent of glutamate receptor-mediated excitotoxicity (5, 18), offering provocative new evidence for ASICs as potential new targets for stroke therapy (4, 5, 19–21).
In the brain, energy is stored mainly in the form of the high energy phosphate compound ATP. In normoxia, ATP is predominantly produced by oxidative phosphorylation of glucose (22). However, under ischemic conditions, a decrease in tissue PO2 results in mitochondrial dysfunction and inhibition of pyruvate oxidation. As a result, the majority of ATP is produced by the glycolytic pathway, converting pyruvate to lactate and H+, resulting in a gradual decrease in overall brain pH (1, 23, 24). During ischemia, extracellular pH can drop to ∼6.2 and even below 6.0 in hyperglycemic animals (2, 25, 26). In contrast to the decrease in extracellular pH with focal ischemia, increases of intracellular pH in some regions of the brain have been reported (27–32). A stable intracellular pH is critical for normal cellular function, and most biological processes are markedly sensitive to pH (26, 33). Even small changes in intracellular pH affect the properties of various ion channels, plasma membrane excitability, and cellular metabolism (33–37).
Investigations of the role of ASICs in ischemic brain injury have focused on the role of extracellular pH in ASIC activation. By contrast, intracellular pH has received little attention. This is a significant knowledge gap because the ASIC response to extracellular pH is known to be modulated by intracellular pH, i.e. activation of these channels by extracellular protons is paradoxically enhanced by intracellular alkalosis (38). We used pharmacological tools to manipulate intracellular pH and to assess the resulting changes in ASIC activity. Quinine is a widely used agent for intracellular alkalization, whereas addition of propionate or NH4Cl withdrawal induces intracellular acidification (36). These agents do not directly activate ASIC currents in either cultured cortical neurons or ASIC1a-expressing Chinese hamster ovary (CHO) cells by themselves, but they modulate ASIC activity induced by extracellular acidosis (38). This suggests that the function of ASICs is modulated not only by extracellular pH but also intracellular pH.
Using in vitro (oxygen-glucose deprivation, OGD) and in vivo (a transient middle cerebral artery occlusion, MCAO) ischemia models, combined with electrophysiological, biochemical, and molecular biological approaches, here we determined whether changing intracellular pH affects ischemic brain injury. We also determined whether the modulation of ASICs by intracellular pH (pHi) is dependent on specific ASIC subunits. Finally, the potential domains and/or site(s) involved in the modulation of ASICs by intracellular pH were examined.
Results
Changing Intracellular pH Alters OGD-induced Injury of Cultured Mouse Cortical Neurons
Intracellular alkalization potentiates, whereas intracellular acidification inhibits, neuronal injury induced by extracellular acidosis (38). To determine whether changing intracellular pH also affects ischemic neuronal injury, we measured lactate dehydrogenase (LDH) release following OGD plus acidosis, a commonly used in vitro model of brain ischemia. Cultured mouse cortical neurons were divided into different groups and subjected to 1 h of OGD in an anaerobic chamber. MK-801 (10 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 20 μm), and nimodipine (5 μm) were added in all groups to eliminate a potential secondary activation of glutamate receptors and voltage-gated Ca2+ channels (5). We first compared the combined OGD and acid-induced neuronal injury in the absence or presence of quinine or propionate. As anticipated, LDH release was increased by lowered extracellular pH (0.5 ± 0.05) but reduced by propionate (0.2 ± 0.04) compared with control (pH 7.4) (Fig. 1B). In contrast to propionate, quinine (1 mm) increased the acid-induced injury (0.6 ± 0.2), but PcTX1 (10 nm) blocked the enhancement of LDH release by quinine (1 mm) (0.3 ± 0.05; Fig. 1C). Similar to LDH measurements, fluorescein diacetate and propidium iodide staining of alive and dead neurons taken at 6 h following the 1 h of OGD treatment in the absence or presence of quinine or propionate revealed that increasing intracellular pH (by quinine) potentiates and intracellular acidification (by propionate) inhibits OGD-induced neuronal injury (Fig. 1A). These data indicate that intracellular pH modulates ischemic injury in neurons.
FIGURE 1.

Effect of quinine/propionate on OGD-induced injury of cultured mouse cortical neurons. A, fluorescein diacetate (green) and propidium iodide (red) staining of live and dead neurons taken at 6 h following 1 h of OGD treatment in the absence or presence of quinine or propionate shows that increasing intracellular pH (quinine) potentiates, whereas decreasing intracellular pH (propionate) inhibits OGD-induced neuronal injury. B, summary bar graph showing relative LDH release induced by OGD plus acidosis in the absence and presence of propionate. The LDH was increased by lowered extracellular pH, but reduced by propionate (10 mm) (one-way ANOVA, effect of treatment, F(2,9) = 43.55, p < 0.0001. Tukey's multiple comparisons test, **, p < 0.01 compared with pH 7.4; ##, p < 0.01 compared with pH 6.0 alone). C, summary bar graph showing that PcTX1 (10 nm) blocked enhancement of LDH release by quinine (1 mm) (one-way ANOVA, effect of treatment, F(2,9) = 9.5, p = 0.0061. Tukey's multiple comparisons test, **, p < 0.01 compared with quinine alone).
Effect of Changing Intracellular pH on Ischemia-induced Injury in Vivo
We also determined the effects of manipulating intracellular pH on ischemic brain injury in vivo. Ischemia (60 min) was induced using the MCAO model. A total of 2 μl of aCSF alone or aCSF-containing quinine (1 mm) or propionate (10 mm) was injected intracerebroventricularly 30 min before and after the ischemia. Infarct volume was determined by TTC staining (39) at 24 h following ischemia. As shown in Fig. 2A, large infarct area (representative image) and volume (bar graph) were observed in brains from aCSF (44 ± 6%, n = 6) and quinine-injected mice (44 ± 7%, n = 6), although much smaller infarct area (representative image) and volume (bar graph) were characteristic of brains from propionate-injected mice (25 ± 9%, n = 5; Fig. 2A). Note that injury was not potentiated by quinine in vivo, which is consistent with the report that neurons already demonstrate intracellular alkalosis after ischemia. Another possibility is that MCAO for 60 min already induced a maximal brain injury that could not be further potentiated by quinine. Similar to our previous findings (5), ischemia-induced infarct volume was significantly reduced in PcTX1-injected animals (25 ± 7%, n = 6; Fig. 2B). Furthermore, in the presence of PcTX1-injected animals, the infarct volume was not changed by the addition of the intracellular alkalizing agent quinine (21 ± 7%, n = 6) or the acidifying agent propionate (27 ± 7%, n = 6; Fig. 2B), supporting the involvement of ASIC1a in the modulation of infarct volume by intracellular pH-modifying agents. These results show that reducing intracellular pH protects from ischemia-induced cell injury in vivo.
FIGURE 2.

Neuroprotection by intracellular acidifying agent propionate against brain ischemia in vivo. A, TTC-stained brain sections (image, left) and summary data (bar graph, right) showing increased infarct area (infarct volume) in brains from quinine-injected mice but reduction in infarct area (infarct volume) in brains from propionate-injected mice (one-way ANOVA, effect of treatment, F(2,14) = 9.36, p = 0.0026. Tukey's multiple comparisons test, **, p < 0.01 compared with aCSF; ##, p < 0.01 compared with quinine). B, summary data showing significantly reduced ischemia-induced infarct volume in PcTX1-injected mice. The infarct volume was not different in PcTX1 plus quinine or PcTX1 plus propionate-injected mice compared with PcTX1-alone injected mice (one-way ANOVA, F(3,15) = 8.13, p = 0.0019. Tukey's multiple comparisons test, **, p < 0.01; ***, p < 0.001 compared with aCSF).
Subunit-dependent Potentiation of ASICs by Intracellular Alkalization
Our previous studies in primary cultured mouse cortical neurons have shown that intracellular alkalization (e.g. with bath perfusion of quinine) potentiated the peak amplitude of ASIC currents, shifted pH dose-response and steady-state inactivation curves, and accelerated the recovery of the channels from desensitization (38). ASICs in native neurons are composed of different ASIC subunits with distinct electrophysiological or pharmacological properties and likely functional implications. Therefore, it is important to know which subunit is involved in the modulation of the ASIC responses by pHi.
Using the pH-sensitive membrane-impermeable dye 2′,7′-bis(2-carboxyethyl)-5-(and 6)-carboxyfluorescein acetoxymethylester (BCECF, 100 μm) (36, 40), we also confirmed that addition of quinine or propionate, or withdrawal of NH4Cl, altered intracellular pH in CHO cells. Similar to previous reports for neurons (36, 41, 42), bath perfusion of 1 mm quinine increased intracellular pH, whereas NH4Cl withdrawal decreased intracellular pH in CHO cells (data not shown). To examine the possibility that modulation of ASICs by pHi depends on the presence of a specific ASIC subunit, we studied the effects of intracellular pH-modifying agents on the current mediated by different ASIC subunits expressed in CHO cells. Individual ASIC currents were activated at pH values close to their pH50 values (see below).
