

Abstract
Although numerous biological functions of the activating transcription factor 4 (ATF4) have been identified, a direct effect of ATF4 on alcoholic liver steatosis has not been described previously. The aim of our current study is to investigate the role of ATF4 in alcoholic liver steatosis and elucidate the underlying mechanisms. Here, we showed that the expression of ATF4 is induced by ethanol in hepatocytes in vitro and in vivo, and liver-specific ATF4 knock-out mice are resistant to ethanol-induced liver steatosis, associated with stimulated hepatic AMP-activated protein kinase (AMPK) activity. Furthermore, adenovirus-mediated AMPK knockdown significantly reversed the suppressive effects of ATF4 deficiency on ethanol-induced liver steatosis in mice. In addition, ethanol-fed ATF4 knock-out mice exhibit AMPK-dependent inhibition of fatty acid synthase and stimulation of carnitine palmitoyltransferase 1 (CPT1) in the liver. Moreover, hepatic Tribbles homolog 3 (TRB3) expression was stimulated by ethanol in an ATF4-dependent manner, and adenovirus-mediated TRB3 knockdown blocked ATF4-dependent ethanol-induced AMPK inhibition and triglyceride accumulation in AML-12 cells. Finally, TRB3 directly interacted with AMPK to suppress its phosphorylation. Taken together, these results identify the ATF4-TRB3-AMPK axis as a novel pathway responsible for ethanol-induced liver steatosis.
Keywords: AMP-activated kinase (AMPK), fatty acid synthase (FAS), lipid, liver injury, liver metabolism, ATF4, Alcoholic liver steatosis, CPT1, TRB3
Introduction
People consuming excessive amounts of alcohol for prolonged periods suffer from alcoholic liver disease, which encompasses fatty liver, hepatic inflammation and necrosis, progressive fibrosis, and hepatocellular carcinoma (1). Alcoholic fatty liver is the early stage of alcoholic liver disease, characterized by increased accumulation of fat in the liver, which is usually caused by increased free fatty acid (FFA)4 uptake, augmented de novo lipogenesis, decreased β-oxidation, and/or impaired triglyceride (TG) export (2). Studies have identified some important regulators for ethanol-induced liver steatosis, including sirtuin 1 (SIRT1) (3), sterol regulatory element-binding protein 1 (SREBP-1) (4), peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) (5), and 5′-adenosine monophosphate-activated protein kinase (AMPK) (6–8). Among them, AMPK is one of the most important regulators (6–8). AMPK is a heterotrimeric complex consisting of a catalytic α subunit and two regulatory β/γ subunits. It acts as a key metabolic “switch” by phosphorylating target enzymes involved in lipid metabolism, such as acetyl-CoA carboxylase (ACC) (9). Malonyl-CoA, the product of ACC, is both a precursor for fatty acid biosynthesis and a potent inhibitor of β-oxidation at the step regulated by carnitine palmitoyltransferase I (10). AMPK activity is suppressed under ethanol exposure (6) and activation of AMPK by its activator (5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside) or metformin prevents ethanol-induced liver steatosis (11, 12). However, the molecular mechanisms underlying ethanol suppression of AMPK activity remain largely unknown.
Activating transcription factor 4 (ATF4) is a transcription factor that belongs to the family of basic zipper-containing proteins (13), which is constitutively expressed in a wide variety of tissues, including white adipose tissue, brown adipose tissue, and liver (14). ATF4 is involved in the regulation of many biological processes, including osteoblast differentiation (15) and oxidative stress response (16). Recent studies have also demonstrated an important role of ATF4 in glucose and lipid metabolism (17–21). For example, deletion of ATF4 protects mice from high-carbohydrate diet- or high-fat diet-induced liver steatosis (17, 18) and high-fructose diet-induced hypertriglyceridemia (21). A role of ATF4 in ethanol-induced liver steatosis, however, has not previously been studied. Interestingly, ATF4 expression is induced by ethanol exposure in human hepatocytes (22), bone marrow stromal cells (23), and rat livers (24). Furthermore, ATF4 inhibits the activity of hypothalamic AMPK (20), a kinase that plays a central role in ethanol-induced liver steatosis (6–8). We therefore hypothesized that ATF4 may play a role in ethanol-induced liver steatosis. The aim of our current study is to investigate this possibility and elucidate the underlying mechanisms.
In our current study, we showed that ATF4 liver-specific knock-out (ALKO) mice are resistant to ethanol-induced liver steatosis via activation of AMPK. Furthermore, ATF4 regulates AMPK activity via Tribbles homolog 3 (TRB3), a direct target of ATF4 (25), which could bind directly to AMPK and suppress its phosphorylation. Taken together, our results identify the ATF4-TRB3-AMPK axis as a novel pathway responsible for ethanol-induced liver steatosis.
Results
Ethanol Treatment Induces ATF4 Expression in Hepatocytes in Vitro and in Vivo
As predicted, Atf4 mRNA and protein were significantly increased following ethanol treatment compared with control cells (Fig. 1, A and B). Similar results were obtained in the livers of mice fed an ethanol diet for 4 weeks compared with mice maintained on a pair-fed control diet (Fig. 1, C and D).
FIGURE 1.
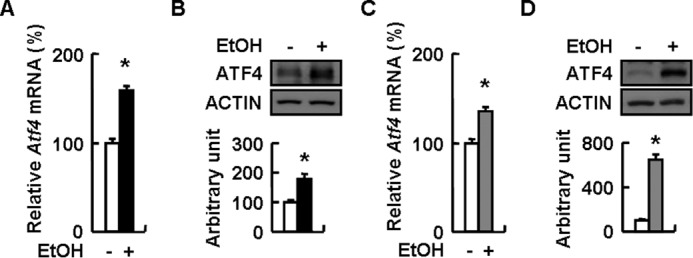
Ethanol induces ATF4 expression in hepatocytes in vitro and in vivo. A and B, mouse primary hepatocytes were treated without (−EtOH) or with 100 mm ethanol (+EtOH) for 24 h. C and D, WT mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. Means ± S.E. (error bars) shown are representative of at least three independent in vitro experiments or at least two independent in vivo experiments, with the number of mice included in each group in each experiment indicated (n = 5–7). Statistical significance was determined by Student's t test for the effect of with versus without ethanol treatment (*, p < 0.05). A and C, Atf4 mRNA; B and D, ATF4 protein (top, Western blotting; bottom, quantitative measurements of ATF4 relative to actin).
ALKO Mice Are Resistant to Ethanol-induced Liver Steatosis
The elevated expression of ATF4 in response to ethanol exposure indicates a possible involvement of ATF4 in ethanol-induced liver steatosis. To test this possibility, we fed ALKO or WT mice with a control or an ethanol diet for 4 weeks and examined whether hepatic ATF4 knock-out could prevent ethanol-induced liver steatosis. ALKO mice were generated as described previously (26) and exhibited decreased Atf4 mRNA levels only in the liver and not in muscle or white adipose tissue, with similar body weight, liver weight and food intake compared with ATF4flox/flox (ATF4f/f) mice under a normal chow diet.
