

Abstract
Collisions between DNA replication complexes (replisomes) and barriers such as damaged DNA or tightly bound protein complexes can dissociate replisomes from chromosomes prematurely. Replisomes must be reloaded under these circumstances to avoid incomplete replication and cell death. Bacteria have evolved multiple pathways that initiate DNA replication restart by recognizing and remodeling abandoned replication forks and reloading the replicative helicase. In vitro, the simplest of these pathways is mediated by the single-domain PriC protein, which, along with the DnaC helicase loader, can load the DnaB replicative helicase onto DNA bound by the single-stranded DNA (ssDNA)-binding protein (SSB). Previous biochemical studies have identified PriC residues that mediate interactions with ssDNA and SSB. However, the mechanisms by which PriC drives DNA replication restart have remained poorly defined due to the limited structural information available for PriC. Here, we report the NMR structure of full-length PriC from Cronobacter sakazakii. PriC forms a compact bundle of α-helices that brings together residues involved in ssDNA and SSB binding at adjacent sites on the protein surface. Disruption of these interaction sites and of other conserved residues leads to decreased DnaB helicase loading onto SSB-bound DNA. We also demonstrate that PriC can directly interact with DnaB and the DnaB·DnaC complex. These data lead to a model in which PriC acts as a scaffold for recruiting DnaB·DnaC to SSB/ssDNA sites present at stalled replication forks.
Keywords: DNA helicase, DNA replication, DNA-protein interaction, nuclear magnetic resonance (NMR), protein-protein interaction, DNA replication restart, DnaB helicase, PriC, single-stranded DNA-binding protein
Introduction
Replication of circular chromosomes found in many bacteria is initiated by sequence-specific binding of the DnaA initiator protein to the origin of replication, oriC, which promotes duplex DNA melting (1–4). Single-stranded DNA (ssDNA)4 exposed by DnaA unwinding is rapidly bound by the ssDNA-binding protein (SSB). DnaA, along with the helicase loader DnaC, then directs loading of the replicative helicase, DnaB, onto the SSB-coated ssDNA (4–7). The remaining replication proteins are recruited through protein interactions to form the full replication complex, termed the replisome (8–12). With each round of replication, two replisomes are loaded at oriC to replicate bidirectionally around the chromosome until converging at the terminator region (13).
Replisomes assembled at oriC frequently encounter physical barriers, such as damaged DNA or genome-bound protein complexes (e.g. transcription machinery), that can stall and/or prematurely dissociate the replisome from the DNA template (14). Estimates from studies in Escherichia coli suggest that very few replisomes translocate to the replication terminus without dissociating at least once during each replication cycle (15). Because unrepaired premature termination events lead to incomplete replication, genome instability, and cell death, DNA replication restart mechanisms that reload replisomes onto abandoned replication forks are essential in bacteria (16). Due to the sporadic nature of replication failure, DNA replication restart pathways must recognize abandoned replication forks in a structure-specific and sequence-independent manner to enable reloading of the replicative helicase.
E. coli encodes three genetically defined DNA replication restart pathways that rely on distinct subsets of proteins: PriA/PriB/DnaT, PriC/Rep, and PriA/PriC (17). In vitro reconstitution of the PriA/PriB/DnaT pathway shows that it relies on a complex multiprotein hand-off mechanism to reload the DnaB helicase (18, 19). PriC, in contrast, is able to mediate DnaB loading without a requirement for Rep or other restart proteins in vitro (20). Although PriC is not well conserved among bacterial species, the simplicity of PriC-mediated DnaB loading makes it an excellent system for probing the minimal requirements for replication restart.
Given that DnaB loading at oriC is heavily regulated, reloading of the helicase at stalled forks is also likely to be a regulated process to ensure that the replisome does not assemble at improper sites. Accordingly, replication restart proteins are activated by recognition of appropriate replication fork substrates (20). Recent advances in defining the mechanisms underlying PriC-mediated replication restart have provided insights into how fork recognition and remodeling occur. Interaction between PriC and SSB is required for both fork recognition and remodeling, whereas an interaction between PriC and ssDNA is predicted to play a role in mediating fork recognition (21–23). Despite these recent studies, a deeper understanding of PriC replication restart mechanisms has been hampered by the limited amount of structural information on PriC and the lack of insights into how DnaB is recruited to PriC-bound replication forks.
To better define the mechanisms of DNA replication restart, we have determined the NMR structure of PriC from Cronobacter sakazakii. PriC consists of a compact bundle of five α-helices with residues that mediate interactions with SSB and ssDNA clustering together on the protein surface in adjacent binding sites. A biochemical study of PriC variants with altered conserved surface residues confirms the critical contribution of the SSB and ssDNA binding sites for in vitro DnaB loading. Moreover, conserved regions outside of these binding sites were found to be essential for PriC function in vitro and in vivo. Finally, we demonstrate that PriC directly binds to DnaB and the DnaB·DnaC complex. Taken together, these data support a model in which PriC acts as a scaffold that recruits DnaB to SSB/ssDNA present at stalled replication forks.
Results
In Vitro and in Vivo Functions of CsPriC
To determine the structure of full-length PriC, we initially attempted to crystallize PriC proteins from several bacterial species. Although crystals were not obtained for any of these targets, C. sakazakii PriC (CsPriC; 41% identical, 55% similar to EcPriC (Fig. 1A)) demonstrated better solubility characteristics than other PriC homologs, making it a potential target for structure determination by NMR. The activity of CsPriC was therefore examined to determine whether it retained the in vitro and in vivo activities expected for a bona fide PriC.
FIGURE 1.

In vitro and in vivo analysis of CsPriC. A, sequence alignment of EcPriC (top line) and CsPriC (bottom line). Secondary structural elements are indicated above the sequence. Residues tested in this study as well as those involved in SSB and ssDNA binding are highlighted. B, ITC analysis of CsPriC·SSB-Ct complex formation. Shown are heat-evolved (top) and binding isotherm (bottom) from titration of the SSB-Ct peptide into a solution of CsPriC. The dissociation constant was derived from a fit of the data to a single-site model. C, fluorescence polarization analysis of CsPriC and EcPriC binding to 5 nm fluorescein-labeled dT15 in the presence of 0 or 75 mm NaCl. Data are the mean polarization values from triplicate experiments with one S.D. value of the mean shown as error (error bars). D, autoradiogram (bottom) of a DnaB-loading assay testing CsPriC activity. The positions of the substrate and product are indicated to the left. The asterisk marks the location of the 32P label. Shown is a quantification (top) of the percentage of product unwound for each condition. The grid (middle) indicates which reaction components were included (+) or omitted (−) in each lane. Data are the mean of three replicates with one S.D. shown as error. E, co-transduction analysis demonstrating that CsPriC can complement a priC deletion in E. coli. Data are the number of co-transductants (ΔpriB TetR) versus the total number of TetR colonies tested. Linkages are obtained from 2–4 individual transduction experiments. **, p < 0.001 in a χ2 test using the transduction frequency for the wild type as the expected value.
We first measured CsPriC binding to a peptide comprising the SSB C terminus (SSB-Ct) and to ssDNA, both of which are activities that have been reported for EcPriC (21, 23). Isothermal titration calorimetry (ITC) analysis showed that CsPriC bound to the SSB-Ct peptide with a Kd of 2.1 ± 0.4 μm (Fig. 1B), closely matching the Kd for the EcPriC·SSB-Ct complex measured under the same conditions (3.7 ± 0.6 μm (23)). A fluorescence polarization-based ssDNA binding assay showed that CsPriC also bound a 5′-fluorescein-labeled dT15 oligonucleotide with Kd values of <5 nm in the absence of NaCl and 25.9 ± 0.9 nm in the presence of 75 mm NaCl (Fig. 1C). EcPriC bound the same DNA with Kd values of 9.2 ± 2.6 and 52.8 ± 3.7 nm, respectively, under the same conditions (Fig. 1C).
