

Abstract
Maintenance of triplet decoding is crucial for the expression of functional protein because deviations either into the −1 or +1 reading frames are often non-functional. We report here that expression of huntingtin (Htt) exon 1 with expanded CAG repeats, implicated in Huntington pathology, undergoes a sporadic +1 frameshift to generate from the CAG repeat a trans-frame AGC repeat-encoded product. This +1 recoding is exclusively detected in pathological Htt variants, i.e. those with expanded repeats with more than 35 consecutive CAG codons. An atypical +1 shift site, UUC C at the 5′ end of CAG repeats, which has some resemblance to the influenza A virus shift site, triggers the +1 frameshifting and is enhanced by the increased propensity of the expanded CAG repeats to form a stem-loop structure. The +1 trans-frame-encoded product can directly influence the aggregation of the parental Htt exon 1.
Keywords: aggregation, Huntington disease, translation, translation regulation, trinucleotide repeat disease, frameshifting, seeding
Introduction
Homopolymeric amino acid runs are frequent in the human genome (>2000 protein-coding genes contain at least one repeat) (1). These runs, although indispensable as structural elements for the functional dynamics of protein domains, are highly susceptible to genomic rearrangements and mutational expansions that are causative for several human pathologies, including triplet repeat expansion diseases (2), lupus antigenic afflictions, and leukemia (1). Many of these mutation-expanded repeats are often bidirectionally transcribed, e.g. their translation involves non-canonical repeat-associated non-ATG initiation (3) or frameshifting (that is, a shift of the reading frame backwards (−1 frameshifting) or forward (+1 frameshifting)), which generate diverse additional trans-frame products (4). These various products substantially alter aggregation of the zero-frame product (5, 6) and can even be a central component of toxicity for some neurodegenerative disorders (6, 7). Repeat sequence frameshifting also occurs in human DNA tumor viruses, Kaposi sarcoma-associated herpesvirus, and Epstein-Barr virus (8) or at mutation-expanded G strings in herpesvirus (9), suggesting that recoding events might also be frequent in the expression of other genes with repeat runs.
Expanded CAG repeat stretches, implicated in CAG repeat or polyglutamine (polyQ)4 diseases (2), mediate −1 translational frameshifting, although the underlying mechanisms differ depending on the context surrounding repeat runs (5, 6, 10). Our earlier work shows that pathological expansion of the CAG repeats (>35 consecutive CAG codons) in huntingtin (Htt) exon 1, implicated in Huntington disease, involves stochastic −1 frameshifting within the CAG stretch that is triggered by limitation of the charged cognate glutaminyl-tRNAGlnCUG, although uncharged tRNAGlnCUG is plentiful (5). This is conceptually similar to the frameshifting characterized earlier at tandem rare codons in Escherichia coli that is caused by cognate tRNA depletion (11, 12). In partial contrast, for the stochastic −1 frameshifting in decoding the expanded CAG run in the SCA3 gene associated with spinocerebellar ataxia type 3, an mRNA structure formed by the CAG repeats has been proposed as a major stimulatory component of the −1 frameshifting. A depletion of glutaminyl-tRNAGlnCUG is likely to be involved, as alternating bicodon CAA CAG repeats read by different tRNAs but with not too distant propensity to form secondary structure do not exhibit −1 frameshifting (6, 10).
Post-mortem analyses of Huntington disease-affected brain tissues revealed traces of both polyalanine (polyA) or polyserine (polyS) proteins in the CAG-encoded polyQ aggregates of Htt (13). Although our early work has shown that polyA proteins derive from −1 frameshifting in the expanded CAG run of Htt exon 1 (5), the origin of the polyS proteins remained undefined. +1 shift in the decoding frame of the polyCAG repeat would result in a polyACG stretch encoding serines. Here we set to define whether decoding of the Htt exon 1 also involves a +1 frameshift in the zero CAG frame, which would result in synthesis of +1 trans-frame encoded polyS segments. We report that, unlike the −1 frameshifting that occurs at any position, stochastically along the CAG repeat in Htt exon 1 with expanded pathological repeats (5), +1 frameshifting is initiated at a sequence (UCC UUC C) upstream of the first CAG codon and produces only one polyS-containing species. The +1 trans-frame polyS protein alters the aggregation of the zero-frame CAG protein and may contribute to the aggregation heterogeneity.
Results
A Slippery Site Upstream of the CAG Repeat Initiates +1 Frameshifting in Expanded Htt Exon 1
To investigate the formation of +1 frame-encoded protein(s), we fused the coding sequence of YFP in the +1 reading frame 3′ of the Htt exon 1 with a CAG repeat in the pathological (Htt51Q(+1)YFP), non-pathological polyQ repeat length (Htt22Q or Htt7Q(+1)YFP) and with Ala repeat Htt51A(+1)YFP (Fig. 1A). HttQ(+1)YFP constructs were N-terminally tagged with an HA tag to monitor the total expression with the start in the zero-frame (Fig. 1B). Transient expression of Htt51Q(+1)YFP in murine neuroblastoma cells (N2a) revealed a single band that was YFP-positive on immunoblots (Fig. 1B), i.e. corresponds to +1 frame protein(s). +1 frame products were immunoprecipitated using their YFP moiety and immunostained with anti-HA antibodies (Fig. 1C). The HA-detected product migrates as a 46-kDa protein (Fig. 1C), the same size as the YFP-immunodetected protein (Fig. 1B), suggesting that this product has both a zero frame-encoded N terminus and a +1 frame-encoded C terminus, i.e. trans-frame encoded. Note that this approach detects products with a start in the zero frame; possible repeat-associated non-ATG-initiated products remain undetected by this approach.
FIGURE 1.