Our results show that the potentiation of ASICs by the intracellular alkalizing agent quinine depends on the presence of the ASIC1a and ASIC2a subunits. As shown in Fig. 3, A and B, 2 min following the bath perfusion of 1 mm quinine, ASIC1a currents (activated by decreasing pH from 7.4 to 6.0) were significantly potentiated (186 ± 53% of the control, n = 8). Similarly, the ASIC2a currents (activated by decreasing pH from 7.4 to 5.0) were almost equally potentiated (173 ± 21% of the control, n = 8). In contrast to ASIC1a and ASIC2a, currents mediated by ASIC1b (activated by the pH drop from 7.4 to 6.0) and ASIC3 (activated by the pH drop from 7.4 to 6.5) were not potentiated by the same concentration of quinine. In the presence of 1 mm quinine, the peak amplitude of the ASIC3 current was 100 ± 2% of the control (n = 7), and the amplitude of the ASIC1b current was 95 ± 5% of the control (n = 7).
FIGURE 3.

Subunit-dependent modulation of ASICs by intracellular pH. A, representative traces show that the potentiation of ASIC currents by intracellular alkalizing agent quinine depends on the presence of the ASIC1a and ASIC2a subunits. B, summary data showing the potentiation of ASIC current mediated by different ASIC subunits by 1 mm quinine. In the presence of 1 mm quinine, ASIC1a and ASIC2a currents were significantly increased. Currents mediated by ASIC1b and ASIC3 were not potentiated by the same concentration of quinine (one-way ANOVA, effect of treatment, F(4,33) = 22.04, p < 0.0001. Dunnett's multiple comparisons test, **, p < 0.01). C and D, representative traces and summary data showing the inhibition of ASIC currents by the intracellular acidifying agent propionate in cells expressing different ASIC subunits. In the presence of 10 mm propionate, the peak amplitude of the ASIC current mediated by ASIC1a and ASIC2a was significantly reduced. However, the amplitude of ASIC1b and ASIC3 was not affected by bath perfusion of 10 mm propionate (one-way ANOVA, effect of treatment, F(2,9) = 43.55, p < 0.0001. Dunnett's multiple comparisons test, **, p < 0.01).
Subunit-dependent Inhibition of ASICs by Intracellular Acidification
To determine whether the inhibition of ASICs by intracellular acidifying agents is also subunit-dependent, we studied the effects of propionate application and NH4Cl withdrawal, two commonly used methods for inducing intracellular acidification (43, 44), on ASIC currents mediated by different ASIC subunits. As shown in Fig. 3, C and D, the ASIC currents mediated by ASIC1a and ASIC2a were decreased by bath perfusion of 10 mm propionate. After 2–3 min application of propionate, the peak amplitude of the ASIC current mediated by ASIC1a was 66 ± 5% of the control (n = 8). Similarly, the peak amplitude of the ASIC2a current was 72 ± 15% of the control (n = 8). In contrast, the amplitude of ASIC current mediated by ASIC1b and ASIC3 was not affected by bath perfusion with 10 mm propionate (ASIC1b, 95 ± 5% of the control, n = 8; ASIC3, 97 ± 3% of the control, n = 8). Consistent with propionate addition, perfusion of NH4Cl (15 mm, for 3–4 min) followed by its withdrawal reduced the amplitude of ASIC1a and ASIC2a currents but not the currents mediated by ASIC1b and ASIC3 (data not shown).
To confirm that the potentiation of ASIC1a and ASIC2a currents was indeed due to intracellular alkalization, we performed the same experiments using pH well buffered intracellular solutions with 40 mm HEPES. As shown in Fig. 4, significantly reduced effects by quinine and propionate on ASIC1a and ASIC2a currents were observed with the intracellular solutions containing 40 mm HEPES. Significantly smaller potentiation of ASIC1a by quinine was observed in 40 mm HEPES compared with 10 mm HEPES intracellular solutions (n = 8–9, p < 0.05). Similarly, ASIC2a current amplitude was 144 ± 12% of the control with the intracellular solution containing 40 mm HEPES (n = 8, p < 0.01 compared with 10 mm HEPES intracellular solution, n = 7; Fig. 4, A and C). Reduced inhibition in the amplitude of ASIC1a and ASIC2a currents by 10 mm propionate was observed in 40 mm HEPES pipette solution. The current amplitude of ASIC1a was 66 ± 5% of the control (n = 10, p < 0.01 compared with 10 mm HEPES intracellular solution, n = 8; Fig. 4B). Similarly, the ASIC2a current amplitude was 88 ± 9% of the control (n = 8, p < 0.05 compared with 10 mm HEPES intracellular solution, n = 8; Fig. 4D). Taken together, these data show that ASIC1a and ASIC2a subunits are responsible for modulation of ASIC currents by intracellular pH.
FIGURE 4.

Increasing HEPES concentration in the intracellular solution reduces the modulation of ASICs by the intracellular pH-modifying agents. A, summary data showing the reduced effects of ASIC1a current by intracellular pH-modifying agents. 1 mm quinine potentiates the peak amplitude of the ASIC1a current in the pipette solution containing 40 mm HEPES, which is significantly smaller than its effect with 10 mm HEPES in the pipette solution (n = 8–9, t(15) = 2.9, unpaired t test, *, p < 0.05). B, ASIC1a current amplitude is inhibited by 10 mm propionate with 40 mm HEPES in the pipette solution, which is significantly smaller than its effect with 10 mm HEPES in the pipette solution (n = 8–10, t(16) = 6.5, unpaired t test, **, p < 0.01). C, 1 mm quinine potentiates the peak amplitude of the ASIC2a current with 40 mm HEPES in the pipette solution, which is significantly different from its effect with standard (10 mm) HEPES in the pipette solution (n = 7–8, t(13) = 3.2, unpaired t test, **, p < 0.01). D, summary bar graph showing that ASIC2a current amplitudes were inhibited by 10 mm propionate with 40 mm HEPES in the pipette solution, which is significantly different from its effect with standard (10 mm) HEPES in the pipette solution (n = 8, t(14) = 2.4, unpaired t test, *, p < 0.05).
H+ Dose-response Curves for ASIC1a and ASIC2a Are Shifted by Intracellular Alkalization
Next, we determined whether changing pHi also affects the pH-dependent activation of ASICs in a subunit-dependent manner. H+ dose-response curves were constructed for ASIC1a and ASIC2a expressed in CHO cells before and after bath perfusion of quinine. As shown in Fig. 5A, the pH-dependent activation curve of ASIC1a channels showed a leftward shift after bath perfusion of 1 mm quinine. The half-maximum activation of pH (pH50) was shifted from 6.03 ± 0.1 to 6.24 ± 0.1 in the presence of quinine (n = 6, p < 0.05; Fig. 5A). No significant change in Hill coefficient was observed (before quinine, 1.21 ± 0.05; after quinine, 1.29 ± 0.03, p > 0.05). Similarly, the activation curve of ASIC2a channels was shifted toward less acidic values by quinine. The pH50 was 4.7 ± 0.2 and 5.1 ± 0.1 in the absence and presence of 1 mm quinine (n = 6, p < 0.01; Fig. 5B). Likewise, there was no significant change in Hill coefficient (before quinine, 1.3 ± 0.1; after quinine, 1.2 ± 0.1, p > 0.05). These data indicate that changing intracellular pH alters the affinity of ASIC1a and ASIC2a to extracellular H+.
FIGURE 5.

Effects of changing intracellular pH on pH dose-response curve for ASIC1a and ASIC2a subunits. A, representative traces and summary data showing the shift in activation curve of ASIC1a currents by the intracellular alkalizing agent quinine. pH50 values were significantly increased in the presence of quinine (n = 6, t(5) = 2.9, paired t test, p < 0.05). B, representative traces and summary data showing the shift in pH50 of ASIC2a currents by quinine. Intracellular alkalization by quinine significantly increased pH50 (n = 6, t(5) = 5.2, paired t test, p < 0.01).
To provide additional evidence that the levels of pHi regulate the activities of ASICs, the pH-dependent activation of ASIC currents was recorded with intracellular solutions directly buffered at different pH values. The pH dose-response curves of ASIC1a and ASIC2a were constructed. Similar to the effect of quinine, increasing pHi to 8.5 produced a leftward shift in the pH50 of ASIC1a (pH50 = 6.2 ± 0.1 at 8.5, n = 6; pH50 = 5.8 ± 0.2 at 7.3, n = 7, p < 0.01; Fig. 6A). Similarly, the pH50 value for ASIC2a-mediated currents was shifted from 4.7 ± 0.2 at pHi 7.3 (n = 6) to 5.1 ± 0.1 at pHi 8.5 (n = 6, p < 0.01; Fig. 6B).
FIGURE 6.

Dose-response curves of ASIC1a and ASIC2a were shifted by intracellular solutions directly buffered at alkalizing values. A, left, representative traces showing the ASIC1a currents recorded with intracellular solutions directly buffered at pH 7.3 and 8.5. Right, summary data showing the pH50 of ASIC1a was shifted by intracellular solution buffered at pHi 8.5 compared with pHi 7.3 (n = 6–7, unpaired t test, t(11) = 4.9, p < 0.01). B, left, representative traces showing the ASIC2a currents recorded with intracellular solutions directly buffered at pH 7.3 and 8.5. Right, summary data showing the pH50 of ASIC2a was shifted by intracellular solution buffered at pHi 8.5 compared with pHi 7.3 (n = 6, unpaired t test, t(10) = 3.9, p < 0.01).
