

Abstract
Investigations on cellular protein interaction networks (PINs) reveal that proteins that constitute hubs in a PIN are notably enriched in Intrinsically Disordered Proteins (IDPs) compared to proteins that constitute edges, highlighting the role of IDPs in signaling pathways. Most IDPs rapidly undergo disorder-to-order transitions upon binding to their biological targets to perform their function. Conformational dynamics enables IDPs to be versatile and to interact with a broad range of interactors under normal physiological conditions where their expression is tightly modulated. IDPs are involved in many cellular processes such as cellular signaling, transcriptional regulation, and splicing; thus, their high-specificity/low-affinity interactions play crucial roles in many human diseases including cancer. Prostate cancer (PCa) is one of the leading causes of cancer-related mortality in men worldwide. Therefore, identifying molecular mechanisms of the oncogenic signaling pathways that are involved in prostate carcinogenesis is crucial. In this review, we focus on the aspects of cellular pathways leading to PCa in which IDPs exert a primary role.
Keywords: inflammation, intrinsically disordered proteins, prostate cancer
INTRODUCTION
Prostate cancer (PCa) is the second most frequently diagnosed cancer and the sixth leading cause of cancer deaths in males.1,2 In recent years, several targets have been identified and current treatment methods for PCa include radical prostatectomy or radiation, androgen deprivation therapy (ADT), and chemotherapy. However, most patients treated with ADT develop castrate-resistant PCa (CRPC).3 Hence, there is a great interest in unveiling the molecular events that are crucial for the development of the disease and in introducing new agents for the treatment of its advanced stages. Targeting the androgen receptor (AR) signaling pathway is the current standard treatment for hormone-naive CRPC. After 2–3 years, however, AR signaling is reactivated leading to castrate-resistant PCa.4 Abiraterone acetate and enzalutamide represent the next-generation of antiandrogen agents and are routinely used in the clinical treatment compared to flutamide or cyproterone acetate which are on a significant decline in the chemotherapy.5,6,7 Cabazitaxel was approved for CRPC after docetaxel relapse.8 Recently, radium-223 chloride (radium-223) was approved for the treatment of metastatic CRPC.9 Table 1 provides an overview of drugs currently approved for treatment of CRPC and their molecular targets as well as the overall survival (OS) and the progression-free survival (PFS) rates. These data underscore the dire need for new and effective therapeutics to treat and manage PCa.10
Table 1.
Drugs currently approved for metastatic CRPC
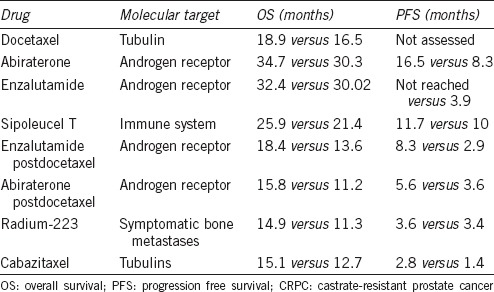
Recent investigations on protein interaction networks (PINs) revealed that proteins that constitute hubs in a PIN are more disordered compared to proteins that constitute edges (schematic representations of selected PINs are presented in Figure 1). These proteins are known as intrinsically disordered proteins (IDPs)11,12,13,14,15,16,17,18,19,20,21,22 or hybrid proteins possessing both structured domains and disordered protein regions (IDPRs). Their structural plasticity and conformational adaptability to changes in their environment, binding promiscuity and unique capability to fold differently while interacting with different binding partners23,24 define a wide set of functional advantages of IDPs/IDPRs over fully ordered proteins. These factors determine the abundant involvement of IDPs/IDPRs in various signaling, regulatory, and recognition processes. Furthermore, deregulation of disordered proteins leads to protein misfolding, misidentification and inaccurate signaling promoting numerous human diseases such as cancer, cardiovascular disease neurodegenerative diseases, and diabetes.25,26,27 Several cellular mechanisms such as chromosomal translocations, aberrant splicing, altered expression, posttranslational modifications, aberrant proteolytic degradation, and defective trafficking are some of the factors that induce pathogenic transformations of IDPs.28 The misfolding of many proteins is often accompanied with protein aggregation causing several human diseases that originate from the deposition of protein aggregates formed from specific proteins or protein fragments, which accumulate in a variety of organs and tissues.29,30,31,32,33,34,35,36,37 In contrast, several diseases are caused by misfolding and, therefore, dysfunctional proteins.17,38,39
Figure 1.