We next utilized an in vitro assay to test whether CsPriC could load EcDnaB onto a synthetic replication fork structure as has been previously observed for EcPriC (20, 23). In this assay, a radiolabeled forked DNA substrate is prebound by SSB, which blocks spontaneous loading of DnaB from the DnaB·DnaC helicase·loader complex. The addition of EcPriC relieves this blockage and stimulates DnaB loading, as scored by DnaB-mediated DNA unwinding. As expected for a bona fide PriC, the addition of CsPriC to this reaction facilitated DnaB loading onto the DNA (Fig. 1D).
Finally, CsPriC in vivo activity was assessed by determining its ability to allow deletion of the priB gene from an E. coli ΔpriC strain. Ordinarily, the simultaneous deletion of both priC and priB is lethal in E. coli, because this combination eliminates all replication restart pathways (17). However, the presence of a functional priC gene on a plasmid confers viability in chromosomal priC priB double mutant strains (23). Expression plasmids encoding CsPriC or EcPriC (positive control) or lacking a priC gene (negative control) were transformed into a priC303::kan E. coli strain, where a kanamycin resistance marker had been inserted to disrupt the priC locus. These strains then underwent P1 transduction with a ΔpriB donor carrying a linked tetracycline resistance marker (TetR). TetR colonies were screened for the successful co-transduction of the ΔpriB allele, which would indicate that the plasmid-borne PriC is functional in vivo and can support the priB deletion. As expected, 0 of the TetR colonies co-transduced the ΔpriB mutation in the negative control (empty vector), whereas ∼56% of the TetR colonies co-transduced the ΔpriB mutation in the positive control (EcPriC) (Fig. 1E). Consistent with its function in vivo, ∼24% of the TetR transductants with the CsPriC-encoding plasmid carried the ΔpriB mutation (Fig. 1E). Although this co-transduction frequency is lower than that of the positive control, the fact that multiple colonies carried the priB deletion indicates that CsPriC can complement EcPriC function. Taken together, these data demonstrate canonical PriC function for CsPriC, making it an appropriate target for structure determination.
NMR Structure of Full-length CsPriC
We next used heteronuclear 1H/13C/15N NMR data and residual dipolar coupling measurements to determine the NMR structure of CsPriC. The low energy bundle of NMR structures had a backbone root mean square deviation of 0.62 Å in well folded regions. A relatively high number of unambiguously assigned NOEs per residue (14.0) and the use of residual dipolar coupling constraints contributed to the high precision of the final coordinates (Table 1).
TABLE 1.
Summary of structure quality factors
RMSD, root mean square deviation; RMS, root mean square; PDB, Protein Data Bank.

a Residues with sum of φ and ψ order parameters > 1.8 Ordered residue ranges: 5-23 Å,31-40 Å,45-97 Å,100-104 Å,108-168 Å.
b Residues selected based on user-defined residues. Selected residue ranges: 5-20 Å,46-65 Å,69-94 Å,108-135 Å,139-168 Å.
c With respect to mean and S.D. for a set of 252 x-ray structures < 500 residues, of resolution ≤ 1.80 Å, R-factor ≤ 0.25, and R-free ≤ 0.28; a positive value indicates a “better” score.
The CsPriC structure consists of five α-helices arranged in a compact bundle, with an extended 20-residue loop connecting α1 and α2 (Figs. 1A and 2). A search for proteins that share structural similarity with CsPriC using the DALI server (24) revealed over 4,000 related folds within other proteins or protein domains. This large number most likely arises from the high frequency with which helical bundle folds are found in proteins. Surprisingly, a previously reported NMR structure of an N-terminal fragment of EcPriC (22, 25) was not identified in the DALI search due to significant differences in the arrangement of helices between the two structures (Fig. 3). In the N-terminal EcPriC fragment structure, the N-terminal-most helix, α1, substitutes for the position of α4 in the full-length structure. The position of this helix may shift to compensate for the absence of α4 in the N-terminal fragment structure.
FIGURE 2.

Structure of full-length CsPriC. A, stereo image of the 20 lowest energy NMR structures of CsPriC. B, schematic diagram (top), electrostatic surface representation (middle), and conservation analysis (bottom) of full-length PriC from C. sakazakii. Scales for electrostatic charge and conservation are shown. Known and predicted binding sites for SSB and DNA are indicated. Residues involved in SSB and ssDNA binding are shown (schematic diagram, top).
FIGURE 3.
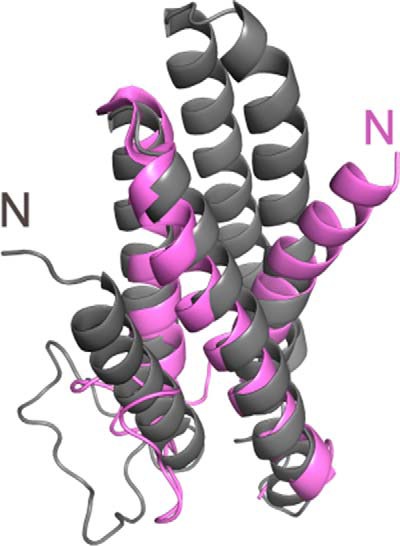
Comparison of full-length PriC structure with truncated PriC N-terminal domain structure. Alignment of the full-length structure (gray) with the previously solved N-terminal domain structure (purple) (25) (Protein Data Bank entry 2RT6). The second and third helices align well between the two structures; however, the placement of the first helix differs dramatically. In the truncated structure, the N-terminal helix is located where the fourth helix packs in the full-length protein.
Structural Insights into Protein and DNA Interaction Sites in PriC
Previous studies have identified two residues in EcPriC, Arg-121 and Arg-155 (Arg-121 and Arg-151 in CsPriC), that are essential for stabilizing its interaction with SSB (Table 2 (23)). These residues were found in adjacent α-helices in the CsPriC structure, with the helical arrangement placing the two basic side chains in close proximity (Figs. 2 and 4A). Additional EcPriC residues that have been implicated in mediating the SSB interaction (Phe-118, Arg-129, and Tyr-152 in EcPriC (21); Tyr-118, Arg-129, and Leu-148 in CsPriC) are also localized to this region (Table 2 and Figs. 2 and 4A). The electrostatics of the PriC SSB-Ct binding site resemble those observed in other SSB-associated proteins in which basic residues bind to the α-carboxyl group of the C-terminal Phe and to the Asp side chains within the SSB-Ct element (26–30) (Fig. 2B). The PriC SSB binding site is also evolutionarily well conserved (Fig. 2B), consistent with the essential nature of the PriC/SSB interaction for PriC-mediated DNA replication restart in vivo (23). Interestingly, the SSB-Ct binding site in PriC differs from SSB binding sites in other proteins in that it is much “flatter” than those previously observed. Analysis by the program EPOSBP (31) failed to identify pockets on the surface of CsPriC, whereas SSB-Ct binding pockets in other binding proteins range from 330 (exonuclease I (29)) to 700 Å3 (PriA helicase (30)) in the absence of SSB-Ct binding. It is possible that a structural rearrangement takes place in PriC to form the pocket needed for accommodating the SSB-Ct element. A similar SSB-Ct dependent rearrangement was observed for E. coli ribonuclease HI, which lacks an apparent binding pocket in isolation but forms a 480-Å3 pocket in the ribonuclease HI·SSB-Ct complex (26). The addition of an SSB-Ct peptide to high concentrations of CsPriC causes the protein to precipitate, supporting the possibility of an SSB-induced structural rearrangement but also precluding NMR studies of the CsPriC·SSB-Ct complex.
TABLE 2.
