

Abstract
The LDL receptor-related protein 1 (LRP1) is a large endocytic receptor that binds and mediates the endocytosis of numerous structurally diverse ligands. Currently, the basis for ligand recognition by LRP1 is not well understood. LRP1 requires a molecular chaperone, termed the receptor-associated protein (RAP), to escort the newly synthesized receptor from the endoplasmic reticulum to the Golgi. RAP is a three-domain protein that contains the following two high affinity binding sites for LRP1: one is located within domains 1 and 2, and one is located in its third domain. Studies on the interaction of the RAP third domain with LRP1 reveal critical contributions by lysine 256 and lysine 270 for this interaction. From these studies, a model for ligand recognition by this class of receptors has been proposed. Here, we employed surface plasmon resonance to investigate the binding of RAP D1D2 to LRP1. Our results reveal that the high affinity of D1D2 for LRP1 results from avidity effects mediated by the simultaneous interactions of lysine 60 in D1 and lysine 191 in D2 with sites on LRP1 to form a bivalent D1D2-LRP1 complex. When lysine 60 and 191 are both mutated to alanine, the binding of D1D2 to LRP1 is ablated. Our data also reveal that D1D2 is able to bind to a second distinct site on LRP1 to form a monovalent complex. The studies confirm the canonical model for ligand recognition by this class of receptors, which is initiated by pairs of lysine residues that dock into acidic pockets on the receptor.
Keywords: chaperone, low density lipoprotein (LDL), mutagenesis, receptor endocytosis, surface plasmon resonance (SPR), LRP1, RAP, ligand
Introduction
The LDL receptor-related protein 1 (LRP1) is a member of the LDL receptor family and is a highly efficient endocytic and signal-transducing receptor that plays an important role in vascular development, lipoprotein metabolism, and inflammation (1–3). Originally identified as the hepatic receptor responsible for the removal of α2-macroglobulin (α2M)3-protease complexes (4), we now know that LRP1 recognizes numerous ligands, including lipoproteins, matrix proteins, growth factors (1, 2, 5, 6), and extracellular proteases (7–11). Deletion of the Lrp1 gene in mice results in early embryonic lethality at E13.5 (12, 13) due to extensive hemorrhaging resulting from a failure to recruit and maintain vascular smooth muscle cells and pericytes in the vessels. Selective deletion of LRP1 in vascular smooth muscle cells (smLRP1−/− mice) reveals that LRP1 protects against the development of atherosclerosis by attenuating PDGF receptor activation (14, 15) and prevents formation of aneurysms (11, 16) in part by regulating the levels of proteases that are known to degrade matrix components (11).
The efficient delivery of LRP1 and certain other members of the LDL receptor family to the cell surface require their association with a 39-kDa protein termed the receptor-associated protein (RAP). RAP was initially identified when it co-purified with LRP1 (17, 18). Subsequent work demonstrated that RAP binds tightly to LRP1 and prevents ligands from associating with this receptor (19, 20). In the endoplasmic reticulum (ER), RAP functions as a molecular chaperone to assist in the efficient folding and delivery of LRP1 and other family members to the cell surface (18, 19, 21–24). After escorting these receptors to the Golgi, RAP dissociates in the lower pH environment of the Golgi via activation of a histidine switch mechanism located in D3 (25). RAP is then retrieved back to the ER via the recognition of an HNEL carboxyl-terminal sequence on RAP by ERD2 (26), a receptor that functions in the retrieval of ER resident proteins from the secretory pathway. Genetic deletion of RAP results in ineffective delivery of LRP1 and other family members to the cell surface, reducing the amount of functional receptor in organs such as the liver and brain
LRP1 is composed of a modular structure made up of complement-type repeats (CR), EGF repeats, and β-propeller domains. The CR modules are organized into clusters, termed clusters I–IV, which are highly conserved regions where most LRP1 ligands bind. LRP1 binds between 2 and 3 molecules of RAP via interactions involving the CR modules located in clusters II–IV (19, 27). RAP is organized into three independent domains, D1, D2, and D3, each of which are composed of a three-helical bundle (25, 28, 29). These domains are connected by flexible linkers allowing RAP to adopt various conformations when interacting with LDL receptor family members. There are two major binding sites for LRP1 located on RAP (30, 31). The first binding site is located within D1D2, and its binding to LRP1 prevents certain ligands, such as activated forms of α2-macroglobulin, from binding to LRP1 (27, 32). The second site, located within D3, not only functions to block ligands from binding to LRP1 but is also responsible for escorting LRP1 from the ER to the Golgi (25, 27).
Information regarding specific amino acid residues located in D3 that are responsible for interacting with LRP1 resulted from the studies of Migliorini et al. (32), who employed random mutagenesis experiments of D3 and identified Lys-256 and Lys-270 as critical lysine residues on D3 that are necessary for high affinity binding of D3 to LRP1. When a crystal structure of the D3 domain of RAP in complex with the 3rd and 4th CR module of the LDL receptor (termed CR34) was solved, it was discovered that the ϵ-amino groups of Lys-256 and Lys-270 formed salt bridges with carboxylates of aspartic acid residues located within CR34 (33). These interactions are complemented by van der Waals interactions resulting from aromatic residues (phenylalanine and tryptophan) located on CR34 interacting with the aliphatic portion of the lysine residue that is docked in the acidic pocket. Even though RAP binds poorly to the LDL receptor and CR34 of the LDL receptor, this model is supported by structural studies of fragments derived from several different LDL receptor family members in complex with various ligands and from the structure of the LDL receptor ectodomain (34–40).
In contrast to the studies on the D3 domain of RAP, little information is available regarding the binding of D1D2 and its individual D1 and D2 domains to LRP1, and to date studies have investigated the binding of D1 or D2 to fragments of LRP1 (31, 37, 41). These studies reveal that both D1 and D2 appear capable of binding CR56 of LRP1, although rather weakly (37, 41). Additionally, the studies found that mutation of lysine 60 located in D1 reduced the affinity of D1 for CR56 substantially, suggesting that this residue might be important in the binding of D1 to LRP1 (37). Finally, Bloem et al. (42) employed a novel chemical footprinting approach that not only confirmed the involvement of Lys-60, but also implicated lysine 191 in D2 as an important residue for the interaction of D1D2 to LRP1.
The objectives of this current study were to derive quantitative information on the binding of D1D2 and its individual domains to full-length LRP1. The results of this work indicate that the high affinity of D1D2 for LRP1 results from avidity effects mediated by the simultaneous interactions of lysine 60 in D1 and lysine 191 in D2 with sites on LRP1 to form a bivalent D1D2-LRP1 complex. Our studies further show that when these two residues are mutated to alanine, the binding of D1D2 to LRP1 is ablated. Finally, the data reveal that additional lysine residues present on the flexible loop connecting D1 with D2 (Lys-93/Lys-94) and a pair of lysine residues located within D2 (Lys-123/Lys-125) contribute to formation of the bivalent D1D2-LRP1 complex.
Results
Bivalent Binding of RAP D1D2 to LRP1
Prior work employing an ELISA revealed that RAP D1D2 binds to LRP1 with high affinity; however, in the same experiment, no binding of either D1 or D2 to LRP1 was observed (30). In contrast to these findings, subsequent studies demonstrated that individual D1 and D2 fragments competed for the binding of D1D2 to LRP1 (28). To resolve these apparent contradictions, we set out to examine the binding of D1D2 and its individual domains to full-length LRP1. Initially, to confirm that recombinant D1, D2, and D1D2 are appropriately folded, we measured their circular dichroism (CD) spectra and temperature-induced denaturation by monitoring the changes in the CD spectra. The CD spectra of D1, D2, and D1D2 all displayed negative ellipticity at 222 nm, indicative of the presence of α-helix (Fig. 1A). We next used circular dichroism spectroscopy measurements at 222 nm to investigate the thermally induced unfolding of D1, D2, and D1D2. These experiments revealed that D1, D2, and D1D2 all undergo a cooperative transition upon heating, confirming that these domains are folded (Fig. 1B).
FIGURE 1.