Steady-state Inactivation Curves of ASIC1a and ASIC2a Are Shifted by Intracellular Alkalization
We also determined whether changing pHi affects steady-state inactivation of ASICs in a subunit-dependent manner. The steady-state inactivation curves were constructed as described in our previous study (38). Briefly, CHO cells expressing ASIC1a or ASIC2a channels were perfused with extracellular solutions at various conditioning pH values (e.g. 7.8, 7.6, 7.4, 7.2, 7.0, and 6.8) for ∼4 min, and the ASIC1a or ASIC2a currents were then activated by dropping extracellular pH to 6.0 or 5.0. The amplitude of the ASIC currents recorded with different conditioning pH levels was normalized to that recorded with the conditioning pH of 7.8 (no inactivation) and then plotted against the conditioning pH values to generate the steady-state inactivation curve. The half-maximum inactivation of pH (pH50) of the homomeric ASIC1a current was 7.3 ± 0.1 with a Hill coefficient of 2.2 ± 0.2. The steady-state inactivation curve was shifted to a more acid value after application of 1 mm quinine; the pH50 was changed from 7.3 ± 0.1 to 7.1 ± 0.2 (n = 6, p < 0.01; Fig. 7A). No change in Hill coefficient was observed (2.2 ± 0.2 before quinine and 2.5 ± 0.3 after quinine, p > 0.05). In addition to ASIC1a, similar experiments were performed with ASIC2a expressed in CHO cells. Because homomeric ASIC2a has a low H+ sensitivity and cannot be activated with a pH drop from 7.4 to above 5.5, more acidic conditioning pH values were used to study the steady-state inactivation of this channel. Cells were exposed to conditioning pH values between 6.8 and 6.2 for ∼4 min before the ASIC2a current was activated by a pH drop to 4.5. The pH50 value for steady-state inactivation was 6.3 ± 0.1 in the absence of quinine. However, it became 6.1 ± 0.1 in the presence of 1 mm quinine (n = 5, p < 0.05; Fig. 7B). There was no significant change in the Hill coefficient (before quinine, 1.47 ± 0.1; after quinine, 1.51 ± 0.03, p > 0.05). These results suggest that changes in steady-state desensitization could contribute to the effects of the intracellular pH on ASIC-mediated responses.
FIGURE 7.

Effects of changing pHi on steady-state inactivation of ASICs in cells expressing ASIC1a and ASIC2a subunits. A, left, representative traces showing steady-state inactivation of ASIC1a current before and after bath perfusion of quinine. Right, summary data showing the shift in the steady-state inactivation curve of ASIC1a channels by the intracellular alkalizing agent quinine. The pH50 of the inactivation curve was significantly shifted by quinine (n = 6, paired t test, p < 0.01). B, representative traces and summary data showing the shift in the steady-state inactivation curve of ASIC2a channels by the intracellular alkalizing agent quinine (n = 5, paired t test, p < 0.05).
Increasing Intracellular pH Accelerates the Recovery of the Homomeric ASIC1a Channels from Desensitization
We then determined whether quinine affects the recovery of ASIC1a and ASIC2a from desensitization. Pairs of low pH pulses (e.g. from 7.4 to 6.0 for ASIC1a) were applied to activate ASIC currents at various time intervals (e.g. 1, 3, 5, 15, 25, 50, and 100 s) between the end of the first and the beginning of the second acid exposure. The peak amplitude of the second current was normalized to the first one and then plotted against the time intervals between the two acid exposures (38). The time constant of recovery (τ) was calculated by fitting the curves using either mono-exponential (for ASIC1a) or double exponential (for ASIC2a) functions, as described by a recent study (45). As reported earlier (46), homomeric ASIC1a channels have a significantly increased time constant of recovery (time constant τ = 31 ± 16 s, n = 5) than homomeric ASIC2a channels (τ = 1.1 ± 0.2 s, n = 5; pH from 7.4 to 5.0). Bath application of 1 mm quinine dramatically accelerated the recovery from desensitization for ASIC1a current (τ was reduced to 6 ± 2 s, n = 5, p < 0.01) but had little effect on the recovery of ASIC2a current when extracellular pH changed from 7.4 to 5.0 (τ = 1.0 ± 0.1 s, n = 5, t(8) = 0.8, unpaired t test, p > 0.05). To confirm this result, we also activated ASIC2a-mediated currents at pH 4.0. The time constant of recovery was calculated as described above, and the time interval (x axis) was displayed on a log scale to better demonstrate the entire time course. Similarly, the recovery rate was significantly changed by quinine for ASIC1a-mediated (p < 0.01; Fig. 8A), but not ASIC2a-mediated currents (p > 0.05; Fig. 8B). These results further support the notion that increasing intracellular pH accelerates the recovery from desensitization of ASIC1a channels.
FIGURE 8.

Effects of changing pHi on the recovery rate from desensitization of ASIC1a and ASIC2a channels. A, left, representative traces showing the time-dependent recovery of ASIC1a current from desensitization in the absence and presence of 1 mm quinine. Right, peak current amplitude activated by the second acid application was normalized to the amplitude of the first current in each pair and plotted against the time interval before and after quinine application. The solid lines are single exponential fits to the data points. Intracellular alkalization by quinine significantly accelerates the time constants of recovery (control: 31 ± 16 s, n = 5; quinine: 6 ± 2 s, n = 5, t(8) = 3.3, unpaired t test, p < 0.01). B, representative current traces (left) and summary data (right) showing the unchanged recovery rate from desensitization of the ASIC2a channel by intracellular alkalizing agent quinine (control: 1.3 ± 0.2 s, n = 8; quinine: 2.0 ± 0.2 s, n = 6, t(12) = 0.9, unpaired t test, p > 0.05). Right, solid lines are double exponential fits to the data points. Note that x axis is log scale.
Changing pHi Affects the Desensitization of ASICs
ASICs, especially ASIC1a channels, desensitize rapidly during persistent acid incubation, raising the question of whether these channels can play an important role in brain ischemia where the extracellular pH (pHo) drop is generally believed to be persistent. To determine whether changing pHi affects the desensitization of ASIC channels, the effect of quinine on the desensitization time constant of ASICs was examined. The desensitization time constant was measured by fitting the decay phase of the current with a single exponential function. At pHo 4.5, the decay time constant (τd) of the ASIC1a-mediated currents was increased by quinine (0.5 ± 0.1 s in the absence of quinine and 0.9 ± 0.2 s in the presence of quinine, p < 0.001; Fig. 9A and Table 1B). The sustained (steady state) currents were not significantly changed by quinine (Table 1, part B). At pHo 5.0, however, both decay time constant and steady-state currents were increased by quinine (Table 1, part A). Similar results were observed in ASIC2a-mediated currents at pHo 4.0 (Fig. 9B and Table 1, part C). The slow desensitization of ASICs in the presence of quinine suggests that the activity of ASICs is longer lasting in the region where alkalizing pHi exists, thus potentiating the extracellular acidosis-induced Ca2+ increase and neuronal injury.
FIGURE 9.
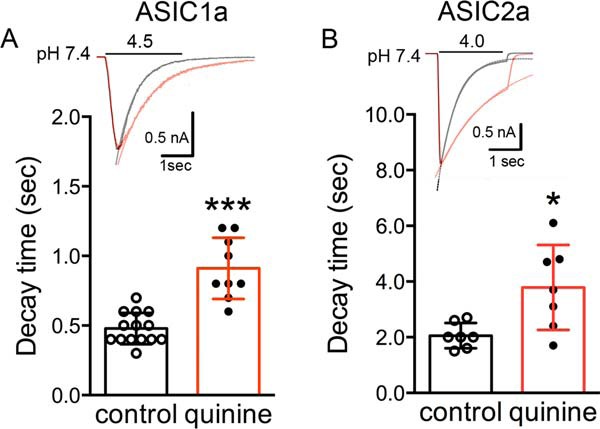
Quinine increases decay constants of ASIC currents in ASIC1a and ASIC2a subunits. A and B, upper, representative ASIC1a- and ASIC2a-mediated currents are shown with a superimposed exponential curve. The two traces are shown superimposed after scaling with an indication of the time constant (τ) to emphasize differences in deactivation. Black traces (control) and red traces (quinine). Bottom left, summary data showing quinine increases the decay of ASIC1a-mediated current activated by pH 4.5 (0.9 ± 0.2 s compared with control 0.5 ± 0.1 s, n = 9–14, unpaired t test, t(21) = 6.2, ***, p < 0.001, left). Bottom right, summary data showing quinine increases the decay of ASIC2a mediated current activated by pH 4.0 (3.8 ± 1.5 s compared with control 2.1 ± 0.5 s, n = 7, unpaired t test, t(12) = 2.8, *, p < 0.05).
TABLE 1.