Schematic representation of PIN (protein interaction networks) of several proteins investigated in this review: (a) SOCS1, IRS1: insulin receptor substrate 1, IL4R: interleukin 4 receptor, JAK2Janus kinase 2, RELAv-rel: reticuloendotheliosis viral oncogene homolog A (avian), TCEB1: transcription elongation factor B (SIII), (b) IRF1: interferon regulatory factor 1, ITGB3: integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61), ITGAV: integrin, alpha V, TP53: tumor protein p53, UBC: ubiquitin C, (c) IGFBP1: insulin-like growth factor binding protein 1, IGFBP4: insulin-like growth factor binding protein 4, IGFBP5: insulin-like growth factor binding protein 5, IGF1R: insulin-like growth factor 1 receptor, IGFBP3: insulin-like growth factor binding protein 3, (d) PAK6p21 protein, (Cdc42/Rac)-activated kinase 6, PXN: paxillin, RAC3: ras-related C3 botulinum toxin substrate 3.
Several well-known cancer-related proteins with experimentally confirmed IDPRs include p53,40 BRCA1,41 HPV protein,42 PTEN,43 and an overwhelming majority of the Cancer/Testis Antigens (CTAs).44 Bioinformatic approaches allow investigators to predict the presence of IDRs in natural proteins by assembling specific datasets of proteins associated with a given disease and by computationally analyzing these datasets using a number of disorder predictors.38 This approach has revealed that the majority of proteins involved in eukaryotic signal transduction are IDRs, and further, 79% of cancer-associated and 66% of cell-signaling proteins contain predicted regions of disorder of thirty residues or longer.19,23 Similarly as many as 40%–50% of all eukaryotic genes are predicted to encode proteins containing lengthy disordered segments (>forty residues).45,46 These proteins are usually involved in molecular recognition and assembly, protein modification (e.g., phosphorylation, acetylation, methylation) and entropic chain activities (e.g., linkers, springs, and spacers). In contrast to globular proteins, IDPs usually are constituted by only single continuous segments designated molecular recognition elements (MoRE/MoRF) or linear motifs, whereas the binding sites of ordered proteins are more segmented.25 These motifs are short, conserved within larger protein segments that function as sites of regulation, and many are posttranslationally modified.
Chromosomal translocation causes several forms of cancer, such as acute myelogenous leukemia (AML), acute lymphoblastic leukemia (ALL), chronic myelogenous leukemia (CML), chronic myelomonocytic leukemia (CMML), primary myelofibrosis (PMF), anaplastic large cell lymphoma, non-Hodgkin's lymphoma, Ewing's sarcoma (EWS), colorectal cancer (CRC), nonsmall cell lung cancer, lung adenocarcinoma, and sporadic and radiation-associated papillary thyroid carcinomas. Computational analysis of the 406 translocation-related human proteins revealed that these cancer-related proteins are enriched in disordered regions, with the translocation breakpoints mostly located outside the functional domains.47 In this scenario, although the flexibility of Alternative Splicing (AS) represents an evolutionary advantage for higher eukaryotes, it also represents a risk. Indeed, defective AS regulation correlates with the onset and progression of human cancers, and many cancer-associated genes are regulated through AS suggesting a significant role of this posttranscriptional regulatory mechanism in the production of oncogenes and tumor suppressors.48,49,50 Most proteins resulting from aberrant splicing are enriched in intrinsic disorder, such as those involved in apoptosis51 and in spliceosome assembly.7,52,53
Some of the crucial proteins affected by AS in PCa (e.g., AR, zinc finger transcription factor, Kruppel-like-factor 6, Bcl-x, and cyclin D1)54 are known to contain IDPRs. A case of extensive AS of the TMPRSS2-ERG gene fusion represents an important illustration of the combined effects of chromosomal translocation and AS in cancer progression.54,55 ETS-related gene (ERG), which is a member of the erythroblast transformation-specific (ETS) transcription factor family, is typically expressed at very low levels in benign prostate epithelial cells but, when fused with the androgen-responsive transmembrane protease serine 2 (TMPRSS2), generates a PCa oncogene. Furthermore, this fusion-derived gene undergoes AS and generates multiple mRNA variants encoding both full-length ERG proteins and isoforms lacking the ETS domain. Notably, an increase in the abundance of transcripts encoding full-length ERG was shown to correlate with less favorable outcomes in PCa patients.55 Thus, it follows that a plethora of cellular events are responsible for initiation and progression of PCa, but most of them remain poorly understood. Here, we focus on some of the important molecular mechanisms associated with oncogenic signaling pathways that are involved in prostate carcinogenesis namely, inflammation, insulin-like growth factor axis, and androgen receptor signaling pathway.