RAP D1 and D2 domains bind weakly to LRP1. A, CD spectroscopy was used to measure changes in molar ellipticity as an indication of helical content in RAP D1D2 (black), RAP D1 (blue), and RAP D2 (green). B, CD spectroscopy was used to measure the thermally induced denaturation of RAP D1D2 (black), RAP D1 (blue), and RAP D2 (green) at 222 nm. Surface plasmon resonance measurements of RAP D1 (C) and RAP D2 (D) (each at 8, 16, 31, 63, 125, 250, 500, and 1000 nm) binding to LRP1 are shown. Req values were determined by fitting the data to a pseudo-first order process (blue lines). Response units at equilibrium were plotted against RAP domain concentration (insets) to determine equilibrium binding constants by fitting the data to a single class of sites using non-linear regression analysis.
Next, we performed experiments to quantify the binding of D1 and D2 to full-length LRP1. Because ELISA approaches failed to detect binding of these domains to LRP1, we measured their potential to bind to LRP1 using surface plasmon resonance experiments, an approach capable of detecting weak interactions and also able to measure the association and dissociation rates. In these experiments, full-length LRP1 was first immobilized on the SPR chip surface, and increasing concentrations of D1 or D2 were flowed over the surface. The results of these experiments reveal that the binding of both D1 and D2 to LRP1 is characterized by rapid association and dissociation rates from LRP1 (Fig. 1, C and D). Analysis of the data at equilibrium (Fig. 1, C and D, insets) revealed KD values of 53 ± 5 and 238 ± 56 nm for D1 and D2, respectively. Kinetic analysis of the data confirmed rapid association rates as well as rapid dissociation rates for both D1 and D2 (Table 1). The rapid dissociation of both D1 and D2 domains from LRP1 reveals why no binding of these domains to LRP1 was detected in ELISA-based approaches, as this technique requires extensive washing procedures. The KD values for the binding of D1 and D2 to LRP1 are much weaker than the KD values of D1D2 binding to LRP1. This observation, along with previously published data revealing that both D1 and D2 are able to compete for the binding of D1D2 to LRP1 (28), reveals that the high affinity binding of D1D2 to LRP1 occurs from avidity effects arising from determinants located on both D1 and D2.
TABLE 1.
Kinetic and equilibrium constants for the binding of RAP D1 and RAP D2 to LRP1
Kinetic data were derived by global fitting of the association and dissociation data to a 1:1 binding interaction. The experiments were performed in duplicate and values shown are averages ± S.E.
| RAP fragment | ka | kd | t1/2 | KDa |
|---|---|---|---|---|
| m−1 s−1 | s−1 | s | nm | |
| D1 | 7.65 ± 0.17 × 105 | 0.04 ± 0.004 | 17 | 53 ± 5 |
| D2 | 1.58 ± 0.50 × 106 | 0.27 ± 0.06 | 2.5 | 238 ± 56 |
a Data were determined from equilibrium measurements.
Model for Bivalent Binding of RAP D1D2 to LRP1
To characterize the binding of D1D2 with LRP1, we employed a simple model for bivalent binding in which a determinant located on D1D2 (designated a and b) associates with a binding site on LRP1 (A_B) to form complex I (Fig. 2B, Scheme I). This initial association is followed by the interaction of a second determinant on D1D2 with a second site on LRP1 to form the bivalent complex (complex II, Fig. 2B, Scheme I). When we attempted to fit our experimental SPR data to Scheme I (Fig. 2B), however, we noted that as the concentration of D1D2 increased, the fits of the experimental data to this model dramatically declined. This was due to the appearance at higher D1D2 concentrations of a binding component that contained a rapid dissociation rate (see Fig. 3A). Thus, we also incorporated a second scheme into the model in which D1D2 is also able to bind to a second distinct site on LRP1 to form a monovalent complex (complex III, Fig. 2B, Scheme II). To simplify the model, we assumed that the ka1 and kd1 in Scheme II is identical to the first step in Scheme I (Fig. 2B). This assumption is validated by observing that the equilibrium data for the binding of the D1 domain or the D2 domain to LRP1 was well described by a model containing a single class of sites (see Fig. 1, C and D). When our experimental SPR data were fit to a model containing both Scheme I and Scheme II (Fig. 2B), an excellent fit was obtained (Fig. 3A), which displayed minimal deviations in the residual plots (Fig. 3B). The kinetic parameters along with calculated equilibrium constants derived from this fit are summarized in Table 2. At low concentrations of D1D2, most of its binding to LRP1 occurred via Fig. 2B, Scheme I (bivalent binding mode, see Fig. 7A). However, modeling experiments using the parameters in Table 2 revealed that the amount of complex III formed increased as the concentration of D1D2 was increased, reaching a plateau of ∼50% of the complex species (Fig. 3C). Furthermore, modeling experiments reveal that at all concentrations of D1D2, very little amounts of complex I are present due to the rapid formation of complex II.
FIGURE 2.
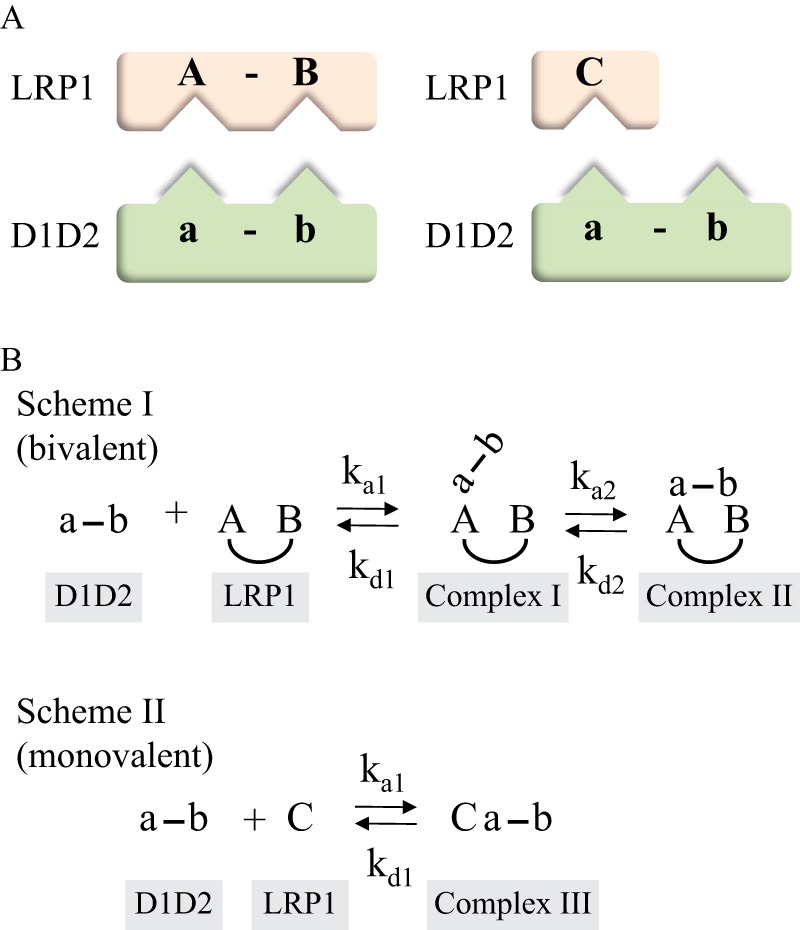
Bivalent model for the binding of RAP D1D2 to LRP1. A, schematic diagram of model in which LRP1 contains two binding sites for RAP D1D2; one containing two determinants on LRP1 (left) and a second containing only one determinant on LRP1 (right). B, scheme showing kinetic constants. Binding of D1D2 at site 1 on LRP1 allows for bivalent binding in which determinants on both the D1 domain and D2 domain interact with corresponding regions on LRP1 (Scheme I). In the model, LRP1 also contains a second site that can only interact with one determinant on D1D2 to form a monovalent complex (Scheme II).
FIGURE 3.