The +1 frame product is the result of a +1 translational frameshift. A, nucleotide and amino acid sequence of the HttQ(+1)YFP reporter with Gln7, Gln22, or Gln51 and YFP in the +1 frame. The amino acid sequence of the putative +1 trans-frame product is depicted under the zero-frame product. B, Western blot analysis (IB) of N2a cells transiently expressing Htt51Q(+1)YFP. Mock denotes cells transfected with empty plasmid. Anti-GFP antibodies also recognize the YFP moiety. C, immunoprecipitation (IP) of Htt51Q(+1)YFP expressed in N2a/Htt103Q-CFP cells with anti-GFP antibodies and subsequent detection by immunoblotting with anti-HA antibodies. The input used for the immunoprecipitation is immunostained with anti-GPF antibodies. The asterisk indicates the +1 trans-frame product, and # designates Htt103-CFP, whose CFP moiety is also recognized by anti-GFP antibodies. The expression of the positive, in-frame control Htt51S-YFP in N2a/Htt103Q-CFP is detected by immunostaining with anti-HA antibody. D, MALDI-TOF analysis in linear mode of Htt51Q(+1)YFP expressed in N2a/Htt103-CFP for 24 h (blue spectrum) compared with the reference spectra of Htt51S-YFP (green) and YFP (red). Inset, peaks marked with an asterisk designate the periodic difference of 17 Da resulting from dissociation of a hydroxyl group from serine side chains. The table contains the theoretical amino acid sequence of the identified peaks 1–3. Note that peak 2 represents an incomplete peptide derived from 1 and peak 3 a typical peptide resulting from tryptic digestion of YFP. a.i., arbitrary intensity. E, amino acid sequence of the detected peptides in reflector mode. F, percentage of N2a cells (gray columns) or N2a/Htt103Q-CFP (white columns) with the detectable +1 trans-frame product of those expressing the zero frame protein. For comparison the percentage of cells detectable with the −1 trans-frame product (5) are shown. Values are mean ± S.E. (n = 5).
The immunoprecipitated +1 trans-frame products using their YFP moiety (Fig. 1C) were also subjected to mass spectrometry analysis (Fig. 1D). For detection reasons, we expressed Htt51Q(+1)YFP in N2a/Htt103Q-CFP cells (14), as they produce larger amounts of the +1 trans-frame-encoded protein than N2a cells (Fig. 1, B and C). Tryptic digested product was analyzed by MALDI-TOF in linear and reflector mode. Repeat proteins commonly deliver poor spectra, as they dwell poorly in the gas phase; some of them, polyQ proteins, are undetectable by this approach. MALDI-TOF analysis revealed the products SFS49NSR and S49NSR (linear mode; Fig. 1D, inset and table) in which the CAG repeat encoding polyQ is completely converted to a +1-frame-recoded polyS product. The k.AFESLK.s peptide (Fig. 1E) originated from the N-terminal region of the intact Htt exon 1 in zero frame and was identified in reflector mode (Fig. 1A). We did not detect any product of +1-frame decoding in the region 5′ upstream of the CAG stretch. Also, the two amino acids (SF) in the SFS49NSR peptide specified by zero-frame decoding of the two codons adjacent to the polyQ stretch imply that the site of frame transition is immediately 5′ of the repeat CAG codons. The periodical peak pattern in the spectrum is typical for fragments with multiple amino acids containing hydroxyl groups (Fig. 1D, inset).
To mimic a frameshifting event prior to the CAG repeat, an “in-frame” construct was made with a single nucleotide deletion 5′ adjacent to the CAG repeat to bring serine codons into the zero frame (Htt51S-YFP). The product of Htt51S-YFP co-migrated with the trans-frame product of Htt51Q(+1)YFP (Fig. 1C), and MALDI-TOF analysis yielded the same pattern of tryptic digested peaks, resulting from Htt51S-YFP being more pronounced (Fig. 1D).
We next sought to determine the frequency of frameshifting. In most studies of reading frame transitions, the ratio of trans-frame-encoded product is compared with the product of standard zero-frame decoding (mainly by electrophoresis-based approaches) to calculate the frameshift frequency. However, the full-length zero-frame product of Htt51Q is highly insoluble and readily forms SDS-insoluble aggregates (5, 15) that cannot be separated electrophoretically to monomers, thus precluding reliable quantitation. We adopted an approach used previously to determine −1 frameshifting of other polyQ-containing aggregation-prone proteins, e.g. ataxin-3 (10) and Htt (5). We calculated the fraction of YFP-positive cells (which reports on the presence of +1 trans-frame product) from all HA-Htt51Q-expressing cells (i.e. immunostained positively for the HA tag). Note that this approach does not report on the fraction of trans-frame product from the zero-frame protein. Approximately 10% of N2a cells expressing Htt51Q(+1)YFP and ≈20% of Htt103Q-CFP cells are detectably YFP-positive (Fig. 1F). Compared with our previous results, the frequency of the +1 frameshifting is much lower than that of −1 frameshifting measured with YFP fused in the −1 frame to the polyCAG reading frame, Htt51Q(−1)YFP (5) (Fig. 1F).
Is the Identity of the A Site Codon Significant for Frameshifting?
As deduced from the mass spectrometry analysis (Fig. 1, D and E), the site of frame transition is 5′ adjacent to the repeat CAG codons run, i.e. at the sequence UUC CAG CAGC50 (last zero frame, UUC, and first +1 codon, AGC). UUC C resembles to some extent the +1 slippery site UUU C in influenza A virus (16, 17). However, in the virus context, the UUU C mutation to UUC C (which would be similar to the sequence in Htt) mediates less ribosomal frameshifting than UUU C (16). To explore whether a similar effect occurs in the Htt context, the wild-type UUC CAG of HttQ was changed to UUU CAG (underlining denotes a mutated nucleotide). This mutation increased the frequency of +1 frameshifting from 10% for wild-type Htt51Q with the UUC CAG sequence to 40% with UUU CAG (Fig. 2A).