INFLAMMATION
SOCS proteins in JAK-STAT cellular pathway
There are several types of cells in the prostate, but nearly all PCas start in the gland cells. This kind of cancer is known as adenocarcinoma.45,46 Currently, other than grading by Gleason Score, there are no effective biomarkers to discern patients with indolent disease from those with aggressive disease which needs to be treated immediately. Investigations by several labs indicate that chronic inflammation is a known contributor to several forms of human cancer, with an estimated 20% of adult cancers attributable to chronic inflammatory conditions caused by infectious agents, chronic noninfectious inflammatory diseases and or other environmental factors. Thus, chronic inflammation is now regarded as an ‘enabling characteristic’ of human cancer.56,57 In PCa, chronic inflammation has been postulated to be a trigger for epithelial to mesenchymal transition (EMT), a process that leads to increased CRPC and drug resistant disease. In fact, infiltration of the tumor tissue with M2 macrophages has been shown to induce an EMT, just as these wound-healing macrophages do in normal cells after a cut. This process is visible on histological examination by white blood cells, increased Gleason Score, and certain markers; however easily assayable early detection of EMT in PCa has remained elusive.
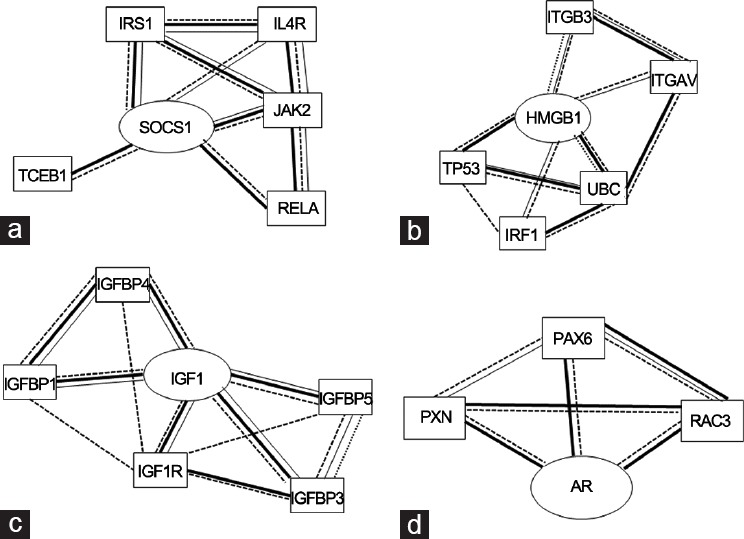
Since the Janus kinases/signal transducer and activator of transcription factors (JAK-STAT) pathway play a key role in the inflammatory reaction in cancer,58 particular attention is focused on the role of the negative regulators and suppressor of cytokine signaling proteins (SOCSs) of STATs. The SOCS family comprises eight members, SOCS-1 to -7 and CIS. SOCS family members share the central Src homology 2 domain and SOCS box in the carboxy-terminal which play a crucial role in proteasomal degradation of binding partners59,60,61,62 (Figure 2a). The N-terminal domains of SOCS proteins vary in length and amino acid sequence, and only SOCS-1 and SOCS-3 possess a kinase inhibitory region (KIR, 17 residues) immediately upstream of the central SH2 domain.63 SOCS proteins attenuate cytokine signal transduction by binding through their SH2 domains to phosphorylated tyrosine residues on signaling intermediates, such as receptor subunits and JAKs. Thus, the binding of SOCSs to JAK kinases blocks further signaling in a negative feedback loop.64 The structures of SOCS-3 in complex with a phosphotyrosine-containing peptide from the interleukin-6 (IL-6) receptor related to the signaling subunit gp130,65 with JAK266 and those of ternary complexes of SOCS-2, -3 and -4 associated with elongin B and elongin C67,68 have been reported. However, the structure and function of the N-terminal domains of the SOCS proteins remain poorly characterized. However, they have been suggested to mediate the ubiquitination of substrate proteins bound to their N-terminal domains and peptide, and peptidomimetics covering these unstructured regions revealed potential anti-inflammatory properties.69,70,71,72 Furthermore, the N-terminal domains of SOCS-4 and -5 play an important role in mediating interactions with the epidermal growth factor receptor