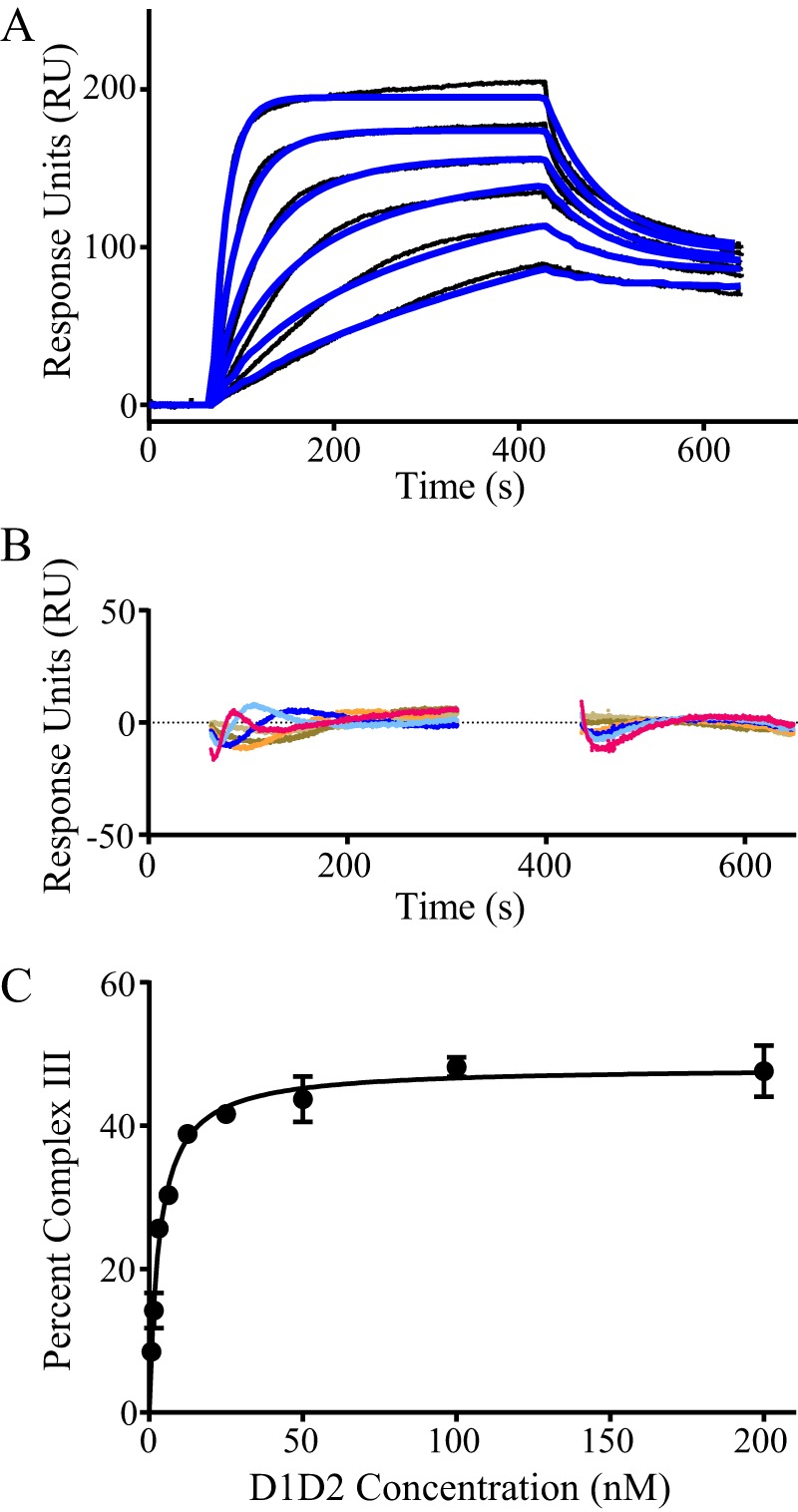
High affinity interaction of RAP D1D2 to LRP1 occurs via bivalent binding to ligand. A, surface plasmon resonance measurements of RAP D1D2 (1.6, 2.1, 6.3, 12.5, 25, and 50 nm) binding to LRP1. The raw data (black lines) were fit (blue lines) using the model incorporating Schemes 1 and 2. The fit parameters from this model are summarized in Table 2. B, residual plot comparing the experimental data to the fit data. C, using the fit parameters summarized in Table 2, the percent of complex III (monovalent complex) present at 400 s as a function of D1D2 concentration is shown.
TABLE 2.
Kinetic and equilibrium constants for the binding of RAP D1D2 and mutants to LRP1
Kinetic constants were obtained by fitting the data to a model containing Scheme I and Scheme II (Fig. 2B). These experiments were performed in triplicate, and the values shown are the average ± S.E.
| Protein | ka1 | kd1 | ka2 | kd2 | KD1a | KD2b | % Scheme Ic |
|---|---|---|---|---|---|---|---|
| m−1 s−1 | s−1 | m−1 s−1 | s−1 | m | m | ||
| WT | 1.1 ± 0.1 × 106 | 0.028 ± 0.002 | 3.2 ± 1.1 × 105 | 0.0017 ± 0.0001 | 1.1 ± 0.1 × 10−16 | 2.6 ± 0.1 × 10−8 | 52–92d |
| K60A | 2.6 ± 0.4 × 106 | 0.074 ± 0.013 | 6.7 ± 3.3 × 105 | 0.0041 ± 0.0003 | 4.0 ± 1.5 × 10−16 | 2.6 ± 0.4 × 10−8 | 10–20 |
| K191A | 4.1 ± 0.9 × 106 | 0.029 ± 0.005 | 4.4 ± 2.8 × 105 | 0.0033 ± 0.0003 | 2.6 ± 1.6 × 10−16 | 7.3 ± 0.3 × 10−9 | 38 |
a The equilibrium binding constant KA1 was calculated using the following equation: KA1 = (ka1/kd1)·(1 + (ka2/ kd2) and KD1 was calculated as KD1 = 1/KA1.
b The equilibrium binding constant KA2 was calculated using the following equation: KA2 = ka1/kd1 and KD2 was calculated as KD2 = 1/KA2.
c Percent of complex II that forms (bivalent binding, Fig. 2B, Scheme I) is shown.
d For WT D1D2, the amount of complex II that forms is dependent upon D1D2 concentration (see Fig. 3).
FIGURE 7.
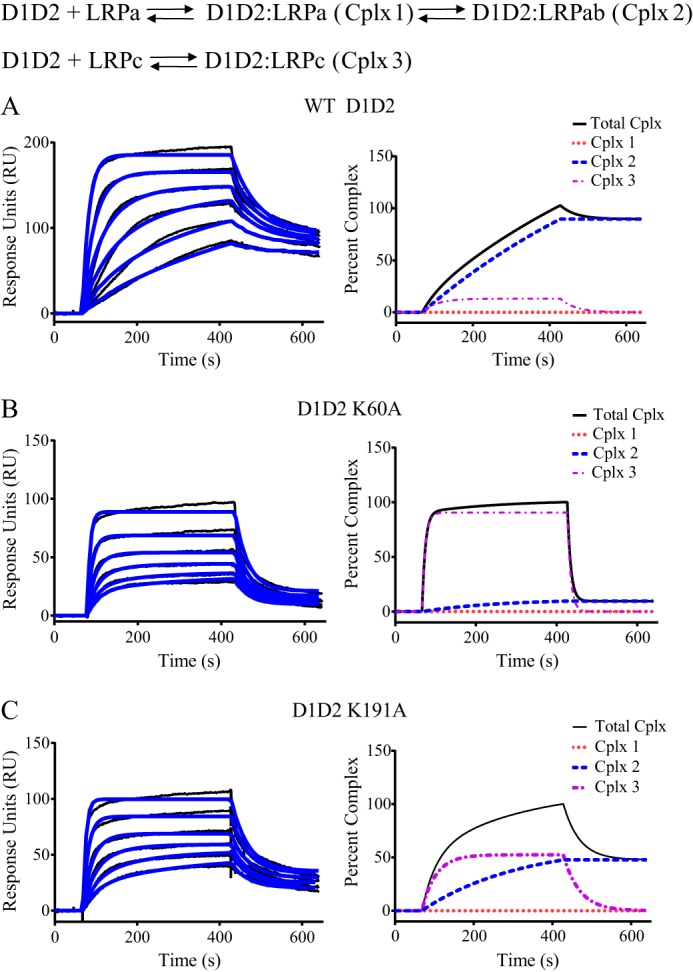
Mutagenesis of lysine 60 and lysine 191 in RAP D1D2 impacts the mode of binding to LRP1. Surface plasmon resonance measurements of WT RAP D1D2 (A), RAP D1D2 K60A (B), and RAP D1D2 K191A (C) (1.6, 2.1, 6.3, 12.5, 25, and 50 nm) binding to LRP1. The data were fit to the model incorporating Schemes 1 and 2, and the fit curves are shown in blue. In the right panels, the percent of total complex (black), complex I (red dotted line), complex 2 (blue dashed line), and complex 3 (purple dashed line) are shown as a function of time from the fit data obtained at 1.6 nm D1D2.