As predicted, ethanol elevated hepatic Atf4 mRNA levels in WT mice, but not in ALKO mice, compared with corresponding control diet mice (Fig. 2A). However, there was no significant difference in body weight and food intake among the four groups of mice, except that liver weight showed an increased tendency in ethanol-fed WT mice but not in ALKO mice. Histopathological analysis revealed that ethanol caused microvesicular and macrovesicular steatosis in the livers of WT mice compared with control-fed WT mice (Fig. 2B). In contrast, ALKO mice substantially alleviated lipid accumulation in livers when fed an ethanol diet (Fig. 2B). Accordingly, lower levels of hepatic and serum TG were detected in ALKO mice compared with WT mice when challenged with ethanol (Fig. 2, C and D). However, no difference was observed in ethanol-increased hepatic or serum total cholesterol (TC), as well as serum FFAs, between WT and ALKO mice (Fig. 2, C and D). Also, ethanol increased serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in WT mice, but not in ALKO mice, compared with corresponding control-fed mice (Fig. 2E).
FIGURE 2.

ALKO mice are resistant to ethanol-induced liver steatosis. A–E, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. Means ± S.E. (error bars) shown are representative of at least two independent in vivo experiments, with the number of mice included in each group in each experiment indicated (n = 8). Statistical significance was determined by Student's t test for the effect of with versus without ethanol feeding in WT or ALKO mice (*, p < 0.05) or ALKO mice versus WT mice under ethanol diet or control diet (#, p < 0.05). A, Atf4 mRNA; B, pictures of mouse livers, representative Oil Red O staining, and H&E staining of liver tissues (magnification, ×10); C and D, hepatic or serum TG, TC, and FFA levels; E, serum ALT and AST levels.
ALKO Mice Suppress Ethanol-induced Liver Steatosis via Activation of AMPK
It has been reported that AMPK is an important regulator for ethanol-induced liver steatosis (6) and AMPK activity is inhibited by ATF4 activation in the hypothalamus of mice (20). These results suggest that ALKO mice may resist ethanol-induced liver steatosis via the activation of AMPK. To test this possibility, we examined phosphorylation of hepatic AMPK and its direct downstream target ACC (27) in WT and ALKO mice fed a control or ethanol diet. As expected, ethanol significantly decreased hepatic p-AMPK and p-ACC in WT mice compared with control-fed WT mice, whereas the suppressive effects of ethanol were largely abolished in ALKO mice (Fig. 3A).
FIGURE 3.

ALKO mice are resistant to ethanol-induced liver steatosis via activation of AMPK. A, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. B–F, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 4 days, and then these mice were injected with Ad-GFP (−Ad-DN AMPK) or Ad-DN AMPK (+Ad-DN AMPK), followed by ethanol feeding for another 17 days. Means ± S.E. (error bars) shown are representative of at least two independent in vivo experiments, with the number of mice included in each group in each experiment indicated (n = 8). Statistical significance was determined by Student's t test or one-way analysis of variance followed by the Student-Newman-Keuls test for the effect of with versus without ethanol feeding in WT or ALKO mice (*, p < 0.05), ALKO mice versus WT mice under ethanol feeding (#, p < 0.05), or Ad-DN AMPK versus Ad-GFP in ALKO mice under ethanol feeding (†, p < 0.05). A and B, p-AMPK, AMPK, p-ACC, and ACC proteins (top, Western blotting; bottom, quantitative measurements of p-AMPK, AMPK, and p-ACC relative to their total proteins or actin). C, pictures of mouse livers, representative Oil Red O staining, and H&E staining of liver tissues (magnification, ×10). D and E, hepatic or serum TG, TC, and FFA levels. F, serum ALT and AST levels.
To confirm a role of AMPK in ATF4 regulation of ethanol-induced liver steatosis, we injected ethanol-fed ALKO mice with adenovirus expressing a dominant negative AMPK variant (Ad-DN AMPK) or control green fluorescent protein (Ad-GFP). Functional validation of Ad-DN AMPK was demonstrated by its increased expression and its ability to reduce ethanol-increased p-ACC in the livers of ALKO mice injected with Ad-DN AMPK (Fig. 3B). No significant difference in body weight and liver weight was observed in ALKO mice following injection of Ad-DN AMPK compared with Ad-GFP injection. In contrast, Ad-DN AMPK significantly reversed the protective effects of ATF4 deficiency on ethanol-induced liver steatosis, as demonstrated by histological changes, as well as the contents of hepatic TG and serum TG, although it had no significant effects on contents of hepatic TC and serum TC and FFAs (Fig. 3, C–E). Interestingly, Ad-DN AMPK decreased hepatic FFAs in ethanol-fed ALKO mice (Fig. 3D). Ad-DN AMPK also reversed the suppressed serum AST and ALT levels in ALKO mice following ethanol exposure (Fig. 3F).
ALKO Mice Exhibit AMPK-dependent Inhibition of FAS and Stimulation of CPT1 in Liver following Ethanol Feeding
A disorder in lipid metabolism is one of the major causes for liver steatosis (28). We therefore examined the effects of ATF4 on genes and proteins related to lipid metabolism in the livers of ALKO and WT mice fed a control or ethanol diet. Genes involved in lipogenesis including Srebp1c (sterol regulatory element-binding protein 1c), Chrebp (carbohydrate-responsive element-binding protein), PPARγ (peroxisome proliferator-activated receptor γ), Acc (acetyl-CoA carboxylase), Acl (ATP citrate lyase), Fas (fatty acid synthase), and Scd1 (stearoyl-CoA desaturase); genes involved in fatty acid oxidation including Pparα (peroxisome proliferator-activated receptor α) and Cpt1α (carnitine palmitoyltransferase 1a); genes involved in TG secretion including ApoB (apolipoprotein B) and ApoE (apolipoprotein E); and genes involved in fatty acid uptake including Cd36 (cluster of differentiation 36) and Fabp (fatty acid binding protein). Expression of Srebp1c, Acc, and Fas was significantly induced, but expression of Pparα and Cpt1α was reduced, in the livers of WT mice fed an ethanol diet compared with WT mice maintained on a control diet, whereas these ethanol-induced changes in WT mice livers were abolished in ALKO mice (Fig. 4A). No significant differences in other genes were observed in the livers of WT and ALKO mice fed an ethanol diet (Fig. 4A). Furthermore, Ad-DN AMPK significantly reversed the expression of Srebp1c, Acc, Fas, Pparα, and Cpt1α in the livers of ethanol-fed ALKO mice compared with Ad-GFP-injected ALKO mice (Fig. 4B). As with the changes of mRNA levels, the protein levels of hepatic FAS and CPT1 were also reversed by Ad-DN AMPK injection (Fig. 4, C and D).
