

Abstract
Amyloid fibril deposits found in Alzheimer disease patients are composed of amyloid-β (Aβ) protein forming a number of hydrophobic interfaces that are believed to be mostly rigid. We have investigated the μs-ms time-scale dynamics of the intra-strand hydrophobic core and interfaces of the fibrils composed of Aβ1–40 protein. Using solid-state 2H NMR line shape experiments performed on selectively deuterated methyl groups, we probed the 3-fold symmetric and 2-fold symmetric polymorphs of native Aβ as well as the protofibrils of D23N Iowa mutant, associated with an early onset of Alzheimer disease. The dynamics of the hydrophobic regions probed at Leu-17, Leu-34, Val-36, and Met-35 side chains were found to be very pronounced at all sites and in all polymorphs of Aβ, with methyl axis motions persisting down to 230–200 K for most of the sites. The dominant mode of motions is the rotameric side chain jumps, with the Met-35 displaying the most complex multi-modal behavior. There are distinct differences in the dynamics among the three protein variants, with the Val-36 site displaying the most variability. Solvation of the fibrils does not affect methyl group motions within the hydrophobic core of individual cross-β subunits but has a clear effect on the motions at the hydrophobic interface between the cross-β subunits, which is defined by Met-35 contacts. In particular, hydration activates transitions between additional rotameric states that are not sampled in the dry protein. Thus, these results support the existence of water-accessible cavity recently predicted by molecular dynamics simulations and suggested by cryo-EM studies.
Keywords: Alzheimer disease, amyloid, amyloid-β (AB), protein dynamic, solid state NMR, solvation
Introduction
One of the characteristics of Alzheimer disease is the presence of neurotoxic deposits in brain tissue, which are largely made up of a short, 39–42-amino acid-long peptide referred to as amyloid β (Aβ).2 Αβ peptide plays a major role in Alzheimer disease pathogenesis (1). The deposits (plaques) are an active subject of investigation in many research centers in an attempt to design preventative and therapeutic approaches to the disease (2–14). The molecular structures and morphologies of the fibrils comprising the plaques have been studied at various levels with a variety of imaging and spectroscopic techniques including neutron diffraction, atomic force microscopy, cryo-electron microscopy, magnetic resonance spectroscopy, and fluorescence (8, 11–13, 15–22).
A typical characteristic of the fibrils is the presence of ribbon-like β-sheets with the strands close to perpendicular to the axis of the fibril, whereas the hydrogen bonds between different strands run parallel to the axis. In vitro studies of the full-length fibrils (that comprise 40–42-residue peptides) demonstrate multiple morphological possibilities for fibril structures that are very much dependent on the details of the growth conditions (23–25). However, the characteristics of molecular structures are largely preserved (11). For example, in the wild-type sequence, only structures that contain parallel β-sheets are observed (Fig. 1) (9, 24–28). By contrast, antiparallel β-sheet structures can found in the D23N (Iowa) mutant (6, 13, 29, 30), which is associated with an early onset of the neurodegeneration process (6). The polymorphs of D23N with the antiparallel β-sheet structures are “protofibrils”; i.e. they are relatively short and curved fibril-like intermediates. They are metastable and eventually convert to mature D23N fibrils, which have parallel β-sheet structures that are very similar to wild-type Aβ fibrils (13, 22, 29).
FIGURE 1.
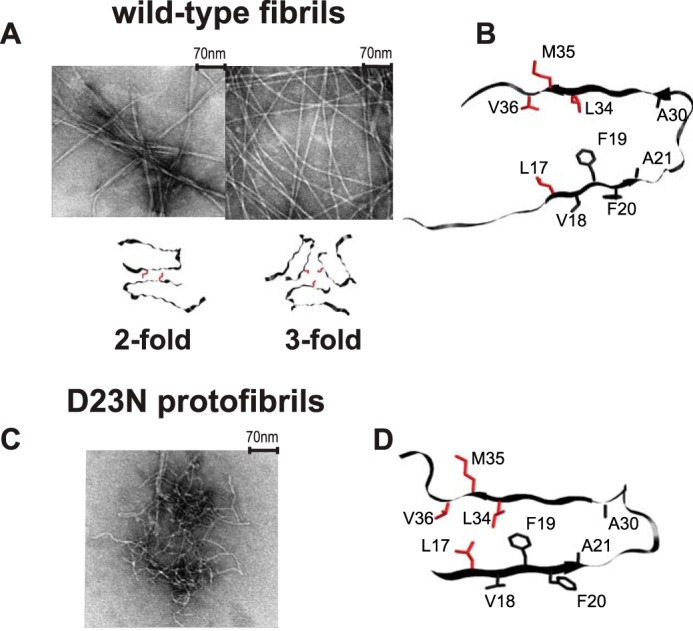
A, examples of transmission electron microscopy images of the wild-type fibrils composed of Aβ1–40 and the corresponding quaternary structures in which the position of Met-35 side chains are color-coded in red. The following polymorphs are shown: striated-ribbon/2-fold symmetric polymorph of native Aβ1–40 (PDB ID 2LMN) and twisted/3-fold symmetric polymorph of native Aβ1–40 (PDB ID 2LMP). B, a ribbon diagram of the monomer corresponding to the wild-type Aβ1–40 in the 2-fold morphology with the side chains of Leu-17, Leu-34, Val-36, and Met-35 investigated in this work shown in red. The 3-fold morphology has a very similar monomeric unit. C, an example of transmission electron microscopy image of the D23N mutant Aβ1–40 protofibrils with antiparallel β-sheet structure. D, a ribbon diagram of the D23N protofibrils with the side chains of Leu-17, Leu-34, Val-36, and Met-35 investigated in this work shown in red (PDB ID 2LNQ).
Although both parallel and antiparallel structures can display cytotoxicity (13), the level of toxicity varies greatly depending on the morphological form (11, 31). These polymorphs are often viewed as twisted or parallel shapes using transmission electron microscopy (11, 25, 32, 33). The antiparallel β-sheet structure represents perhaps the most extreme variability in morphology among polymorphs identified up to date. Effective drug development has not been achieved so far, and one of the possible reasons is the complexity of the polymorphs and interconversions between them as well as the presence of conformational diversity within each polymorph.
The intrinsic conformational diversity of the Aβ monomer is well known. In solution, Aβ lacks regular α-helical or β-stranded structure and is recognized as an intrinsically disordered protein (33–36). However, studies of dynamics in the fibril forms at site-specific level are relatively sparse (37). The most common view is that the hydrophobic core of the fibrils is rigid, complemented by other relative flexible regions (33, 38–40). The flexibility of non-core regions has been shown by multiple techniques such as nuclear magnetic resonance (9, 24, 25, 41–43), electron paramagnetic resonance, (28) hydrogen-deuterium exchange probed by mass-spectrometry (44–46), and x-ray crystallography (47). Based on x-ray, cryo-EM studies (16, 48), and molecular dynamics simulations (49, 50), it has also been suggested that some of the polymorphs can have a hollow hydrophobic core cavity that can be relatively water-accessible. A recent study based on solid-state NMR and modeling has also explored the role of cavities in the interaction between Aβ and a non-steroidal anti-inflammatory drug (51, 52). In this respect the relative orientation and hydrophobic contact between methionine 35 residues belonging to different cross-β subunits are particularly important in defining the structure of the cavities (Fig. 1).
Two main lines of inquiry are pursued in this work. First, we challenge the view that the hydrophobic core of Aβ is rigid. In principle, rigidity/flexibility can be either thermodynamic or kinetic in nature or both. Our experiments allow for distinguishing among these scenarios and emphasize specifically the role of kinetic factors in the μs-ms motions of the methyl-bearing side chains. Furthermore, we investigate the effect of solvation on the dynamics of the hydrophobic core and hydrophobic interfaces of Aβ. One piece of evidence for the flexibility of the core region was obtained by Adler et al. (53) on the basis of solid-state 13C,1H order parameters of several non-polar side chains in fibrils composed of Aβ1–40. They have probed the side chains of Val-18, Phe-20, Ala-21, and Gly-33 in the native form and with several systematic mutations involving the Phe-19–Leu-34 hydrophobic contact (9, 23, 25) and found relatively large amplitude motions for the solvent-exposed side chains of Val-18 and Phe-20. In addition, Fawzi et al. (54) utilized solution NMR dark-exchange saturation transfer measurements of methyl groups to characterize the ensemble of states that can be sampled on the surfaces of protofibrils, which likely has implications for the flexibility of the Aβ1–40 peptide in the mature fibril forms.