Lysine Residues on D1D2 Contribute to the Binding of D1D2 to LRP1
To identify the specific amino acids that contribute to the binding of D1D2 to LRP1, we initiated chemical modification studies. Because previous studies have implicated an important contribution of lysine residues to the binding of ligands to LRP1 (32, 33), we chemically modified all lysine residues of D1D2 with Sulfo-NHS-acetate, which blocks the primary amines found in lysine side chains by forming stable, covalent amide bonds. Circular dichroism measurements revealed that the CD spectra for native and chemically modified D1D2 were virtually identical, with both displaying negative ellipticity at 222 nm, which was reduced upon heating to 90 °C revealing a loss of α-helical structure at higher temperatures (Fig. 4A). Furthermore, the overall stability of each fragment was identical as revealed from thermally induced unfolding using CD measurements at 222 nm (Fig. 4B). We conclude from these studies that the overall structure of chemically modified D1D2 is very similar to that of the unmodified protein. To measure the impact of lysine modification on the binding of D1D2 to LRP1, SPR measurements were performed, and no binding was observed when 200 nm chemically modified D1D2 was injected over the LRP1-coated chip (Fig. 4C) revealing that chemical modification of the lysine residues in D1D2 completely abolished binding. To further confirm this observation, we investigated the ability of chemically modified D1D2 to compete for the binding of 125I-labeled D1D2 to microtiter wells coated with LRP1. The results of these studies, shown in Fig. 4D, demonstrate that in contrast to WT D1D2, lysine-modified D1D2 was unable to compete for the binding of this molecule to LRP1. Thus, these studies confirm an important role for lysine residues in the interaction of D1D2 with LRP1.
FIGURE 4.
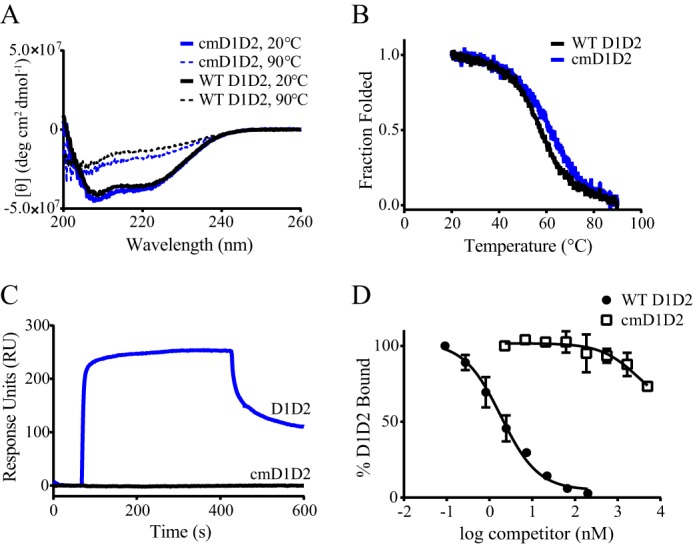
Lysine residues in RAP D1D2 are required for binding to LRP1. A, CD spectroscopy was used to measure the changes in molar ellipticity as an indication of helical content in WT RAP D1D2 (black) and chemically modified RAP D1D2 (blue) at 20 °C (solid lines) and 90 °C (dashed lines). B, CD spectroscopy measured at 222 nm was used to measure the heat-induced denaturation of RAP D1D2 (black) and chemically modified RAP D1D2 (blue). C, 200 nm WT RAP D1D2 (blue) or chemically modified RAP D1D2 (black) was injected on an LRP1-coupled SPR chip, and their binding was measured. D, LRP1 immobilized in microtiter wells was incubated with 1 nm 125I-RAP D1D2 in the presence of increasing concentrations of unlabeled RAP D1D2 (closed circles) or chemically modified RAP D1D2 (open squares), and the amount of 125I-RAP D1D2 bound to LRP1 was detected. Data are presented as mean ± S.D. from duplicate wells.
Lysine 60 on D1 and Lysine 191 on D2 Are Critical for the Binding of D1D2 to LRP1
To identify specific amino acid residues involved in the binding of D1D2 to LRP1, we used site-directed mutagenesis to prepare several mutants of D1D2, including the following mutants: K60A, K191A, and K60A/K191A. Circular dichroism studies were used to determine whether the structure of the mutant proteins was preserved. Although the spectra of the K60A mutant overlapped that of the WT, interestingly, the K191A mutant as well as the K60A/K191A mutant had slightly more negative ellipticity at 222 and 208 nm, suggestive of increased helical content (Fig. 5A). The characteristic negative ellipticity at 222 nm was reduced when the spectra were recorded at 90 °C (Fig. 5B) revealing the expected loss of α-helical structure. Thermal unfolding experiments revealed similar melting profiles for the WT and three mutant proteins (Fig. 5C). We conclude from these experiments that these mutations do not substantially alter the structure of D1D2.
FIGURE 5.

RAP D1D2 K60A, K191A, and K60A/K191A mutations do not significantly alter the structure or stability of the molecule. CD spectroscopy was used to measure the molar ellipticity as an indicator of helical content at 20 °C (A) and 90 °C (B) for WT RAP D1D2 (black), RAP D1D2 K60A (blue), RAP D1D2 K191A (green), and RAP D1D2 K60A/K191A (purple). C, thermally induced denaturation of WT RAP D1D2 (black), RAP D1D2 K60A (blue), RAP D1D2 K191A (green), and RAP D1D2 K60A/K191A (purple) was measured using CD spectroscopy at 222 nm.
We next measured the ability of the K60A and the K191A mutant D1D2 molecules to compete for the binding of WT D1D2 to LRP1. The data reveal that both the K191A and K60A mutants inhibited the binding of 125I-labeled D1D2 to LRP1 with KI values of 19 and 640 nm, respectively (Fig. 6A). In contrast, the K60/K191A mutant was not very effective at inhibiting the binding of D1D2 to LRP1 (Fig. 6B), and SPR measurements confirmed very little binding of this mutant protein to LRP1 when compared with WT D1D2 (Fig. 6C).
FIGURE 6.
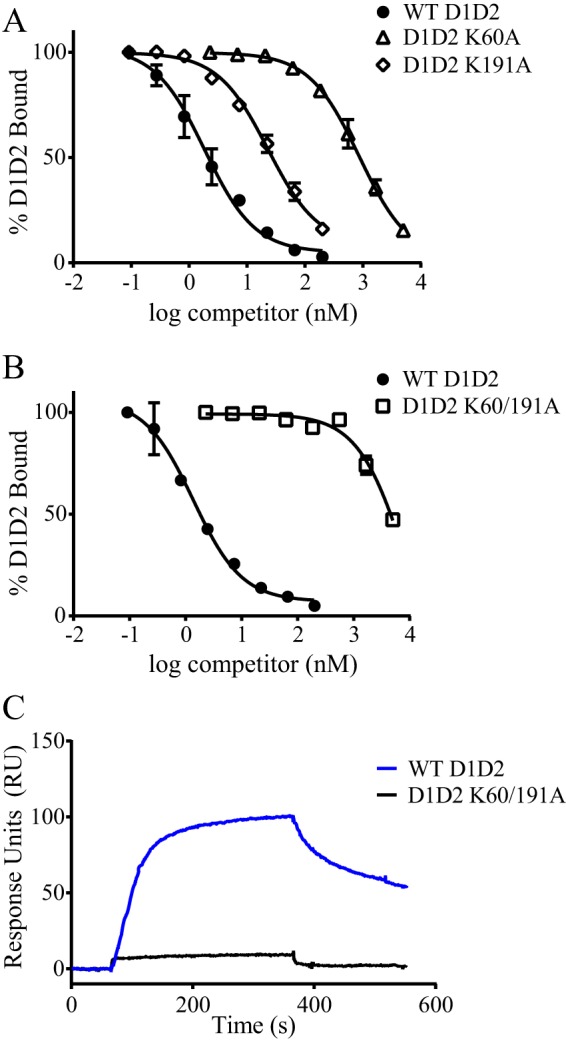
Mutation of both Lys-60 and Lys-191 is necessary to eliminate the binding of RAP D1D2 to LRP1. LRP1 was immobilized in microtiter wells and incubated with 1 nm 125I-RAP D1D2 (A) or 5 nm 125I-RAP D1D2 (B) in the presence of increasing concentrations of unlabeled WT RAP D1D2 (closed circles), RAP D1D2 K60A (open triangles, A), RAP D1D2 K191A (open diamonds, A) or RAP D1D2 K60A/K191A (open squares, B). Following incubation and washing, the amount of bound 125I-D1D2 was detected, and data are represented as mean ± S.D. of duplicates. C, 200 nm WT RAP D1D2 (blue) or RAP D1D2 K60A/K191A (black) were injected on an LRP1-coupled SPR chip and their binding measured.