FIGURE 4.

ALKO mice exhibit AMPK-dependent inhibition of FAS and stimulation of CPT1 in liver following ethanol feeding. A and C, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. B and D, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 4 days, and then these mice were injected with Ad-GFP (−Ad-DN AMPK) or Ad-DN AMPK (+Ad-DN AMPK), followed by ethanol feeding for another 17 days. Means ± S.E. (error bars) shown are representative of at least two independent in vivo experiments, with the number of mice included in each group in each experiment indicated (n = 8). Statistical significance was determined by Student's t test or one-way analysis of variance followed by the Student-Newman-Keuls test for the effect of with versus without ethanol feeding in WT or ALKO mice (*, p < 0.05), ALKO mice versus WT mice under ethanol feeding (#, p < 0.05), or Ad-DN AMPK versus Ad-GFP in ALKO mice under ethanol feeding (†, p < 0.05). A and B, Srebp1c, Chrebp, Pparγ, Acc, Acl, Fas, Scd1, Pparα, Cpt1α, ApoB, ApoE, Cd36, and Fabp mRNAs. C and D, FAS and CPT1 proteins (top, Western blotting; bottom, quantitative measurements of FAS and CPT1 relative to actin).
ATF4 Inhibits AMPK Activity and Promotes TG Accumulation via Increasing TRB3 Expression in Vitro under Ethanol Treatment
TRB3 is a direct target gene of ATF4 (29, 30) and induced by ethanol in rat liver (31), suggesting the possible involvement of loss of TRB3 in mediating ATF4 deletion on stimulated AMPK activity following ethanol exposure. Consistent with this possibility, ethanol increased hepatic TRB3 expression in WT mice, and this effect was abolished in ALKO mice (Fig. 5A). Furthermore, adenovirus-mediated TRB3 knockdown increased AMPK and ACC phosphorylation in mouse primary hepatocytes (Fig. 5B), and overexpression of TRB3 had the opposite effects in mouse primary hepatocytes (Fig. 5C). AMPK and ACC phosphorylation induced by human tumor suppressor liver kinase B1 (LKB1) and calmodulin-activated kinase kinase (CaMKK2), the two well known stimulators for AMPK activity (32), were also suppressed by TRB3 overexpression in mouse primary hepatocytes (Fig. 5, D and E).
FIGURE 5.

TRB3 regulation of AMPK phosphorylation in mouse primary hepatocytes. A, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. B–E, mouse primary hepatocytes were infected with Ad-scrambled (−Ad-shTRB3) or Ad-shTRB3 (+Ad-shTRB3) for 72 h or transfected with control vector (−MYC-TRB3) or Myc-TRB3 (+MYC-TRB3) at different doses, without (−HA-LKB1 or −HA-CaMKK2) or with HA-tagged LKB1 (+HA-LKB1) or CaMKK2 (+HA-CaMKK2) for 48 h. Means ± S.E. (error bars) shown are representative of at least three independent in vitro experiments or at least two independent in vivo experiments, with the number of mice included in each group in each experiment indicated (n = 8). Statistical significance was determined by Student's t test for the effect of with versus without ethanol in WT or ALKO mice in A or Ad-shTRB3, Myc-TRB3, HA-LKB1, or HA-CaMKK2 versus corresponding controls in B–E (*, p < 0.05); ALKO versus WT under the same treatment (with or without ethanol) in A, with versus without Myc-TRB3 in the presence of HA-LKB1 or HA-CaMKK2 in D and E (#, p < 0.05). A–E, TRB3, p-AMPK, AMPK, p-ACC, and ACC proteins (top or left, Western blotting; bottom or right, quantitative measurements of TRB3, p-AMPK, and p-ACC relative to their total proteins or actin).
A role of TRB3 in mediating ethanol inhibition of AMPK was then tested in a mouse hepatocyte cell line AML-12 infected with adenovirus expressing small hairpin RNAs against TRB3 (Ad-shTRB3) or scrambled adenovirus (Ad-scrambled) in the presence or absence of ethanol. As predicted, ethanol-induced AMPK and ACC inhibition, as well as TG accumulation, were blocked by TRB3 knockdown (as demonstrated by decreased Trb3 mRNA and protein) in AML-12 cells compared with control cells (Fig. 6, A and B). The possibility that TRB3 is a downstream effector of ATF4 under ethanol exposure was demonstrated by the fact that ethanol-induced TRB3 expression was blocked by adenovirus expressing a dominant negative ATF4 variant (Ad-DN ATF4) in AML-12 cells (Fig. 6, C and D). Furthermore, ATF4 knockdown-induced stimulation of AMPK and ACC phosphorylation and inhibition of TG accumulation under ethanol treatment was largely reversed by adenovirus-mediated overexpression of TRB3 in AML-12 cells (Fig. 6, E and F).
FIGURE 6.

ATF4 inhibits AMPK activity and promotes TG accumulation via increasing TRB3 expression in vitro under ethanol treatment. A–E, AML-12 cells were infected with Ad-scrambled (−Ad-shTRB3) or Ad-shTRB3 (+Ad-shTRB3), Ad-GFP (−Ad-DN ATF4) or Ad-DN ATF4 (+Ad-DN ATF4), or Ad-GFP (−Ad-TRB3) or Ad-TRB3 (+Ad-TRB3) as indicated for 48 h, followed without (−EtOH) or with 100 mm ethanol treatment (+EtOH) for 24 h. Means ± S.E. (error bars) shown are representative of at least three independent in vitro experiments. Statistical significance was determined by Student's t test for the effect of Ad-shTRB3 or Ad-DN ATF4 versus corresponding control under the same treatment (with or without ethanol) (*, p < 0.05); with versus without ethanol treatment in the presence of the same adenovirus infection in A–D or with versus without Ad-TRB3 in the presence of Ad-DN ATF4 in E and F (#, p < 0.05). A, C, and E, Atf4 and/or Trb3 mRNAs and TRB3, p-AMPK, AMPK, p-ACC, ACC, and ATF4 proteins (left, Western blotting; right, quantitative measurements of TRB3, p-AMPK, and p-ACC relative to their total proteins or actin); B, D, and F, TG contents.