We evaluate the flexibility of the core by site-specific studies of key methyl groups in mature fibrils composed of Aβ1–40 peptide. We investigate the core within each cross-β subunit by using Leu-17, Leu-34, and Val-36 as probes as well as the hydrophobic interface between different cross-β subunits using the Met-35 side chains as probes. We probe the effect of morphology on the dynamics of these sites by looking at fibrils in two distinctly different polymorphs for which the structural models are available (11, 13). The examples of transmission electron microscopy images and basic structural representations for these two morphologies, referred to as the “twisted/3-fold symmetric” and “striated ribbon/2-fold symmetric,” are displayed in Fig. 1. The twisted polymorph is composed of trimers, whereas the striated ribbon morphology has a 2-fold symmetry. We note that other investigated polymorphs of Aβ have the same arrangement of side chains, although other aspects of the structures are different (25). Furthermore, we investigate the effect of differences in the secondary structures by looking at the D23N mutant with the antiparallel β-sheet structure. The D23N protofibrils are the only toxic intermediate for which a detailed molecular structural model exists. This form is not thermodynamically stable, and it has been shown to spontaneously convert to the parallel β-sheet structure upon quiescent incubation of the mixture for 30 days at room temperature (29). Structural models indicate that the hydrophobic core of the D23N mutant is somewhat smaller compared with the wild-type in both the protofibrils and the mature forms (13, 22). In this respect probing the dynamics at the Val-36 position, which is not part of the hydrophobic core in the D23N protofibrils, is particularly interesting, as it can be correlated with the structural differences.
We measure the motions on the time scale of 103-108 s−1 over a very wide temperature range between 310 and 150 K. Static solid-state deuteron NMR line shape analysis is our main experimental tool, which is sensitive to motions on the order of the quadrupolar coupling constant (55). Deuterium is a particularly sensitive probe of dynamics and renders a relative ease of data interpretation, as the spectra are dominated by one single-particle mechanism; that is, the interaction of the nuclear electric quadrupole moment with the electric field gradient at the site of the nucleus (55). We observe hydrophobic core flexibility, which is driven by the kinetic factors for both the wild-type fibrils and the D23N protofibrils at all sites and morphologies. Furthermore, we characterize the differences in the dynamics between these Aβ polymorphs as well the dependence of the dynamics on solvation. Although the methyl group dynamics in the core of the individual cross-β subunits are not dependent on solvation, the flexibility of the hydrophobic interface between the cross-β subunits, as probed by Met-35 contacts, clearly depends on hydration.
Results
Line Shape Data Indicates Hydrophobic Core Mobility Down to 200–190 K
In our work we use a so-called “single-labeling” approach, in which deuterium labels are introduced in only one selected residue. The details of the labeling patterns are described under “Experimental Procedures.” For every morphology, four samples have been prepared with methyl groups deuterated at either Leu-17, Leu-34, Met-35, or Val-36 side chains. Deuterium NMR line shape analysis on each sample thus reflects the dynamics at each individual side chain. 2H line shapes are governed by the quadrupolar interaction, which is modulated by internal fluctuations. In the absence of motions, a characteristic Pake pattern is obtained. Fast methyl rotation narrows the widths of the pattern by a factor of ∼⅓ (Fig. 2C). Motions on the time scale of the quadrupolar coupling constant affect both the width and the shape of the spectrum.
FIGURE 2.

Experimental 2H NMR static line shapes for Leu-17, Leu-34, and Val-36 (A) and Met-35 (B) in the native Aβ1–40 2-fold (blue) and 3-fold (red) symmetric polymorphs and in the D23N mutant protofibrils (black). C, a simulated Pake-pattern for a methyl group undergoing fast methyl rotations in the absence of μs-ms motions. The experimental spectra were collected twice to ensure reproducibility. Additionally, the following replicate samples were prepared and analyzed to ensure reproducibility: Leu-34-labeled in the 2- and 3-fold polymorphs, Leu-17-labeled in the 3-fold polymorph, and Met-35 in the 3-fold polymorph.
The first significant result is that motional narrowing is present to a considerable extent at temperatures as low as 230–190 K for all sites and all three fibril forms investigated here (Fig. 2 and supplemental Fig. S1); that is, the wild-type Aβ with either the 2-fold or the 3-fold morphologies and the D23N mutant with the antiparallel β-sheet structure. This result indicates hydrophobic core mobility persisting to low temperatures and contrasts with data obtained for a globular protein, villin headpiece, for which μs-ms motions were quenched as the temperature was lowered to 250–240 K (56). We note that the narrow peak at zero frequency originates from liquid HOD molecules present in the sample at higher temperatures.
Dependence of Line Shapes on Morphology and Molecular Structure
For the core residues (Leu-17, Leu-34, and Val-36) there are modest but clear differences in the line shapes and their temperature dependences between the 2-fold and 3-fold morphologies of the wild-type Aβ (Fig. 2), indicating that the dynamics are sensitive to the morphological state. The Val-36 site appears most sensitive to changes in morphology. At room temperature, the 3-fold morphology line shapes are less motionally narrowed compared with the 2-fold morphology for all of the three core sites. Although this trend persists for all temperatures for the Val-36 side chain, it is switched for the lowest temperatures for Leu-17 and Leu-34 side chains, reflecting variations in the temperature dependence of the dynamics. A comparison between the wild-type and the D23N mutant reveals that the dynamics of the mutant are indistinguishable from the 2-fold wild-type variant for the Leu-34 site. For the Leu-17 side chain, the line shape data for the mutant does not overlay perfectly with the 2-fold wild-type data, but it is clear that it is much closer to the wild-type 2-fold morphology in comparison to that of the 3-fold morphology. The mutant protein is much more mobile at the Val-36 site compared with the wild-type protein.
For the Met-35 side chain defining the hydrophobic interface between different cross-β subunits (Fig. 1), the line shapes are indistinguishable for the 2-fold and 3-fold morphology for both the hydrated and dry states (Fig. 2 and supplemental Fig. S1), indicating that the motions on the time scales of the quadrupolar coupling constant (53 kHz) are the same in the two forms. The differences in the line shapes between the wild-type and D23N mutant proteins became more pronounced at temperatures below ∼260 K.
Dependence of the Line Shapes on Solvation
The line shapes for the core residues of Leu-17, Leu-34, and Val-36 are identical in the hydrated state (200% water content by weight) and dried lyophilized state (Fig. 3). This is in striking contrast with results for hydrophobic core residues of globular proteins for which the dynamics on a μs-ms time scale are strongly dependent on the hydration state (56–59). Unlike the core residues, Met-35 line shapes display strong hydration dependence for both polymorphs (Fig. 3 and supplemental Fig. S1), with the hydrated state showing much more motionally narrowed line shapes at high (physiological) temperatures and wider line shapes at lower temperatures. Thus, the presence of water has a profound effect on the dynamics of the hydrophobic cavity defined by the Met-35 contacts.
FIGURE 3.
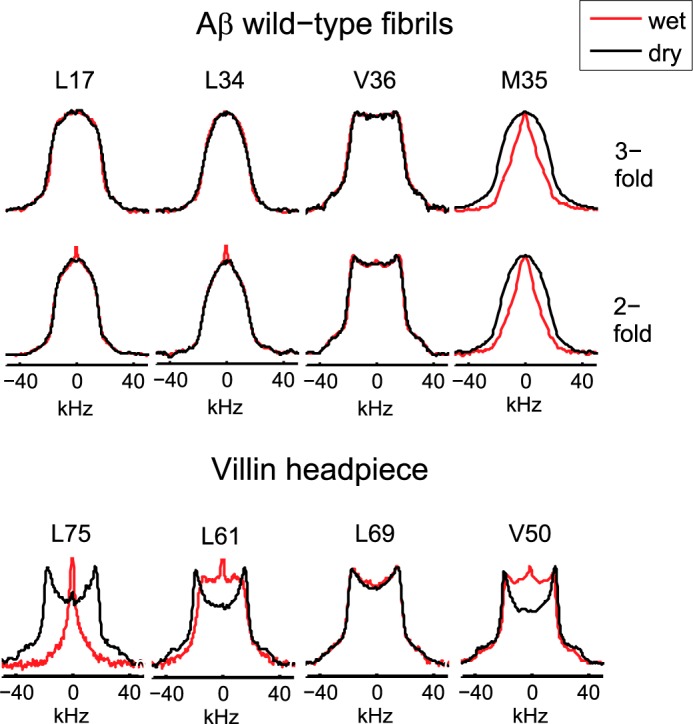
Experimental line shapes for the dry (black) and hydrated (red) states. Upper panel, Aβ1–40 wild-type fibrils at 294 K for Leu-17, Leu-34, and Val-36 and at 310 K for Met-35. Lower panel, globular villin headpiece protein hydrophobic core residues at 298 K (56).