We next used SPR measurements to compare the binding of K60A and K191A mutant proteins with WT D1D2 by fitting the SPR data to the model in Fig. 2. The results of this analysis are shown in Fig. 7, and the kinetic constants derived from these fits are summarized in Table 2. Fig. 7 also shows the distribution of complexes I–III as a function of time modeled from the fit parameters at low concentrations of D1D2 (1.6 nm) for each of these molecules. These data reveal that in the case of WT D1D2 (Fig. 7A), greater than 90% of the D1D2 molecules bind to LRP1 via a bivalent interaction (Scheme I, Fig. 2). In contrast, in the case of the D1D2 K60A mutant (Fig. 7B), ∼80–90% of the molecules bind to LRP1 via a monovalent binding interaction (Scheme II, Fig. 2) confirming the importance of lysine 60 for formation of a bivalent complex. In the case of the K191A mutant (Fig. 7C), the situation is more complex, with monovalent binding accounting for ∼62% of the reaction pathway. These data suggest that upon mutation of Lys-191, other lysine residues can substitute for Lys-191 to form a bivalent complex, albeit not as effectively as Lys-191 itself.
Lysine Residues 93, 94, 123, and 125 Impact Formation of the Binary Complex with LRP1
To gain insight into any additional contributions of lysine residues within D2 that might contribute to the interaction of this domain with LRP1, we sequentially mutated every lysine residue in D2 to alanine, and we measured the impact on binding to LRP1. The results of this analysis are summarized in Table 3 and identify two additional pairs of lysine residues (Lys-93/Lys-94 as well as Lys-123/Lys-125), whose mutation substantially increases the KD1 value derived from the fit of the data to the model shown in Fig. 2.
TABLE 3.
KD values for the binding of RAP D1D2 and mutants located in D2 to LRP1
Experiments were performed in duplicate. Boldface entries identify large changes in KD1 relative to WT D1D2.
| Protein | KD1 fold change | KD2 fold change | % complex IIa |
|---|---|---|---|
| RAP D1D2 | 1.0 | 1.0 | 87 |
| RAP D1D2 K93A | 5.5 | 0.8 | 76 |
| RAP D1D2 K94A | 7.2 | 0.7 | 65 |
| RAP D1D2 K93A/K94A | 23.6 | 0.9 | 70 |
| RAP D1D2 K119A | 3.5 | 0.3 | 58 |
| RAP D1D2 K123A | 9.4 | 0.8 | 80 |
| RAP D1D2 K125A | 4.5 | 0.3 | 61 |
| RAP D1D2 K119A/K123A | 1.2 | 0.4 | 50 |
| RAP D1D2 K123A/K125A | 33.8 | 0.4 | 71 |
| RAP D1D2 129A | 3.3 | 0.8 | 75 |
| RAP D1D2 137A | 2.9 | 0.4 | 63 |
| RAP D1D2 K146A | 4.0 | 0.8 | 80 |
| RAP D1D2 K148A | 2.3 | 0.5 | 57 |
| RAP D1D2 K146A/K148A | 2.1 | 0.4 | 71 |
| RAP D1D2 K179A | 1.0 | 0.4 | 67 |
| RAP D1D2 K191A | 2.3 | 0.3 | 47 |
| RAP D1D2 K193A | 7.5 | 0.3 | 58 |
| RAP D1D2 K191A/K193A | 3.8 | 2.9 | 50 |
| RAP D1D2 K148A/K193A | 1.3 | 0.7 | 72 |
a Percent of complex II present in the reaction with 1.6 nm D1D2 is shown.
Discussion
The three-dimensional structure of the RAP D3 domain in complex with CR34 from the LDL receptor provided insight into a potential canonical model for ligand recognition by LDL receptor family members (33). In this model, critical lysine residues on the ligand dock into acidic pockets present in the CR modules similarly spaced on the receptors. It should be noted, however, that RAP binds poorly to the LDL receptor, and the affinity of D3 for CR34 of the LDL receptor is extremely weak in comparison with its binding to LRP1. This is consistent with the finding from the D3/CR34 structure of a relatively small contact surface between the critical lysine residues on D3 and CR34 of the LDL receptor. Clearly, additional interactions are present in the D3-LRP1 complex that contribute to the high affinity interaction observed for this complex. Indeed, the use of tandem mass tags in a chemical footprinting experiment further identified an important contribution for Lys-305 and Lys-306 in the interaction of D3 with cluster II of LRP1 (42), whereas mutational analysis of D3 suggests minor contributions of Lys-253 and perhaps Lys-289 to the interaction of D3 with CR56 of LRP1 (Fig. 8B) (43). Additional structural studies are required to define more precisely the nature of the interaction between D3 and LRP1 resulting in high affinity interactions.
FIGURE 8.
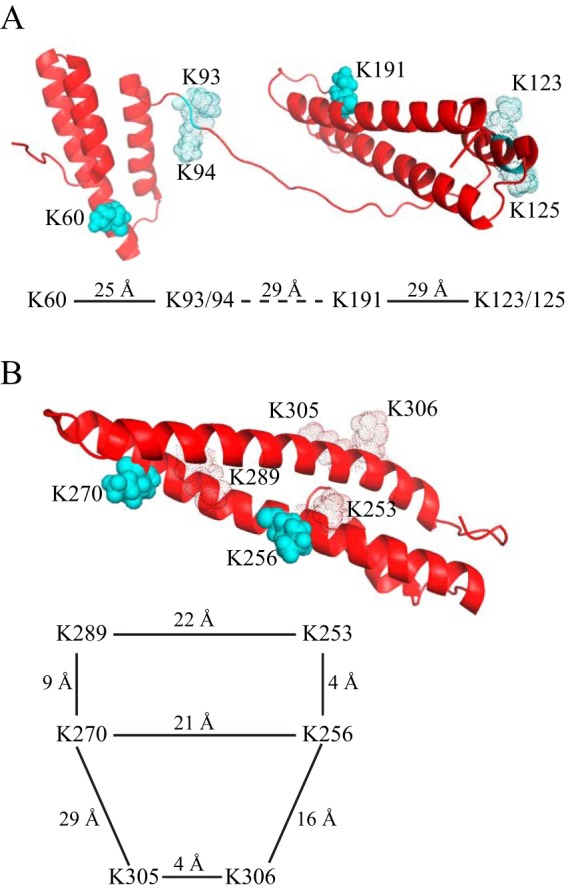
Structure of RAP D1D2 (A) and D3 (B) showing key lysine residues interacting with LRP1. The ribbon diagram shows the critical lysine residues 60 and 191 in D1D2 and critical lysine residues 253 and 270 in D3 as solid cyan spheres. Additional lysine residues that contribute to binding are shown in silhouette. The distances between these residues are diagrammed. Figures are from Protein Data Bank structure 2P03.
In contrast to D3, amino acids involved in the interaction of D1D2 with LRP1 were not well defined until this investigation. In our study, we observed the critical importance of a pair of lysine residues, Lys-60 in D1 and Lys-191 in D2 that are required for the high affinity binding of D1D2 to LRP1. This binding occurs via avidity effects involving the interaction of these two RAP domains with sites on LRP1. It is important to note that the binding of D1D2 to LRP1 was ablated only when both Lys-60 and Lys-191 were mutated to alanine residues. Remarkably, mutating only Lys-60 or Lys-191 in D1D2 generated a molecule that still bound to LRP1, although with a weaker affinity. Kinetic analysis revealed that the major impact of the K60A mutation was to largely prevent the bivalent binding of D1D2 to LRP1, whereas the K191A mutation simply reduced the amount of bivalent complex that formed. Our data identified additional lysine residues present on the flexible loop connecting D1 with D2 (Lys-93/Lys-94) and a pair of lysine residues located within D2 (Lys-123/Lys-125) that also contribute to formation of the bivalent D1D2-LRP1 complex.