Quinine on kinetics of desensitization of CHO cells expressing ASIC1a and ASIC2a subunits
Data are presented as mean ± S.D. *, p < 0.05; ***, p < 0.001 compared with control; Ipeak, peak currents; Isustained, steady-state currents; % desen, percentage desensitization.
| τdesen | Ipeak | Isustained | % desen | n | |
|---|---|---|---|---|---|
| s | nA | nA | |||
| A, ASIC1a at pH0 5.0 | |||||
| Control | 0.7 ± 0.1 | −0.6 ± 1.0 | −0.02 ± 0.01 | 97 ± 3 | 14 |
| Quinine | 1.4 ± 0.3*** | −1.6 ± 1.2* | −0.06 ± 0.04* | 96 ± 2 | 9 |
| B, ASIC1a at pH0 4.5 | |||||
| Control | 0.5 ± 0.1 | −1.1 ± 0.7 | −0.03 ± 0.01 | 96 ± 1.6 | 14 |
| Quinine | 0.9 ± 0.2*** | −2.8 ± 2.0* | −0.03 ± 0.02 | 98 ± 1.4 | 9 |
| C, ASIC2a at pH0 4.0 | |||||
| Control | 2.1 ± 0.5 | −6.9 ± 1.2 | −1.2 ± 0.5 | 83 ± 6 | 7 |
| Quinine | 3.8 ± 1.5* | −10.0 ± 3.5* | −2.6 ± 1.2* | 74 ± 10 | 7 |
Heteromeric ASIC1a + ASIC2a Channels Are Also Modulated by Changing Intracellular pH
In addition to homomeric ASIC1a and ASIC2a, a large percentage of ASIC currents in native neurons are mediated by heteromeric ASIC1a + ASIC2a channels (46–48). We therefore performed experiments to test the effects of changing pHi on the current mediated by heteromeric ASIC1a + ASIC2a channels. To ensure that the currents were mediated by heteromeric ASIC1a + ASIC2a channels, two criteria were employed (49). First, the ASIC current was induced by a drop of extracellular pH from 7.4 to 6.5, which only activates homomeric ASIC1a or heteromeric ASIC1a + ASIC2a but not homomeric ASIC2a channels. Second, the current can be potentiated by 100–300 μm zinc but not inhibited by PcTX1. Similar to homomeric ASIC1a and homomeric ASIC2a channels, the current mediated by heteromeric ASIC1a + ASIC2a channels was potentiated by the intracellular alkalizing agent quinine and inhibited by the intracellular acidifying agent propionate (Fig. 10, A and B). The peak amplitude of the currents mediated by ASIC1a + ASIC2a was increased to 181 ± 33% of the control in the presence of quinine (n = 10, p < 0.01; Fig. 10A). By contrast, the peak amplitude of ASIC1a + ASIC2a was decreased to 81 ± 6% of the control in the presence of propionate (n = 5, p < 0.01; Fig. 10B). In addition to the current amplitude, 1 mm quinine significantly shifted the activation curve of ASIC1a + ASIC2a to a less acidic value. The pH50 values before and after quinine were 4.9 ± 0.2 and 5.7 ± 0.2, respectively (n = 6, p < 0.01; Fig. 10C).
FIGURE 10.

Modulation of heteromeric ASIC1a + ASIC2a currents by pHi. A, representative traces (left) and bar graph (right) showing the potentiation of ASIC1a + ASIC2a heteromeric currents by the intracellular alkalizing agent quinine (n = 10, t(9) = 7.8, paired t test, **, p < 0.01). B, representative traces and bar graph showing the inhibition of ASIC1a + ASIC2a heteromeric currents by intracellular acidifying agent propionate (n = 5, t(4) = 7.2, paired t test, **, p < 0.01). C, representative traces and summary data showing the shift in pH50 of ASIC1a + ASIC2a heteromeric channels induced by intracellular alkalizing agent quinine. Increasing intracellular pH by quinine significantly increases the pH50 values (n = 6, t(5) = 5.3, paired t test, p < 0.01).
Effect of Intracellular pH on ASIC Currents Carried by ASIC1b/1a Chimeras
Next, we determined the intracellular regions and/or amino acid residues that are involved in the modulation of the ASIC1a and ASIC2a currents by pHi. Because ASIC1a and ASIC1b only differ by 176 amino acids at their N termini (50), the finding that changing intracellular pH modulates the activity of ASIC1a but not ASIC1b channel suggests that the N terminus of the ASIC1a channel, particularly the intracellular part of the N terminus, is critical for the modulation by pHi. To test this hypothesis, chimeric channels that exchanged part of the ASIC1a N terminus with the ASIC1b N terminus were then constructed, and the effect of changing intracellular pH on the chimeric ASIC currents was examined (Fig. 11A). Bath perfusion of quinine still potentiated the current carried by an ASIC1b (N1–42 aa)/ASIC1a (44 aa-C terminus) chimeras (183 ± 8% of the control, n = 7; Fig. 11B) but failed to potentiate the current carried by an ASIC1b (N1–52 aa)/ASIC1a (54 aa-C terminus) chimeras (98 ± 8% of the control, n = 8; Fig. 11C). These data point to the amino acids between 42 and 54 of the N terminus as critical for intracellular pH sensitivity of the ASIC1a channel.
FIGURE 11.

Effects of changing intracellular pH on ASIC1b/ASIC1a chimeras. A, schematic representation of ASIC1b (N1–42 aa)/1a (44 aa-C terminus) and ASIC1b (N1–52 aa)/1a (54 aa-C terminus) chimeric constructions. B, representative traces showing the effect of quinine on ASIC currents mediated by ASIC1b(N1–42 aa)/1a(44 aa-C terminus) chimera. Bath perfusion of 1 mm quinine still potentiated current amplitude mediated by ASIC1b(N1–42 aa)/1a(44 aa-C terminus) chimera (n = 7, t(13) = 0.5, unpaired t test, p > 0.05 compared with ASIC1a wild type, n = 8). C, representative traces and summary data showing the effect of quinine on current mediated by chimera ASIC1b (N1–52 aa)/1a (54 aa-C terminus) chimera. Quinine failed to potentiate current amplitude mediated by this mutant (n = 8, t(14) = 3.8, unpaired t test, **, p < 0.01 compared with ASIC1a wild type, n = 8). ns, no significant difference.
Effect of Intracellular pH on ASIC Currents Carried by ASIC1a Point Mutations
Finally, we aimed to identify specific amino acid(s) that may be involved in the modulation of ASIC1a current by pHi. When the sequence of ASIC1a from 42 to 54 was aligned with the corresponding sequence of ASIC1b and ASIC2a, the analysis revealed that three amino acids were identical in ASIC1a and ASIC2a but not in ASIC1b as follows: Arg-43, Gly-52, and Ser-53 in ASIC1a, and Arg-42, Gly-51, and Ser-52 in ASIC2a (Fig. 12A). Site-directed mutageneses of ASIC1a (R43Q, G52I, and S53A) were carried out with these amino acids. Mutants were screened by measuring the potentiation of ASIC currents in response to 1 mm quinine. However, quinine still potentiated the amplitude of the current carried by ASIC1a-R43Q (173 ± 57% of the control, n = 7), ASIC1a-G52I (122 ± 11% of the control, n = 10), and ASIC1a S53A (128 ± 16% of the control, n = 7). A significantly reduced potentiation by quinine was observed for currents carried by ASIC1a-G52I, ASIC1a-S53A, and the triple mutation of ASIC1a-R43Q/G52I/S53A compared with wild type (Fig. 12F). Similarly, a significantly reduced inhibition by propionate was observed for currents carried by ASIC1a-S53A, ASIC1a-G52I, and ASIC1a triple mutants (Fig. 12G). To provide additional evidence that Gly-52 and Ser-53 residues in ASIC1a are involved in the modulation of ASICs by intracellular pH, the dose-response curves of ASIC1a-G52I/S53A were constructed using intracellular solutions directly buffered at pH 8.5 and pH 7.3. The pH50 value was not different between pHi 8.5 (6.08 ± 0.1, n = 8) and pHi 7.3 (6.08 ± 0.1, n = 7, t(13) = 0.08, unpaired t test, p > 0.05). These data suggest that amino acids Gly-52 and Ser-53 in ASIC1a are likely sites for modulation of ACISs by intracellular pH.
FIGURE 12.

Effects of changing pHi on currents mediated by ASIC1a mutants. A, amino acids of ASIC1a are aligned with corresponding sequence of ASIC1b and ASIC2a. B–E, representative traces showing the effects of quinine on ASIC current mediated by ASIC1a wild type and mutants. F, comparison of potentiation of ASIC currents by intracellular alkalizing agent quinine for ASIC1a wild type and mutants. Reduced potentiation by quinine was seen for currents mediated by ASIC1a-G52I, S53A, and triple mutant ASIC1a R43Q/G52I/S53A (one-way ANOVA, effect of treatment, F(4,34) = 7.01, p = 0.0003. Dunnett's multiple comparisons test, *, p < 0.05; **, p < 0.0; ***, p < 0.001). G, comparison of inhibition of ASIC currents by intracellular acidifying agent propionate for ASIC1a wild type and mutants. Reduced inhibition by propionate was seen for currents mediated by ASIC1a-G52I, S53A, and triple mutant ASIC1a R43Q/G52I/S53A (one-way ANOVA, effect of treatment, F(4,36) = 12.69, p < 0.0001. Dunnett's multiple comparisons test, *, p < 0.05; **, p < 0.0; ***, p < 0.001).