FIGURE 2.

The expanded CAG stretch stimulates +1 translational frameshifting. A, percentage of cells with +1 trans-frame species of Htt51Q(+1)YFP or Htt51A(+1)YFP in N2a (gray columns) or N2a/Htt103Q-CFP cells (white columns). Nucleotides mutated in the wild-type Htt51Q(+1)YFP sequence, UUC CAG, are underlined. Values represent the percentage of cells with the +1 trans-frame product of those expressing the zero frame protein. Data are mean ± S.E. (n = 5). Differences between variants were evaluated with chi-square test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. B, frameshifting frequency of Htt51Q(+1)YFP in N2a/Htt103Q-CFP cells upon partial down-regulation of tRNAGlnCUG (-tRNAGlnCUG) compared with a control with scrambled shRNA. C, expression of Htt51S-YFP in N2a and N2a/Htt103Q-CFP cells in the absence or presence of 10 μm MG-132. The detection was carried out with anti-HA antibodies recognizing the N-terminal HA tag fused to Htt51S-YFP. D, +1 trans-frame species of Htt22Q(+1)YFP and Htt7Q(+1)YFP in either N2a or N2a/Htt103Q-CFP cells were not detectable (n.d.). E, quantitative real-time PCR of Htt51S-YFP mRNA expression in N2a and N2a/Htt103Q-CFP cells. Data are mean ± S.E. (n = 3) and presented as -fold change to the expression of actin.
The frameshifting at the UUU C slippery sequence in influenza A virus is stimulated by a rare codon in the ribosomal A site, and its slow-to-decode nature is important for +1 frameshifting (16). The counterpart A site codon in Htt51Q is the first CAG of the CAG repeat. Although CAG is a common codon, translation of expanded CAG runs depletes the cognate charged glutaminyl-tRNAGlnCUG, rendering CAG slow to decode (5). Increasing the amount of simultaneously translated CAG codons by expressing Htt51Q(+1)YFP in N2a/Htt103Q-CFP cells increased the number of cells with +1 frameshifting by 2-fold compared with expression in N2a cells (Fig. 1E). (N2a/Htt103Q-CFP cells stably express Htt103Q, in which glutamines are encoded by alternating CAG CAA codons (14), i.e. 51 CAG codons per mRNA copy.) To investigate whether increased consumption of glutaminyl-tRNAGlnCUG by decoding consecutive CAG codons correlates with +1 frameshifting, we down-regulated tRNAGlnCUG using a specific shRNA probe. Notably, partial knockdown of tRNAGlnCUG did not change the frameshifting frequency in N2a/Htt103Q-CFP cells expressing the Htt51Q(+1)YFP reporter and was comparable with the control with scrambled shRNA (Fig. 2B). This suggests that +1 frameshifting in Htt51Q is independent of tRNAGlnCUG.
Further proof that an induced hungry codon downstream of the slippery site did not play a role in +1 frameshifting came from the following experiment. Variants with mutated UUC CAG (Gln) to UUC GAG (Glu) or UUC AAG (Lys) yielded a level of +1 frame product comparable with that of the wild-type UUC CAG sequence in both N2a or N2a/Htt103Q-CFP cells (Fig. 2A). Also, we detected a similar approximate increase of +1 frameshifting with UUC AAG or UUC GAG in N2a/Htt103Q-CFP compared with N2a, which we observed with the wild-type UUC CAG sequence in N2a/Htt103Q-CFP compared with N2a (Fig. 2A).
Furthermore, we reasoned that the higher amount of trans-frame product in N2a/Htt103Q-CFP cells compared with N2a cells (Fig. 1F) might be due to differences in degrading it in both cells. To prove this, we expressed Htt51S-YFP with serine codons into the zero frame (Htt51S-YFP) in N2a cells and when co-expressed with a polyQ-protein in N2a/Htt103Q-CFP cells. Notably, Htt51S-YFP accumulated in detectably higher amounts in N2a/Htt103Q-CFP than in N2a (Fig. 2C, top panels, compare the 4- and 6-h expression time points). Homopolymeric polyQ peptides impair proteasome function (18), which most likely results in higher amount of Htt51S-YFP in N2a/Htt103Q-CFP cells (Fig. 2C, bottom panels), although the Htt51S-YFP mRNA amount is equal in both N2a and N2a/Htt103Q-CFP cells (Fig. 2E). We further determined the expression level of Htt51S-YFP in both cell lines in the presence of the proteasomal inhibitor MG-132 (Fig. 2C, right panels). The rationale behind this comparison is that if Htt103Q-CFP protein has already inhibited the proteasomal function, then the proteasomal inhibitor MG-132 would not additionally stabilize the Htt51S-YFP in N2a/Htt103Q-CFP cells. Indeed, MG-132 stabilized Htt51S-YFP only in N2a cells and marginally in N2a/Htt103Q-CFP cells (Fig. 2C, compare ±MG-132 for each cell line). Together, these results suggest that the higher frameshifting frequency in N2a/Htt103Q-CFP cells is a result of the higher stability of the +1 trans-frame product in these cells.
CAG Expansion Acts as a Stimulator for +1 Frameshifting
mRNA structure 3′ of productively utilized +1 frameshift sites can stimulate frameshifting (19, 20). Experimental evidence shows that elongated CAG runs exhibit a high propensity to form elongated stem-loop structures (21). This raised the question of whether the structural propensity of the CAG repeat influences the +1 frameshifting in Htt exon 1. Thus, we generated two additional variants with shorter repeat lengths in the non-pathological range, Htt22Q(+1)YFP and Htt7Q(+1)YFP, for which secondary structures are predicted (Fig. 3, A–C), but their stability is much lower and they do not persist as experimentally shown earlier (21). For both Htt22Q(+1)YFP and Htt7Q(+1)YFP, we did not detect any +1 trans-frame products when expressed in either N2a cells or N2a/Htt103Q-CFP cells (Fig. 2D), although they expressed at comparable levels (Fig. 3E). Moreover, no YFP-positive cells were detected even when the more shift-prone site UUU C was introduced in Htt22Q(+1)YFP (Fig. 2D), suggesting that the stability of the 22-CAG run is not enough to stimulate +1 frameshifting.