and interleukin-4 receptor. N-terminal domains have been predicted and experimentally demonstrated as internally disordered regions (Figure 2).73 In Figure 2a, a schematic representation of SOCSs structure and the profile of disorder prediction for the whole human sequences of SOCS-1 and -3 (using PONDR-FIT74) (Figure 2b and 2c) are reported. Our results appear similar to those related to other disorder predictors (such as VSL245). The altered expression of SOCS-1 and -3 in PCa75,76 was recently confirmed by analyzing the expression and localization of STAT and SOCS-1 proteins using immunohistochemistry on 150 Formalin-fixed and paraffin-embedded human prostate tissues of different grades; fundamental differences in major oncogenic signaling cascades between benign and malignant form of prostate tissue were observed. A critical association between altered expression of STAT-3 and -5 with SOCS-1 suggested for the latter a potential role as a negative regulator independent of JAK-STAT pathway in tumorigenic transformation of prostate tissue.77 The growth-inhibitory effects of SOCS-1 in human PCa cell lines were also demonstrated by the use of the SOCS-1 mimetic peptide Tkip that acted as a negative growth regulator of both DU-145 and LNCaP cells.69 These studies unveiled that the SOCS-1 inhibitory effect is mediated through negative regulation of STAT3 phosphorylation and that its down-regulation increased expression of cyclins D1, cyclins E, cdk 2 and cdk 4, which drive cell cycle progression to S phase. Differential regulation of SOCS-1 in PCa cell lines may be explained by either the presence of AR mutations or insufficient expression of a coactivator, which is critical for SOCS-1 expression. At present, it could be hypothesized that SOCS-1 down-regulation affects growth regulation by androgens in PCa.78 Further SOCS-1, if expressed in PCa cells, has a growth-regulatory role in this malignancy. Although the presence of both SOCS-1 mRNA and protein was detected in all tested cell lines, expression levels decreased in samples taken from patients undergoing hormonal therapy but increased in specimens from patients who failed this therapy. In LNCaP-interleukin-6 PCa cells, SOCS-1 was upregulated by interleukin-6 and in PC3-AR cells by androgens; such upregulation was also found to impair cell proliferation. In contrast, down-regulation of SOCS-1 expression caused a potent growth stimulation of PC3, DU-145, and LNCaP-IL-6 cells that were associated with the increased expression levels of cyclins D1 and E as well as cyclin-dependent kinases 2 and 4. Other studies demonstrated that the down-regulation of SOCS-3 causes cell death of PCa cells through activation of the extrinsic and intrinsic apoptosis pathways.79 The underlying mechanism is that SOCS-3 antagonizes the proliferative and migratory ability of PCa cells by inhibition of p44/p42 MAPK signaling.80 In addition, SOCS-3 inhibits the signal transducer and activator closely related to PCa cell proliferation and invasiveness, such as STAT3.81
Figure 2.
(a) Schematic representation of SOCS proteins modular structures, IDRs are indicated as stars shape. Prediction of disorder tendency of (b) SOCS-1, (c) SOCS-3 sequences with PONDR-FIT.