Discussion
In this study, we investigated how intracellular pH exerts its effects on ASICs activated by extracellular protons. We demonstrated that ASIC currents and acidosis or ischemia-mediated cell injury are modulated by changes in intracellular pH in both in vitro and in vivo models. Furthermore, we determined that the modulation of ASIC currents by intracellular pH is ASIC subunit-dependent. Finally, we identified the domain and amino acids in ASIC1a that are likely involved in the modulation of this subunit by intracellular pH.
Provocative evidence has demonstrated that ASICs, which are known to be activated by extracellular H+, are novel targets for ischemic stroke therapy (51–53). In contrast to the decrease in extracellular pH with focal ischemia, intracellular pH changes and specifically an increase of intracellular pH have been reported in many studies (27, 29, 31, 32, 54). A stable intracellular pH is critical for a normal cellular function, and most biological processes are markedly pH-sensitive (33, 42, 55). Small changes of intracellular pH affect various ion channels, membrane excitability, and cellular metabolism (33–35, 37). Our data in in vitro and in vivo ischemia models revealed that increasing intracellular pH (by quinine) potentiates and decreasing intracellular pH (by propionate) inhibits acidosis- or ischemia-induced neuronal injury. These data provide evidence that ASIC activity is modulated by changes in not only extra- but also intracellular pH. Notably, intracellular pH is known to play a critical role in ischemic brain injury.
ASIC channels in native neurons are composed of different subunits, and the channels formed by different configurations of ASIC subunits have distinct physiological and pathological functions (15, 17, 49, 56, 57). The potentiation/inhibition of ASIC currents by intracellular alkalizing/acidifying agents is observed in cells expressing homomeric ASIC1a, ASIC2a, or heteromeric ASIC1a + ASIC2a channels but not in those expressing ASIC1b and ASIC3 channels. To further investigate the potential mechanism(s) underlying the modulation of ASIC1a and ASIC2a channels by intracellular pH, we studied the effects of changing pHi on the activation, inactivation, and desensitization of ASIC currents mediated by ASIC1a and ASIC2a channels. We show that increasing intracellular pH (e.g. by bath perfusion of 1 mm quinine) shifts the pH dose-response curve of both ASIC1a and ASIC2a channels toward less acidic pH values. This indicates that intracellular alkalization increases the sensitivity of ASIC1a and ASIC2a channels to proton. In addition, the steady-state inactivation curve was shifted to more acidic pH values by the intracellular alkalizing agent quinine, which may explain the potentiation of the current amplitude. However, unlike in native neurons, intracellular alkalization accelerated the recovery rate (as shown by reduced recovery time constant) only in engineered cells expressing the ASIC1a channel. In contrast, the time constant of recovery from desensitization of ASIC2a current was not affected by quinine even at pHo 4.0, which triggers about the same response with and without quinine. Similar to our previous findings in cortical neurons (38), the intracellular alkalizing agent quinine significantly reduces desensitization of ASIC1a-mediated currents. Desensitization is critical to ASIC function. It has been reported that desensitization is responsible for regulating sustained currents through ASICs in the central neurons during ischemia (58). In agreement with this, our previous findings in cortical neurons and that of this study in ASIC1a-transfected CHO cells show that both desensitization and sustained currents are changed by quinine at pHo 6.0 or 5.0. At extreme pHo (i.e. 4.5), however, the desensitization is reduced, whereas the sustained currents are not changed by quinine (Table 1, part B). One possible explanation might be that quinine affects sustained current of ASIC1a in a pHo-dependent manner. At mild pHo, fewer channels are open. The channels are more sensitive to quinine; therefore, more drastic increases occur in peak (Ipeak) and sustained currents (Isustained) of ASIC1a. At more acidic pHo (i.e. 4.5), ASIC1a channels are close to saturation due to high extracellular proton concentrations. Quinine is less effective at opening more channels and sustains their open configuration, thus producing less increase in Ipeak and minimal change in Isustained.
An increase in recovery rate and a decrease in desensitization of ASICs in the presence of intracellular alkalization suggest that the activity of ASICs is enhanced in the region where alkalizing pHi exists, thus potentiating the extracellular acidosis-induced Ca2+ increase and neuronal injury. Because the Ca2+-permeable ASIC1a subunit is an important player in ischemic damage (5), regulation of ASIC1a activation and desensitization kinetics by intracellular pH could dramatically alter the neuronal responses to ischemia-induced acidosis.
The ASIC1a channel is uniquely permeable to Ca2+ and selectively inhibited by PcTX1 (5). PcTX1 reduces both ischemia-induced (5) and OGD-induced neuronal injury (Figs. 1 and 2). In focal ischemia, pharmacological blockade or genetic deletion of ASIC1a protects the brain from injury, suggesting that targeting ASIC1a may lead to novel therapeutic strategies for stroke. Although the homomeric ASIC1a, ASIC2a, and heteromeric ASIC1a + ASIC2a channels are all influenced by intracellular pH, in this study we focused on the ASIC1a subunit.
One of the characteristics of ASIC1a is tachyphylaxis, a phenomenon of the gradual decrease of ASIC1a current amplitude with repeated activation of the channels. A previous study (59) in oocytes suggested that permeating protons is one of mechanisms underlying tachyphylaxis of ASIC1a. Consistent with proton inhibition of ASIC1a from intracellular site(s), our study shows that decreasing the intracellular pH, which is the same as increasing intracellular proton concentration, decreases ASIC1a-mediated currents, whereas decreasing intracellular proton concentration by alkalizing agent quinine potentiates ASIC1a currents. In this study, we did not attempt to study the effects of quinine or propionate on tachyphylaxis of ASIC1a. Instead, quinine or propionate was applied after stable ASIC currents were established (at least 10 min after establishing the whole-cell recording). In this case, no further tachyphylaxis was observed (data not shown). Future studies will explore whether intracellular pH-modifying agents have any effect on the initial tachyphylaxis of ASIC1a.
Previous studies have demonstrated that the arginine residue 43 (Arg-43) is located in the channel pore. The finding that the ASIC current mediated by the ASIC1a-R43C mutant was inhibited by methanethiosulfonates (e.g. (2-aminoethyl)-methanethiosulfonate) indicated that the first transmembrane domain (TM1) underwent conformational changes during channel gating (60). We hypothesized that changing the intracellular pH would not affect ASIC function in this mutant. However, the mutation of 43R in ASIC1a had no effect on the potentiation of ASIC1a by quinine, suggesting that the 43rd amino acid residue of ASIC1a is not involved in the modulation of ASIC activity by intracellular pH. However, a reduced potentiation/inhibition of ASICs by quinine/propionate was observed in ASIC1a-G52I and ASIC1a-S53A mutants. We also find that intracellular pH-modifying agents fail to potentiate, or inhibit, ASIC currents carried by the triple mutation in ASIC1a (i.e. ASIC1a-R43Q/G52I/S53A). These findings suggest that the amino acid 52 or 53 in ASIC1a is one of the likely action sites that are involved in the modulation of ASICs by intracellular pH. Gly-52 and Ser-53 residues located in the first transmembrane domain of ASIC1a may be involved in forming the channel/pore for ions. It is noted that glycine and serine are not the ideal receiver or provider of protons. However, these amino acids could play an important role in protein folding. The point mutants of G52I and S53A may cause the conformational change of ASIC1a, which may in turn affect the activity of ASIC1a. Altogether, this study identified new candidate molecular targets for development of novel therapeutics, such as small peptide inhibitors.
In addition to the N terminus of ASIC1a, other amino acids may also be involved in the activity change in ASIC1a during intracellular pH changes (61, 62). For example, histidine (His) residues in many proteins serve as a regulatory site and have been shown to be critical for gating of the ASIC channels (63–65). Histidine has a basic side chain that is protonated at even slightly acidic conditions (pH just below 7.0), and this can change properties of the amino acid and the polypeptide as a whole. There are two His residues in the intracellular N-terminal domain (His-28/His-32), two in the transmembrane domain (His-72/His-73), two in the extracellular domain (His-163 and His-173), and four in the C-terminal intracellular domain of ASIC1a. Based on the alignment of His residues in ASIC1a with the corresponding sequence of ASIC1b, three His residues are unique in ASIC1a: His-72, His-110, and His-173. Future studies are necessary to identify whether His residues found in ASIC1a, but not ASIC1b, are also involved in the modulation of ASICs by intracellular pH.
Ischemic stroke is a chronic problem, and the cellular mechanisms underlying ischemic brain injury are of great clinical significance. Previous studies have demonstrated an important role for ASICs in acidosis-mediated ischemic brain injury, and ASICs represent novel therapeutic targets for stroke/brain ischemia (5, 18). As discussed before, Ca2+-permeable ASIC1a plays an important role in glutamate-independent acidosis-induced neuronal injury. Established ASIC blockers PcTX1 and amiloride are neuroprotective in stroke/ischemia, but they have various limitations that prevent their use in stroke patients. ASICs are widely distributed in the nervous system and contribute to a range of functions (15–17, 20, 66). For that reason, a general inhibition of ASICs may have serious undesirable side effects.
It is well accepted that stroke and ischemia lead to changes in extracellular pH and thus activation of ASIC channels. In contrast, intracellular pH has received less attention, although intracellular pH is known to modulate the ASIC response to extracellular pH and can be altered in a complex time-dependent fashion in a range of stroke/ischemia models (27–32). Intracellular pH also plays a role in extracellular acidosis-induced cell injury. For example, buffering intracellular pH has been shown to be neuroprotective in vitro (38). This study is consistent with these findings and demonstrates that reducing intracellular pH attenuates neuronal damage in both in vivo (MCAO) and in vitro (OGD) models. A better understanding of this modulation could direct a design of molecules that specifically block the cellular effects triggered by pH changes after stroke.