Summary of PriC protein variant activities
Variants with a >75% decrease in activity relative to wild type are defined as not having in vitro activity in this table. ND, no data; smFRET, single-molecule FRET; FP, fluorescence polarization; Y2H, yeast two-hybrid; GS, gel shift.
| Residue variant | SSB binding | ssDNA binding | In vitro activity | In vivo activity | Additional information | Source |
|---|---|---|---|---|---|---|
| R33E | Yes (ITC) | Yes (FP) | No | No | Interacts with DnaB (Y2H) | This study |
| W74A | Yes (ITC) | Yes (FP) | Partial | Yes | Interacts with DnaB (Y2H) | This study |
| R88A | Yes (Y2H) | ND | ND | ND | Ref. 23 | |
| E89A | Yes (ITC) | Yes (FP) | No | No | Interacts with DnaB (Y2H) | This study |
| R96A | Yes (ITC, Y2H) | Yes (FP) | Yes | No | Interacts with DnaB (Y2H) | This study, Ref. 23 |
| K104A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R107A | ND | ∼2-fold reduced (GS) | ND | ND | Ref. 21 | |
| W108A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R110A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| K111A | Yes (ITC) | 2-Fold reduced (FP, GS) | Very low | Yes | Interacts with DnaB (Y2H) | This study, Ref. 21 |
| R112A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| F118A | Weakened (ITC) | ∼2-Fold reduced (GS) | ND | ND | Ref. 21 | |
| R120A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R121A | No (ITC, Y2H) | ∼2-Fold reduced (GS) | No | No | Refs. 21 and 23 | |
| R123A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R129A | No (ITC) | Yes (GS) | ND | ND | Ref. 21 | |
| R130A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R132A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R135A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R147A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| Y152A | Weakened (ITC) | Yes (GS) | ND | ND | Ref. 21 | |
| R155A | No (ITC, Y2H) | Yes (GS, smFRET) | No | No | Refs. 21 and 23 | |
| R158A | Yes (Y2H) | Yes (GS) | ND | ND | Refs. 21 and 23 | |
| H161A | Yes (ITC) | Yes (FP) | No | Yes | Interacts with DnaB (Y2H) | This study |
| K165A | ND | ∼2-Fold reduced (GS) | ND | ND | Ref. 21 | |
| R169A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R172A | ND | Yes (GS) | ND | ND | Ref. 21 | |
| R175A | Yes (ITC) | Yes (FP) | No | Yes | Interacts with DnaB (Y2H) | This study |
| NTD (residues 1–97) | ND | No | ND | ND | Ref. 22 | |
| CTD (residues 96–175) | ND | Yes (GS) | ND | ND | Ref. 22 | |
| V149S/L156S/L163S/L170S | ND | ND | ND | ND | Does not oligomerize, very unstable construct | Ref. 22 |
FIGURE 4.

Known and putative binding sites on PriC. A, view of the SSB-Ct binding site. Residues that are essential (Arg-151 and Arg-121 in CsPriC) or important (Arg-129, Leu-148, and Tyr-118 in CsPriC) for binding SSB-Ct are shown in orange. B, view of the putative ssDNA-binding site. Residues implicated in binding to ssDNA (Arg-107, Arg-111, and Arg-161 in CsPriC) are shown in cyan. SSB-Ct-binding residues Tyr-118 and Arg-121 (orange) have also been implicated in ssDNA binding. C, view of the putative oligomerization region. Residues suggested to play a role in oligomerization (Ile-145, Leu-152, Ile-159, and Leu-166 in CsPriC) are shown in green.
Previous mutagenesis studies have also identified PriC residues with roles in ssDNA binding, including Arg-107, Lys-111, and Lys-165 (Table 2 (21)). The equivalent residues in CsPriC (Arg-107, Arg-111, and Arg-161) and other basic residues on helices 2, 4, and 5 form a highly electropositive groove on the surface of PriC that is adjacent to the SSB-Ct binding site (Figs. 2B and 4B). Conservation of residues along helix 2 is low relative to those on helix 4 and 5. DNA binding roles for EcPriC residues Phe-118 and Arg-121 have been suggested as well (21), and these residues are adjacent to the previously identified basic cluster. It is possible that ssDNA could extend across the surface of PriC.
It has also been reported that PriC can oligomerize and that three Leu residues and a Val within the C-terminal helix mediate self-association (Table 2 (22)). Mutation of these residues leads to a predominantly insoluble protein (22). Our structure shows that the side chains of two of these residues, Val-149 and Leu-156 (Ile-145 and Leu-152 in CsPriC), form part of the hydrophobic core of the protein, packing against α2 and α4, suggesting that these residues are not likely to mediate oligomerization directly. The side chains of the remaining residues, Leu-163 and Leu-170 (Ile-159 and Leu-166 in CsPriC), are more surface-exposed and could potentially be involved in oligomerization (Fig. 4C).
In addition to supporting data from previous studies, the structure also highlights evolutionarily conserved PriC surfaces that have not yet been investigated for their contribution to activity (Fig. 2B, bottom row). One of these regions is the extended loop, which includes the surface-exposed side chain of Arg-33. Additionally, there are conserved residues located on the C-terminal end of helix 3 (including Glu-89 and Arg-96) that could also be important for PriC function. The effects of altering these sites are explored further below.
E. coli PriC Interacts with DnaB and the DnaB·DnaC Complex
In addition to binding SSB and ssDNA, PriC must recruit the replicative helicase (DnaB) in complex with the helicase loader (DnaC) to abandoned replication forks, but the link connecting PriC to DnaB has not been established. A proteomic study has shown that EcPriC co-purifies with affinity-tagged DnaB, along with DnaC, suggesting that PriC may interact with DnaB and/or the DnaB·DnaC complex (32). To test the possible interaction between PriC and DnaB or DnaC, we first used a yeast two-hybrid approach. Plasmids expressing the Gal4 DNA binding domain fused to the N terminus of PriC (pGBD-PriC (23)) and the Gal4 activation domain fused to the N terminus of DnaB or DnaC (pGAD-DnaB, pGAD-DnaC) were co-transformed into an S. cerevisiae strain in which expression of HIS3 and ADE2 is under control of the GAL1 and GAL2 promoters, respectively. Interaction between PriC and DnaB or DnaC would support expression of the HIS3 and ADE2 reporter genes, allowing growth of the strain on His- and Ade-deficient media. Consistent with a direct interaction between PriC and DnaB, the strain transformed with pGBD-PriC and pGAD-DnaB was able to grow on selective media (Fig. 5A). Control transformations with plasmid pairs lacking either EcPriC or EcDnaB failed to support growth. In addition, co-transformation of pGBD-PriC with pGAD-DnaC did not support growth, suggesting that EcPriC interacts with DnaB but not EcDnaC (Fig. 5A).
FIGURE 5.