Computational Modeling of the Data Indicates Rotameric Jumps with Low Activation Barriers
The Motional Models
Leucine and Valine
The leucine side chain has two torsional angles (χ1 and χ2) that allow for existence of multiple rotameric states (Fig. 4). Therefore, rotameric jumps are the main mechanism of large-amplitude motions in these side chains. The motional model of rotameric interconversions involving both χ1 and χ2 angles for leucine side chains has been used for a variety of systems from free amino acids to proteins to peptides adsorbed on surfaces (60–62). Thus, the use of such a model allows for comparison between different types of systems. As described in detail in the supplemental information, all rotamers of leucine side chains can be reduced to four magnetically non-equivalent sites. We thus take interconversions between four rotameric states as the basis of our model (Fig. 4). Several works have used a two-state approximation (60, 61); however, we found that the four-state model provides a significant improvement over the two-state scenario. We assumed that the four states consist of one major conformer and three non-equivalent minor conformers, such that the populations can be modeled in w:1:1:1 ratio, where w > 1 stands for the relative weight of a single major conformer. Introduction of non-equivalent populations of the minor conformers did not lead to significant improvement of simulated data. Besides the population parameter w, the rate of jumps between the rotameric states, krot, is another parameter of the model. The side chain of valine allows for rotations around one dihedral angle only (χ1), leading to the existence of three rotameric states modeled with the w:1:1 ratio. The full description of the model and its implementation can be found in supplemental Figs. S2 and S3). We note that the additional librations inside the rotameric well, which manifest themselves as an effective asymmetry, were not visible across the entire temperature range and, therefore, were not included in the model for the hydrophobic core methyl groups in Aβ. In contrast, in the hydrophobic core of the globular villin headpiece protein the asymmetry was evident at temperatures below 260–240 K (56, 62).
FIGURE 4.

A, motional models of leucine. Rotameric jumps around χ1 and χ2 angle are represented by four magnetically non-equivalent conformers (out of nine possible configurations) pointing toward the corners of a tetrahedron. White spheres correspond to the Cδ position, blue spheres correspond to Cγ, and black spheres are for Cβ and Cα, the positions of which are not changed during the rotameric inter-conversions. B, valine. Rotameric jumps around the χ1 angle are represented by three conformers. C, methionine. Arc motion mode (motions within rotameric potential) is used for all states and all temperatures. Rotameric motions mode involving only the χ3 angle is used for the dry state at all temperatures and for the hydrated state at T < 250 K. For the hydrated state at T > 250 K, rotameric motions (involving in reality all three χ1, χ2, and χ3 angles) are approximated by four artificial symmetrical conformers. Positions of the 2H labels at the methyl groups of the side chains are shown in red.
Methionine
Methionine has the longest side chain with three dihedral angles, thus displaying the most complex dynamics. We have first attempted to fit the entire temperature range postulating that the only motional mode is due to rotations around the χ3 dihedral angle leading to three conformers in analogy to the valine model (Fig. 4). An additional improvement in the quality of the fits was obtained by introducing the motions inside the rotameric well. The need for such a mode is evident from low temperature spectra, which display an additional asymmetry. Based on our previous work on globular proteins (62), this type of motion is modeled well by diffusion of the methyl axes along an arc (Fig. 4).
This approach was sufficient for the dry methionine-labeled sample but has failed to produce an adequate fit to the data for the hydrated samples at temperatures above 250 K (supplemental Fig. S4 A). We have then attempted to fit the data using two additional conformers with respect to the χ2 angle listed as statistically most populated conformers in the DYNEOMICS database (63). This approach has not provided significant improvement, thus indicating that at high temperatures almost all rotameric conformers are activated. Rather than specifying all possible geometries, populations, and rates around all of the three dihedral angles, which would be a clear overfitting of the data, we have chosen to approximate all of these multiple possibilities by the symmetrical scheme analogous to the leucine case: four directions of the S-Cϵ bond stemming from the sulfur atoms toward the four corners of the tetrahedron (Fig. 4). This approach has adequately represented the high temperature regime (approximately higher than 250 K) but produced a significantly worse quality of the fit for low temperatures (supplemental Fig. S4A) compared with the simpler model with rotameric transition around the χ3 angle only. If this model is nonetheless forced over the entire temperature regime, the resulting temperature dependence of populations (ln w) starts to deviate from linearity around 240–250 K (supplemental Fig. S4B). Thus, the data for the hydrated samples pointed toward a change in the motional regime around 250 K; below this temperature only rotamers corresponding to rotations around the χ3 angle are significantly populated, whereas above this temperature additional rotameric transitions are activated.
Rotameric Jump Rate Constants and Energy Differences between the Rotameric States
The main parameters of the model are the weights of the major rotamer w and the rate of rotameric interconversions krot. The physical meaning of the essential parameters discussed in this section is summarized in Table 1, and examples comparing experimental and fitted spectra are shown in Fig. 5. The details of the modeling procedure, additional examples of the quality of the fits, and the resulting fitting parameters can be found in the supplemental material, Figs. S5 and S6, and Table S1).
TABLE 1.
Main parameters and their physical meaning
| Parameter | Meaning |
|---|---|
| krot | Rate constant for jumps between different rotameric states of the side chains (kinetic). |
| w | Population of the dominant rotameric state relative to all other states (thermodynamic). |
| ΔE | Energy difference between the dominant rotameric state and all other states, which are assumed to be degenerate (thermodynamic). |
| Ea | Activation energy for the transitions between the rotameric states (kinetic). |
| O2 | Methyl axis order parameter, signifying the extent of fluctuations from the dominant methyl axes orientation (thermodynamic). |
FIGURE 5.
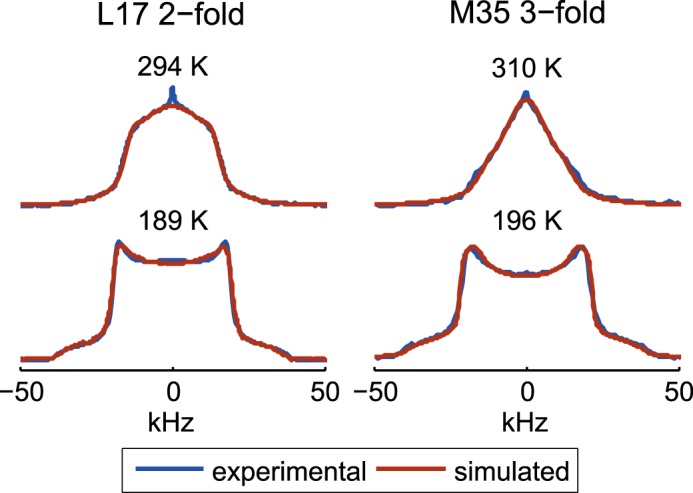
Examples of overlays of the experimental (blue) and fitted spectra (brown line) shown for the Leu-17 site in the 2-fold morphology and the Met-35 site in the 3-fold morphology in the hydrated state.
The temperature dependence of the fitted values of w (supplemental Fig. S5) indicates that, in general, they should not be constrained to follow the Boltzmann law because a non-zero y-intercept has been observed for several cases (supplemental Table S1, denoted by b). The temperature dependence of the populations yields the energy differences between the major and minor rotameric states ΔE (Fig. 6), and the temperature dependence of the rate constants (supplemental Fig. S6) is used to calculate the activation energy of the rotameric jumps Ea (Fig. 7) according to the Arrhenius law. Note that for the methionine-labeled samples two different fits were performed for the ranges below and above 250 K, corresponding to the two different regimes, as elaborated in the above section.