In summary, we have characterized the modulation of ASIC currents and acid-mediated cell injury by intracellular pH, and we have defined the sites within ASIC1a that are involved in its modulation by intracellular pH. This study proposes molecular targets for the development of new therapeutics for stroke/brain ischemia that will prevent the pathological potentiation of ASIC currents without blocking the normal function of these channels. Our present studies have the potential to fill the gap created by the failure of clinical trials using glutamate antagonists and result in the development of the next generation of stroke therapeutics.
This study is focused on understanding how pathological changes in intracellular pH modulate the normal function of ASICs. By further understanding of how extra- and intracellular pH alters the function of ASICs after stroke, better therapeutics can be designed that will selectively target the pathological changes associated with ischemia. This may lead to specific inhibitors that will not affect the ASIC channel function as a whole but instead selectively target the acidosis-induced potentiation of ASIC currents that, in turn, induces cellular damage following stroke/brain ischemia.
Experimental Procedures
All procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the Oregon Health and Science University and Morehouse School of Medicine.
Primary Culture of Mouse Cortical Neurons
Mouse cortical neurons were cultured as described previously (67). Timed pregnant Swiss mice (embryonic day 16) were anesthetized with halothane followed by cervical dislocation. Brains of fetuses were removed rapidly and immediately placed in Ca2+/Mg2+-free ice-cold PBS. Cerebral cortices were dissected under a dissection microscope and incubated with 0.05% trypsin-EDTA for 10 min at 37 °C, followed by trituration with fire-polished Pasteur glass pipettes. Cells were counted and plated in poly-l-ornithine-coated 24-well plates at a density of 2 × 105 cells per well. Neurons were cultured with NeurobasalTM medium supplemented with B27 serum-free supplement and were maintained at 37 °C in a humidified 5% CO2 incubator. Medium was replaced twice a week. Cells were used for injury assays after 2–3 weeks in culture.
Ischemia-induced Injury in Vitro
Ischemia-induced injury in vitro was modeled by OGD with acidosis, as described previously (5). Briefly, neurons grown in 24-well culture plates were washed three times with extracellular fluid (ECF; pH 7.4) and randomly divided into different treatment groups. Dizocilpine (MK801) (10 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (20 μm), and nimodipine (5 μm) were added in all groups to eliminate potential activity of glutamate receptors and voltage-gated Ca2+ channels. Cells were pretreated with ECF at pH 7.4 or 6.0 in the presence or absence of intracellular pH-modifying agents for 15 min and then subjected to OGD. OGD was induced by replacing normal ECF with glucose-free ECF and placing cultures in an anaerobic chamber. The chamber was flushed with anaerobic gas containing 85% N2 + 5% CO2 + 10% H2 for 1 h at 35 °C. OGD was terminated after 1 h by replacing glucose-free ECF with normal ECF and placing cultures in a conventional cell culture incubator at 37 °C. Ischemic injury of the cells in different groups was assessed at 24 h by cell injury assays, LDH measurements and live/dead staining (5).
Ischemia-induced Cell Injury in Vivo
Ischemia-induced injury in vivo was assessed by transient MCAO (5). Transient focal ischemia was induced by suture occlusion of the middle cerebral artery in male adult C57BL6 mice (8–10 weeks old, Charles River Laboratories) anesthetized using 1.5% isoflurane, 70% N2O, and 28.5% O2 with intubation and ventilation. Rectal and temporalis muscle temperatures were maintained at 37 ± 0.5 °C with a thermostatically controlled heating pad and lamp. Cerebral blood flow was monitored by transcranial LASER Doppler. Animals with cerebral blood flow not reduced below 20% by the suture occlusion were excluded. Animals were killed with an overdose of isofluorane 24 h after ischemia. Brains were rapidly removed, sectioned coronally at 1-mm section thickness, and stained by immersion in vital dye (2%) TTC. Mouse intraventricular injection was performed by stereotaxic technique using a microsyringe pump with cannula inserted stereotactically at 0.5 mm posterior to bregma, 1.0 mm lateral to midline, and 2.5 mm ventral to the dura. All manipulations and analyses were performed by individuals blinded to treatment groups. A total of 2 μl of aCSF alone, aCSF-containing quinine (1 mm), propionate (10 mm), or psalmotoxin 1 (PcTX1, 1 μm), or PcTX1 plus quinine or propionate was injected intracerebroventricularly 30 min before and after the ischemia. Infarct volume was determined by TTC staining.
Transfection of Chinese Hamster Ovary (CHO) Cells
ASIC subunits were transiently expressed in CHO cells, which have no endogenous ASIC channels. CHO cells were cultured in F-12 medium supplemented with 10% fetal bovine serum in a 37 °C incubator. At ∼50% confluence, cells were transfected with cDNAs encoding channels bearing ASIC1a, ASIC2a, ASIC1b, ASIC3, or ASIC1a + ASIC2a using FuGENE 6 transfection reagents (Roche Applied Science). 36–48 h after transfection, whole-cell patch clamp recordings were performed.
Chimeric Channels Construction
The construction of chimeras between rat ASIC1a and ASIC1b was carried out using overlap extension technique (68) and was fully verified by sequencing. Briefly, the different domains were amplified with primers containing the sequence of the desired junctions between ASIC1a and ASIC1b and were joined by a second recombinant PCR with primers flanking the entire open reading frame. CHO cells at 50% confluence were transfected with pcDNA3 encoding a chimeric channel bearing ASIC1b (N1–52 aa)/ASIC1a (54 aa-C terminus) and ASIC1b (N1–42 aa)/ASIC1a (44 aa-C terminus at 1.8 μg per dish). pcDNA3.1 encoding GFP (0.2 μg per dish) was co-transfected with ASIC chimeras. 36–48 h after transfection, whole-cell patch clamp recordings were performed. GFP-positive cells were viewed under a fluorescent microscope and selected for the recording of ASIC currents.
Creation of Point Mutations
Point mutations of ASIC1a were created in pcDNA3.1 plasmid using the QuikChange site-directed mutagenesis kit (Agilent Technologies, 200519-5). Primers with the desired nucleotide changes were designed by using the guidelines provided with the kit. New plasmids with the desired mutation were created by using PCR with a Pfu polymerase. The residual unmutated template plasmid was digested by the DpnI enzyme. Mutations were verified by sequencing. 36–48 h after transfection, whole-cell patch clamp recordings were performed. The following primers were used for PCR: pcDNA ASIC1a R43Q forward, 5′-CGG CTG TCT CTG AAG CAG GCA CTG TGG GCC-3′, and reverse, 5′-GGC CCA CAG TGC CTG CTT CAG AGA CAG CCG-3′; ASIC1a S53A forward, 5′-CTG TGC TTC CTG GGT GCG CTG GCC GTC CTG-3, and reverse 5′-CAG GAC GGC CAG CGC ACC CAG GAA GCA CAG-3′; and ASIC1a G52I/S53A forward, 5′-C CTG TGC TTC CTG ATT GCG CTG GCC GTC CTG C-3′, and reverse, 5′-G CAG GAC GGC CAG CGC AAT CAG GAA GCA CAG G-3′. For the pcDNA ASIC1a R43Q/G52I/S53A mutants, pcDNA ASIC1a R43Q was served as template.
Solutions and Chemicals
Extracellular solution contained (in mm) the following: 140 NaCl, 5.4 KCl, 20 HEPES, 10 glucose, 2.0 CaCl2, 1.0 MgCl2. pH was adjusted with NaOH or HCl, and osmolarity was maintained at 320–330 mosm. Intracellular solution contained (in mm) the following: 140 CsF, 2.0 MgCl2, 1.0 CaCl2, 10 HEPES, 11 EGTA, 2.0 tetraethylammonium chloride, pH 7.3, osmolarity of 290–300 mosm. Unless otherwise stated, chemicals including quinine, propionate, and NH4Cl were purchased from Sigma. PcTX1 was purchased from Spider Pharm Inc. (Yarnell, AZ).
Whole-cell Patch Clamp Recording
Acid-induced currents were activated by lowering extracellular pH and recorded using whole-cell patch clamp techniques. Patch pipettes were pulled by a two-step puller (PP83, Narishige, Tokyo, Japan) using thin-wall borosilicate glass (1.5 mm diameter, WPI, Sarasota, FL). The pipettes had a resistance of 3–4 megohms when filled with intracellular solution. Whole-cell recordings were performed with Axopatch 200B amplifier, Digidata 1322A DAC unit, and pClamp 9.0 software (Molecular Devices, LLC Sunnyvale, CA). Data were low-pass filtered at 2 kHz and digitized at 5–10 kHz for on-line and later off-line analysis using pClamp 9.0 and SigmaPlot 11 (Systat Software Inc., San Jose, CA) or Prism 6.0 (Graphpad Software Inc., La Jolla, CA). Unless otherwise stated, ASIC currents were activated every 2 min to achieve a complete recovery from desensitization. A multibarrel perfusion system (SF-77, Warner Instruments, Hamden, CT) was used to achieve a rapid exchange of extracellular solutions (within 50 ms). During each experiment, series resistance was compensated by 70–80%. A voltage step of −10 mV from the holding potential was applied periodically to monitor cell capacitance and access resistance. Recordings in which access resistance or capacitance changed by more than 15% during the experiment were excluded for data analysis. Quinine or propionate was applied after the baseline stable currents were established. The effects of quinine or propionate on ASICs were determined by comparing the current amplitude right before the drug application to that after the stable drug effects.