TRB3 Binds to AMPKα Directly and Inhibits Its Phosphorylation
It is shown that TRB3 inhibits AKT phosphorylation by directly binding to AKT/PKB (33). However, little is known regarding the mechanisms of TRB3 inactivation of AMPK. Interestingly, using the multiple sequence alignment tool DNAman, we found that amino acid sequence around Thr-172 of AMPKα is highly similar to the sequence around Thr-308 of AKT/PKB, which was exactly the domain that binds to TRB3 (33) (Fig. 7A). This resemblance suggested that TRB3 may directly bind to AMPK and inhibit its phosphorylation. Indeed, co-immunoprecipitation (co-IP) experiments in HEK-293T cells transfected with Myc-TRB3 and HA-AMPKα revealed that HA-AMPKα could interact with TRB3 (Fig. 7B). Furthermore, GST pull-down assays also showed a direct binding of AMPKα and TRB3 (Fig. 7C).
FIGURE 7.

TRB3 directly binds to AMPKα and inhibits its phosphorylation. A, alignment of the amino acid sequences between AMPK alpha and AKT. B–E, a series of constructs of control vector (−MYC-TRB3 or −GST-TRB3) or Myc-tagged (+MYC-TRB3) or GST-tagged total TRB3 (+GST-TRB3) or deletion mutants (as indicated) were cotransfected with the plasmid expressing control vector (−HA −AMPKα) or HA-tagged AMPKα (+HA −AMPKα) or deletion mutants (as indicated) into HEK-293T cells for 24 h. F, WT and ALKO mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. G and H, primary hepatocytes infected with Ad-DN ATF4 (+Ad-DN ATF4) and transfected with plasmids expressing Myc-TRB3/WT or Myc-TRB3/Δm as indicated for 48 h, followed by treatment with 100 mm ethanol (+EtOH) for 24 h. I and J, WT mice were fed a control (−EtOH) or ethanol-containing diet (+EtOH) for 28 days. Means ± S.E. (error bars) shown are representative of at least three independent in vitro experiments or two independent in vivo experiments, with the number of mice included in each group in each experiment indicated (n = 8). Statistical significance was determined by Student's t test for the effect of Myc-TRB3/WT or Myc-TRB3/Δm versus control plasmid in G and H or with versus without ethanol treatment in I (*, p < 0.05). A, alignment of the amino acid sequences possibly mediating the interaction between AMPKα and AKT; B and E, co-IP assay for the interaction between AMPKα and TRB3; C, GST pull-down assay for the interaction between AMPKα and TRB3; D, schematic representation of TRB3 and AMPKα deletion mutants; F, binding of AMPK and TRB3 in WT and ALKO mouse livers exposed to ethanol or not; G, p-AMPK, AMPK, p-ACC, and ACC protein levels (left, Western blotting; right, quantitative measurements of p-ACC or p-AMPK relative to their total proteins); H, TG contents; I, p-AKT, AKT, and TRB3 protein levels (left, Western blotting; right, quantitative measurements of p-AKT and TRB3 relative to their total protein or actin); J, binding of AKT with TRB3.
To identify the region mediating the interaction between AMPKα and TRB3, we constructed different HA-tagged AMPKα fragments, including residues 287–552 (AMPKα/ΔN), 1–278 (AMPKα/ΔC), and full-length (AMPKα/WT) (Fig. 7D), and co-transfected these plasmids with Myc-TRB3 in HEK-293T cells. A co-IP assay revealed that the region mediating the interaction between AMPKα and TRB3 was located in residues 1–278 of the kinase domain of AMPKα (Fig. 7E). Further deletion mutant analysis showed that an activation loop (residues 157–183) was necessary for AMPK interaction with TRB3 (Fig. 7E). TRB3 has the N-terminal kinase domain (residues 1–113), the kinase-like domain (residues 114–315), and the C-terminal domain (residues 316–355). Truncated mutant analysis showed that neither the N- nor the C-terminal domain of TRB3 was necessary for its interaction with AMPKα (Fig. 7E). In contrast, deletion mutant analysis revealed that the central region (residues 238–266) within the kinase-like domain of TRB3 was required for its interaction with AMPKα (Fig. 7E). Interestingly, we also observed direct binding between TRB3 and AMPK in the livers of WT mice, and this binding was augmented by ethanol exposure and abolished in ALKO mice (Fig. 7F). Furthermore, we found that the effect of TRB3 on AMPK and ACC phosphorylation and TG accumulation depends on the central region within the kinase-like domain of TRB3 (Fig. 7, G and H). Consistent with previous reports showing that TRB3 can bind to and inhibit AKT phosphorylation (33), we also found that the AKT activity is inhibited by ethanol feeding-induced TRB3 activation in the livers of WT mice (Fig. 7I), and the binding between AKT and TRB3 is increased in livers of mice following ethanol treatment (Fig. 7J).
Discussion
Although it has been shown that ATF4 expression is induced by ethanol (22, 23, 24, 34), a direct effect of ATF4 on alcoholic liver steatosis has not been described previously. Furthermore, the knowledge about tissue-specific function of ATF4 is very limited, except for a study showing that ATF4 expressed in the osteoblast or liver is important for blood glucose control (36). In this study, we showed that knock-out of ATF4 in the liver has no effect on hepatic TG accumulation under a pair-fed control diet, but it significantly prevented ethanol-induced liver steatosis. Consistent with our work, other studies showed that CYP2E1 (cytochrome P450 2e1) regulates ethanol-induced liver steatosis, associated with corresponding changes in ATF4 expression (22). In addition, we believe that the effect of ATF4 is a hepatocyte cell-autonomous response because similar results were obtained in our in vitro study using AML-12 cells. Furthermore, we did not observe any difference in body weight and fat mass between ALKO and control mice under an ethanol diet. Thus, our current study demonstrates a novel function of hepatic ATF4 in the regulation of ethanol-induced liver steatosis, which should shed light on the causes of ethanol-induced liver steatosis. Moreover, elevated ATF4 expression is also observed in other ethanol-induced liver injury (37), suggesting that ATF4 might be involved in the development of other ethanol-induced liver metabolic diseases, which need to be studied in the future.
In recent years, accumulated evidence has identified several important targets of ethanol action in the liver, including SIRT1 (3), SREBP-1 (8), PGC-1α (5), and AMPK (6–8, 38). Among those molecules, AMPK appears to be the most important target of ethanol in liver. AMPK activity is impaired by ethanol exposure and then stimulates downstream effectors, for example, lipin 1 (39), resulting in subsequent inhibition of fatty acid β-oxidation and stimulation of lipogenesis that contribute to accumulation of TG in vitro or in vivo (7, 40, 41). Except for SIRT1 (3), FGF21 (fibroblast growth factor 21) (41), and adiponectin (7), very little is known about the molecular mechanisms leading to the impairment of AMPK activity under ethanol exposure. A previous study (20) has shown that AMPK activity is regulated by ATF4 in the hypothalamus, whereas no study has tested whether it may mediate ATF4 downstream effects. As observed in the hypothalamus (20), we showed that ethanol-fed hepatic AMPK activity was stimulated in ALKO mice. The importance of the activation of AMPK in ALKO mouse resistance to ethanol-induced liver steatosis was demonstrated by the reversal effects of Ad-DN AMPK on the protective effects of ATF4 knock-out. Consistent with these results, we also provided evidence showing that the protective role of ATF4 knock-out and the reversal effects of AMPK knockdown are associated with the changes in genes and proteins related to β-oxidation and lipogenesis, as shown previously (6, 8). These results not only demonstrate an important role of AMPK in ATF4 regulation of ethanol-induced liver steatosis but also provide evidence for a novel upstream signaling to AMPK inhibition during ethanol exposure.