In addition, SOCS-3 inhibits PCa cell growth and Liver X receptors (LXRs) agonists may inhibit the carcinogenesis of PCa via the SOCS-3. In cells treated with control-siRNA, indeed the inhibitor GW3965 enhanced SOCS-3 expression and inhibited the phosphorylation of STAT3, NF-κB and AP1 expression, accompanied by dramatically reduced cellular proliferation rate, immigration and invasion of cultured cells.82,83
Interestingly, when the interaction between the tumor-selective apoptosis inducer tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and SOCS-3 was investigated, it was observed that SOCS-3 appeared as one of the proteins which influence the ability of TRAIL and resveratrol to cause programmed cell death in PCa.84
HMG1: a proinflammatory cytokine
HMGB1 is secreted by immune cells (like macrophages, monocytes and dendritic cells) through leaderless secretory pathway.85 HMGB1 is also a DNA binding protein involved in its replication and repair process86,87 but is secreted by activated macrophages and monocytes as a cytokine mediator of inflammation.88 HMGB1-RAGE axis plays a major role in inflammation induced by carcinogenesis:89,90 in ovarian cancer and PCa, different isoforms of the human HMGB1, encoded by the HMGB1 gene, have been reported91 and other genes (HMGB-2, -3 and -4) encoding similar, although less studied HMGB proteins, are present in the human genome.
HMGB1 is highly expressed in PCa cells92 and targeting HMGB1 disrupts tumor progression by inhibiting activation of T-cells and reducing infiltration of macrophages which are considered to be key inflammatory cells in promoting variety of cancers including PCa.93 Interestingly, androgen deprivation caused HMG1's secretion in prostatic stromal cells supporting the notion that deprivation therapy may upregulate the expression of HMGB1 leading to either hormone resistance or metastatic disease.94
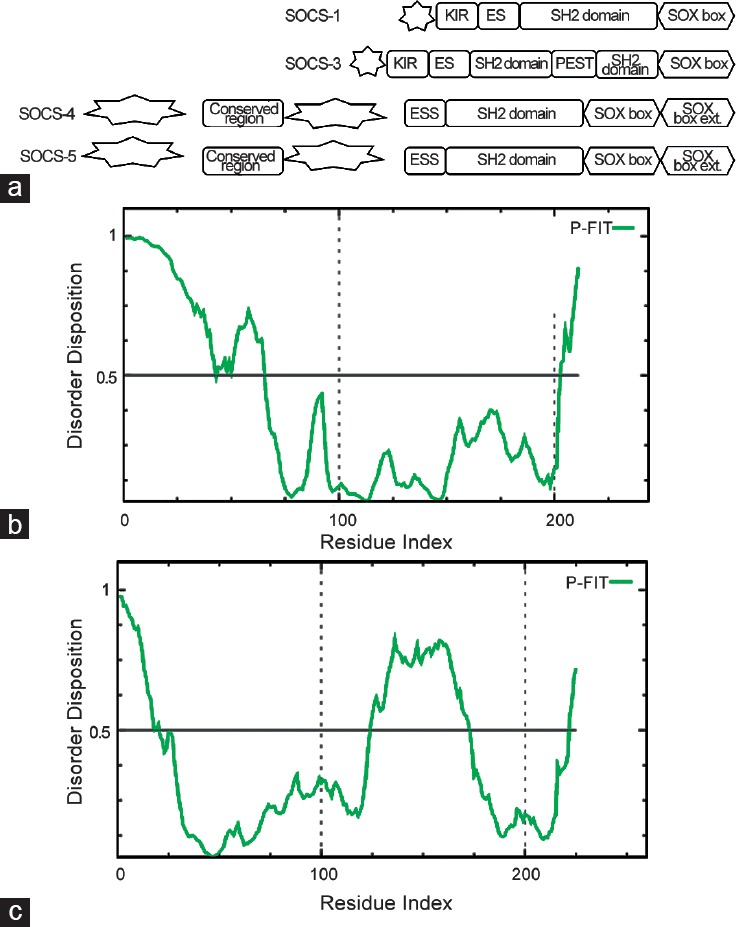
HMGB1 is the most studied member among the human HMGB protein family. It has many different functions that depend on its redox state and posttranslational modifications, including acetylation, which determine its cellular or extracellular localization. HMGB1 is polyacetylated near its nuclear-localization sequences (NLSs) and this modification blocks the interaction with nuclear importer proteins.95 In addition, HMGB proteins have a common modular structure, reported in Figure 3a with two positively charged DNA binding domains, HMG A-box and HMG B-box, folded in the characteristic L-shaped architecture. Each domain is formed by three alpha helix-stretches.