FIGURE 3.

The expanded CAG run forms an elongated stem-loop. A–D, the predicted secondary structure of the CAG repeat in Htt51Q (A), Htt22Q (B), Htt7Q (C), and Htt51A (D) using Vienna RNA package 2.0. The slippery UUC codon and the first nucleotide of the first CAG codon is marked in green. The calculated energy of the uniform polyCAG stem-loops, including the first 17 amino acids, are −70.7 kcal/mol for Htt51Q, −35.7 kcal/mol for Htt22Q, −18.7 kcal/mol for Htt7Q, and −68.6 kcal/mol for Htt51A. E, mRNA expression analysis of HttQ(+1)YFP constructs using quantitative real-time PCR. Data are mean ± S.E. (n = 3) and are presented as -fold change to the expression of actin.
Together, these data imply that pathological expansion of the CAG repeat stabilizes the secondary structure (21) so that it acts as a +1 frameshifting stimulatory element. To further address the implication that the secondary structure is a major determinant of +1 frameshifting, we tested another construct without glutamine codons, Htt51A(+1)YFP, which displays very similar secondary structure stability as Htt51Q(+1)YFP. Its slippery sequence, UUC C, is intact (Fig. 3D). Our reasoning was to use a construct with identical nucleotide sequence to the polyCAG tract to ensure the same stability of the hairpin, but, unlike Htt51Q, it has a lower aggregation propensity (22, 23). Indeed, the expression Htt51A(+1)YFP displayed a similar frequency of cells with +1 trans-frame products (Fig. 2A). Cumulatively, our results suggest that the secondary structure is likely the major determinant of +1 frameshifting.
The +1 trans-Frame Product Predominantly Localizes in the Nucleus and Alters polyQ Aggregation
To determine whether the frameshift-derived product with 51 serines, Htt51S, interacts with the parental Htt51Q protein and thus potentially modulates its aggregation, we first analyzed the intracellular localization of Htt51S inclusions by confocal fluorescence microscopy. The YFP-positive trans-frame product localized almost exclusively in the nucleus; only very rarely (less than 5%) did we find a cell with additional cytoplasmic inclusions (Fig. 4A). The Htt51S-YFP control also exclusively localized in the nucleus (Fig. 4B). Nuclear +1 trans-frame protein was diffusively distributed with small fluorescently denser loci, whereas the rare cytoplasmic counterpart formed larger, ring-shaped structures surrounding the core aggregates composed of Htt103Q-CFP (Fig. 4A). The mobility of nuclear and cytoplasmic +1 trans-frame products differed markedly (Fig. 4C). The nuclear loci were composed of mobile species, as their fluorescence rapidly recovered after photobleaching, whereas the cytoplasmic structures contained species with reduced mobility, as they displayed much slower recovery (Fig. 4C). The nuclear +1 trans-frame products seem to not alter the nuclear membranes; the continuous lamin B1 rims are indicative of an intact nuclear envelope (Fig. 4, A and B).
FIGURE 4.

The +1 trans-frame product predominantly localizes in the nucleus and influences polyQ aggregation at high concentrations. A, representative images of immunostained N2a or N2a/Htt103Q-CFP (CFP) cells transiently expressing Htt51Q(+1)YFP (YFP) for 24 h with nuclear and cytoplasmic +1 trans-frame products. The rare cytoplasmic Htt103Q-CFP inclusions co-localizing with the +1 trans-frame product are marked with a white arrow. Scale bars = 5 μm. B, confocal fluorescence images of N2a cells expressing Htt51S-YFP for 24 h. Nuclei were counterstained with DAPI and the nuclear envelope with lamin-B1 antibody. Scale bars = 10 μm. C, fluorescence recovery after photobleaching analysis of nuclear (Nu) and cytoplasmic (Cy) aggregates of the frameshifted species of Htt51Q(+1)YFP transiently expressed in N2a cells for 24 h. The white squares on the prebleach image mark the area of photobleaching. Scale bars = 2 μm. The bottom panel represents the quantification of the total fluorescence intensity within the bleached area. a.i., arbitrary intensity.
The co-localization of Htt51S with Htt103Q in the rare cytoplasmic aggregates raised the question of whether the polyS-containing product derived from +1 frameshifting can influence the aggregation of the poly-glutamine protein. To test this, we added Htt51S in different concentrations at the onset of Htt52Q aggregation and monitored aggregation by light scattering at 90° and a filter retardation assay; the latter captures only large detergent (SDS)-resistant aggregates. Htt51S accelerated Htt52Q aggregation only at high concentration (Fig. 5A). The lag phase of Htt51Q aggregation was markedly shortened, suggesting an impact on the initial aggregation phase, whereas the exponential stage of aggregates growth remained unaltered (Fig. 5A). Notably, the SDS-resistant Htt52Q aggregates contained Htt51S (Fig. 5B), indicating that the two proteins co-aggregated. When individually subjected to aggregation, Htt51S exhibited no clear sigmoidal aggregation signature as observed for Htt52Q (Fig. 5C) and, unlike Htt52Q, formed no SDS-resistant aggregates within the time frame of the experiments (Fig. 5D). Importantly, the lack of SDS-resistance of Htt51S aggregates alone correlated with the observed motility of the nuclear aggregates composed solely of Htt51AGC-YFP. Detergent-labile inclusions are highly mobile, whereas amyloid SDS-resistant aggregates display restricted mobility (24). Most likely, the cytoplasmic aggregates with restricted mobility (Fig. 4C) are amyloid Htt51Q/Htt103Q-CFP assemblies that can recruit Htt51S-YFP (monomer and/or preformed aggregates).