TRB3, a known direct target of ATF4 (25, 29, 30), is induced by ethanol treatment in cells (33, 42) and livers, as shown in our current study. However, the role of TRB3 in alcoholic liver steatosis has not been studied previously. Consistent with previous results (31), we found that TRB3 expression was induced by ethanol in hepatocytes in an ATF4-dependent manner. We further provided evidence demonstrating that TRB3 functions as a downstream signal of ATF4 in the regulation of ethanol-induced inhibition of AMPK and ACC phosphorylation and stimulation of TG accumulation in AML-12 cells. Interestingly, we found that overexpression of TRB3 significantly decreased mRNA levels of ATF4 in AML-12 cells, which is possibly due to a feedback regulation, as shown previously (29). We also noticed that there was a difference in TRB3 protein levels between the livers of mice fed an ethanol diet for 28 days and the AML-12 cells treated with ethanol for 24 h. We speculated that the difference might be caused by the difference in the duration of ethanol treatment and the sensitivity to ethanol stimulation in mice livers and AML-12 cells.
In this study, we not only demonstrated an important role of TRB3 in mediating ATF4 regulation of ethanol-induced response, including inhibition of AMPK and ACC activity and stimulation of TG accumulation, but also provided evidence showing that TRB3 could directly regulate AMPK activity in mouse primary hepatocytes. Interestingly, we found that TRB3 overexpression not only inhibited basal AMPK and ACC activity but also blocked LKB1- or CaMKK2-stimulated AMPK and ACC activation in mouse primary hepatocytes. In contrast to our results, there are other studies showing that decreased TRB3 is associated with decreased AMPK activity in LKB1 muscle knock-out mice (43), or TRB3 does not affect AMPK activity in HepG2 cells (44). The differences in the effects of TRB3 on AMPK activity could be due to the involvement of different signals or treatment under different experimental conditions.
We further explored the possible mechanisms underlying TRB3 control of AMPK activity. A previous study (33) has shown that TRB3 directly binds to AKT and blocks its phosphorylation at Thr-308. It has also been shown that ethanol increases cytosolic TRB3 protein levels, and TRB3 binds to AKT and prevents its plasma membrane association (42). Using multiple sequence alignment tools, we found that AKT and AMPKα have conserved amino acid sequence, especially for the amino acids around Thr-172 of AMPK and Thr-308 of AKT. We therefore hypothesized that TRB3 binds to AMPKα and prevents its phosphorylation in a similar manner to how TRB3 binds to AKT. This speculation was then confirmed by GST pull-down and co-IP assays, which revealed that TRB3 could bind directly to AMPKα. Deletion mutant analysis showed that the interaction region of AMPKα was located in the activation loop. Interestingly, the central region (residues 238–266) of the kinase-like domains of TRB3, which binds to AKT (33), was not only responsible for the interaction between TRB3 and AMPKα but also indispensable for its inhibition of AMPK and ACC phosphorylation. Endogenous co-IP indicated that ethanol treatment promoted the interaction between TRB3 and AMPKα. Because ethanol increases TRB3 expression and the increase is blocked by ATF4 knock-out, we speculate that the altered binding between TRB3 and AMPK in vivo might be determined by the altered TRB3 levels. All of these results implied that TRB3 is a novel important suppressor of AMPK via directly binding to it. We have to note that our study represents the first time that a direct binding of TRB3 to AMPK has been demonstrated. Because TRB3 is also a suppressor of AKT (33), and the AKT activity is decreased and the binding between AKT and TRB3 is increased in the livers of ethanol-feeding mice, AKT might also be involved in ATF4-regulated ethanol-induced liver steatosis. However, activation of AKT promotes fatty acid synthesis (45); therefore, given the effects of ethanol on stimulating TG accumulation, we believe that the inactivated AMPK plays a more significant role than AKT following ethanol treatment.
It is well known that ATF4 is an endoplasmic reticulum stress induced transcription factor (46). Alcohol-induced endoplasmic reticulum stress has recently been established as an important mechanism for ethanol-induced liver diseases (47); therefore, whether endoplasmic reticulum stress is involved in ATF4-regulated ethanol-induced liver steatosis needs to be studied in the future.
In summary, we have shown that ethanol induces hepatic ATF4 and TRB3 expression, which subsequently inhibits AMPK activity and results in liver steatosis. Therefore, hepatic ATF4 knock-out mice were resistant to ethanol-induced liver steatosis. Furthermore, we demonstrate TRB3 as a direct upstream regulator of AMPK. These results highlight the ATF4-TRB3-AMPK axis as a major signaling route in the pathogenesis of alcoholic liver steatosis and also a potential therapeutic target for the treatment of alcoholic liver steatosis and possibly other related liver diseases.
Experimental Procedures
Animal Studies and Chronic Ethanol Model
The 8–10-week-old male C57/BL6 background liver-specific ALKO mice were bred by crossing ATF4f/f mice with albumin-Cre mice (26). ALKO and ATF4f/f mice were divided into four groups: 1) ATF4f/f mice pair-fed a control diet (Bio-Serv, Flemington, NJ); 2) ATF4f/f mice fed an ethanol diet (Bio-Serv); 3) ALKO mice pair-fed a control diet; and 4) ALKO mice fed an ethanol diet. The animal feeding protocol was described previously (48). Briefly, all mice were fed a control diet for 5 days to adapt to liquid diet feeding, and on day 6, ethanol-fed mice group were changed to the isocaloric alcoholic diet containing 5% ethanol. All mice were then fed each liquid diet for 4 weeks. For the ethanol feeding period, food intake of the ethanol-fed mice was recorded, and the weight-matched control feeding mice were pair-fed with the same volume of the control diet, so that ethanol- and pair-fed mice consumed equal amounts of diet and ingested the same energy. All experiments were conducted in accordance with the guidelines of the institutional animal care and use committee of the Institute for Nutritional Sciences, Shanghai Institute for Biological Sciences, Chinese Academy of Sciences. Animals were housed individually in a room with controlled temperature (20–22 °C), humidity (55–65%), and lighting (on at 7 a.m. and off at 7 p.m.).