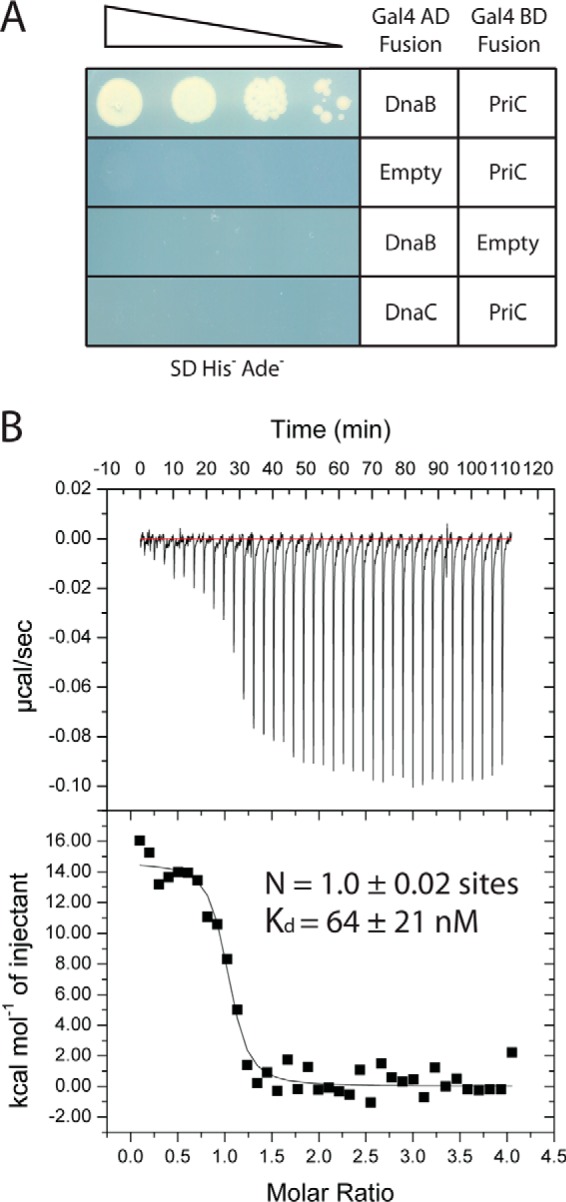
PriC interacts with DnaB within the DnaBC complex. A, yeast two-hybrid assay in which the indicated plasmids were co-transformed and spot-plated in a dilution series on selective medium. Growth on the selective medium is dependent upon protein interactions. B, ITC analysis of the PriC·DnaB·DnaC complex. Shown are heat-evolved (top) and binding isotherms (bottom) from titration of the DnaB·DnaC complex into a solution of PriC. Fitting of the data to a single-site model provided the stoichiometry (N) and dissociation constant for the interaction.
Next, ITC was used to determine whether a direct interaction between PriC and DnaB could be detected in vitro and to measure the stability and stoichiometry of the complex. We first tested PriC binding to the DnaB·DnaC complex because this best represents the relevant cellular complex that PriC would be expected to recruit to replication forks. PriC bound to the DnaB·DnaC complex with a Kd of 64 ± 21 nm and a stoichiometry of 1.0 ± 0.02 molecule of PriC per molecule of DnaB (Fig. 5B). We next tested for binding between PriC and DnaB or DnaC individually. Consistent with the yeast two-hybrid results, no interaction was detected between PriC and DnaC (data not shown). We were able to observe an interaction between PriC and DnaB; however, the heat of dilution of DnaB alone was so large that it prevented the reliable measurement of binding parameters (data not shown). These data are consistent with PriC binding directly to DnaB within the DnaB·DnaC complex. Given that the stoichiometry of the DnaB·DnaC complex is 6:6 (33) and PriC directly interacts with DnaB (Fig. 5A), PriC appears to be able to bind to each DnaB subunit within the DnaB·DnaC complex.
Mutagenesis Screening of the Surface of PriC
Examination of the PriC structure showed that residues tested for activity thus far are clustered near the SSB- and ssDNA-binding sites, whereas the rest of the structure remains underexplored. Moreover, contributions of the ssDNA-binding region to PriC-mediated replication restart have not yet been characterized. To expand our understanding of the roles of different surfaces of PriC and to more precisely define the importance of residues in proximity to known binding sites, we created a panel of single-site EcPriC variants that alter conserved surface residues for examination in vitro and in vivo (Fig. 6A). This panel included PriC variants that alter residues near the ssDNA binding site (K111A, H161A, and R175A) and others that map to regions away from previously established binding surfaces on PriC (R33E, W74A, E89A, and R96A). Each of the PriC variants bound SSB with approximately wild-type affinity, indicating that any observed defects in DnaB loading are not due to the disruption of the SSB interaction or to gross misfolding of the variant (Table 3). In addition, the variants all bound to ssDNA with an affinity similar to that of wild type PriC (Table 3 and Fig. 6 (B and C)). Under conditions lacking NaCl, all variants had Kd values of <10 nm. The probe concentration in these experiments was 5 nm, so our analysis of variant DNA binding could only provide an upper limit for many of the Kd values. Under conditions with 75 mm NaCl, ssDNA binding affinities were weakened relative to the NaCl-free conditions and were similar to that of wild type EcPriC. Only one variant, K111A, exhibited a modest 2-fold weaker ssDNA binding affinity compared with wild type EcPriC (Table 3 and Fig. 6 (B and C)). This suggested that although this region plays a role in ssDNA binding, single residue changes do not drastically alter the ability of PriC to bind ssDNA.
FIGURE 6.

Functionality of EcPriC single-site variants. A, surface rendering of CsPriC with the residues tested for DNA binding, SSB binding, and DnaB loading highlighted in orange. Residue labels reflect the EcPriC sequence. B and C, binding curves generated from incubating increasing amounts of the EcPriC variants with 5 nm fluorescein-labeled dT15 in the presence of 0 mm NaCl (B) or 75 mm NaCl (C). Data are the normalized mean polarization values from triplicate experiments with one S.D. value shown as error (error bars). D, autoradiogram (bottom) of a representative DnaB-loading assay comparing wild-type EcPriC and EcPriC variants. Shown is a quantification (top) of the percentage of product unwound for each condition. The black line indicates background levels in the absence of PriC. The positions of the substrate and product are indicated to the left. The asterisk marks the location of the 32P label. The grid (middle) indicates which reaction components were included (+) or omitted (−) in each lane. Data are the mean of three replicates with one S.D. value shown as error. E, yeast two-hybrid analysis of EcPriC variants interacting with DnaB.
TABLE 3.
In vitro and in vivo activities of structure-directed PriC variants
*, p < 0.01; **, p < 0.001 in a χ2 test using the transduction frequency for the wild type as the expected value.
| dT15 affinity |
SSB-Ct affinity | del(priB)302 linkage to zif-599::Tn10 | ||
|---|---|---|---|---|
| 0 mm NaCl | 75 mm NaCl | |||
| nm | μm | |||
| Wild-type | 9.2 ± 2.6 | 52.8 ± 3.7 | 3.7 ± 0.6 | 25/45 (56%) |
| R33E | <5 | 38.9 ± 3.4 | 5.2 ± 2.8 | 0/24 (0%)** |
| W74A | 5.2 ± 1.0 | 35.4 ± 6.1 | 3.7 ± 0.6 | 3/20 (15%) |
| E89A | <5 | 32.1 ± 3.1 | 7.7 ± 3.8 | 0/13 (0%)* |
| R96A | 6.5 ± 0.5 | 84.6 ± 6.2 | 3.6 ± 0.8 | 0/14 (0%)* |
| K111A | 6.8 ± 0.6 | 118.3 ± 6.6 | 1.85 ± 0.65 | 8/16 (50%) |
| H161A | <5 | 84.0 ± 13.3 | 2.1 ± 0.4 | 6/11 (55%) |
| R175A | <5 | 79.7 ± 9.1 | 3.5 ± 0.5 | 9/15 (60%) |
| Empty vector | 0/33 (0%)** | |||
We next tested the PriC variants in the reconstituted DnaB loading assay. Interestingly, all but two of the EcPriC variants displayed at least a 2-fold reduction in DnaB loading activities (Fig. 6D). The decreased DnaB loading ability of the K111A variant and others within the putative DNA binding tract could suggest that DNA binding is important in PriC-mediated replication restart, although the impact of the mutation on DnaB loading is clearly greater than that on DNA binding affinity. The defects observed with the R33E and E89A variants indicate that previously uncharacterized regions outside of the SSB and ssDNA binding sites are also important in DnaB loading.
Because all of the variants tested retained their ability to bind to SSB and ssDNA but demonstrated diminished abilities to load DnaB in vitro, we sought to determine whether the variants interfered with PriC binding to DnaB. Mutations were made in the Gal4-binding domain-PriC fusion plasmid (pGBD-PriC (23)) to express each of the single-site PriC variants along with the Gal4 activation domain-DnaB fusion protein in our two-hybrid assay. All of the variants were able to support growth when co-transformed with pGAD-DnaB, indicating that these individual residue changes do not abolish DnaB/PriC complex formation (Fig. 6E). Thus, the reduced in vitro DnaB loading abilities of PriC variants are not due to their inability to bind DnaB as measured by the two-hybrid assay.