FIGURE 5.
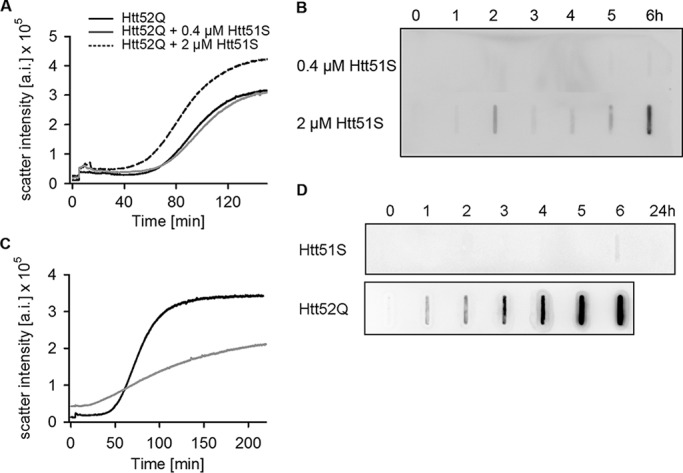
The +1 trans-frame product influences polyQ aggregation at high concentrations. A and B, different concentrations of Htt51S were added at the onset of Htt52Q (4 μm), and the aggregation was monitored by light scattering at 90° (A) or filter retardation assay (B). The aggregation curve of Htt52Q alone is included in A as a control. Retained Htt51S in the co-aggregation reaction is immunostained with anti-His antibody. C, Htt51S alone forms SDS-labile aggregates. Aggregation kinetics of purified Htt51S (4 μm, gray) and Htt52Q (4 μm, black) were monitored by light scattering at 90°. Note the significantly shorter (almost absent) lag phase of Htt51S compared with Htt52Q. D, kinetics of formation of SDS-resistant aggregates of Htt51S (4 μm) and Htt52Q (3 μm) monitored by filter retardation assay and visualized by immunostaining with anti-His (for Htt51S) and anti-HA (for Htt52Q) antibody. a.i., arbitrary intensity.
Together, these results suggest that the +1 trans-frame Htt51S protein aggregated by itself and formed rather detergent-labile aggregates. When present in high concentration, it also co-aggregated with the amyloid Htt52Q aggregates, although it is unclear whether Htt51S was stably incorporated into the Htt52Q fibrils or rather co-assembles at the edges of Htt52Q aggregates.
Discussion
The CAG-encoded polyQ aggregates of Huntington disease-affected brain tissues contain traces of both polyA and polyS proteins (25). Although, in earlier work, we have shown that polyA proteins derive from −1 frameshifting in the expanded CAG repeat of Htt exon 1 (5), here we show that polyS proteins originate from +1 frameshifting guided by a slippery sequence, UUC C, upstream of the CAG repeat. Both −1 and +1 frameshifting are detectable only in Htt variants with pathological CAG lengths, but their underlying mechanisms clearly differ. Partial depletion of glutaminyl-tRNAGlnCUG stimulates −1 frameshifting in HttQ (5), whereas +1 frameshifting is tRNAGlnCUG-independent (Fig. 2B) and is greatly enhanced by the stem-loop structure of the elongated CAG repeat.
Translational recoding with a shift of the canonical triplet periodicity movement of the ribosomes occurs more frequently than initially thought and is beneficial either for regulatory purposes and/or to expand the coding capacities of the genome (26, 27). A typical recoding cassette involves a shift-prone site and a discrete enhancing signal 5′ or 3′ or both to the frameshift site (26–33). The mechanisms of +1 frameshifting are less defined than for −1 frameshifting, in part because −1 frameshift sites are easier to recognize, and their stimulatory elements are more idiosyncratic. Typically, −1 frameshifting is stimulated by secondary structures adjacent to the slippery site (29, 31, 34–37). +1 frameshifting in the expression of Htt with expanded CAG repeats resembles this pattern. A slippery sequence, UUC C, redirects linear readout from the 0 to the +1 frame and is enhanced by the stem-loop structure of the elongated CAG repeat (21) compared with its counterparts with a CAG length in the non-pathological range. The shift site in Htt resembles that of the influenza A virus ribosomal frameshift site, UUU C, but is a rather atypical sequence with substantially lower frameshifting efficiency (16, 17). The secondary structure downstream of the slippery sequence in Htt is greatly stabilized by the CAG expansion; exclusively expanded CAG repeats stimulate +1 frameshifting. This secondary structure most likely pauses some of the translating ribosomes and allows for repairing of tRNAPheAAG reading the UUC codon. It is unknown whether peptidyl-tRNA anticodon dissociates from the zero-frame codon UUC and repairs to the overlapping UCC or whether +1 frameshifting proceeds without its repairing to mRNA. However, if repairing to mRNA after realignment takes place, it would involve the non-Watson-Crick rule (38) and include an A:C apposition at the central and most important position of the tRNAPheAAG anticodon (39). It shares some similarities with the +1 frameshifting utilized in yeast retrotransposon decoding: Saccharomyces cerevisiae Ty3 +1 frameshifting has been proposed to occur in the absence of complete dissociation of peptidyl-tRNA anticodon loop pairing (40). Also, a hexanucleotide −1 frameshifting occurs in the apparent absence of peptidyl-tRNA repairing (41), and a low level of translational bypassing resumption can occur in the absence of peptidyl-tRNA pairing (42).