We conclude from the studies examining the interaction of D1D2 and D3 with LRP1 that the canonical model for high affinity ligand binding to LRP1 (and other LDL receptor family members) occurs via avidity effects in which at least two or more critically spaced lysine residues on ligands dock into the acidic pockets of appropriately spaced CR modules on LRP1. These initial interactions are no doubt complemented by additional interactions that contribute to the high affinity binding noted for these molecules. It is important to highlight that in the instance of D1D2, simply mutating a single lysine residue was not sufficient to prevent its binding to LRP1. Similar results were found when examining the binding of D3 to CR56 from LRP1 (43) and by measuring the ability of LRP1 to endocytose mutants of D3 (44). Together, these studies concluded that mutation of both Lys-256 and Lys-270 is required to ablate the binding of D3 to LRP1.
Examination of the structure of RAP reveals that most of the critical lysine residues identified to participate in the binding interactions in both D1D2 and D3 are spaced at intervals ranging from 21 to 29 Å (Fig. 8). Because D1 is connected to D2 via a flexible linker, it is not possible to measure the distance between the Lys-60 and Lys-191 located in D1 and D2, respectively. In α2-macroglobulin, a single lysine residue (Lys-1370) located in the receptor binding domain of this molecule is essential for its binding to LRP1, and mutation of Lys-1370 to alanine totally blocks binding of activated forms of α2M to LRP1 (45, 46). Because human α2M is composed of four identical subunits, Lys-1370 located on each subunit of α2M is capable of docking into appropriately spaced CR modules on LRP1 to form multivalent interactions, which likely accounts for the extremely high affinity observed for the binding of this ligand to LRP1 (47). Interestingly, cell-based experiments reveal that the LRP1-mediated uptake of α2M requires CR modules from both cluster I and cluster II (48). Structural analysis of the dimeric receptor binding domain from rat α2M reveals that the critical lysine residues are located 18 Å apart (49), implying that CR modules in LRP1 that have this spacing are likely binding sites.
Currently, very little information is available regarding the structure of LDL receptor family members, and in most cases, only the structure of individual domains has been solved. However, a structure for the ectodomain of the LDL receptor at endosomal pH has been solved, and at this pH the distances between the CR2 and CR7 modules range from 20 to 43 Å (34). Interestingly, the distance between CR3 and CR4 in this structure of 32 Å is quite different from the distance of 21 Å measured between CR3 and CR4 in the RAP D3/CR34 structure (33) confirming flexibility in distances between the CR repeats of this receptor family. Finally, the structure of CR56 of LRP1 reveals a distance of 22 Å between the acidic pockets (37).
Although additional structural studies on LDL receptor family members are certainly required, it might be possible to identify critical lysine residues located on ligands that participate in the binding interaction with LDL receptor family members from structural analysis of the ligand by searching for pairs of exposed surface lysine residues that are located between 18 and 43 Å apart. For certain LRP1 ligands, such as fVIII, identification of lysine pairs that contribute to LRP1 binding might be beneficial, as fVIII is used as a clinical product to treat hemophilia, and delaying its LRP1-mediated hepatic clearance would likely generate a molecule with a longer circulating half-life. Using hydrogen deuterium exchange experiments along with mutational analysis and SPR binding analysis, van den Biggelaar et al. (50) concluded that the interaction between fVIII and cluster II of LRP1 occurs over an extended surface composed of multiple lysine residues and that no individual lysine residues dominate the binding interaction. However, if fVIII follows the same trend as RAP D1D2 and RAP D3, then mutation of two or more critical lysine residues would be required to sufficiently reduce its affinity for LRP1.
In summary, this study has examined the requirement for specific lysine residues in D1D2 that are responsible for interacting with LRP1. The data obtained confirm the canonical model for ligand recognition by this class of receptors, and the data stress the fact that mutational analysis of at least two lysine residues are often required to substantially reduce the affinity of the ligand for LRP1. Finally, the studies stress that additional structural information is required, and obtaining a structure of ligand in complex with receptor will give additional important insight into the mode of ligand recognition by LRP1 and other LDL receptor family members.
Experimental Procedures
Site-directed Mutagenesis, Protein Expression, and Purification of LRP1, RAP Fragments, and RAP Fragment Mutants
LRP1 was purified from human placenta as described (4). Mutagenesis of RAP D1D2 was performed using the GENEART site-directed mutagenesis system (Invitrogen) using RAP D1D2 pGex2T (19, 30) as a template. Primer pairs were designed to introduce the desired mutation(s) of interest. All mutations were confirmed by forward and reverse sequencing. RAP D1(1–99), RAP D2(100–216), and RAP D1D2(1–216) proteins were expressed in Escherichia coli and purified as described previously (19, 30).
Chemical Modification of RAP D1D2
Chemical modification of RAP D1D2 to block the primary amines in lysine side chains was performed using Sulfo-NHS-acetate (ThermoFisher Scientific). Sulfo-NHS-acetate was dissolved in H2O to make a 10 mm solution. 1 mg of RAP D1D2 was incubated with an equal mass amount of the Sulfo-NHS-acetate in Hepes-buffered saline for 1 h at room temperature. The modified RAP D1D2 protein was dialyzed into Hepes-buffered saline to remove the excess Sulfo-NHS-acetate. The resulting protein was run on a 4–12% BisTris gel to confirm protein purity and molecular weight.
Circular Dichroism
CD spectra were recorded on a Jasco-715 spectropolarimeter with a Peltier PFD-350S unit for temperature control. Proteins were dialyzed in 10 mm phosphate buffer, pH 7.5, for all spectra. Spectra were collected at 20 and 90 °C from 260 to 190 nm in a 1-mm cell, with data recorded every 0.1 nm. For melting curves, thermal stability was measured at a constant wavelength of 222 nm, from either 5 or 20 °C up to 90 °C in steps of 0.1 °C using a 1-mm path length cell.
Surface Plasmon Resonance
Purified LRP1 was immobilized on a CM5 sensor chip surface to the level of 10,000 response units, using a working solution of 20 μg/ml LRP1 in 10 mm sodium acetate, pH 4, according to the manufacturer's instructions (BIAcore AB). An additional flow cell was activated and blocked with 1 m ethanolamine without protein to act as a control surface. All binding experiments were performed in HBS-P buffer (0.01 m Hepes, 0.15 m NaCl, 0.005% surfactant P, 1 mm CaCl2), pH 7.4. All experiments were performed on a BIAcore 3000 instrument, using a flow rate of 20 μl/min at 25 °C, with binding and dissociation occurring for 5 min each, using protein concentrations from 200 nm down to 0.78 nm for RAP D1D2 and its mutants and 1 μm to 7.81 nm for RAP D1 and RAP D2 fragments. Sensor chip surfaces were regenerated by 15-s injections of 100 mm phosphoric acid at a flow rate of 100 μl/min.
Kinetic Analysis of Surface Plasmon Resonance Data
The data were simultaneously fit to Scheme I and Scheme II (see Fig. 2) using numerical integration algorithms available in BIAevaluation software as shown in Schemes 1 and 2, where A represents D1D2; B1 indicates binding site 1 on LRP1; B2 indicates binding site 2 on LRP1; AB1, indicates the LRP1-D1D2 complex at site 1; AB2 indicates the LRP1-D1D2 complex at site 2; and ABx indicates the LRP1-D1D2 bivalent complex.
SCHEME 1.

SCHEME 2.
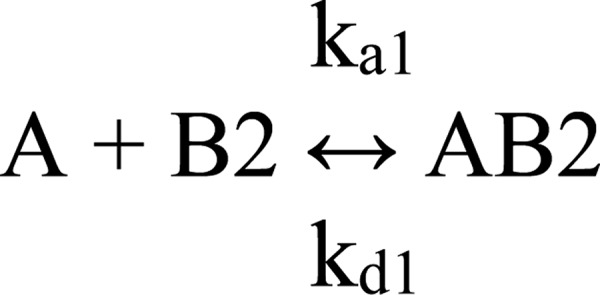
For numerical integrations, the following were used: A = concentration; B1[0] = Rmax1; B2[0] = Rmax2; AB1[0] = 0; AB2[0] = 0; Bx[0] = 0; dB1/dt = −(ka1·A·B1 − kd1·AB1). dB2/dt = − (ka1·A·B2 − kd1−AB2); dAB1/dt = (ka1·A·B1 − kd1·AB1) − (ka2·AB1 − kd2·ABx); dAB2/dt = (ka1·A·B2 − kd·AB2); dABx/dt = (ka2·AB1 − kd2·ABx). The total response is as follows: AB1 + AB2 + ABx. The overall equilibrium binding constant for Scheme 1(KA1) is calculated as described Equation 1 (51, 52),
The equilibrium binding constant for Scheme 2 (KA2) is calculated as shown in Equation 2,
To facilitate the fitting process, initial estimates for kd1 and kd2 were first obtained by fitting the dissociation data globally to a two exponential decay model. The values obtained from these fits were then used to constrain kd1 and kd2 in the fit of the experimental data to the model depicted in Fig. 2. During this process, ka1 and ka2 were fit globally, whereas Rmax1 and Rmax2 were fit locally.