FIGURE 6.
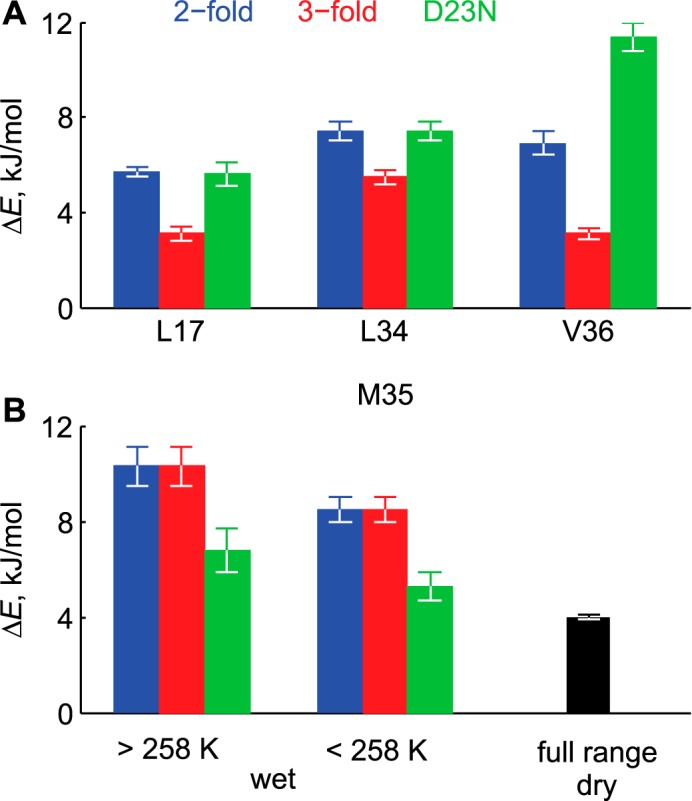
Energy differences ΔE between the major and minor rotamers. A, hydrophobic core residues. The values for the dry and hydrated samples are identical. B, Met-35 site. The values for the 2-fold and 3-fold polymorphs are identical in both the dry and hydrated states.
FIGURE 7.
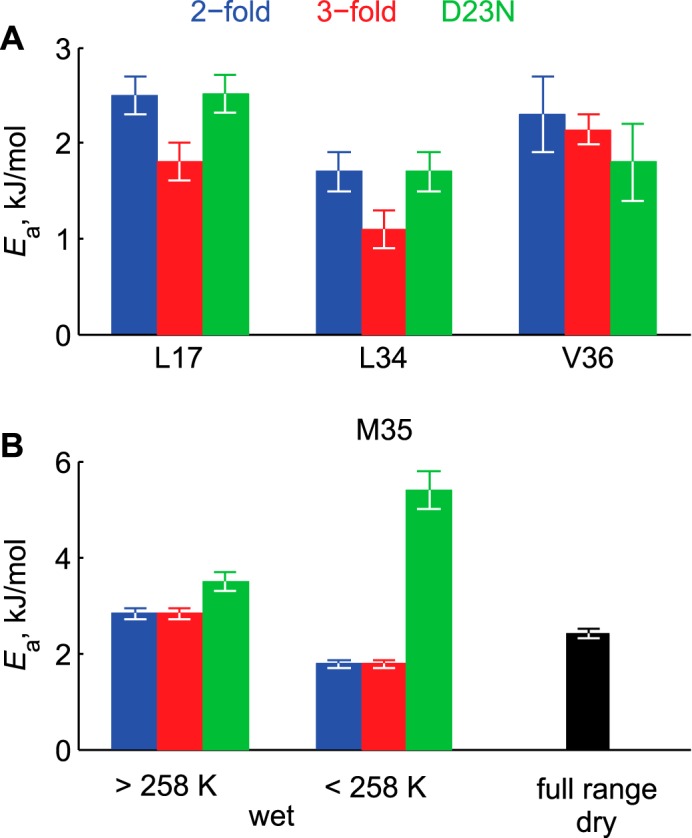
Activation energies Ea of rotameric jumps. A, hydrophobic core residues. The values for the dry and hydrated samples are identical. B, Met-35 site. The values for the 2-fold and 3-fold polymorphs are identical in both the dry and hydrated states.
For the hydrophobic core residues of Leu-17, Leu-34, and Val-36, the ΔE values (Fig. 6) are smaller for the 3-fold morphology of the wild-type Aβ compared with the 2-fold by a factor of 1.3–2.3. The largest effect is seen for the Val-36 site. The values of ΔE for the D23N are indistinguishable from the 2-fold wild-type morphology for Leu-17 and Leu-34 but is larger for the Val-36 by a factor of 1.7 and is smaller for Met-35 by a factor of 1.5. In general, the values of ΔE range between 3 to 10 kJ/mol and are comparable to what we have previously observed for the globular protein, villin headpiece. (56). Comparison between the hydrated Met-35-labeled protein and the dry one indicates that the value of ΔE changes from 8.5 kJ/mol (hydrated) to 4 kJ/mol (dry), indicating that hydration increases the separation of energy levels of the rotameric states even at the low temperature regime when the bulk water is frozen.
The values of the activation energies Ea for the hydrophobic core region are somewhat smaller (by a factor up to 1.5) for the 3-fold morphology of the wild-type in comparison to the 2-fold wild-type Aβ (Fig. 7). The fits for the Leu-17 side chain in D23N indicate that its value of Ea is indistinguishable from the values obtained for the 2-fold morphology of the wild-type samples. Thus, the observed slight differences in the line shapes for the Leu-17 samples in the native 2-fold and antiparallel D23N states are not due the differences in the activation energies but, rather, are due to the differences in the Arrhenius prefactors. This suggests variations in the width of the potentials rather than their heights. The difference in the prefactor is also evident between the 2-fold and 3-fold variants for the Val-36 site (supplemental Table S1).
Comparison of the hydrated and dry proteins' activation energies at the Met-35 site (Fig. 7, supplemental Table S1) in the low temperature region indicates both a smaller activation energy and a smaller prefactor in the hydrated sample, again emphasizing that the effect of solvation persists beyond the temperatures at which the bulk water freezes. The D23N mutant activation energy is somewhat larger for the high temperature regime (3.5 kJ in the mutant compared with 2.8 kJ/mol in the wild-type), and the prefactor is a factor of 10 larger, indicating large differences in the widths of the potential. The activation energy is significantly larger for the mutant sample (5.4 kJ/mol) in the low temperature regime. Thus, at low temperatures the dynamics appear to be no less sensitive to the details of the local environment compared with high temperatures. An important overall conclusion is that for all of the samples, activation energies of the rotameric jump modes are very low, under 3.5 kJ/mol, with the exception of Met-35 residue in D23N sample in the low temperature regime.
Order Parameters for Methyl Axes Fluctuations
Whereas computational modeling of the data within the framework of a specific motional model is useful in providing mechanistic insights into the dynamic processes and their driving forces, analysis of the data within the framework of order parameters facilitates comparison between different proteins and different techniques. For the case of rotameric jumps, order parameters for the methyl axes fluctuations reflect the distribution over multiple rotameric states and, thus, provide a thermodynamic picture of the mobility. Details regarding calculation of the order parameters are presented in the supplemental material.
The values of the squared order parameters O2 as well as their temperature dependence are shown in Fig. 8. For the highest temperatures of 310 K and 295 K, the values of O2 are in the 0.44–0.69 range for all leucine and valine side chains with the exception of the D23N Val-36 sample, for which the order parameter is much lower with the value of 0.24. All of the order parameters for methyl axes motion are comparable to or smaller than the value of 0.72 measured for the solvent-exposed Val-18 side chain by Huster and co-workers (53) using dipolar 13C,1H experiments. Although the location of the probe and the choice of the experiment can affect the resulting value of the order parameters, in this case it is clear that the core residues are no less flexible than this solvent-exposed side chain.
FIGURE 8.

A, the values of the squared order parameters of methyl axes O2 as a function of temperature T. The solid lines represent linear fits. B, the values of dO2/dT.