Cell Culture and Treatment
The mouse AML-12 hepatocyte cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured as described previously (8). For ethanol treatment in cells, the incubator was added with water containing the same concentration of ethanol to prevent ethanol volatilization as described before (48). Primary hepatocytes were prepared by collagenase perfusion as described previously (49) and transfected with plasmid using an Effectene transfection reagent (Qiagen, Hilden, Germany) following the manufacturer's instructions. HEK-293T cells were transfected with plasmid using Lipofectamine 2000 (Invitrogen). Myc-TRB3 plasmid was amplified from mouse cDNA and cloned into Myc-tagged plasmid. Plasmid containing AMPKα coding sequence was a gift from Dr. Jia Li (Shanghai Institute of Materia Medica, Chinese Academy of Sciences) and cloned into HA-tagged plasmid. HA-LKB1 and HA-CaMKK2 were kindly provided by Dr. Hongbin Ji (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences) and Dr. Hao Ying (Shanghai Institute of Nutritional Sciences, Chinese Academy of Sciences), respectively.
Generation and Administration of Recombinant Adenovirus
The recombinant adenovirus expressing a dominant negative ATF4 variant (Ad-DN ATF4) was generated using the AdEasyTM vector system (Qiagen, Hilden, Germany). Adenovirus expressing a dominant negative AMPK variant (Ad-DN AMPK) was kindly provided by Dr. Jia Li. Adenovirus expressing TRB3 (Ad-TRB3) was purchased from Hanbio (Shanghai, China). Adenovirus expressing scrambled or short hairpin RNA for mouse TRB3 was generated as reported previously (50). Viruses were diluted in PBS and administered at a dose of 1.5 × 107 plaque-forming units/well in a 12-well plate or through tail vein injection using 5 × 108 plaque-forming units/mouse.
Biochemical Analysis
Hepatic and cellular lipids were extracted with chloroform/methanol (2:1), as described previously (51, 52). TG, TC, FFAs, ALT, and AST were determined using a TG kit (BHKT Clinical Reagents, Beijing, China), a TC kit (SSUF-C, Shanghai, China), an FFA kit (Wako Pure Chemical Industries, Osaka, Japan), and an AST or ALT kit (ShenSuoYouFu, Shanghai, China), respectively. All of these assays were performed according to the manufacturer's instructions.
Histopathological Analysis
Liver sections were stained with hematoxylin and eosin as reported previously. Frozen sections of liver were stained with Oil Red O (Sigma).
RNA Isolation and Quantitative RT-PCR
RNA isolation and RT-PCR were performed as described previously (19). GAPDH was used as an internal control for each gene of interest. The sequences of primers used in this study is shown in Table 1.
TABLE 1.
Primers for quantitative RT-PCR
| Primer | Sequence |
|---|---|
| Acc-F | 5′-TGACAGACTGATCGCAGAGAAAG-3′ |
| Acc-R | 5′-TGGAGAGCCCCACACACA-3′ |
| Acl-F | 5′-GCCAGCGGGAGCACATC-3′ |
| Acl-R | 5′-CTTTGCAGGTGCCACTTCATC-3′ |
| Apob-F | 5′-CGTGGGCTCCAGCATTCTA-3′ |
| Apob-R | 5′-TCACCAGTCATTTCTGCCTTTG-3′ |
| Apoe-F | 5′-GCTGGGTGCAGACGCTTT-3′ |
| Apoe-R | 5′-TGCCGTCAGTTCTTGTGTGACT-3′ |
| Atf4-F | 5′-CCTGAACAGCGAAGTGTTGG-3′ |
| Atf4-R | 5′-TGGAGAACCCATGAGGTTTCAA-3′ |
| Cd36-F | 5′-TGGAGCTGTTATTGGTGCAG-3′ |
| Cd36-R | 5′-TGGGTTTTGCACATCAAAGA-3′ |
| Chrebp-F | 5′-CTGGGGACCTAAACAGGAGC-3′ |
| Chrebp-R | 5′-GAAGCCACCCTATAGCTCCC-3′ |
| Cpt1α-F | 5′-TGGCATCATCACTGGTGTGTT-3′ |
| Cpt1α-R | 5′-GTCTAGGGTCCGATTGATCTTTG-3′ |
| Fas-F | 5′-GGAGGTGGTGATAGCCGGTAT-3′ |
| Fas-R | 5′-TGGGTAATCCATAGAGCCCAG-3′ |
| Fabp-F | 5′-GCTGCGGCTGCTGTATGA-3′ |
| Fabp-R | 5′-CACCGGCCTTCTCCATGA-3′ |
| Pparα-F | 5′-CTGCAGAGCAACCATCCAGAT-3′ |
| Pparα-R | 5′-GCCGAAGGTCCACCATTTT-3′ |
| Pparγ-F | 5′-GAGAAGCTGTTGGCGGAGAT-3′ |
| Pparγ-R | 5′-GCTCGCAGATCAGCAGACTCT-3′ |
| Scd1-F | 5′-CCGGAGACCCCTTAGATCGA-3′ |
| Scd1-R | 5′-TAGCCTGTAAAAGATTTCTGCAAACC-3′ |
| Srebp-1c-F | 5′-GGAGCCATGGATTGCACATT-3′ |
| Srebp-1c-R | 5′-GGCCCGGGAAGTCACTGT-3′ |
| Trb3-F | 5′-CAGGAAGAAACCGTTGGAGTT-3′ |
| Trb3-R | 5′-CCAAAAGGATATAAGGCCCCAGT-3′ |
Western Blotting
Western blotting was performed as described previously (17). Primary antibodies (anti-ATF4, anti-TRB3, and anti-Myc tag (Santa Cruz Biotechnology, Inc., Dallas, TX); anti-AMPK, anti-p-AMPK (Thr-172/183), anti-ACC, anti-p-ACC (Ser-79), anti-AKT, and anti-p-AKT (Thr-308) (Cell Signaling Technology, Danvers, MA); and anti-HA and anti-β-actin (Sigma)) were incubated overnight at 4 °C, and specific proteins were visualized by ECL Plus (GE Healthcare).
Co-IP and GST Pull-down Assays
Co-IP and GST pull-down assays were conducted as described previously (35). For the GST pull-down assay, 293T cells were cotransfected with the plasmids expressing the GST-His8-tagged TRB3 and AMPKα.
Statistics
All data are expressed as means ± S.E. Significant differences were assessed by two-tailed Student's t test or one-way analysis of variance followed by the Student-Newman-Keuls test. p < 0.05 was considered statistically significant.