Data Analysis
pH dose-response curves and steady-state inactivation curves were fitted using the following equation: I = a/(1 + (C50/pH)n), where a is the amplitude of ASIC current; C50 is the pH at which half-maximal response is induced, and n is the Hill coefficient. The time constant (τ) of desensitization and recovery from desensitization was determined using either mono- or double-exponential fitting (45, 67). For those double-exponential fits, we report the weighted time constants as described in our previous study (69). All recordings were made in extracellular solution at a room temperature of 24–25 °C.
Statistical Analysis
Statistical tests were performed with SigmaPlot or GraphPad Prism. All data are expressed as mean ± S.D. Paired or unpaired Student's t test was used in two group comparisons. One-way ANOVA followed by multiple comparison tests were used when more than two group comparisons were appropriate to determine statistical significance. The criterion of significance was set at p < 0.05 (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Author Contributions
M.-H. L. provided partial funding, designed the project, conducted the experiments, analyzed the results, and wrote the paper. T. L. conducted the experiments. X.-C. F. conducted chimeric channel construction and ASIC point mutations. T. Y. conducted the experiments of ischemia-induced cell injury in vivo. R. P. S. provided partial funding and revised the paper. Z.-G. X. helped to design the project, provided partial funding, and revised the paper.
Acknowledgment
We thank Dr. Agnieszka Balkowiec for critical reading of the manuscript.
This work was supported by American Heart Association Grant 13SDG14590005 (to M. H. L.) and National Institutes of Health Grants R01NS066027 and U54NS08932 (to Z. G. X.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ASIC
- acid-sensing ion channel
- PcTX1
- psalmotoxin
- ANOVA
- analysis of variance
- ECF
- extracellular fluid
- OGD
- oxygen-glucose deprivation
- LDH
- lactate dehydrogenase
- MCAO
- middle cerebral artery occlusion
- aCSF
- artificial cerebrospinal fluid
- aa
- amino acid
- TTC
- 2,3,5-triphenyltetrazolium hydrochloride.
References
- 1. Mutch W. A., and Hansen A. J. (1984) Extracellular pH changes during spreading depression and cerebral ischemia: mechanisms of brain pH regulation. J. Cereb. Blood Flow Metab. 4, 17–27 [DOI] [PubMed] [Google Scholar]
- 2. Katsura K., Ekholm A., Asplund B., and Siesjö B. K. (1991) Extracellular pH in the brain during ischemia: relationship to the severity of lactic acidosis. J. Cereb. Blood Flow Metab. 11, 597–599 [DOI] [PubMed] [Google Scholar]
- 3. Nedergaard M., Goldman S. A., Desai S., and Pulsinelli W. A. (1991) Acid-induced death in neurons and glia. J. Neurosci. 11, 2489–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yermolaieva O., Leonard A. S., Schnizler M. K., Abboud F. M., and Welsh M. J. (2004) Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U.S.A. 101, 6752–6757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xiong Z. G., Zhu X. M., Chu X. P., Minami M., Hey J., Wei W. L., MacDonald J. F., Wemmie J. A., Price M. P., Welsh M. J., and Simon R. P. (2004) Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698 [DOI] [PubMed] [Google Scholar]
- 6. Waldmann R., Champigny G., Bassilana F., Heurteaux C., and Lazdunski M. (1997) A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177 [DOI] [PubMed] [Google Scholar]
- 7. Waldmann R., Champigny G., Lingueglia E., De Weille J. R., Heurteaux C., and Lazdunski M. (1999) H+-gated cation channels. Ann. N.Y. Acad. Sci. 868, 67–76 [DOI] [PubMed] [Google Scholar]
- 8. Krishtal O. (2003) The ASICs: signaling molecules? Modulators? Trends Neurosci. 26, 477–483 [DOI] [PubMed] [Google Scholar]
- 9. Krishtal O. (2015) Receptor for protons: First observations on acid sensing ion channels. Neuropharmacology 94, 4–8 [DOI] [PubMed] [Google Scholar]
- 10. Alvarez de la Rosa D., Zhang P., Shao D., White F., and Canessa C. M. (2002) Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc. Natl. Acad. Sci. U.S.A. 99, 2326–2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alvarez de la Rosa D., Krueger S. R., Kolar A., Shao D., Fitzsimonds R. M., and Canessa C. M. (2003) Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J. Physiol. 546, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Price M. P., Lewin G. R., McIlwrath S. L., Cheng C., Xie J., Heppenstall P. A., Stucky C. L., Mannsfeldt A. G., Brennan T. J., Drummond H. A., Qiao J., Benson C. J., Tarr D. E., Hrstka R. F., Yang B., et al. (2000) The mammalian sodium channel BNC1 is required for normal touch sensation. Nature 407, 1007–1011 [DOI] [PubMed] [Google Scholar]
- 13. Price M. P., McIlwrath S. L., Xie J., Cheng C., Qiao J., Tarr D. E., Sluka K. A., Brennan T. J., Lewin G. R., and Welsh M. J. (2001) The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 14. Ugawa S., Yamamoto T., Ueda T., Ishida Y., Inagaki A., Nishigaki M., and Shimada S. (2003) Amiloride-insensitive currents of the acid-sensing ion channel-2a (ASIC2a)/ASIC2b heteromeric sour-taste receptor channel. J. Neurosci. 23, 3616–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wemmie J. A., Chen J., Askwith C. C., Hruska-Hageman A. M., Price M. P., Nolan B. C., Yoder P. G., Lamani E., Hoshi T., Freeman J. H. Jr., and Welsh M. J. (2002) The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477 [DOI] [PubMed] [Google Scholar]
- 16. Wemmie J. A., Coryell M. W., Askwith C. C., Lamani E., Leonard A. S., Sigmund C. D., and Welsh M. J. (2004) Overexpression of acid-sensing ion channel 1a in transgenic mice increases acquired fear-related behavior. Proc. Natl. Acad. Sci. U.S.A. 101, 3621–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wemmie J. A., Askwith C. C., Lamani E., Cassell M. D., Freeman J. H. Jr., and Welsh M. J. (2003) Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J. Neurosci. 23, 5496–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pignataro G., Simon R. P., and Xiong Z. G. (2007) Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain 130, 151–158 [DOI] [PubMed] [Google Scholar]
- 19. Xiong Z. G., Chu X. P., and Simon R. P. (2007) Acid sensing ion channels–novel therapeutic targets for ischemic brain injury. Front. Biosci. 12, 1376–1386 [DOI] [PubMed] [Google Scholar]
- 20. Wemmie J. A., Price M. P., and Welsh M. J. (2006) Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends Neurosci. 29, 578–586 [DOI] [PubMed] [Google Scholar]
- 21. Benveniste M., and Dingledine R. (2005) Limiting stroke-induced damage by targeting an acid channel. N. Engl. J. Med. 352, 85–86 [DOI] [PubMed] [Google Scholar]
- 22. Ereciśnka M., and Silver I. A. (1989) ATP and brain function. J. Cereb. Blood Flow Metab. 9, 2–19 [DOI] [PubMed] [Google Scholar]
- 23. von Hanwehr R., Smith M. L., and Siesjö B. K. (1986) Extra- and intracellular pH during near-complete forebrain ischemia in the rat. J. Neurochem. 46, 331–339 [DOI] [PubMed] [Google Scholar]
- 24. Katsura K., Asplund B., Ekholm A., and Siesjö B. K. (1992) Extra- and intracellular pH in the brain during ischaemia, related to tissue lactate content in normo- and hypercapnic rats. Eur. J. Neurosci. 4, 166–176 [DOI] [PubMed] [Google Scholar]
- 25. Tombaugh G. C., and Sapolsky R. M. (1990) Mechanistic distinctions between excitotoxic and acidotic hippocampal damage in an in vitro model of ischemia. J. Cereb. Blood Flow Metab. 10, 527–535 [DOI] [PubMed] [Google Scholar]
- 26. Siesjö B. K. (1992) Pathophysiology and treatment of focal cerebral ischemia. Part II: mechanisms of damage and treatment. J. Neurosurg. 77, 337–354 [DOI] [PubMed] [Google Scholar]
- 27. Back T., Hoehn M., Mies G., Busch E., Schmitz B., Kohno K., and Hossmann K. A. (2000) Penumbral tissue alkalosis in focal cerebral ischemia: relationship to energy metabolism, blood flow, and steady potential. Ann. Neurol. 47, 485–492 [PubMed] [Google Scholar]
- 28. Mabe H., Blomqvist P., and Siesjö B. K. (1983) Intracellular pH in the brain following transient ischemia. J. Cereb. Blood Flow Metab. 3, 109–114 [DOI] [PubMed] [Google Scholar]
- 29. Svichar N., Esquenazi S., Chen H. Y., and Chesler M. (2011) Preemptive regulation of intracellular pH in hippocampal neurons by a dual mechanism of depolarization-induced alkalinization. J. Neurosci. 31, 6997–7004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Blomqvist P., Mabe H., and Siesjö B. K. (1982) Transient ischemia leads to intracellular alkalosis in the brain. Acta Physiol. Scand. 116, 103–104 [DOI] [PubMed] [Google Scholar]