D23N Val-36 site displays the steepest temperature dependence with the derivative dO2/dT of 5.7 × 10−3 K−1. All other temperature dependence coefficients for valine and leucine side chains are in the 1.6 × 10−3 K−1 to 3.6 × 10−3 K−1 range, which is similar to those observed for villin headpiece subdomain and other proteins based on solution NMR relaxation data (64–66). We also note that the temperature dependence of the order parameters for valine and leucine side chains is 1.3–1.9× smaller in the 3-fold morphology compared with that of the 2-fold morphology.
As expected based on the long and flexible methionine side chains, their order parameters are relatively small at 310 K, ranging from 0.18 to 0.33 for the hydrated samples. Interestingly, despite the onset of the additional rotameric modes above around 250 K, the temperature dependence of the order parameters remains roughly linear, with the temperature coefficients of 4.2 ·10−3 K−1 for the hydrated wild-type protein, and 2.6 × 10−3 K−1 for the mutant protein. The wild-type dry protein has a significantly lower temperature coefficient of 2.5 × 10−3 K−1 compared with the hydrated protein, again underlying the role of solvent. These temperature coefficients are within the ranges reported for methionine in other systems (67). Around the transition region between the two regimes (238 and 258 K points), larger deviations from the linear trend were observed, indicating the imperfection of fitting the line shapes with two different models.
Discussion
Low Activation Energies Are the Driving Force for the Persistence of the Dynamics at Low Temperatures
Our 2H NMR studies have shown that the hydrophobic core and interfaces of amyloid fibrils are highly dynamic, with μs-ms motions persisting to temperatures of 230–190 K. This result contrasts with our previous finding for a globular protein (56) for which the dynamics on this time scale were already frozen at 240 K for all hydrophobic core leucine and valine residues. Modeling and comparison with the villin headpiece data indicates that low activation energies for rotameric jumps are the driving force for the persistence of the dynamics at low temperatures. The activation energies range from 1.1 to 3.5 kJ/mol for almost all of the samples in Aβ, whereas in villin headpiece some of the methyl-bearing side chains exhibited activation energy barriers as high as 37 kJ/mol.
Comparison of the core dynamics of Aβ fibrils and villin headpiece at room temperature indicates markedly larger values of rotameric rate constants for Aβ; the median value is 2.3 × 104 s−1 for hydrophobic core villin headpiece and 1.4 × 105 s−1 for all wild-type Aβ sites probed here. The values of w are somewhat larger at room temperatures for Aβ (median of 11.8) compared with the villin headpiece (median of 6.5).
A comparison of the observed ranges of methyl axes order parameters for the hydrophobic core Aβ with the data for villin headpiece subdomain and other globular proteins in solution state supports the notion that amplitudes/statistical weights and their temperature dependence is relatively similar in these systems. A direct comparison between the rotameric rate for globular proteins obtained by means of solution NMR with the values obtained here for Aβ is difficult due to the fact that the solid-state experiments are sensitive to slow ms-μs time scales, which are masked by overall molecular tumbling in laboratory frame solution NMR relaxation measurements. The main conclusion is that the kinetics, rather than the statistical weights of the conformers, drive the main differences between the dynamics in the fibrils compared with globular proteins, as emphasized in Table 2.
TABLE 2.
Comparison of Aβ mobility (in the hydrated state) with globular proteins
| Thermodynamic | Kinetic |
|---|---|
| ΔE; similar | krot at room temperature; much larger for Aβ (higher mobility for Aβ) |
| Temperature dependence of O2; similar | Ea; much smaller for Aβ (higher mobility for Aβ) |
Sensitivity to Morphological Forms
The dynamics in the core region are moderately sensitive to morphological form. The twisted/3-fold morphology has a tendency to have somewhat smaller values of activation energies as well as smaller values of energy differences between the rotameric states. The differences in the line shapes are most pronounced for the Val-36 side chain. The dynamics of the D23N mutant protein with the antiparallel β-sheet structure are very similar to the 2-fold variant for both the Leu-17 and Leu-34 sites but are clearly different (more mobile) for the Val-36 site. This result is consistent with the structural data that indicated that in the D23N protofibrils, Val-36 is outside the hydrophobic core region, has limited hydrophobic contacts compared with Leu-17 and Leu-34 side chains, and lacks interstrand hydrogen bonding that is present in the wild-type polymorphs of Aβ (13, 22).
Met-35 side chains display the most complex dynamics characterized by rotameric modes, which can include either one or all three dihedral angles depending on the temperature and solvation. In addition, methionine side chains display methyl axis motions inside the rotameric wells. Met-35 line shapes are identical for the 2-fold and 3-fold morphologies, which is surprising based on the differences in the structural models. Further NMR relaxation measurements can shed light on possible differences for motions at a different range of time scales. Prade et al. (52) have noted that despite the structural differences in the contact interface within the 2-fold and 3-fold polymorphs, the polarity of the cavities do not differ. For the mutant protein at the Met-35 position, the differences in the line shapes from the wild-type morphologies are most pronounced at temperatures below 250–260 K, at which the motions around the χ3 angle start to become predominant. Thus, the monomeric nature of the protofibrils appears to have the largest effect at low temperatures for the Met-35 side chain. The single subunit with antiparallel β-sheet structure definitely affects the local environment of the Met-35 methyl axes. It is also of note that, according to the structural models (13), Met-35 is right at the edge of the β-sheet core region in the protofibrils, whereas the core extends to the C-terminal residues in the wild-type Aβ.
Lee and Wand (64) emphasized the role of tri-modal distribution for the methyl-axes order parameters. They are statistically clustered in three bands, the low band with the average of 0.3 at room temperature, the middle band with the average of 0.6, and the upper band with the average of 0.8. The dynamics of the lowest band correspond to large scale fluctuations and are most coupled to the solvent modes according to the hierarchical view of the dynamics proposed by Frauenfelder et al. (68). The dynamics of the middle and upper bands involve interconversions between substates depending on the internal protein modes. Within this definition, hydrophobic core side chains of Leu-17 and Leu-34 in all morphological forms as well as the side chain Val-36 in the 3-fold form fall into the middle band. Val-36 in the 2-fold form with the order parameter of 0.75 at 295 K is within the upper band, and the D23N variant of Val-36 is the only site that falls into the lower band for hydrophobic core residues. The methionine side chains fall into the lower band in all three morphological forms of the fibrils.
Hydrophobic Core Methyl Group Dynamics Do Not Depend on Hydration, Whereas the Met-35 Site Is Hydration-dependent
An overview of hydration and morphology dependence of Aβ1–40 is summarized in Table 3. Solvation can be essential in changing the overall plasticity/rigidity of the protein, and multiple experimental and computational efforts have been devoted to describing the role of water in defining protein dynamics on various time scales (59, 68). The role of water on the dynamics and the presence of water-accessible cavities can play an important role in rational drug design. A very important and unexpected result of this work is the effect of solvation on the motions of methyl axes. The dynamics in the hydrophobic core region (Leu-17, Leu-34, and Val-36) sites are not at all dependent on the hydration. Thus, this distinguishes the core of the fibrils from the core of globular proteins, for which methyl axes dynamics show strong dependence on water content. In contrast to the fibril core, there is a pronounced hydration dependence at the hydrophobic interface defined by Met-35 contacts spanning different strands/units. Specifically, above ∼250–260 K, solvation activates transitions between additional rotameric states, which are not significantly populated in the absence of water. It is also interesting that the effect of solvation on the dynamics persists across the entire temperature range, well beyond the point at which the bulk solvent is frozen. These results support the existence of a water-accessible cavity predicted by the recent molecular dynamics studies using the 3-fold structure (49, 50) as well as with the work by McDonald et al. (16) based on x-ray diffraction measurements, thus further emphasizing the role of this cavity in targeted drug design. Based on the fact that both the 3-fold and the 2-fold polymorphs show identical dynamics on μs-ms time scale as well as identical hydration dependence, our experiments support the notion that the Met-35 contacts defining the cavity are along the fibril axis rather than across the axis, in agreement to what has been proposed based on the molecular dynamics studies (49).
TABLE 3.