A large body of functional studies has been based on the assumption that single proteins are expressed from any gene associated with trinucleotide repeat disorders. The emerging evidence for alternative products generated through repeat-associated non-ATG translation (3, 43), by translational −1 frameshifting (5, 6, 10), or, as revealed here, by translational +1 frameshifting suggests that dynamic reprogramming of translation to generate alternative products from one gene is more common than previously appreciated. The toxicity of HttQ proteins is mainly attributed to nuclearly localized species (44, 45). The almost exclusive localization of the +1 trans-frame-encoded polyS in the nucleus in our cellular model system and their higher dynamic mobility compared with the cytoplasmic aggregates suggest a likely contribution to toxicity at the cellular level. The estimation for the frequency of +1 frameshifting in brain tissues is difficult, as the steady-state concentration of endogenous Htt mRNA is not known. However, the presence of polyS proteins that are the +1 trans-encoded product of Htt in Huntington disease-affected brain tissues (13) provides evidence for the likelihood of +1 frameshifting of endogenous Htt with an expanded CAG repeat in neuronal tissues, which may contribute to aggregation onset or even toxicity at the cellular level. Along this line, in ataxin 3, in which the CAG expansion is causative of spinocerebellar ataxia type 3, evidence has been presented suggesting that neither the expanded mRNA nor the zero-frame polyQ translation product are directly responsible for the cellular toxicity in Drosophila and mammalian neurons but, rather, that the −1-trans-frame-encoded product is a key (6).
Conceptually, the +1 frameshifting in Htt with pathological expansion of the CAG repeat shows resemblance with repeat recoding in some human DNA tumor viruses (8). The common mechanistic signature between frameshifting in CAG repeat sequences and the examples of repeat recoding in DNA viruses imply that frameshifting might be commonly associated with translation of highly repetitive sequences that are frequently found in coding sequences of eukaryotic genomes.
Experimental Procedures
Constructs and Their Expression in Cell Culture
HA-Htt51Q(+1)YFP, HA-Htt22Q(+1)YFP, and HA-Htt7Q(+1)YFP were cloned into the pTRE2hyg Tet-off plasmid and transfected in N2a using JetPrime reagent (PeqLab). Both plasmids were used as a backbone to introduce various mutations in HttQ(+1)-YFP. N2a cells stably expressing Htt103Q-CFP were cultured as described previously (14). For imaging, cells were grown on poly-l-lysine-precoated coverslides, fixed in 4% paraformaldehyde in two different buffers (0.1 m PIPES (pH 6.5), 2 mm MgCl2, 5 mm EGTA, and 0.1 m sodium borate), permeabilized with 0.1% Triton X-100 (Sigma) at room temperature, and immunostained with monoclonal anti-HA antibody (HA.11, Covalance) or polyclonal anti-lamin B antibody, followed by Alexa 568-labeled anti-rabbit or anti-mouse secondary antibody (Invitrogen). Cells were imaged on an Axiovert confocal microscope (Zeiss) or DMi8 (Leica) equipped with a ×63 oil objective lens, and images were taken at 405-, 514-, and 561-nm laser wavelengths for CFP-, YFP-, and Alexa 568-labeled antibodies, respectively.
More than 1000 cells from several independent biological replicates were imaged, and the frameshifting frequency was determined as a fraction of cells expressing YFP (which reports on +1 frameshifting) from all HA-Htt51Q-expressing cells (i.e. stained positively for the HA tag using anti-HA antibodies). Because the frameshift frequency per cell differs, we set up a threshold of YFP fluorescence to consider a cell with frameshifting over the background fluorescence. The threshold value of 0.48 was determined by averaging the integrated background fluorescence of 50 N2a cells that do not express HA-Htt51Q(+1)YFP. For the siRNA experiments, cells cotransfected (1:1 ratio, 1 μg of total DNA) with the plasmid for expression of Htt51Q(+1)YFP and pSuper bearing the shRNA against tRNAGlnCUG or scrambled control were imaged to determine the frameshifting frequency (5). Unless stated otherwise, results are expressed as mean ± S.E. of n replicates. Differences between groups were evaluated using chi-square test and considered statistically significant when p < 0.05. In the proteasome inhibition experiment, to cells expressing different constructs for 24 h, 10 μm MG-132 was added, and samples were withdrawn at various times.
For fluorescence recovery after photobleaching analysis, transfected cells were grown in in 35-mm CellviewTM dishes with glass slides (Greiner), and an area of 1.13–1.37 μm2 was photobleached for 6 s with a 514-nm laser wavelength at 100% power, and single images were collected before and every 1 s after photobleaching with an interval of 100 s.
Immunoblotting and Mass Spectrometry
A total of 250,000 cells were harvested and directly dissolved in 250 μl of formic acid and incubated at 37 °C for 40 min to maximally dissolve aggregates, and expression was analyzed either by SDS-PAGE or spotted directly onto nitrocellulose membrane in a slot-blot manifold. The detection was carried out by immunostaining with HA or GFP antibody (Roche).
For mass spectrometry, ∼30 × 106 cells were lysed in 1× MNT buffer (30 mm Tris-HCl, 20 mm MES, 100 mm NaCl, and 1% (w/v) Triton-X100), immunoprecipitated with anti-GFP antibodies (MicroBeads, Miltenyi), and subsequently fractionated by two-dimensional gel (pH 3–11) electrophoresis. Protein spots were excised from the gel and digested with 30 ng/μl trypsin (sequencing grade, Roche) at 37 °C overnight. The resulting peptides were eluted with acetonitrile and formic acid and analyzed by MALDI-TOF-MS (microflex LRF, Bruker Daltonics) in linear mode.
RNA Isolation and Quantitative Real-time PCR
Total RNA was isolated from cultures expressing different HttQ variants using TRI reagent (Sigma). The integrity of the RNA was analyzed using 1% formaldehyde gel electrophoresis. 0.5 μg of RNA was treated with DNase I (Thermo Scientific) and used for cDNA synthesis with oligo(dT) primers and Revert Aid reverse transcriptase (Thermo Scientific). The cDNA in 1:10 dilution was used in a quantitative real-time PCR (SYBR Green-based approach, Qiagen). Each reaction was performed in duplicates, and each run included control samples containing either no template or no reverse-transcribed transcript. Actin mRNA was used for normalization. Results are represented as mean ± S.E.