Figure 3.
(a) Schematic representation of HMGB1 modular structure, IDRs are represented by a star shape, (b) Prediction of disorder tendency of HMG1 sequence with PONDR-FIT.
In HMGB1, the HMG A-box includes amino acids 1–79, and the HMG B-box spanning amino acids 89–163. The acidic C-terminal domain (186–215) is negatively charged with an extended and flexible sequence (as bioinformatics analysis in Figure 3b), which constitutes the intrinsically disordered region96 and can interact with residues within and between the two HMG boxes97 although it has the highest affinity for the HMG B-box.98 Different functions of HMGB1 are associated with redox changes making it a master redox sensor. This function depends on three cysteine residues (at 23, 45 and 106 positions): Cys23 and Cys45 easily form an intramolecular disulfide bridge, while Cys106 remains reduced (the semi-oxidized HMGB1 form). The formation of this disulfide bond is thermodynamically favored; thus, a significant fraction of HMGB1 is in the semi-oxidized form within cells.99 The proximity of the Cys residues to residues required for DNA binding100 highlights the importance of redox-regulated conformational changes in HMGB1, which may modulate their affinity for DNA. Thus, redox changes affect the interaction with other proteins and modify their biological functions. The structural analysis of HMGB1 A-box reveals that Cys23 and Cys45 are located at the center of helix I and helix II, respectively, opposing each other and at a distance that allows the formation of a disulfide bond under appropriate oxidative conditions. Two NLSs, rich in lysine residues and spanning 28–44 and 179–185 regions, respectively, can be specified.101 Although Cys106 is not located within the NLS, thiol may participate in nuclear transport through nuclear pore complex binding, ubiquitination, or transporter interaction102 and is important to preserve the nuclear functions of these proteins. Taken together, these data suggest that the intrinsically disordered HMG protein may represent novel targets in PCa.
IGF AXIS SIGNALING
Interest in insulin-like growth factors (IGFs) and their effect on carcinogenesis has increased recently because high serum concentrations of IGF1 are associated with an increased risk of breast, prostate, colorectal and lung cancers. Physiologically, IGF1 is not only the major mediator of the effects of the growth hormone strongly influencing cell proliferation and differentiation, but it is also a potent inhibitor of apoptosis.103
IGFs are proteins with high sequence similarity to insulin and are part of a complex system that cells use to communicate with their physiologic environment. This system, “IGF axis” consists of two cell-surface receptors (IGF1R and IGF2R), two ligands IGF-I and IGF-2, a family of six high-affinity IGF-binding proteins (IGFBP-1 to IGFBP-6), as well as associated IGFBP degrading enzymes, referred to collectively as proteases.104 The IGF axis is one of the most investigated pathways in cancer and mediates critical physiological processes, including cellular growth, cell differentiation, apoptosis, development, as well as glucose and lipid metabolism.105 Moreover, the IGF axis plays a fundamental role in regulating embryonic growth and IGFs also exert growth stimulating and pro-survival effects on tumor cells where they have been shown to mediate carcinogenesis, angiogenesis, malignant cell proliferation and metastatic growth of a variety of tumors.106 To date, more than 100 agents have been developed to target the receptors of the IGF axis and many are currently being evaluated in preclinical and clinical studies. These investigations have demonstrated that the IGF axis modulates the development and progression of PCa. Indeed, IGF1 signaling is elevated in PCa compared to benign prostate tissue and exerts a role in tumor progression.107 Data obtained from mouse cells with a disrupted IGF1R gene suggest that this receptor is a prerequisite for cells to undergo oncogene-induced transformation. Consistently, inhibition of IGF1R shows therapeutic efficiency in a number of preclinical models of hormone-dependent tumors, including those of the prostate and breast.108 Epidemiological data suggest that high circulating IGF1 levels are associated with a moderately increased risk for PCa development. Pharmacological inhibition strategies include growth factor entrapping monoclonal antibodies, employing kinase defective mutant receptors, antisense oligonucleotides, growth factor sequestrating IGFBPs, soluble forms of the receptor, receptor neutralizing antibodies and small-molecule tyrosine kinase inhibitors of IGF1R and INSR.109,110,111
The solution structure of IGF1 was investigated with a combination of nuclear magnetic resonance and restrained molecular dynamics methods. The results show that it is quite similar to that of insulin, but minor differences exist. In detail resonance assignments were hampered by the lack of spectral dispersion and broad line widths, arising from conformational averaging and the tendency of IGF-I to aggregate.112,113 The regions homologous to insulin are well-defined, well conserved and confined in the first 27 amino acids of the sequence as reported in Figure 4a. Clearly, the N-terminal domain is the most important to its function, while the remainder of the molecule exhibits greater disorder and has been demonstrated to contain the sites for interaction with a number of physiologically important proteins.114,115
Figure 4.