Summary of essential results on hydration and morphology dependence
Ν/Α, not applicable.
| Regions | Hydration dependence (for native polymorphs of Aβ only) | Morphology dependence (includes D23N protofibrils) |
|---|---|---|
| Hydrophobic core of Aβ (Leu-17, Leu-34, Val-36) | None | Moderate |
| Met-35 of Aβ | Strong | None |
| Hydrophobic core of villin headpiece subdomain | Strong | N/A |
Experimental Procedures
Preparation of Aβ1–40 Peptide and D23N Mutant
The native and D23N mutant proteins were prepared using solid-state peptide synthesis (performed by Thermo Fisher Scientific Co., Rockford, IL). Fluorenylmethyloxycarbonyl (Fmoc)-leucine-5,5,5-d3 and Fmoc-valine-d8 were purchased from Cambridge Isotopes Laboratories (Andover, MA), and Fmoc-metionine-d3 was from CDN Isotopes, Inc. (QC, Canada). The native sequence is DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV. The peptides were purified by reversed-phase HPLC, and their identity and purity were confirmed by mass spectrometry and reversed-phase HPLC. The resulting peptides had isotopic labels in only one chosen residue. For leucine- and methionine-labeled peptides this procedure leads to 50% labeling for each of the two prochiral methyl groups in the protein. The details of the labeling patterns are shown in Fig. 4.
Preparation of the Fibrils
Wild-type Protein
Fibrils of the wild-type Aβ1–40 for both the twisted/3-fold and striated ribbon/2-fold morphologies were produced using established seeded growth protocols under quiescent conditions (11, 24). For preparation of the bulk samples, lyophilized peptides were initially dissolved at 5 mm concentration in dimethyl sulfoxide (DMSO) and immediately transferred to the solution of 10 mm monosodium phosphate buffer at the pH of 7.4, also containing seeds in 1:10 ratio by weight. The fibrils were allowed to grow for 48–72 h at room temperature with sonication performed at 12 and 24 h during the growth of the bulk samples; aliquots corresponding to 10% of the bulk volume were taken out of solution and sonicated for 120 s using a probe sonicator, cooled for 15 min, and then added back to the bulk solution. The peptide concentration of the bulk sample was 1 mg/ml. Sonication patterns were different for the preparation of seeds; for the 2-fold symmetric polymorph, sonication was performed every 3–4 h, and the growth occurred under agitated conditions. For the 3-fold symmetric variant, sonication was performed at 24 h with the growth occurring under quiescent conditions. The bulk fibrils were pelleted by centrifuging at 300,000 g for 3 h followed by resuspending the pellets in the buffer and the second round of centrifugation. The washing procedure was done to bring the amount of any signal due to the natural abundance of 2H in the DMSO solvent, used for dissolving the peptide, under 0.1%. Fibril pellets were resuspended in deionized water, rapidly frozen with liquid nitrogen, and lyophilized. Typical yields of lyophilized fibrils were 50–70% that of the mass of the initially dissolved peptide. Lower yields are possibly due to the fact that the bulk sample preparation required larger amounts of protein compared with what is typically used for structural measurements, and thus, the growth has been performed in larger/different vessels. It is possible that longer growth times in excess of 3 days could be beneficial for larger amounts of Aβ for room temperature growth conditions. The resulting morphologies were confirmed with transmission electron microscopy imaging (Fig. 1 and supplemental Fig. S7). To sample the extent of homogeneity in the samples, 24 images per sample have been collected. The representative spread in the morphologies is shown in supplemental Fig. S7.
D23N Protein
Preparation of the D23N protofibrils with anti-parallel β-sheet structure utilized a two-step seeding/filtration cycle that takes advantage of the differences in fibril formation rate between the parallel and anti-parallel structures (13). The formation of the bulk sample in the final step took around 5–7 days at 4 °C under quiescent conditions. For this type of sample we have confirmed that a growth time under 5 days is clearly detrimental to the yield. The peptide concentration of the bulk sample was 0.5 mg/ml. Centrifugation for pelleting was performed at 425,000 × g at 4 °C.
Transmission Electron Microscopy Imaging
Samples were stored at room temperature (native Aβ) or at 4 °C (D23N) and diluted to ∼0.2 mg/ml with hydro, Millipore-filtered water. Samples were negatively stained using the drop method (69) in the following manner; 4-μl aliquots of sample, Millipore-filtered water, and 2% uranyl acetate aqueous stain were applied sequentially to a freshly glow-discharged (70) Formvar carbon-coated 300 mesh copper grid for the following incubation times: 2 min, 1min, and 1 min. Each 4-μl droplet was removed with a 5-s filter paper blot before the addition of the next droplet. After staining, the grid was air-dried for 3 min. Images were collected under low dose conditions at a magnification of 43,750× on a Philips CM-12 electron microscope (Philips Electron Optics, Eindhoven, The Netherlands) with a Teitz 1K × 1K CCD camera (TVIPS, Gauting, Germany).
Hydration and Preparation of NMR Sample
A hydrated state with water content of 200% by weight was achieved by either one of the following two ways, and we have confirmed that the results do not depend on the chosen strategy. The first strategy involved exposing lyophilized powder to water vapor in a sealed chamber at 25 °C until the water content reached saturating levels corresponding to ∼40% by weight (∼12–16 h was sufficient to achieve this) followed by pipetting the remaining water using deuterium-depleted H2O. The second strategy involved pipetting in all water as deuterium-depleted H2O and allowing the sample to equilibrate for 12–36 h. The samples were packed in 5-mm NMR tubes (cut to 21 mm length) using Teflon tape to center the sample volume in the coil of the NMR probe. The amount of material packed varied from 8 to 15 mg for leucine and valine-labeled peptides to 22–32 mg for methionine-labeled peptides.
Deuteron Solid-state NMR Spectroscopy
Experiments were performed on a 17.6 T spectrometer equipped with a static probe operating at temperatures between 140 and 380 K. Line-shape experiments were performed with a quadrupole echo pulse sequence based on an eight-step phase cycle (55), with a delay of 31 μs between 90° pulses. Additional experimental details are given in the supplemental material.
Author Contributions
L. V. conceived and coordinated the project, designed and performed the NMR experiments, and wrote the paper with input from all the authors. M. A. C. and I. B. F. prepared the wild-type fibril samples and performed some NMR measurements. D. O. performed modeling with input from L. V. D. G. performed the microscopy measurements. W. Q. prepared the D23N mutant fibril samples. G. L. H. provided NMR resources and technical assistance. All authors discussed the results and participated in the final presentation of the work.
Supplementary Material
Acknowledgment
We are grateful to Prof. Robert Tycko for providing fibril seeds, assistance with sample preparation, and for a critical review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant (NIH)1R15 GM111681-02 (to L. V.) as well as by an Institutional Development Award (IDeA) under NIH Grant P20GM103395. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental information, Figs. S1–S7, and Table S1.
- Aβ
- amyloid-β.