To determine the in vivo functionality of the PriC variants, each was screened for its ability to complement a priC deletion and allow the deletion of priB as described earlier (24). Surprisingly, R96A was found to be non-functional, despite demonstrating wild-type levels of DnaB loading in vitro (Table 3 and Fig. 6D). Of the variants that displayed a decreased ability to load DnaB in vitro, only R33E and E89A were also unable to complement in vivo (Table 3). This indicates that surfaces outside of the previously defined SSB and ssDNA binding sites are essential for PriC function in vivo and that some variants with reduced in vitro activity still retain in vivo functionality. In addition, the inability of PriC variants with normal in vitro functions to complement priC cellular phenotypes suggests that current models accounting for the activities required for PriC functions in DNA replication restart are incomplete.
Discussion
DNA replication restart is an essential process in bacteria, and PriC is unique among the restart proteins in its ability to load DnaB from DnaB·DnaC complexes onto SSB-coated DNA substrates without assistance from additional replication factors (20). This property makes PriC a model for defining the minimal requirements needed for abandoned DNA replication fork recognition and remodeling, as well as for DnaB reloading. To better define the structural mechanisms underlying PriC-mediated replication restart, we have determined the NMR structure of the full-length C. sakazakii PriC. Our structure reveals a compact monomeric fold for PriC that is defined by five interacting α-helices. A biochemical analysis of PriC revealed a direct interaction with the replicative helicase, DnaB, in isolation and in DnaB·DnaC complexes. Our analysis also identified conserved residues in previously unexplored PriC surfaces that are essential for cellular function.
Previous proteolytic mapping studies suggested that EcPriC could be a two-domain protein, comprising N-terminal (residues 1–97) and C-terminal (residues 98–175) domains (22). However, the NMR structure of full-length CsPriC is consistent with the protein forming a compact single domain with a continuous hydrophobic core (Fig. 2). The arrangement of helices observed in an earlier solution structure of the EcPriC N-terminal region differs significantly from that observed in the full-length CsPriC structure (Fig. 3) (25). In the N-terminal EcPriC fragment, the N-terminal-most helix, α1, substitutes for the position of α4 in the full-length structure. This alters the tertiary structure and topology of the fragment to the extent that similarity between the two structures is not recognized by the DALI structural search algorithm. The position of this helix may shift to compensate for the absence of α4 in the N-terminal fragment. However, it remains possible that the differences between the two structures reflect structural flexibility in PriC.
The full-length CsPriC NMR structure resolves the protein's binding sites for both SSB and ssDNA (21–23). Residues involved in binding to these molecules map to two adjacent regions on PriC. The SSB-Ct binding site, which is formed predominantly by residues from α4 and α5, shares electrostatic similarities with other known SSB-Ct binding sites. These similarities include conserved hydrophobic residues surrounded by basic side chains that bind hydrophobic and electronegative SSB-Ct elements, respectively, in other complexes (26–30). One distinguishing feature of the PriC SSB binding site is that it is remarkably flat, which leads to the hypothesis that a conformational rearrangement may be necessary to form a pocket for SSB-Ct binding. A similar remodeling occurs in E. coli ribonuclease HI, which lacks an identifiable pocket in the absence of SSB-Ct but creates a cavity to accommodate the peptide in the complex (26).
Residues implicated in ssDNA binding map to an electropositive tract that is adjacent to the SSB binding site. These include Lys-111 (Table 3 and Fig. 6) (21) along with Arg-107 and Arg-161 that were identified in an earlier study (21). Sequence changes within this region lead to decreased DnaB loading activity levels in vitro (Fig. 6D), suggesting that ssDNA binding is important for PriC-mediated DNA replication restart. Slight perturbations of PriC ssDNA binding affinity do not appear to alter the activity of PriC in vivo (Table 3); however, it is possible that PriC variants with more significantly reduced ssDNA binding affinities could have reduced cellular activity. It has been reported that residues Arg-121 and Phe-118, which are important for SSB binding, also contribute to ssDNA binding (21). This could be important for the mechanism of PriC in potentially facilitating a hand-off of substrates, or it could arise from the similar electrostatic characteristics of SSB-Ct and ssDNA binding sites.
Additionally, the structure illuminated additional regions of conservation outside of the characterized ssDNA and SSB binding sites. Mutation of conserved residues within these areas of PriC demonstrated that these regions are important for PriC activity both in vitro and in vivo (Table 3 and Fig. 6). Possible roles for these regions include direct binding to additional cellular factors and/or productive coordination of PriC interactions to facilitate DnaB reloading.
Our investigation further revealed an interaction between PriC and the replicative helicase, DnaB. PriC appears to bind to DnaB both in isolation and in the DnaB·DnaC complex (Fig. 5), the latter being the form that would probably be most relevant for replication restart. A simple model explaining the role of PriC binding to DnaB·DnaC is that the interaction would help in recruitment and loading of helicase·loader complexes at abandoned replication forks. It is also possible that PriC binding to DnaB could stimulate release of DnaC from the DnaB·DnaC complex to allow DnaB to be activated. A similar mechanism has been noted for DnaG primase, in which binding of DnaG stimulates DnaC release from DnaB·DnaC (34).
Integrating the results described here with previous observations leads to a refined model for PriC function (Fig. 7). Bacterial DNA replication restart systems appear to share three core functions: abandoned replication fork recognition, lagging strand remodeling to generate a DnaB loading site, and DnaB reloading. PriC recognizes and remodels stalled forks through a combination of interactions with SSB and ssDNA (21, 23). The structure reported here shows that the respective binding sites for these ligands are adjacent to one another on the PriC surface. This arrangement could facilitate PriC binding to SSB and ssDNA in a coordinated manner. Previous studies examining the interaction between PriC and SSB indicated that complex formation can unwrap ssDNA from SSB by altering the SSB DNA binding mode to expose a potential site for DnaB loading (23). It is also possible that once ssDNA is exposed, PriC no longer binds the SSB-Ct and utilizes both the ssDNA- and SSB-binding sites for coordinating ssDNA binding. Having both SSB and ssDNA binding sites adjacent to one another on PriC may aid in driving this process.
FIGURE 7.
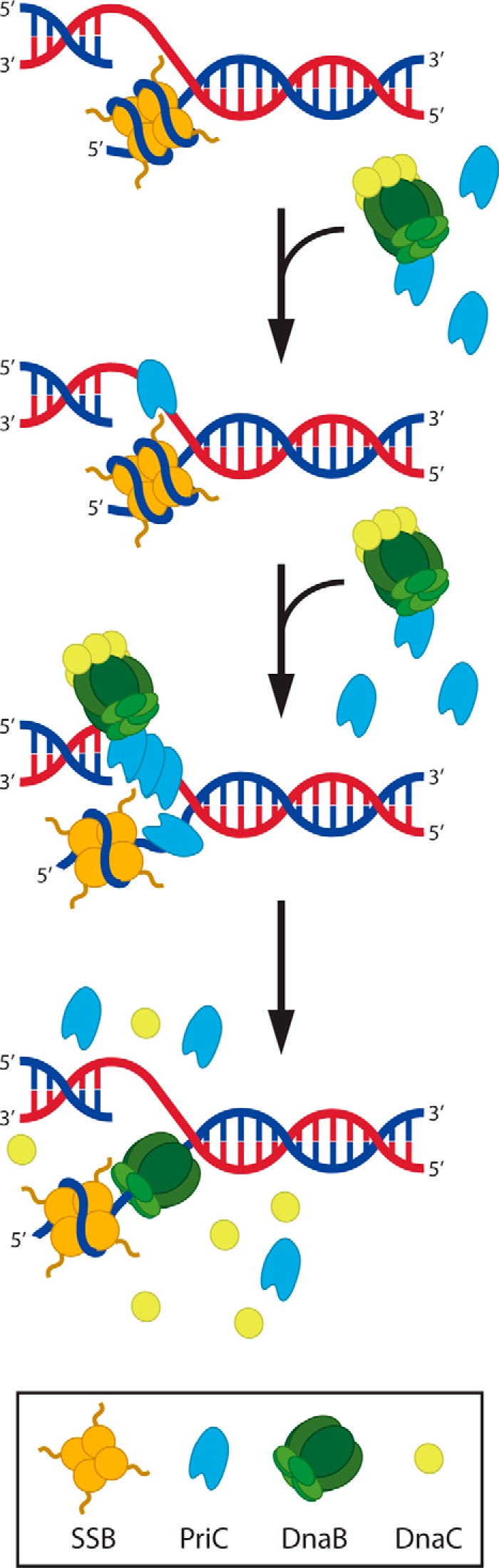
Model of PriC-mediated DnaB helicase loading. PriC recognizes a stalled replication fork through interactions with ssDNA and SSB. Binding of PriC to ssDNA (shown here on a leading strand gap because such structures are efficiently processed by PriC (20)) facilitates binding of additional monomers through oligomerization/cooperative binding (shown here binding to both strands), one monomer of which has the potential to bring with it DnaB·DnaC, thus localizing DnaB to the stalled fork. Additionally, binding of PriC to SSB results in a structural change to the SSB·DNA complex that results in the exposure of a small tract of ssDNA. Once this ssDNA is exposed, DnaB is loaded, and DnaC dissociates, resulting in a fork poised to recruit the remaining replisome components.