In Vitro Aggregation Assays
Proteins were expressed as GST fusions on the pGEX-6P-1 plasmid (GE Healthcare) in E. coli BL21(DE3) and affinity-purified with glutathione-Sepharose 4 Fast Flow (GE Healthcare) as described previously (15). Aggregation was initiated by adding PreScission protease, which removes the GST tag, and monitored by means of a filter retardation assay (15) and static 90° light scattering at 532 nm (Quanta Master 30, PTI).
Author Contributions
P. S. performed most of the in vivo experiments. F. A. conducted the mRNA expression, protein stability, and shRNA experiments. R. S. performed the in vitro aggregation experiments. P. S. and Z. I. analyzed the data, conceived the concepts, planned and designed the experiments, and wrote the manuscript. J. F. A. contributed with significant input to mutation design, interpretation of the data, and manuscript writing.
Acknowledgments
We thank Ai Yamamoto (Columbia University) for the N2a/Htt103Q-CFP cells and Otto Baumann and Jörg Fettke (University of Potsdam) for help with the confocal microscope and mass spectrometry analysis, respectively.
This work was supported by Deutsche Forschungsgemeinschaft Grants FOR 1805 and SFB 740 (to Z. I.). The authors declare that they have no conflicts of interest with the contents of this article.
- polyQ
- polyglutamine
- Htt
- huntingtin
- polyA
- polyalanine
- polyS
- polyserine
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein.
References
- 1. Karlin S., Brocchieri L., Bergman A., Mrazek J., and Gentles A. J. (2002) Amino acid runs in eukaryotic proteomes and disease associations. Proc. Natl. Acad. Sci. U.S.A. 99, 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Orr H. T., and Zoghbi H. Y. (2007) Trinucleotide repeat disorders. Ann. Rev. Neurosci. 30, 575–621 [DOI] [PubMed] [Google Scholar]
- 3. Cleary J. D., and Ranum L. P. (2014) Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr. Opin. Genet. Dev. 26, 6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wojciechowska M., Olejniczak M., Galka-Marciniak P., Jazurek M., and Krzyzosiak W. J. (2014) RAN translation and frameshifting as translational challenges at simple repeats of human neurodegenerative disorders. Nucleic Acids Res. 42, 11849–11864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Girstmair H., Saffert P., Rode S., Czech A., Holland G., Bannert N., and Ignatova Z. (2013) Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 3, 148–159 [DOI] [PubMed] [Google Scholar]
- 6. Stochmanski S. J., Therrien M., Laganière J., Rochefort D., Laurent S., Karemera L., Gaudet R., Vyboh K., Van Meyel D. J., Di Cristo G., Dion P. A., Gaspar C., and Rouleau G. A. (2012) Expanded ATXN3 frameshifting events are toxic in Drosophila and mammalian neuron models. Hum. Mol. Genet. 21, 2211–2218 [DOI] [PubMed] [Google Scholar]
- 7. Gaspar C., Jannatipour M., Dion P., Laganière J., Sequeiros J., Brais B., and Rouleau G. A. (2000) CAG tract of MJD-1 may be prone to frameshifts causing polyalanine accumulation. Hum. Mol. Genet. 9, 1957–1966 [DOI] [PubMed] [Google Scholar]
- 8. Kwun H. J., Toptan T., Ramos da Silva S., Atkins J. F., Moore P. S., and Chang Y. (2014) Human DNA tumor viruses generate alternative reading frame proteins through repeat sequence recoding. Proc. Natl. Acad. Sci. U.S.A. 111, E4342–E4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Griffiths A., Link M. A., Furness C. L., and Coen D. M. (2006) Low-level expression and reversion both contribute to reactivation of herpes simplex virus drug-resistant mutants with mutations on homopolymeric sequences in thymidine kinase. J. Virol. 80, 6568–6574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toulouse A., Au-Yeung F., Gaspar C., Roussel J., Dion P., and Rouleau G. A. (2005) Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum. Mol. Genet. 14, 2649–2660 [DOI] [PubMed] [Google Scholar]
- 11. Gurvich O. L., Baranov P. V., Gesteland R. F., and Atkins J. F. (2005) Expression levels influence ribosomal frameshifting at the tandem rare arginine codons AGG_AGG and AGA_AGA in Escherichia coli. J. Bacteriol. 187, 4023–4032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang G., Fedyunin I., Miekley O., Valleriani A., Moura A., and Ignatova Z. (2010) Global and local depletion of ternary complex limits translational elongation. Nucleic Acids Res. 38, 4778–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davies J. E., and Rubinsztein D. C. (2006) Polyalanine and polyserine frameshift products in Huntington's disease. J. Med. Genet. 43, 893–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto A., Cremona M. L., and Rothman J. E. (2006) Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 172, 719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G. P., Davies S. W., Lehrach H., and Wanker E. E. (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 [DOI] [PubMed] [Google Scholar]
- 16. Firth A. E., Jagger B. W., Wise H. M., Nelson C. C., Parsawar K., Wills N. M., Napthine S., Taubenberger J. K., Digard P., and Atkins J. F. (2012) Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2, 120109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jagger B. W., Wise H. M., Kash J. C., Walters K. A., Wills N. M., Xiao Y. L., Dunfee R. L., Schwartzman L. M., Ozinsky A., Bell G. L., Dalton R. M., Lo A., Efstathiou S., Atkins J. F., Firth A. E., et al. (2012) An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Venkatraman P., Wetzel R., Tanaka M., Nukina N., and Goldberg A. L. (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104 [DOI] [PubMed] [Google Scholar]