For equilibrium binding analysis the association data were fit to a pseudo-first order process to determine Req values which were plotted versus ligand concentration. The KD value was determined by fitting the data to a single class of sites using non-linear regression analysis employing GraphPad Prism 6.0 software.
Competition Assay
Wells in 96-well plates were coated with human LRP1 (4 μg/ml in TBS, 100 μl/well) overnight and then blocked with 300 μl of assay buffer (20 mm Hepes, 0.15 m NaCl, 2 mm CaCl2, 0.1% Tween 80, and 1% BSA) for 1 h at 37 °C. Wells were washed three times with 300 μl of assay buffer before addition of 100 μl of 125I- RAP D1D2 in the presence or absence of a competitor. Binding occurred overnight at 4 °C. Wells were washed, and radioactivity associated with each well was counted to determine 125I-RAP D1D2 bound to LRP1.
Author Contributions
J. M. P. and P. A. Y. designed experiments, collected and analyzed data, and assisted in writing portions of the paper. D. K. S. designed experiments, assisted in analyzing data, and wrote the paper with J. M. P. All authors approved the final version of the manuscript.
Acknowledgments
We thank Selen C Muratoglu and Mary Migliorini for their critical review of this manuscript.
This work was supported in part by National Institutes of Health Grants HL114379 and HL120388 (to D. K. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- α2M
- α2-macroglobulin
- RAP
- receptor-associated protein
- ER
- endoplasmic reticulum
- CR
- complement-type repeat
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- SPR
- surface plasmon resonance.
References
- 1. Herz J., and Strickland D. K. (2001) LRP: a multifunctional scavenger and signaling receptor. J. Clin. Invest. 108, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lillis A. P., Van Duyn L. B., Murphy-Ullrich J. E., and Strickland D. K. (2008) LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 88, 887–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mantuano E., Brifault C., Lam M. S., Azmoon P., Gilder A. S., and Gonias S. L. (2016) LDL receptor-related protein-1 regulates NFκB and microRNA-155 in macrophages to control the inflammatory response. Proc. Natl. Acad. Sci. U.S.A. 113, 1369–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ashcom J. D., Tiller S. E., Dickerson K., Cravens J. L., Argraves W. S., and Strickland D. K. (1990) The human α2-macroglobulin receptor: identification of a 420-kD cell surface glycoprotein specific for the activated conformation of α2-macroglobulin. J. Cell Biol. 110, 1041–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. May P., Herz J., and Bock H. H. (2005) Molecular mechanisms of lipoprotein receptor signalling. Cell. Mol. Life Sci. 62, 2325–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boucher P., and Herz J. (2011) Signaling through LRP1: Protection from atherosclerosis and beyond. Biochem. Pharmacol. 81, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hahn-Dantona E., Ruiz J. F., Bornstein P., and Strickland D. K. (2001) The low density lipoprotein receptor-related protein modulates levels of matrix metalloproteinase 9 (MMP-9) by mediating its cellular catabolism. J. Biol. Chem. 276, 15498–15503 [DOI] [PubMed] [Google Scholar]
- 8. Rozanov D. V., Hahn-Dantona E., Strickland D. K., and Strongin A. Y. (2004) The low density lipoprotein receptor-related protein LRP is regulated by membrane type-1 matrix metalloproteinase (MT1-MMP) proteolysis in malignant cells. J. Biol. Chem. 279, 4260–4268 [DOI] [PubMed] [Google Scholar]
- 9. Yang Z., Strickland D. K., and Bornstein P. (2001) Extracellular MMP2 levels are regulated by the LRP scavenger receptor and thrombospondin 2. J. Biol. Chem. 276, 8403–8408 [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto K., Owen K., Parker A. E., Scilabra S. D., Dudhia J., Strickland D. K., Troeberg L., and Nagase H. (2014) Low density lipoprotein receptor-related protein 1 (LRP1)-mediated endocytic clearance of a disintegrin and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4): functional differences of non-catalytic domains of ADAMTS-4 and ADAMTS-5 in LRP1 binding. J. Biol. Chem. 289, 6462–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muratoglu S. C., Belgrave S., Hampton B., Migliorini M., Coksaygan T., Chen L., Mikhailenko I., and Strickland D. K. (2013) LRP1 protects the vasculature by regulating levels of connective tissue growth factor and HtrA1. Arterioscler. Thromb. Vasc. Biol. 33, 2137–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herz J., Couthier D. E., and Hammer R. E. (1993) Correction: LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 73, 428. [DOI] [PubMed] [Google Scholar]
- 13. Nakajima C., Haffner P., Goerke S. M., Zurhove K., Adelmann G., Frotscher M., Herz J., Bock H. H., and May P. (2014) The lipoprotein receptor LRP1 modulates sphingosine-1-phosphate signaling and is essential for vascular development. Development 141, 4513–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boucher P., Gotthardt M., Li W. P., Anderson R. G., and Herz J. (2003) LRP: Role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332 [DOI] [PubMed] [Google Scholar]
- 15. Basford J. E., Moore Z. W., Zhou L., Herz J., and Hui D. Y. (2009) Smooth muscle LDL receptor-related protein-1 inactivation reduces vascular reactivity and promotes injury-induced neointima formation. Arterioscler. Thromb. Vasc. Biol. 29, 1772–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boucher P., Li W. P., Matz R. L., Takayama Y., Auwerx J., Anderson R. G., and Herz J. (2007) LRP1 functions as an atheroprotective integrator of TGFβ and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS ONE 2, e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Strickland D. K., Ashcom J. D., Williams S., Burgess W. H., Migliorini M., and Argraves W. S. (1990) Sequence identity between the α2-macroglobulin receptor and low density lipoprotein receptor-related protein suggests that this molecule is a multifunctional receptor. J. Biol. Chem. 265, 17401–17404 [PubMed] [Google Scholar]
- 18. Strickland D. K., Ashcom J. D., Williams S., Battey F., Behre E., McTigue K., Battey J. F., and Argraves W. S. (1991) Primary structure of α2-macroglobulin receptor-associated protein. Human homologue of a Heymann nephritis antigen. J. Biol. Chem. 266, 13364–13369 [PubMed] [Google Scholar]
- 19. Williams S. E., Ashcom J. D., Argraves W. S., and Strickland D. K. (1992) A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J. Biol. Chem. 267, 9035–9040 [PubMed] [Google Scholar]
- 20. Herz J., Goldstein J. L., Strickland D. K., Ho Y. K., and Brown M. S. (1991) 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J. Biol. Chem. 266, 21232–21238 [PubMed] [Google Scholar]
- 21. Willnow T. E., Rohlmann A., Horton J., Otani H., Braun J. R., Hammer R. E., and Herz J. (1996) RAP, a specialized chaperone, prevents ligand-induced ER retention and degradation of LDL receptor-related endocytic receptors. EMBO J. 15, 2632–2639 [PMC free article] [PubMed] [Google Scholar]
- 22. Willnow T. E., Armstrong S. A., Hammer R. E., and Herz J. (1995) Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 4537–4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bu G., and Rennke S. (1996) Receptor-associated protein is a folding chaperone for low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 22218–22224 [DOI] [PubMed] [Google Scholar]
- 24. Bu G., Geuze H. J., Strous G. J., and Schwartz A. L. (1995) 39-kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 14, 2269–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee D., Walsh J. D., Mikhailenko I., Yu P., Migliorini M., Wu Y., Krueger S., Curtis J. E., Harris B., Lockett S., Blacklow S. C., Strickland D. K., and Wang Y.-X. (2006) RAP uses a histidine switch to regulate its interaction with LRP in the ER and Golgi. Mol. Cell 22, 423–430 [DOI] [PubMed] [Google Scholar]