The final step of DNA replication restart is DnaB recruitment and loading. One key component required for this activity is a direct physical link connecting PriC with DnaB. We have detected interactions between PriC and DnaB, both in isolation and in DnaB·DnaC complexes. It could be that PriC first binds to abandoned DNA replication forks and then recruits free DnaB·DnaC to the loading site. Alternatively, it is possible that free PriC binds to the DnaB·DnaC complex in cells and that this ternary complex is recruited to abandoned forks through PriC/PriC interactions mediated by oligomerization/cooperative ssDNA binding. Either mechanism could explain how the PriC/DnaB interaction functions in DNA replication restart.
Taken together, our data provide further insight into how replication restart pathways function in the recruitment and loading of DnaB. Although PriC is not well conserved among bacterial species, similar steps could mediate the more well conserved PriA-mediated restart pathways in bacteria. Interactions between DnaB and either PriB or DnaT have not been detected (35)5; however, these proteins form part of a larger PriA·PriB·DnaT complex that may have the ability to bind DnaB. The ability to interact with SSB, ssDNA, and DnaB can be used as a set of characteristics to identify functional homologs that mediate replication restart in diverse bacteria.
Experimental Procedures
Protein Purification
E. coli PriC (EcPriC) and SSB were purified as described previously (23, 36). C. sakazakii PriC (CsPriC) was purified following the same protocol used for EcPriC with the following exceptions. After resuspension of the ammonium sulfate pellet, CsPriC was dialyzed against 10 mm HEPES-HCl, pH 7.0, 0.2 m NaCl, 10% glycerol, 1 mm EDTA, 1 mm DTT. The dialyzed protein was loaded onto an SPFF ion exchange column (GE Healthcare), and CsPriC was eluted using a 0.2–1 m NaCl gradient. For NMR structure determination, CsPriC-expressing cells were grown in M9 medium with [15N] ammonium chloride and [13C]glucose.
For DnaB overexpression, C41 E. coli cells transformed with a T7-inducible overexpression plasmid that encodes E. coli DnaB with an N-terminal His6 tag (pSW022) were grown in Luria broth medium supplemented with 100 μg/ml kanamycin at 37 °C. Cultures were induced to express DnaB by the addition of isopropyl 1-thio-β-d-galactopyranoside to 1 mm at early log phase (A600 nm = 0.3). After 3 h of additional growth, cells were harvested by centrifugation; resuspended in 20 mm HEPES-HCl, pH 7.5, 0.5 m NaCl, 10% glycerol, 10 mm MgCl2, 2 mm PMSF, 2 mm benzamidine; and lysed by sonication on ice. Lysates were clarified by centrifugation at 4 °C, and solid ammonium sulfate was added to the soluble lysate to a final concentration of 0.17 g/ml. Precipitated protein was pelleted by centrifugation and resuspended in Buffer A (20 mm HEPES-HCl, pH 7.5, 0.1 m NaCl, 10% glycerol, 10 mm MgCl2, 0.01 mm ATP, 1 mm 2-mercaptoethanol) and then loaded onto a QFF ion exchange column (GE Healthcare) equilibrated in Buffer A. DnaB was eluted from the column using a 0.1–0.75 m NaCl gradient. Fractions containing DnaB were combined, concentrated, and loaded on a Sephacryl S-300 column (GE Healthcare) equilibrated in Buffer B (20 mm Tris-HCl, pH 8.5, 0.8 m NaCl, 10% glycerol, 5 mm MgCl2, 0.1 mm ATP). Fractions containing pure DnaB were combined and dialyzed against 20 mm Tris-HCl, pH 8.5, 0.5 m NaCl, 5 mm DTT, 50% glycerol, and stored at −20 °C.
For DnaC overexpression, C41 E. coli cells transformed with a T7-inducible overexpression plasmid encoding E. coli DnaC with an N-terminal His6 and maltose-binding protein tag (from James Berger, Johns Hopkins University) were grown in Luria broth medium supplemented with 100 μg/ml ampicillin at 37 °C. Cultures were induced to express DnaC by the addition of isopropyl 1-thio-β-d-galactopyranoside to 1 mm at mid-log phase (A600 nm = 0.5). After an additional 2 h of growth, cells were pelleted by centrifugation; resuspended in 50 mm Tris-HCl, pH 7.5, 1 m KCl, 10% glycerol, 30 mm imidazole, 10 mm MgCl2, 0.01 mm ATP, 1 mm 2-mercaptoethanol, 2 mm PMSF, 2 mm benzamidine; and lysed by sonication on ice. Lysate was clarified by centrifugation, and the soluble lysate was incubated with nickel-nitrilotriacetic acid resin (Qiagen) for 1 h at 4 °C. Resin was washed with 20 column volumes of Buffer C (50 mm Tris-HCl, pH 7.5, 0.5 m KCl, 10% glycerol, 30 mm imidazole, 10 mm MgCl2, 0.01 mm ATP, 1 mm 2-mercaptoethanol), and then DnaC was eluted in Buffer D (50 mm Tris-HCl, pH 7.5, 0.5 m KCl, 10% glycerol, 500 mm imidazole, 10 mm MgCl2, 0.1 mm ATP, 1 mm 2-mercaptoethanol). The elution was concentrated and incubated with 40 units of tobacco etch virus protease at 4 °C overnight to cleave the His6/maltose-binding protein tag. Following cleavage, the solution was diluted to 10 mm NaCl with Buffer E (30 mm Tris-HCl, pH 7.5, 10% glycerol, 10 mm MgCl2, 0.1 mm ATP, 1 mm 2-mercaptoethanol) and loaded onto a QFF ion exchange column equilibrated with Buffer F (30 mm Tris-HCl, pH 7.5, 10% glycerol, 25 mm KCl, 10 mm MgCl2, 0.1 mm ATP, 1 mm 2-mercaptoethanol). At 25 mm KCl, the majority of the purification tag is retained on the column, whereas DnaC flows through. The DnaC eluent was incubated with nickel-nitrilotriacetic acid resin for 1 h at 4 °C to remove any uncleaved protein. The solute was concentrated and loaded onto a Sephacryl S-100 column equilibrated in Buffer G (30 mm Tris-HCl, pH 7.5, 10% glycerol, 0.5 m KCl, 10 mm MgCl2, 0.1 mm ATP, 1 mm 2-mercaptoethanol). Fractions containing pure DnaC were combined and dialyzed against 20 mm HEPES, pH 7.5, 0.5 m KCl, 50% glycerol, and stored at −20 °C.