- 31. Chopp M., Chen H., Vande Linde A. M., Brown E., and Welch K. M. (1990) Time course of postischemic intracellular alkalosis reflects the duration of ischemia. J. Cereb. Blood Flow Metab. 10, 860–865 [DOI] [PubMed] [Google Scholar]
- 32. LaManna J. C., Griffith J. K., Cordisco B. R., Lin C. W., and Lust W. D. (1992) Intracellular pH in rat brain in vivo and in brain slices. Can. J. Physiol. Pharmacol. 70, S269–S277 [DOI] [PubMed] [Google Scholar]
- 33. Busa W. B., and Nuccitelli R. (1984) Metabolic regulation via intracellular pH. Am. J. Physiol. 246, R409–R438 [DOI] [PubMed] [Google Scholar]
- 34. Chesler M. (1990) The regulation and modulation of pH in the nervous system. Prog. Neurobiol. 34, 401–427 [DOI] [PubMed] [Google Scholar]
- 35. Chesler M., and Kaila K. (1992) Modulation of pH by neuronal activity. Trends Neurosci. 15, 396–402 [DOI] [PubMed] [Google Scholar]
- 36. Chu X. P., Zhu X. M., Wei W. L., Li G. H., Simon R. P., MacDonald J. F., and Xiong Z. G. (2003) Acidosis decreases low Ca2+-induced neuronal excitation by inhibiting the activity of calcium-sensing cation channels in cultured mouse hippocampal neurons. J. Physiol. 550, 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Collins A., and Larson M. (2005) Regulation of inward rectifier K+ channels by shift of intracellular pH dependence. J. Cell Physiol. 202, 76–86 [DOI] [PubMed] [Google Scholar]
- 38. Wang W. Z., Chu X. P., Li M. H., Seeds J., Simon R. P., and Xiong Z. G. (2006) Modulation of acid-sensing ion channel currents, acid-induced increase of intracellular Ca2+, and acidosis-mediated neuronal injury by intracellular pH. J. Biol. Chem. 281, 29369–29378 [DOI] [PubMed] [Google Scholar]
- 39. Bederson J. B., Pitts L. H., Germano S. M., Nishimura M. C., Davis R. L., and Bartkowski H. M. (1986) Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17, 1304–1308 [DOI] [PubMed] [Google Scholar]
- 40. Boyarsky G., Ganz M. B., Sterzel R. B., and Boron W. F. (1988) pH regulation in single glomerular mesangial cells. I. Acid extrusion in absence and presence of HCO3. Am. J. Physiol. 255, C844–C856 [DOI] [PubMed] [Google Scholar]
- 41. Dixon D. B., Takahashi K., Bieda M., and Copenhagen D. R. (1996) Quinine, intracellular pH and modulation of hemi-gap junctions in catfish horizontal cells. Vision Res. 36, 3925–3931 [DOI] [PubMed] [Google Scholar]
- 42. Roos A., and Boron W. F. (1981) Intracellular pH. Physiol. Rev. 61, 296–434 [DOI] [PubMed] [Google Scholar]
- 43. Deitmer J. W., and Ellis D. (1980) Interactions between the regulation of the intracellular pH and sodium activity of sheep cardiac Purkinje fibres. J. Physiol. 304, 471–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bonnet U., Leniger T., and Wiemann M. (2000) Moclobemide reduces intracellular pH and neuronal activity of CA3 neurones in guinea-pig hippocampal slices-implication for its neuroprotective properties. Neuropharmacology 39, 2067–2074 [DOI] [PubMed] [Google Scholar]
- 45. MacLean D. M., and Jayaraman V. (2016) Acid-sensing ion channels are tuned to follow high-frequency stimuli. J. Physiol. 594, 2629–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Askwith C. C., Wemmie J. A., Price M. P., Rokhlina T., and Welsh M. J. (2004) Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J. Biol. Chem. 279, 18296–18305 [DOI] [PubMed] [Google Scholar]
- 47. Baron A., Waldmann R., and Lazdunski M. (2002) ASIC-like, proton-activated currents in rat hippocampal neurons. J. Physiol. 539, 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Joeres N., Augustinowski K., Neuhof A., Assmann M., and Gründer S. (2016) Functional and pharmacological characterization of two different ASIC1a/2a heteromers reveals their sensitivity to the spider toxin PcTx1. Sci. Rep. 6, 27647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chu X. P., Wemmie J. A., Wang W. Z., Zhu X. M., Saugstad J. A., Price M. P., Simon R. P., and Xiong Z. G. (2004) Subunit-dependent high-affinity zinc inhibition of acid-sensing ion channels. J. Neurosci. 24, 8678–8689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bässler E. L., Ngo-Anh T. J., Geisler H. S., Ruppersberg J. P., and Gründer S. (2001) Molecular and functional characterization of acid-sensing ion channel (ASIC) 1b. J. Biol. Chem. 276, 33782–33787 [DOI] [PubMed] [Google Scholar]
- 51. Xiong Z. G., Pignataro G., Li M., Chang S. Y., and Simon R. P. (2008) Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Curr. Opin. Pharmacol. 8, 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kweon H. J., and Suh B. C. (2013) Acid-sensing ion channels (ASICs): therapeutic targets for neurological diseases and their regulation. BMB Rep. 46, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Leng T., Shi Y., Xiong Z. G., and Sun D. (2014) Proton-sensitive cation channels and ion exchangers in ischemic brain injury: new therapeutic targets for stroke? Prog. Neurobiol. 115, 189–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kalogeris T., Baines C. P., Krenz M., and Korthuis R. J. (2012) Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 298, 229–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chesler M. (2003) Regulation and modulation of pH in the brain. Physiol. Rev. 83, 1183–1221 [DOI] [PubMed] [Google Scholar]
- 56. Hesselager M., Timmermann D. B., and Ahring P. K. (2004) pH dependency and desensitization kinetics of heterologously expressed combinations of acid-sensing ion channel subunits. J. Biol. Chem. 279, 11006–11015 [DOI] [PubMed] [Google Scholar]
- 57. Johnson M. B., Jin K., Minami M., Chen D., and Simon R. P. (2001) Global ischemia induces expression of acid-sensing ion channel 2a in rat brain. J. Cereb. Blood Flow Metab. 21, 734–740 [DOI] [PubMed] [Google Scholar]
- 58. Yagi J., Wenk H. N., Naves L. A., and McCleskey E. W. (2006) Sustained currents through ASIC3 ion channels at the modest pH changes that occur during myocardial ischemia. Circ. Res. 99, 501–509 [DOI] [PubMed] [Google Scholar]
- 59. Chen X., and Gründer S. (2007) Permeating protons contribute to tachyphylaxis of the acid-sensing ion channel (ASIC) 1a. J. Physiol. 579, 657–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pfister Y., Gautschi I., Takeda A. N., van Bemmelen M., Kellenberger S., and Schild L. (2006) A gating mutation in the internal pore of ASIC1a. J. Biol. Chem. 281, 11787–11791 [DOI] [PubMed] [Google Scholar]
- 61. Krauson A. J., and Carattino M. D. (2016) The thumb domain mediates acid-sensing ion channel desensitization. J. Biol. Chem. 291, 11407–11419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Frey E. N., Pavlovicz R. E., Wegman C. J., Li C., and Askwith C. C. (2013) Conformational changes in the lower palm domain of ASIC1a contribute to desensitization and RFamide modulation. PLoS ONE 8, e71733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baron A., Schaefer L., Lingueglia E., Champigny G., and Lazdunski M. (2001) Zn2+ and H+ are coactivators of acid-sensing ion channels. J. Biol. Chem. 276, 35361–35367 [DOI] [PubMed] [Google Scholar]
- 64. Paukert M., Chen X., Polleichtner G., Schindelin H., and Gründer S. (2008) Candidate amino acids involved in H+ gating of acid-sensing ion channel 1a. J. Biol. Chem. 283, 572–581 [DOI] [PubMed] [Google Scholar]
- 65. Gründer S., and Chen X. (2010) Structure, function, and pharmacology of acid-sensing ion channels (ASICs): focus on ASIC1a. Int. J. Physiol. Pathophysiol. Pharmacol. 2, 73–94 [PMC free article] [PubMed] [Google Scholar]
- 66. Chu X. P., and Xiong Z. G. (2012) Physiological and pathological functions of acid-sensing ion channels in the central nervous system. Curr. Drug Targets 13, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li M., Kratzer E., Inoue K., Simon R. P., and Xiong Z. G. (2010) Developmental change in the electrophysiological and pharmacological properties of acid-sensing ion channels in CNS neurons. J. Physiol. 588, 3883–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 [DOI] [PubMed] [Google Scholar]
- 69. Li M. H., Suchland K. L., and Ingram S. L. (2015) GABAergic transmission and enhanced modulation by opioids and endocannabinoids in adult rat rostral ventromedial medulla. J. Physiol. 593, 217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]