- 26. Bu G., Rennke S., and Geuze H. J. (1997) ERD2 proteins mediate ER retention of the HNEL signal of LRP's receptor- associated protein (RAP). J. Cell Sci. 110, 65–73 [DOI] [PubMed] [Google Scholar]
- 27. Obermoeller L. M., Warshawsky I., Wardell M. R., and Bu G. (1997) Differential functions of triplicated repeats suggest two independent roles for the receptor-associated protein as a molecular chaperone. J. Biol. Chem. 272, 10761–10768 [DOI] [PubMed] [Google Scholar]
- 28. Lee D., Walsh J. D., Migliorini M., Yu P., Cai T., Schwieters C. D., Krueger S., Strickland DK, and Wang Y.-X. (2007) The structure of receptor-associated protein (RAP). Protein Sci. 16, 1628–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nielsen P. R., Ellgaard L., Etzerodt M., Thogersen H. C., and Poulsen F. M. (1997) The solution structure of the N-terminal domain of α2-macroglobulin receptor-associated protein. Proc. Natl. Acad. Sci. U.S.A. 94, 7521–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Medved L. V., Migliorini M., Mikhailenko I., Barrientos L. G., Llinás M., and Strickland D. K. (1999) Domain organization of the 39-kDa receptor-associated protein. J. Biol. Chem. 274, 717–727 [DOI] [PubMed] [Google Scholar]
- 31. Jensen J. K., Dolmer K., Schar C., and Gettins P. G. (2009) Receptor-associated protein (RAP) has two high-affinity binding sites for the low-density lipoprotein receptor-related protein (LRP): consequences for the chaperone functions of RAP. Biochem. J. 421, 273–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Migliorini M. M., Behre E. H., Brew S., Ingham K. C., and Strickland D. K. (2003) Allosteric modulation of the ligand binding properties of LRP by the receptor associated protein (RAP) requires critical lysine residues within its carboxyl-terminal domain. J. Biol. Chem. 278, 17986–17992 [DOI] [PubMed] [Google Scholar]
- 33. Fisher C., Beglova N., and Blacklow S. C. (2006) Structure of an LDLR-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol. Cell 22, 277–283 [DOI] [PubMed] [Google Scholar]
- 34. Rudenko G., Henry L., Henderson K., Ichtchenko K., Brown M. S., Goldstein J. L., and Deisenhofer J. (2002) Structure of the LDL receptor extracellular domain at endosomal pH. Science 298, 2353–2358 [DOI] [PubMed] [Google Scholar]
- 35. Yasui N., Nogi T., Kitao T., Nakano Y., Hattori M., and Takagi J. (2007) Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 9988–9993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Verdaguer N., Fita I., Reithmayer M., Moser R., and Blaas D. (2004) X-ray structure of a minor group human rhinovirus bound to a fragment of its cellular receptor protein. Nat. Struct. Mol. Biol 11, 429–434 [DOI] [PubMed] [Google Scholar]
- 37. Jensen G. A., Andersen O. M., Bonvin A. M., Bjerrum-Bohr I., Etzerodt M., Thøgersen H. C., O'Shea C., Poulsen F. M., and Kragelund B. B. (2006) Binding site structure of one LRP-RAP complex: implications for a common ligand-receptor binding motif. J. Mol. Biol. 362, 700–716 [DOI] [PubMed] [Google Scholar]
- 38. Lee C. J., De Biasio A., and Beglova N. (2010) Mode of interaction between β2GPI and lipoprotein receptors suggests mutually exclusive binding of β2GPI to the receptors and anionic phospholipids. Structure 18, 366–376 [DOI] [PubMed] [Google Scholar]
- 39. Guttman M., Prieto J. H., Handel T. M., Domaille P. J., and Komives E. A. (2010) Structure of the minimal interface between ApoE and LRP. J. Mol. Biol. 398, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dagil R., O'Shea C., Nykjær A., Bonvin A. M., and Kragelund B. B. (2013) Gentamicin binds to the megalin receptor as a competitive inhibitor using the common ligand binding motif of complement type repeats: insight from the NMR structure of the 10th complement type repeat domain alone and in complex with gentamicin. J. Biol. Chem. 288, 4424–4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andersen O. M., Schwarz F. P., Eisenstein E., Jacobsen C., Moestrup S. K., Etzerodt M., and Thøgersen H. C. (2001) Dominant thermodynamic role of the third independent receptor binding site in the receptor-associated protein RAP. Biochemistry 40, 15408–15417 [DOI] [PubMed] [Google Scholar]
- 42. Bloem E., Ebberink E. H., van den Biggelaar M., van der Zwaan C., Mertens K., and Meijer A. B. (2015) A novel chemical footprinting approach identifies critical lysine residues involved in the binding of receptor-associated protein to cluster II of LDL receptor-related protein. Biochem. J. 468, 65–72 [DOI] [PubMed] [Google Scholar]
- 43. Dolmer K., Campos A., and Gettins P. G. (2013) Quantitative dissection of the binding contributions of ligand lysines of the receptor-associated protein (RAP) to the low density lipoprotein receptor-related protein (LRP1). J. Biol. Chem. 288, 24081–24090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van den Biggelaar M., Sellink E., Klein Gebbinck J. W., Mertens K., and Meijer A. B. (2011) A single lysine of the two-lysine recognition motif of the D3 domain of receptor-associated protein is sufficient to mediate endocytosis by low-density lipoprotein receptor-related protein. Int. J. Biochem. Cell Biol. 43, 431–440 [DOI] [PubMed] [Google Scholar]
- 45. Nielsen K. L., Holtet T. L., Etzerodt M., Moestrup S. K., Gliemann J., Sottrup-Jensen L., and Thogersen H. C. (1996) Identification of residues in α-macroglobulins important for binding to the α2-macroglobulin receptor low density lipoprotein receptor-related protein. J. Biol. Chem. 271, 12909–12912 [DOI] [PubMed] [Google Scholar]
- 46. Arandjelovic S., Hall B. D., and Gonias S. L. (2005) Mutation of lysine 1370 in full-length human α-macroglobulin blocks binding to the low density lipoprotein receptor-related protein-1. Arch. Biochem. Biophys. 438, 29–35 [DOI] [PubMed] [Google Scholar]
- 47. Moestrup S. K., and Gliemann J. (1991) Analysis of ligand recognition by the purified α2-macroglobulin receptor (low density lipoprotein receptor-related protein). Evidence that high affinity of α2-macroglobulin-proteinase complex is achieved by binding to adjacent receptors. J. Biol. Chem. 266, 14011–14017 [PubMed] [Google Scholar]
- 48. Mikhailenko I., Battey F. D., Migliorini M., Ruiz J. F., Argraves K., Moayeri M., and Strickland D. K. (2001) Recognition of α2-macroglobulin by the low density lipoprotein receptor-related protein requires the cooperation of two ligand binding cluster regions. J. Biol. Chem. 276, 39484–39491 [DOI] [PubMed] [Google Scholar]
- 49. Xiao T., DeCamp D. L., and Sprang S. R. (2000) Structure of a rat α(1)-macroglobulin receptor-binding domain dimer. Protein Sci. 9, 1889–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van den Biggelaar M., Madsen J. J., Faber J. H., Zuurveld M. G., van der Zwaan C., Olsen O. H., Stennicke H. R., Mertens K., and Meijer A. B. (2015) Factor VIII interacts with the endocytic receptor low-density lipoprotein receptor-related protein 1 via an extended surface comprising “Hot-Spot” lysine residues. J. Biol. Chem. 290, 16463–16476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Krauss G., Riesner D., and Maass G. (1976) Mechanism of discrimination between cognate and non-cognate tRNAs by phenylalanyl-tRNA synthetase from yeast. Eur. J. Biochem. 68, 81–93 [DOI] [PubMed] [Google Scholar]
- 52. Riesner D., Pingoud A., Boehme D., Peters F., and Maass G. (1976) Distinct steps in the specific binding of tRNA to aminoacyl-tRNA synthetase. Temperature-jump studies on the serine-specific system from yeast and the tyrosine-specific system from Escherichia coli. Eur. J. Biochem. 68, 71–80 [DOI] [PubMed] [Google Scholar]