Author Contributions
K. L., C. W., and Y. X. researched data and wrote, reviewed, and edited the manuscript. J. Y., T. X., B. L., Y. G., and J. D. researched data and contributed to discussion. S. C. researched data and provided research material. F. G. directed the project, contributed to discussion, and wrote, reviewed, and edited the manuscript. F. G. and C. W. are the guarantors of this work and, as such, had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
This work was supported by National Natural Science Foundation Grants 81130076, 81325005, 31271269, 81100615, 81390350, 81300659, and 81471076; Basic Research Project of Shanghai Science and Technology Commission Grant 13JC1409000; International S&T Cooperation Program of China (Singapore Grant 2014DFG32470); and the CAS/SAFEA international partnership program for creative research teams. The authors declare that they have no conflicts of interest with the contents of this article.
- FFA
- free fatty acid
- ALKO
- ATF4 liver-specific knockout
- AMPK
- AMP-activated protein kinase
- FAS
- fatty acid synthase
- TG
- triglyceride
- ACC
- acetyl-CoA carboxylase
- TC
- total cholesterol
- ALT
- alanine aminotransferase
- AST
- aspartate transaminase
- LKB1
- liver kinase B1
- CaMKK2
- calmodulin-activated kinase kinase 2
- co-IP
- co-immunoprecipitation
- Ad-DN AMPK
- adenovirus expressing a dominant negative AMPK variant
- Ad-GFP
- adenovirus expressing control green fluorescent protein
- Ad-shTRB3
- adenovirus expressing small hairpin RNAs against TRB3
- Ad-scrambled
- scrambled adenovirus
- p-AMPK and p-ACC
- phosphorylated AMPK and ACC, respectively.
References
- 1. Purohit V., Gao B., and Song B. J. (2009) Molecular mechanisms of alcoholic fatty liver. Alcohol. Clin. Exp. Res. 33, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rasineni K., and Casey C. A. (2012) Molecular mechanism of alcoholic fatty liver. Indian J. Pharmacol. 44, 299–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yin H., Hu M., Liang X., Ajmo J. M., Li X., Bataller R., Odena G., Stevens S. M. Jr., and You M. (2014) Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146, 801–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. You M., Fischer M., Deeg M. A., and Crabb D. W. (2002) Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 277, 29342–29347 [DOI] [PubMed] [Google Scholar]
- 5. Lieber C. S., Leo M. A., Wang X., and Decarli L. M. (2008) Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1α in rats. Biochem. Biophys. Res. Commun. 370, 44–48 [DOI] [PubMed] [Google Scholar]
- 6. You M., Matsumoto M., Pacold C. M., Cho W. K., and Crabb D. W. (2004) The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 127, 1798–1808 [DOI] [PubMed] [Google Scholar]
- 7. Shen Z., Liang X., Rogers C. Q., Rideout D., and You M. (2010) Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G364–G374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu M., Wang F., Li X., Rogers C. Q., Liang X., Finck B. N., Mitra M. S., Zhang R., Mitchell D. A., and You M. (2012) Regulation of hepatic lipin-1 by ethanol: role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology 55, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Winder W. W., and Hardie D. G. (1996) Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am. J. Physiol. 270, E299–E304 [DOI] [PubMed] [Google Scholar]
- 10. Kudo N., Barr A. J., Barr R. L., Desai S., and Lopaschuk G. D. (1995) High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 270, 17513–17520 [DOI] [PubMed] [Google Scholar]
- 11. Tomita K., Tamiya G., Ando S., Kitamura N., Koizumi H., Kato S., Horie Y., Kaneko T., Azuma T., Nagata H., Ishii H., and Hibi T. (2005) AICAR, an AMPK activator, has protective effects on alcohol-induced fatty liver in rats. Alcohol. Clin. Exp. Res. 29, 240S–245S [DOI] [PubMed] [Google Scholar]
- 12. Bergheim I., Guo L., Davis M. A., Lambert J. C., Beier J. I., Duveau I., Luyendyk J. P., Roth R. A., and Arteel G. E. (2006) Metformin prevents alcohol-induced liver injury in the mouse: critical role of plasminogen activator inhibitor-1. Gastroenterology 130, 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karpinski B. A., Morle G. D., Huggenvik J., Uhler M. D., and Leiden J. M. (1992) Molecular cloning of human CREB-2: an ATF/CREB transcription factor that can negatively regulate transcription from the cAMP response element. Proc. Natl. Acad. Sci. U.S.A. 89, 4820–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vallejo M., Ron D., Miller C. P., and Habener J. F. (1993) C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to cAMP response elements. Proc. Natl. Acad. Sci. U.S.A. 90, 4679–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang X., Matsuda K., Bialek P., Jacquot S., Masuoka H. C., Schinke T., Li L., Brancorsini S., Sassone-Corsi P., Townes T. M., Hanauer A., and Karsenty G. (2004) ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry syndrome. Cell 117, 387–398 [DOI] [PubMed] [Google Scholar]
- 16. Lange P. S., Chavez J. C., Pinto J. T., Coppola G., Sun C. W., Townes T. M., Geschwind D. H., and Ratan R. R. (2008) ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J. Exp. Med. 205, 1227–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C., Li H., Meng Q., Du Y., Xiao F., Zhang Q., Yu J., Li K., Chen S., Huang Z., Liu B., and Guo F. (2014) ATF4 deficiency protects hepatocytes from oxidative stress via inhibiting CYP2E1 expression. J. Cell Mol. Med. 18, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H., Meng Q., Xiao F., Chen S., Du Y., Yu J., Wang C., and Guo F. (2011) ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem. J. 438, 283–289 [DOI] [PubMed] [Google Scholar]
- 19. Wang C., Huang Z., Du Y., Cheng Y., Chen S., and Guo F. (2010) ATF4 regulates lipid metabolism and thermogenesis. Cell Res. 20, 174–184 [DOI] [PubMed] [Google Scholar]
- 20. Zhang Q., Yu J., Liu B., Lv Z., Xia T., Xiao F., Chen S., and Guo F. (2013) Central activating transcription factor 4 (ATF4) regulates hepatic insulin resistance in mice via S6K1 signaling and the vagus nerve. Diabetes 62, 2230–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao G., Zhang T., Yu S., Lee S., Calabuig-Navarro V., Yamauchi J., Ringquist S., and Dong H. H. (2013) ATF4 protein deficiency protects against high fructose-induced hypertriglyceridemia in mice. J. Biol. Chem. 288, 25350–25361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Magne L., Blanc E., Legrand B., Lucas D., Barouki R., Rouach H., and Garlatti M. (2011) ATF4 and the integrated stress response are induced by ethanol and cytochrome P450 2E1 in human hepatocytes. J. Hepatol. 54, 729–737 [DOI] [PubMed] [Google Scholar]