NMR Structure Determination
NMR samples containing 0.4–0.6 mm [U-15N,U-13C]CsPriC in 20 mm MES, pH 6.5, 5 mm DTT, 1 mm EDTA, 1× EDTA-free protease inhibitor mixture (Roche Applied Science), 5% D2O were used to acquire isotropic NMR data at 37 °C. Protein 1DNH and 1DCαHα residual dipolar couplings were recorded with 1 mm [U-15N,U-13C]CsPriC in an anisotropic medium containing 3.1:1 1,2-dimyristoyl-sn-glycero-3-phosphocholine/1,2-dihexanoyl-sn-glycero-3-phosphocholine bicelles at 37 °C. 1H-15N HSQC, 1H-13C HSQC, HNCO, HNCACO, HNCACB, CBCA(CO)NH, C(CO)NH, H(CCO)NH, H(C)CH-TOCSY, and three-dimensional 15N NOESY (tmix = 120 ms) and three-dimensional 13C NOESY aliphatic (tmix = 120 ms) spectra were collected on Bruker Avance III 600- and 700-MHz spectrometers equipped with cryogenic probes. [1H-13C]HSQC (aliphatic and aromatic), (HB)CB(CGCD)HD, (HB)CB(CGCDCE)HE, and three-dimensional 13C NOESY aromatic (tmix = 100 ms) spectra were collected on a Varian VNMRS 600-MHz spectrometer equipped with a cryogenic probe. NH and CαHα couplings were measured on Varian VNMRS 900-MHz and a Bruker Avance III 750-MHz spectrometers, respectively, from two-dimensional ARTSY (37) and three-dimensional HCA(CO)N antiphase 1H-coupled in the 13Cα dimension spectra, respectively.
The NMR data were processed and analyzed with NMRPipe software (38). PIPP/STAPP software (39) was used to manually assign the backbone and side-chain resonances. The TALOS+ program (40) was used to provide pairs of ϕ/ψ backbone torsion angle restraints and to identify the secondary structural elements (confirmed by local NOEs). Two distance restraints of 1.9 and 2.9 Å per involved pair of residues were used to represent hydrogen bonds for HN–O and N–O, respectively (41). NOE peak intensities in three-dimensional NOESY spectra were assigned using the PIPP/STAPP package and converted for Xplor-NIH into a continuous distribution of 2,397 approximate interproton distance restraints, with a uniform 40% distance error applied to take into account spin diffusion.
Structure calculations and refinements made use of the torsion angle molecular dynamics and the internal variable dynamics modules of Xplor-NIH (40) to ensure preservation of the correct peptide geometry when applying residual dipolar coupling and distance constraints simultaneously. PyMOL (DeLano Scientific, LLC) and VMD-XPLOR (42) were used to analyze the structures. There were no consistent (i.e. in >40% of the calculated structures) NOE violations larger than 0.5 Å in the 100 calculated structures. A subset of 20 lowest energy structures (of 100) were selected for further refinement using an implicit solvation potential (43). The structure statistical quality indicators and agreement with experimental residual dipolar couplings are found in Table 1.
DnaB Loading Assays
Reactions were performed as described previously (23).
Yeast Two-hybrid Analysis
Assays were performed as described previously (23). Plasmids expressing Gal4 DnaB and DnaC fusion proteins were generated by cloning dnaB and dnaC open reading frames into the pGAD vector backbone to fuse the Gal4 activation domain to the N terminus of each protein to be tested (44).
Isothermal Titration Calorimetry
ITC of the PriC/SSB-Ct interaction was performed as described previously (23). Briefly, PriC variants were concentrated to 8–23 μm in a buffer containing 10 mm HEPES-HCl, pH 7.0, 0.1 m NaCl, and 3% glycerol. SSB-Ct peptide (WMDFDDDIPF) was dissolved in an identical buffer at a concentration of 525 μm. Titrations were performed on a VP-ITC instrument (Microcal) with 20 1.5-μl injections. Data were fit using a single-site model using Origin software (Microcal).
For the PriC·DnaB·DnaC titrations, all proteins were dialyzed against a buffer containing 20 mm Tris-HCl, pH 8.5, 0.2 m NaCl, 5% glycerol, 5 mm MgCl2, 1 mm 2-mercaptoethanol, and 1 mm ATP. PriC was concentrated to 5 μm, and DnaB and DnaC were concentrated to >200 μm. For analysis of the DnaB·DnaC complex with PriC, DnaB and DnaC were combined at final concentrations of 100 μm DnaB and 120 μm DnaC. For analysis of DnaB and DnaC individually with PriC, the final concentration for DnaB was 80 μm, and that of DnaC was 96 μm. Titrations were performed on a VP-ITC instrument (Microcal) with 25–37 1-μl injections at 20 °C. Data were fit using a single-site model using Origin software (Microcal).
DNA Binding Assays
DNA binding reactions were performed in 10 mm HEPES-HCl, pH 7.0, with either 0 or 75 mm NaCl. 5′-Fluorescein-labeled dT15 ssDNA (5 nm) was incubated with the indicated concentrations of PriC for 30 min at 25 °C. Fluorescence polarization was measured at 25 °C using a BioTek Synergy2 plate reader with 490-nm excitation and 535-nm emission wavelengths for three replicates. The average polarization value was plotted with one S.D. value of the mean shown as error. The polarization of dT15 alone was subtracted from each of the data points, and the data were fit with a single-binding site model with Hill coefficient using GraphPad Prism software.
Co-transduction Tests of priC Mutants
Assays were performed as described previously (23).
Author Contributions
S. R. W. and J. L. K. designed the study and wrote the paper. S. R. W. purified all proteins and carried out biochemical and two-hybrid experiments. S. R. W., C. C. C., G. C., K. H., and J. L. M. determined the PriC NMR structure. A. M., M. L., and S. J. S. carried out all complementation experiments. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Ken Satyshur for assistance with binding pocket volume analysis, Tricia Windgassen for assistance with two-hybrid experiments, and members of the Keck laboratory for critical reading of the manuscript. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIGMS, National Institutes of Health, Grant P41GM103399 (old number: P41RR002301). Equipment was purchased with funds from the University of Wisconsin-Madison; National Institutes of Health Grants P41GM103399, S10RR02781, S10RR08438, S10RR023438, S10RR025062, and S10RR029220; National Science Foundation Grants DMB-8415048, OIA-9977486, and BIR-9214394; and the United States Department of Agriculture.
This work was supported by National Institutes of Health Grant GM098885 (to J. L. K. and S. J. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 2NCJ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
NMR data have been deposited in the Biological Magnetic Resonance Bank (http://www.bmrb.wisc.edu) under BMRB accession number 26026.
S. R. Wessel and J. L. Keck, unpublished observation.
- ssDNA
- single-stranded DNA
- SSB
- ssDNA-binding protein
- SSB-Ct
- SSB C terminus
- ITC
- isothermal titration calorimetry.