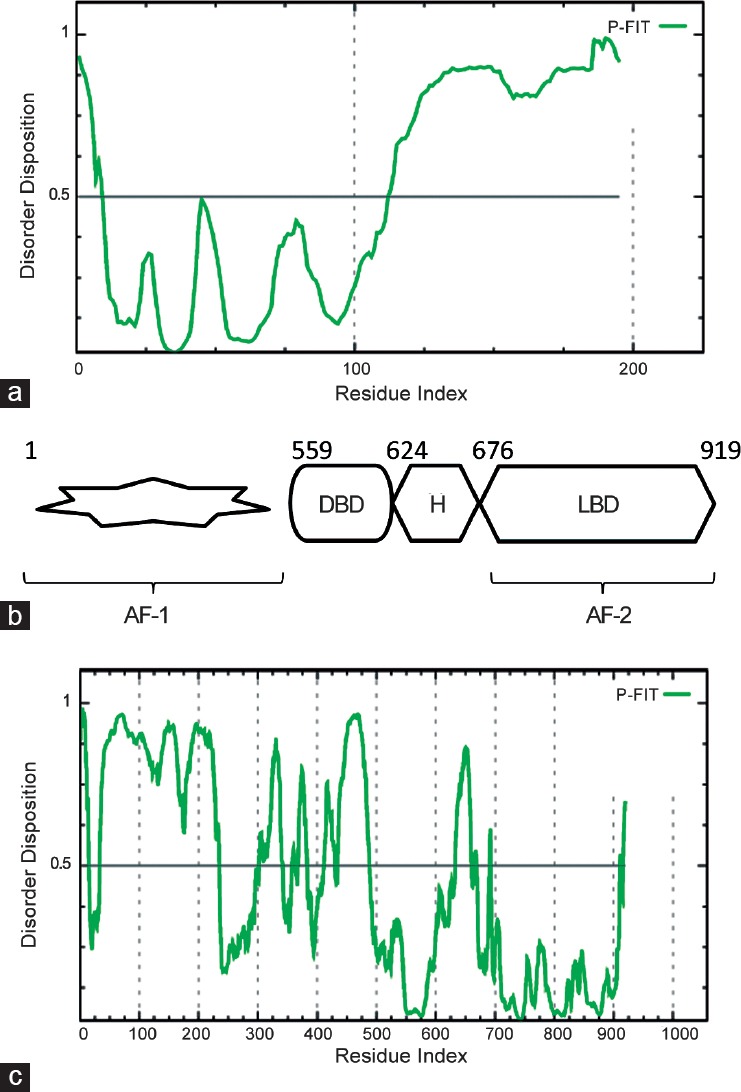
(a) Prediction of disorder tendency of IGF1 sequence with PONDR-FIT, (b) Modular structure of the AR, IDRs are represented by a star shape (c) Prediction of disorder tendency of AR sequence with PONDR-FIT.