- 23. Chen Y., Gao H., Yin Q., Chen L., Dong P., Zhang X., and Kang J. (2013) ER stress activating ATF4/CHOP-TNF-α signaling pathway contributes to alcohol-induced disruption of osteogenic lineage of multipotential mesenchymal stem cell. Cell Physiol. Biochem. 32, 743–754 [DOI] [PubMed] [Google Scholar]
- 24. Sun Q., Zhong W., Zhang W., Li Q., Sun X., Tan X., Sun X., Dong D., and Zhou Z. (2015) Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G757–G766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carraro V., Maurin A. C., Lambert-Langlais S., Averous J., Chaveroux C., Parry L., Jousse C., Ord D., Ord T., Fafournoux P., and Bruhat A. (2010) Amino acid availability controls TRB3 transcription in liver through the GCN2/eIF2α/ATF4 pathway. PLoS One 5, e15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li K., Zhang J., Yu J., Liu B., Guo Y., Deng J., Chen S., Wang C., and Guo F. (2015) MicroRNA-214 suppresses gluconeogenesis by targeting activating transcriptional factor 4. J. Biol. Chem. 290, 8185–8195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kahn B. B., Alquier T., Carling D., and Hardie D. G. (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25 [DOI] [PubMed] [Google Scholar]
- 28. Browning J. D., and Horton J. D. (2004) Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 114, 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jousse C., Deval C., Maurin A. C., Parry L., Chérasse Y., Chaveroux C., Lefloch R., Lenormand P., Bruhat A., and Fafournoux P. (2007) TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J. Biol. Chem. 282, 15851–15861 [DOI] [PubMed] [Google Scholar]
- 30. Ord D., Meerits K., and Ord T. (2007) TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp. Cell Res. 313, 3556–3567 [DOI] [PubMed] [Google Scholar]
- 31. Yao X. H., and Nyomba B. L. (2008) Hepatic insulin resistance induced by prenatal alcohol exposure is associated with reduced PTEN and TRB3 acetylation in adult rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R1797–R1806 [DOI] [PubMed] [Google Scholar]
- 32. Woods A., Dickerson K., Heath R., Hong S. P., Momcilovic M., Johnstone S. R., Carlson M., and Carling D. (2005) Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 [DOI] [PubMed] [Google Scholar]
- 33. Du K., Herzig S., Kulkarni R. N., and Montminy M. (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 34. Chen G., Ma C., Bower K. A., Shi X., Ke Z., and Luo J. (2008) Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J. Neurosci. Res. 86, 937–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu F., Wang Y. L., Chang J. J., Du S. C., Diao L., Jiang N., Wang H. J., Ma D., and Zhang J. (2014) Mammalian sterile 20-like kinase 1/2 inhibits the Wnt/β-catenin signalling pathway by directly binding casein kinase 1ϵ. Biochem. J. 458, 159–169 [DOI] [PubMed] [Google Scholar]
- 36. Yoshizawa T., Hinoi E., Jung D. Y., Kajimura D., Ferron M., Seo J., Graff J. M., Kim J. K., and Karsenty G. (2009) The transcription factor ATF4 regulates glucose metabolism in mice through its expression in osteoblasts. J. Clin. Invest. 119, 2807–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malhi H., and Kaufman R. J. (2011) Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. García-Villafranca J., Guillén A., and Castro J. (2008) Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 90, 460–466 [DOI] [PubMed] [Google Scholar]
- 39. Bi L., Jiang Z., and Zhou J. (2015) The role of lipin-1 in the pathogenesis of alcoholic fatty liver. Alcohol Alcohol. 50, 146–151 [DOI] [PubMed] [Google Scholar]
- 40. Jiang Z., Zhou J., Zhou D., Zhu Z., Sun L., and Nanji A. A. (2015) The adiponectin-SIRT1-AMPK pathway in alcoholic fatty liver disease in the rat. Alcohol. Clin. Exp. Res. 39, 424–433 [DOI] [PubMed] [Google Scholar]
- 41. Zhu S., Ma L., Wu Y., Ye X., Zhang T., Zhang Q., Rasoul L. M., Liu Y., Guo M., Zhou B., Ren G., and Li D. (2014) FGF21 treatment ameliorates alcoholic fatty liver through activation of AMPK-SIRT1 pathway. Acta Biochim. Biophys. Sin. 46, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 42. He L., Simmen F. A., Mehendale H. M., Ronis M. J., and Badger T. M. (2006) Chronic ethanol intake impairs insulin signaling in rats by disrupting Akt association with the cell membrane: role of TRB3 in inhibition of Akt/protein kinase B activation. J. Biol. Chem. 281, 11126–11134 [DOI] [PubMed] [Google Scholar]
- 43. Koh H. J., Arnolds D. E., Fujii N., Tran T. T., Rogers M. J., Jessen N., Li Y., Liew C. W., Ho R. C., Hirshman M. F., Kulkarni R. N., Kahn C. R., and Goodyear L. J. (2006) Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol. Cell Biol. 26, 8217–8227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vara D., Salazar M., Olea-Herrero N., Guzmán M., Velasco G., and Díaz-Laviada I. (2011) Anti-tumoral action of cannabinoids on hepatocellular carcinoma: role of AMPK-dependent activation of autophagy. Cell Death Differ. 18, 1099–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang D., and Sul H. S. (1998) Insulin stimulation of the fatty acid synthase promoter is mediated by the phosphatidylinositol 3-kinase pathway. Involvement of protein kinase B/Akt. J. Biol. Chem. 273, 25420–25426 [DOI] [PubMed] [Google Scholar]
- 46. Whitney M. L., Jefferson L. S., and Kimball S. R. (2009) ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem. Biophys. Res. Commun. 379, 451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ji C. (2014) New insights into the pathogenesis of alcohol-induced ER stress and liver diseases. Int. J. Hepatol. 2014, 513787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bertola A., Mathews S., Ki S. H., Wang H., and Gao B. (2013) Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 8, 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Q., Jiang L., Wang J., Li S., Yu Y., You J., Zeng R., Gao X., Rui L., Li W., and Liu Y. (2009) Abrogation of hepatic ATP-citrate lyase protects against fatty liver and ameliorates hyperglycemia in leptin receptor-deficient mice. Hepatology 49, 1166–1175 [DOI] [PubMed] [Google Scholar]
- 50. Yu J., Xiao F., Guo Y., Deng J., Liu B., Zhang Q., Li K., Wang C., Chen S., and Guo F. (2015) Hepatic phosphoserine aminotransferase 1 regulates insulin sensitivity in mice via Tribbles homolog 3. Diabetes 64, 1591–1602 [DOI] [PubMed] [Google Scholar]
- 51. Folch J., Lees M., and Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 52. Xiao Y., Liu H., Yu J., Zhao Z., Xiao F., Xia T., Wang C., Li K., Deng J., Guo Y., Chen S., Chen Y., and Guo F. (2016) Activation of ERK1/2 ameliorates liver steatosis in leptin receptor-deficient (db/db) mice via stimulating ATG7-dependent autophagy. Diabetes 65, 393–405 [DOI] [PubMed] [Google Scholar]