- 19. Ivanov I. P., and Atkins J. F. (2007) Ribosomal frameshifting in decoding antizyme mRNAs from yeast and protists to humans: close to 300 cases reveal remarkable diversity despite underlying conservation. Nucleic Acids Res. 35, 1842–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Matsufuji S., Matsufuji T., Miyazaki Y., Murakami Y., Atkins J. F., Gesteland R. F., and Hayashi S. (1995) Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell 80, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michlewski G., and Krzyzosiak W. J. (2004) Molecular architecture of CAG repeats in human disease related transcripts. J. Mol. Biol. 340, 665–679 [DOI] [PubMed] [Google Scholar]
- 22. Albrecht A. N., Kornak U., Böddrich A., Süring K., Robinson P. N., Stiege A. C., Lurz R., Stricker S., Wanker E. E., and Mundlos S. (2004) A molecular pathogenesis for transcription factor associated poly-alanine tract expansions. Hum. Mol. Genet. 13, 2351–2359 [DOI] [PubMed] [Google Scholar]
- 23. Oma Y., Kino Y., Sasagawa N., and Ishiura S. (2004) Intracellular localization of homopolymeric amino acid-containing proteins expressed in mammalian cells. J. Biol. Chem. 279, 21217–21222 [DOI] [PubMed] [Google Scholar]
- 24. Hinz J., Lehnhardt L., Zakrzewski S., Zhang G., and Ignatova Z. (2012) Polyglutamine expansion alters the dynamics and molecular architecture of aggregates in dentatorubropallidoluysian atrophy. J. Biol. Chem. 287, 2068–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., and Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 26. Dinman J. D. (2012) Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip. Rev. RNA 3, 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ribas de Pouplana L., Santos M. A., Zhu J. H., Farabaugh P. J., and Javid B. (2014) Protein mistranslation: friend or foe? Trends Biochem. Sci. 39, 355–362 [DOI] [PubMed] [Google Scholar]
- 28. Atkinson J., Dodge M., and Gallant J. (1997) Secondary structures and starvation-induced frameshifting. Mol. Microbiol. 26, 747–753 [DOI] [PubMed] [Google Scholar]
- 29. Brierley I., Gilbert R. J., and Pennell S. (2008) RNA pseudoknots and the regulation of protein synthesis. Biochem. Soc. Trans. 36, 684–689 [DOI] [PubMed] [Google Scholar]
- 30. Liao P. Y., Gupta P., Petrov A. N., Dinman J. D., and Lee K. H. (2008) A new kinetic model reveals the synergistic effect of E-, P- and A-sites on +1 ribosomal frameshifting. Nucleic Acids Res. 36, 2619–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Namy O., Moran S. J., Stuart D. I., Gilbert R. J., and Brierley I. (2006) A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441, 244–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Temperley R., Richter R., Dennerlein S., Lightowlers R. N., and Chrzanowska-Lightowlers Z. M. (2010) Hungry codons promote frameshifting in human mitochondrial ribosomes. Science 327, 301. [DOI] [PubMed] [Google Scholar]
- 33. Taliaferro D., and Farabaugh P. J. (2007) An mRNA sequence derived from the yeast EST3 gene stimulates programmed +1 translational frameshifting. RNA 13, 606–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caliskan N., Katunin V. I., Belardinelli R., Peske F., and Rodnina M. V. (2014) Programmed −1 frameshifting by kinetic partitioning during impeded translocation. Cell 157, 1619–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen J., Petrov A., Johansson M., Tsai A., O'Leary S. E., and Puglisi J. D. (2014) Dynamic pathways of −1 translational frameshifting. Nature 512, 328–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tamm T., Suurväli J., Lucchesi J., Olspert A., and Truve E. (2009) Stem-loop structure of Cocksfoot mottle virus RNA is indispensable for programmed −1 ribosomal frameshifting. Virus Res. 146, 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yan S., Wen J. D., Bustamante C., and Tinoco I. Jr. (2015) Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160, 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satpati P., and Åqvist J. (2014) Why base tautomerization does not cause errors in mRNA decoding on the ribosome. Nucleic Acids Res. 42, 12876–12884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lehmann J., and Libchaber A. (2008) Degeneracy of the genetic code and stability of the base pair at the second position of the anticodon. RNA 14, 1264–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Farabaugh P. J., Zhao H., and Vimaladithan A. (1993) A novel programed frameshift expresses the POL3 gene of retrotransposon Ty3 of yeast: frameshifting without tRNA slippage. Cell 74, 93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Licznar P., Mejlhede N., Prère M. F., Wills N., Gesteland R. F., Atkins J. F., and Fayet O. (2003) Programmed translational −1 frameshifting on hexanucleotide motifs and the wobble properties of tRNAs. EMBO J. 22, 4770–4778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herr A. J., Wills N. M., Nelson C. C., Gesteland R. F., and Atkins J. F. (2004) Factors that influence selection of coding resumption sites in translational bypassing: minimal conventional peptidyl-tRNA:mRNA pairing can suffice. J. Biol. Chem. 279, 11081–11087 [DOI] [PubMed] [Google Scholar]
- 43. Paul J. W., West J. P., and Gitler A. D. (2014) Clogging information flow in ALS. Science 345, 1118–1119 [DOI] [PubMed] [Google Scholar]
- 44. Peters M. F., Nucifora F. C. Jr., Kushi J., Seaman H. C., Cooper J. K., Herring W. J., Dawson V. L., Dawson T. M., and Ross C. A. (1999) Nuclear targeting of mutant Huntingtin increases toxicity. Mol. Cell Neurosci. 14, 121–128 [DOI] [PubMed] [Google Scholar]
- 45. Saudou F., Finkbeiner S., Devys D., and Greenberg M. E. (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66 [DOI] [PubMed] [Google Scholar]