References
- 1. Paravastu A. K., Qahwash I., Leapman R. D., Meredith S. C., and Tycko R. (2009) Seeded growth of β-amyloid fibrils from Alzheimer's brain-derived fibrils produces a distinct fibril structure. Proc. Natl. Acad. Sci. U.S.A. 106, 7443–7448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmed A. B., and Kajava A. V. (2013) Breaking the amyloidogenicity code: methods to predict amyloids from amino acid sequence. FEBS Lett. 587, 1089–1095 [DOI] [PubMed] [Google Scholar]
- 3. Cannon M. J., Williams A. D., Wetzel R., and Myszka D. G. (2004) Kinetic analysis of β-amyloid fibril elongation. Anal. Biochem. 328, 67–75 [DOI] [PubMed] [Google Scholar]
- 4. Chimon S., Jones C., Calero D. C., and Ishii Y. (2007) A missing link in amyloid misfolding: structural insights into amyloid intermediates for Alzheimer's β-amyloid, Aβ(1–40) by solid-state NMR. Biophys. J. S, 195A–195A [Google Scholar]
- 5. Fitzpatrick A. W., Debelouchina G. T., Bayro M. J., Clare D. K., Caporini M. A., Bajaj V. S., Jaroniec C. P., Wang L., Ladizhansky V., Müller S. A., MacPhee C. E., Waudby C. A., Mott H. R., De Simone A., Knowles T. P. (2013) Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 110, 5468–5473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grabowski T. J., Cho H. S., Vonsattel J. P., Rebeck G. W., and Greenberg S. M. (2001) Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 49, 697–705 [DOI] [PubMed] [Google Scholar]
- 7. Kodali R., Williams A. D., Chemuru S., and Wetzel R. (2010) Aβ(1–40) forms five distinct amyloid structures whose β-sheet contents and fibril stabilities are correlated. J. Mol. Biol. 401, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lewandowski J. R., van der Wel P. C., Rigney M., Grigorieff N., and Griffin R. G. (2011) Structural complexity of a composite amyloid fibril. J. Am. Chem. Soc. 133, 14686–14698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., and Riek R. (2005) 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Nuallain B., Shivaprasad S., Kheterpal I., and Wetzel R. (2005) Thermodynamics of Aβ(1–40) amyloid fibril elongation. Biochemistry 44, 12709–12718 [DOI] [PubMed] [Google Scholar]
- 11. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., and Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 12. Qiang W., Kelley K., and Tycko R. (2013) Polymorph-specific kinetics and thermodynamics of β amyloid fibril growth. J. Am. Chem. Soc. 135, 6860–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qiang W., Yau W. M., Luo Y., Mattson M. P., and Tycko R. (2012) Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 109, 4443–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wetzel R. (2006) Amyloid fibrils: common threads in the natural history of proteins. Acc. Chem. Res. 39, 567–567 [DOI] [PubMed] [Google Scholar]
- 15. Kheterpal I., Williams A., Murphy C., Bledsoe B., and Wetzel R. (2001) Structural features of the Aβ amyloid fibril elucidated by limited proteolysis. Biochemistry 40, 11757–11767 [DOI] [PubMed] [Google Scholar]
- 16. McDonald M., Box H., Bian W., Kendall A., Tycko R., and Stubbs G. (2012) Fiber diffraction data indicate a hollow core for the Alzheimer's Aβ 3-fold symmetric fibril. J. Mol. Biol. 423, 454–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pinotsi D., Buell A. K., Dobson C. M., Kaminski Schierle G. S., and Kaminski C. F. (2013) A label-free, quantitative assay of amyloid fibril growth based on intrinsic fluorescence. Chembiochem 14, 846–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Popova L. A., Kodali R., Wetzel R., and Lednev I. K. (2010) Structural variations in the cross-β core of amyloid β fibrils revealed by deep UV resonance Raman spectroscopy. J. Am. Chem. Soc. 132, 6324–6328 [DOI] [PubMed] [Google Scholar]
- 19. Sachse C., Fändrich M., and Grigorieff N. (2008) Paired β-sheet structure of an Aβ(1–40) amyloid fibril revealed by electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 105, 7462–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hou L., Lee H. G., Han F., Tedesco J. M., Perry G., Smith M. A., and Zagorski M. G. (2013) Modification of amyloid-β(1–42) fibril structure by methionine-35 oxidation. J. Alzheimers Dis. 37, 9–18 [DOI] [PubMed] [Google Scholar]
- 21. Schütz A. K., Vagt T., Huber M., Ovchinnikova O. Y., Cadalbert R., Wall J., Güntert P., Böckmann A., Glockshuber R., and Meier B. H. (2015) Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew. Chem. Int. Ed. Engl. 54, 331–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sgourakis N. G., Yau W. M., and Qiang W. (2015) Modeling an in-register, parallel “Iowa” A-β fibril structure using solid state NMR data from labeled samples with Rosetta modeling. Structure 23, 216–227 [DOI] [PubMed] [Google Scholar]
- 23. Paravastu A. K., Leapman R. D., Yau W. M., and Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petkova A. T., Yau W. M., and Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry 45, 498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bertini I., Gonnelli L., Luchinat C., Mao J., and Nesi A. (2011) A new structural model of Aβ(40) fibrils. J. Am. Chem. Soc. 133, 16013–16022 [DOI] [PubMed] [Google Scholar]
- 26. Antzutkin O. N., Leapman R. D., Balbach J. J., and Tycko R. (2002) Supramolecular structural constraints on Alzheimer's β-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry 41, 15436–15450 [DOI] [PubMed] [Google Scholar]
- 27. Benzinger T. L., Gregory D. M., Burkoth T. S., Miller-Auer H., Lynn D. G., Botto R. E., and Meredith S. C. (1998) Propagating structure of Alzheimer's β-amyloid (10–35) is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. U.S.A. 95, 13407–13412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Török M., Milton S., Kayed R., Wu P., McIntire T., Glabe C. G., and Langen R. (2002) Structural and dynamic features of Alzheimer's Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J. Biol. Chem. 277, 40810–40815 [DOI] [PubMed] [Google Scholar]
- 29. Qiang W., Yau W. M., and Tycko R. (2011) Structural Evolution of Iowa mutant β-amyloid fibrils from polymorphic to homogeneous states under repeated seeded growth. J. Am. Chem. Soc. 133, 4018–4029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tycko R., Sciarretta K. L., Orgel J. P., and Meredith S. C. (2009) Evidence for novel β-sheet structures in Iowa mutant β-amyloid fibrils. Biochemistry 48, 6072–6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyer-Luehmann M., Coomaraswamy J., Bolmont T., Kaeser S., Schaefer C., Kilger E., Neuenschwander A., Abramowski D., Frey P., Jaton A. L., Vigouret J. M., Paganetti P., Walsh D. M., Mathews P. M., Ghiso J., Staufenbiel M., Walker L. C., and Jucker M. (2006) Exogenous induction of cerebral β-amyloidogenesis is governed by agent and host. Science 313, 1781–1784 [DOI] [PubMed] [Google Scholar]
- 32. Goldsbury C. S., Wirtz S., Müller S. A., Sunderji S., Wicki P., Aebi U., and Frey P. (2000) Studies on the in vitro assembly of Aβ 1–40: implications for the search for Aβ fibril formation inhibitors. J. Struct. Biol. 130, 217–231 [DOI] [PubMed] [Google Scholar]
- 33. Hubin E., van Nuland N. A., Broersen K., and Pauwels K. (2014) Transient dynamics of Aβ contribute to toxicity in Alzheimer's disease. Cell. Mol. Life Sci. 71, 3507–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hou L., Shao H., Zhang Y., Li H., Menon N. K., Neuhaus E. B., Brewer J. M., Byeon I. J., Ray D. G., Vitek M. P., Iwashita T., Makula R. A., Przybyla A. B., and Zagorski M. G. (2004) Solution NMR studies of the Aβ(1–40) and Aβ(1–42) peptides establish that the met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 126, 1992–2005 [DOI] [PubMed] [Google Scholar]
- 35. Riek R., Güntert P., Döbeli H., Wipf B., and Wüthrich K. (2001) NMR studies in aqueous solution fail to identify significant conformational differences between the monomeric forms of two Alzheimer peptides with widely different plaque-competence, Aβ(1–40)(ox) and Aβ(1–42)(ox). Eur. J. Biochem. 268, 5930–5936 [DOI] [PubMed] [Google Scholar]
- 36. Zhang S., Iwata K., Lachenmann M. J., Peng J. W., Li S., Stimson E. R., Lu Y., Felix A. M., Maggio J. E., and Lee J. P. (2000) The Alzheimer's peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 130, 130–141 [DOI] [PubMed] [Google Scholar]
- 37. Linser R., Sarkar R., Krushelnitzky A., Mainz A., and Reif B. (2014) Dynamics in the solid-state: perspectives for the investigation of amyloid aggregates, membrane proteins and soluble protein complexes. J. Biomol. NMR 59, 1–14 [DOI] [PubMed] [Google Scholar]