References
- 1. Kaguni J. M. (2006) DnaA: controlling the initiation of bacterial DNA replication and more. Annu. Rev. Microbiol. 60, 351–375 [DOI] [PubMed] [Google Scholar]
- 2. Bramhill D., and Kornberg A. (1988) Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell 52, 743–755 [DOI] [PubMed] [Google Scholar]
- 3. Grimwade J. E., Torgue J. J. C., McGarry K. C., Rozgaja T., Enloe S. T., and Leonard A. C. (2007) Mutational analysis reveals Escherichia coli oriC interacts with both DnaA-ATP and DnaA-ADP during pre-RC assembly. Mol. Microbiol. 66, 428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ludlam A. V., McNatt M. W., Carr K. M., and Kaguni J. M. (2001) Essential amino acids of Escherichia coli DnaC protein in an N-terminal domain interact with DnaB helicase. J. Biol. Chem. 276, 27345–27353 [DOI] [PubMed] [Google Scholar]
- 5. Marszalek J., and Kaguni J. M. (1994) DnaA protein directs the binding of DnaB protein in initiation of DNA replication in Escherichia coli. J. Biol. Chem. 269, 4883–4890 [PubMed] [Google Scholar]
- 6. Wahle E., Lasken R. S., and Kornberg A. (1989) The dnaB-dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J. Biol. Chem. 264, 2469–2475 [PubMed] [Google Scholar]
- 7. Wahle E., Lasken R. S., and Kornberg A. (1989) The dnaB-dnaC replication protein complex of Escherichia coli. I. Formation and properties. J. Biol. Chem. 264, 2463–2468 [PubMed] [Google Scholar]
- 8. Kim S., Dallmann H. G., McHenry C. S., and Marians K. J. (1996) Tau couples the leading- and lagging-strand polymerases at the Escherichia coli DNA replication fork. J. Biol. Chem. 271, 21406–21412 [DOI] [PubMed] [Google Scholar]
- 9. Kim S., Dallmann H. G., McHenry C. S., and Marians K. J. (1996) Tau protects β in the leading-strand polymerase complex at the replication fork. J. Biol. Chem. 271, 4315–4318 [DOI] [PubMed] [Google Scholar]
- 10. Kim S., Dallmann H. G., McHenry C. S., and Marians K. J. (1996) Coupling of a replicative polymerase and helicase: a tau-DnaB interaction mediates rapid replication fork movement. Cell 84, 643–650 [DOI] [PubMed] [Google Scholar]
- 11. Fang L., Davey M. J., and O'Donnell M. (1999) Replisome assembly at oriC, the replication origin of E. coli, reveals an explanation for initiation sites outside an origin. Mol. Cell 4, 541–553 [DOI] [PubMed] [Google Scholar]
- 12. Johnson A., and O'Donnell M. (2005) Cellular DNA replicases: components and dynamics at the replication fork. Annu. Rev. Biochem. 74, 283–315 [DOI] [PubMed] [Google Scholar]
- 13. Hill T. M., and Marians K. J. (1990) Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl. Acad. Sci. U.S.A. 87, 2481–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGlynn P., and Lloyd R. G. (2002) Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 3, 859–870 [DOI] [PubMed] [Google Scholar]
- 15. Cox M. M., Goodman M. F., Kreuzer K. N., Sherratt D. J., Sandler S. J., and Marians K. J. (2000) The importance of repairing stalled replication forks. Nature 404, 37–41 [DOI] [PubMed] [Google Scholar]
- 16. Aguilera A., and Gómez-González B. (2008) Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217 [DOI] [PubMed] [Google Scholar]
- 17. Sandler S. J. (2000) Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics 155, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lopper M., Boonsombat R., Sandler S. J., and Keck J. L. (2007) A hand-off mechanism for primosome assembly in replication restart. Mol. Cell 26, 781–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ng J. Y., and Marians K. J. (1996) The ordered assembly of the X174-type primosome. I. Isolation and identification of intermediate protein-DNA complexes. J. Biol. Chem. 271, 15642–15648 [DOI] [PubMed] [Google Scholar]
- 20. Heller R. C., and Marians K. J. (2005) The disposition of nascent strands at stalled replication forks dictates the pathway of replisome loading during restart. Mol. Cell 17, 733–743 [DOI] [PubMed] [Google Scholar]
- 21. Aramaki T., Abe Y., Furutani K., Katayama T., and Ueda T. (2015) Basic and aromatic residues in the C-terminal domain of PriC are involved in ssDNA and SSB binding. J. Biochem. 157, 529–537 [DOI] [PubMed] [Google Scholar]
- 22. Aramaki T., Abe Y., Ohkuri T., Mishima T., Yamashita S., Katayama T., and Ueda T. (2013) Domain separation and characterization of PriC, a replication restart primosome factor in Escherichia coli. Genes Cells 18, 723–732 [DOI] [PubMed] [Google Scholar]
- 23. Wessel S. R., Marceau A. H., Massoni S. C., Zhou R., Ha T., Sandler S. J., and Keck J. L. (2013) PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J. Biol. Chem. 288, 17569–17578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aramaki T., Abe Y., Katayama T., and Ueda T. (2013) Solution structure of the N-terminal domain of a replication restart primosome factor, PriC, in Escherichia coli. Protein Sci. 22, 1279–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petzold C., Marceau A. H., Miller K. H., Marqusee S., and Keck J. L. (2015) Interaction with single-stranded DNA-binding protein stimulates Escherichia coli ribonuclease HI enzymatic activity. J. Biol. Chem. 290, 14626–14636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shereda R. D., Reiter N. J., Butcher S. E., and Keck J. L. (2009) Identification of the SSB binding site on E. coli RecQ reveals a conserved surface for binding SSB's C terminus. J. Mol. Biol. 386, 612–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marceau A. H., Bahng S., Massoni S. C., George N. P., Sandler S. J., Marians K. J., and Keck J. L. (2011) Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 30, 4236–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu D., and Keck J. L. (2008) Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc. Natl. Acad. Sci. U.S.A. 105, 9169–9174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhattacharyya B., George N. P., Thurmes T. M., Zhou R., Jani N., Wessel S. R., Sandler S. J., Ha T., and Keck J. L. (2014) Structural mechanisms of PriA-mediated DNA replication restart. Proc. Natl. Acad. Sci. U.S.A. 111, 1373–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kohlbacher O., and Lenhof H. P. (2000) BALL: rapid software prototyping in computational molecular biology. Biochemicals Algorithms Library. Bioinformatics 16, 815–824 [DOI] [PubMed] [Google Scholar]
- 32. Butland G., Peregrín-Alvarez J. M., Li J., Yang W., Yang X., Canadien V., Hayashi M., Starostine A., Richards D., Beattie B., Krogan N., Davey M., Parkinson J., Greenblatt J., and Emili A. (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537 [DOI] [PubMed] [Google Scholar]
- 33. Arias-Palomo E., O'Shea V. L., Hood I. V., and Berger J. M. (2013) The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 153, 438–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Makowska-Grzyska M., and Kaguni J. M. (2010) Primase directs the release of DnaC from DnaB. Mol. Cell 37, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang Y. H., Lin M. J., and Huang C. Y. (2013) Yeast two-hybrid analysis of prib-interacting proteins in replication restart primosome: a proposed PriB-SSB interaction model. Protein J. 32, 477–483 [DOI] [PubMed] [Google Scholar]
- 36. George N. P., Ngo K. V., Chitteni-Pattu S., Norais C. A., Battista J. R., Cox M. M., and Keck J. L. (2012) Structure and cellular dynamics of Deinococcus radiodurans single-stranded DNA (ssDNA)-binding protein (SSB)-DNA complexes. J. Biol. Chem. 287, 22123–22132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fitzkee N. C., and Bax A. (2010) Facile measurement of 1H-15N residual dipolar couplings in larger perdeuterated proteins. J. Biomol. NMR 48, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 39. Garrett D. S., Powers R., Gronenborn A. M., and Clore G. M. (1991) A common sense approach to peak picking two-, three- and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95, 214–220 [DOI] [PubMed] [Google Scholar]
- 40. Schwieters C. D., Kuszewski J. J., Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 41. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids in Nuclear Magnetic Resonance, Vol. 36, pp 180–182, Royal Society of Chemistry, London [Google Scholar]
- 42. Schwieters C. D., and Clore G. M. (2001) The VMD-XPLOR visualization package for NMR structure refinement. J. Magn. Reson. 149, 239–244 [DOI] [PubMed] [Google Scholar]
- 43. Tian Y., Schwieters C. D., Opella S. J., and Marassi F. M. (2014) A practical implicit solvent potential for NMR structure calculation. J. Magn. Reson. 243, 54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. James P., Halladay J., and Craig E. A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]