- 38. Jan A., Hartley D. M., and Lashuel H. A. (2010) Preparation and characterization of toxic Aβ aggregates for structural and functional studies in Alzheimer's disease research. Nat. Protoc. 5, 1186–1209 [DOI] [PubMed] [Google Scholar]
- 39. Jahn T. R., Makin O. S., Morris K. L., Marshall K. E., Tian P., Sikorski P., and Serpell L. C. (2010) The common architecture of cross-β amyloid. J. Mol. Biol. 395, 717–727 [DOI] [PubMed] [Google Scholar]
- 40. Williams A. D., Portelius E., Kheterpal I., Guo J. T., Cook K. D., Xu Y., and Wetzel R. (2004) Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 335, 833–842 [DOI] [PubMed] [Google Scholar]
- 41. Scheidt H. A., Morgado I., Rothemund S., and Huster D. (2012) Dynamics of amyloid β fibrils revealed by solid state NMR. J. Biol. Chem. 287, 2017–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Olofsson A., Sauer-Eriksson A. E., and Ohman A. (2006) The solvent protection of Alzheimer amyloid-β-(1–42) fibrils as determined by solution NMR spectroscopy. J. Biol. Chem. 281, 477–483 [DOI] [PubMed] [Google Scholar]
- 43. Whittemore N. A., Mishra R., Kheterpal I., Williams A. D., Wetzel R., and Serpersu E. H. (2005) Hydrogen-deuterium (H/D) exchange mapping of A ss(1–40) amyloid fibril secondary structure using nuclear magnetic resonance spectroscopy. Biochemistry 44, 4434–4441 [DOI] [PubMed] [Google Scholar]
- 44. Kheterpal I., Zhou S., Cook K. D., and Wetzel R. (2000) Aβ amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc. Natl. Acad. Sci. U.S.A. 97, 13597–13601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang S. S., Tobler S. A., Good T. A., and Fernandez E. J. (2003) Hydrogen exchange-mass spectrometry analysis of β-amyloid peptide structure. Biochemistry 42, 9507–9514 [DOI] [PubMed] [Google Scholar]
- 46. Kheterpal I., Chen M., Cook K. D., and Wetzel R. (2006) Structural differences in Aβ amyloid protofibrils and fibrils mapped by hydrogen exchange-mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol. 361, 785–795 [DOI] [PubMed] [Google Scholar]
- 47. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., and Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 48. Zhang R., Hu X., Khant H., Ludtke S. J., Chiu W., Schmid M. F., Frieden C., and Lee J.-M. (2009) Interprotofilament interactions between Alzheimer's Aβ(1–42) peptides in amyloid fibrils revealed by cryoEM. Proc. Natl. Acad. Sci. U.S.A. 106, 4653–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miller Y., Ma B., and Nussinov R. (2011) The unique Alzheimer's β-amyloid triangular fibril has a cavity along the fibril axis under physiological conditions. J. Am. Chem. Soc. 133, 2742–2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng J., Jang H., Ma B., Tsai C.-J., and Nussinov R. (2007) Modeling the Alzheimer Aβ(17–42) fibril architecture: tight intermolecular sheet-sheet association and intramolecular hydrated cavities. Biophys. J. 93, 3046–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Prade E., Barucker C., Sarkar R., Althoff-Ospelt G., Lopez Del Amo J. M., Hossain S., Zhong Y., Multhaup G., and Reif B. (2016) Sulindac sulfide induces the formation of large oligomeric aggregates of the Alzheimer's disease amyloid-β peptide which exhibit reduced neurotoxicity. Biochemistry 55, 1839–1849 [DOI] [PubMed] [Google Scholar]
- 52. Prade E., Bittner H. J., Sarkar R., Lopez Del Amo J. M., Althoff-Ospelt G., Multhaup G., Hildebrand P. W., and Reif B. (2015) Structural mechanism of the interaction of Alzheimer Disease Aβ fibrils with the non-steroidal anti-inflammatory drug (NSAID) sulindac sulfide. J. Biol. Chem. 290, 28737–28745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adler J., Scheidt H. A., Krüger M., Thomas L., and Huster D. (2014) Local interactions influence the fibrillation kinetics, structure and dynamics of Aβ(1–40) but leave the general fibril structure unchanged. Phys. Chem. Chem. Phys. 16, 7461–7471 [DOI] [PubMed] [Google Scholar]
- 54. Fawzi N. L., Libich D. S., Ying J., Tugarinov V., and Clore G. M. (2014) Characterizing methyl-bearing side chain contacts and dynamics mediating amyloid β protofibril interactions using C-13(methyl)-DEST and lifetime line broadening. Angew. Chem. Int. Ed. Engl. 53, 10345–10349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vold R. L., and Vold R. R. (1991) Deuterium relaxation in molecular solids. In Advances in Magnetic and Optical Resonance (Warren W., ed.) pp. 85–171, Academic Press, San Diego [Google Scholar]
- 56. Vugmeyster L., Ostrovsky D., Khadjinova A., Ellden J., Hoatson G. L., and Vold R. L. (2011) Slow motions in the hydrophobic core of chicken villin headpiece subdomain and their contributions to configurational entropy and heat capacity from solid-state deuteron NMR measurements. Biochemistry 50, 10637–10646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tamura A., Matsushita M., Naito A., Kojima S., Miura K. I., and Akasaka K. (1996) Dynamics of the three methionyl side chains of Streptomyces subtilisin inhibitor: deuterium NMR studies in solution and in the solid state. Protein Sci. 5, 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krushelnitsky A., Zinkevich T., Mukhametshina N., Tarasova N., Gogolev Y., Gnezdilov O., Fedotov V., Belton P., and Reichert D. (2009) C-13 and N-15 NMR study of the hydration response of T4 lysozyme and αB-crystallin internal dynamics. J. Phys. Chem. B 113, 10022–10034 [DOI] [PubMed] [Google Scholar]
- 59. Hong L., Cheng X., Glass D. C., and Smith J. C. (2012) Surface hydration amplifies single-well protein atom diffusion propagating in the macromolecular core. Phys. Rev. Lett. 108, 238102. [DOI] [PubMed] [Google Scholar]
- 60. Batchelder L. S., Sullivan C. E., Jelinski L. W., and Torchia D. A. (1982) Characterization of leucine side-chain reorientation in collagen fibrils by solid-state H-2 NMR. Proc. Natl. Acad. Sci. U.S.A. 79, 386–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weidner T., Breen N. F., Li K., Drobny G. P., and Castner D. G. (2010) Sum frequency generation and solid-state NMR study of the structure, orientation, and dynamics of polystyrene-adsorbed peptides. Proc. Natl. Acad. Sci. U.S.A. 107, 13288–13293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vugmeyster L., Ostrovsky D., Ford J. J., Burton S. D., Lipton A. S., Hoatson G. L., and Vold R. L. (2009) Probing the dynamics of a protein hydrophobic core by deuteron solid-state nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 131, 13651–13658 [DOI] [PubMed] [Google Scholar]
- 63. van der Kamp M. W., Schaeffer R. D., Jonsson A. L., Scouras A. D., Simms A. M., Toofanny R. D., Benson N. C., Anderson P. C., Merkley E. D., Rysavy S., Bromley D., Beck D. A., and Daggett V. (2010) Dynameomics: a comprehensive database of protein dynamics. Structure 18, 423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee A. L., and Wand A. J. (2001) Microscopic origins of entropy, heat capacity and the glass transition in proteins. Nature 411, 501–504 [DOI] [PubMed] [Google Scholar]
- 65. Sabo T. M., Bakhtiari D., Walter K. F., McFeeters R. L., Giller K., Becker S., Griesinger C., and Lee D. (2012) Thermal coefficients of the methyl groups within ubiquitin. Protein Sci. 21, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Song X. J., Flynn P. F., Sharp K. A., and Wand A. J. (2007) Temperature dependence of fast dynamics in proteins. Biophys. J. 92, L43–L45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Igumenova T. I., Frederick K. K., and Wand A. J. (2006) Characterization of the fast dynamics of protein amino acid side chains using NMR relaxation in solution. Chem. Rev. 106, 1672–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Frauenfelder H., Chen G., Berendzen J., Fenimore P. W., Jansson H., McMahon B. H., Stroe I. R., Swenson J., and Young R. D. (2009) A unified model of protein dynamics. Proc. Natl. Acad. Sci. U.S.A. 106, 5129–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Harris J. R., and Horne J. W. (1991) Negative staining. In Electron Microscopy in Biology (Harris J. R., ed.) pp. 203–228, Oxford, IRL Press at Oxford University Press, Oxford [Google Scholar]
- 70. Dubochet J., Groom M. and Mueller-Neuteboom S. (1982) Mounting of macromolecules for electron microscopy with particular reference to surface phenomena and treatment of support films by glow discharge (Barrer R., and Cosslett V. E., eds) in Advances in Optical and Electron Microscopy, pp 107–135, Academic Press, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.