ANDROGEN RECEPTOR SIGNALING
AR is a member of the nuclear receptor (NR) superfamily of ligand regulated transcription factors that plays an integral role in primary and secondary male sexual development.116 As a key regulator for the growth, terminal differentiation and function of the prostate gland, AR has important functions in regulation of proliferation, cell cycle, apoptosis, angiogenesis and differentiation in PCa.117,118,119 Identified as a primary target for the treatment of PCa, many therapeutic strategies were developed to attenuate AR signaling. While frontline androgen-deprivation therapies targeting either the production or action of androgens usually yield initially favorable responses in PCa patients, a significant number acquire treatment resistance.120 The signaling is restored in castrate-resistant prostate cancer (CRPC) due to many aberrant mechanisms such as AR mutations, amplification, or expression of constitutively active splice-variants. Structural analyses of the AR have identified key surfaces involved in protein–protein interaction with co-regulators that was used to design and develop promising AR-coactivator binding inhibitors for the treatment of PCa. The currently used AR antagonists, such as flutamide, bicalutamide, nilutamide, and enzalutamide (MDV3100) (Table 1), act by binding to the androgen binding site (ABS) of the AR, resulting in conformational changes that prevent its activation. In the absence of ligands, the AR predominantly resides in the cytoplasm where it is associated with heat shock proteins in a transcriptionally inactive form until it is activated by testosterone or the more potent metabolite, 5a-dihydrotestosterone (DHT).121 Upon ligand binding, the AR undergoes a substantial conformational change leading to dissociation from repressor proteins, dimerization, translocation to the nucleus and association to androgen-responsive elements (ARE) in the regulatory regions of target genes.122 Through this regulatory mechanism, the AR regulates the expression of more than a thousand genes including PSA, which is the most well-known biomarker for PCa.
Similar to other nuclear receptors (NRs), the AR displays a modular structure composed of an N-terminal domain (NTD) bearing the activation function (AF-1), a DNA-binding domain (DBD), a connecting hinge region containing a nuclear localization signal (NLS) and a C-terminal ligand-binding domain (LBD) (Figure 4b).123 The LBD is constituted by 12 anti-parallel α-helices with the ABS buried inside which undergoes conformational rearrangement upon agonist binding. The helix 12 (H12) is the most flexible part of the LBD and its repositioning after androgen binding completes the AF-2 binding surface by creating a hydrophobic groove that allows the docking of leucine-rich, LXXLL motif-containing co-regulatory proteins.124 In addition, the intrinsic dipole moment of the co-activator α-helix is matched on the AF-2 by a negatively charged residue E897, on H12, at the N-terminus and a positively charged K720, on H3, at the C-terminus, which form a charge clamp.125 The LXXLL motif, named the NR box, is present in several coactivators such as proteins of the p160 family.126 Mainly formed by residues from helices 3, 4, 5, and 12, the AF-2 surface on the AR-LBD is unique among NRs in preferring to interact with the more bulky hydrophobic motifs F/WXXLF respect to the LXXLL. These interdomain interactions play an important role in selective AR-dependent gene regulation but are disrupted by DNA binding, which in turn would expose the NTD and AF-2 surfaces for interactions with co-regulators.127 Also known as the coactivator binding pocket, the AF-2 is the major protein–protein interaction (PPI) surface used by NRs for coactivator recruitment. While AF-2 is a privileged site to develop inhibitors of AR-protein-protein interactions (PPIs), other sites were also recently exploited.128
The AR-NTD plays an essential role in ligand-dependent AR transcriptional activity but, in addition, has a ligand-independent transactivation function.129 Indeed, C-terminal truncated ARs lacking the LBD retain a constitutive activity and such splice variants can be observed in CRPC.130,131 The AF-1 region located in the NTD contributes to these functions by interacting with other proteins such as components of transcription factors (TF) IIF and IIH and co-activators SRC1-3 and CREB-binding protein (CBP).131 The intact NTD domain (AR1–558) resulted in being able to inhibit both ligand-dependent and independent transcriptional activities by sequestering co-regulatory proteins.132 The NTD is the least conserved domain across steroid receptors with <15% homology and very few disease-associated mutations of the AR have been reported in this domain.133 Thus, the AR- NTD represents a very attractive target to develop inhibitors that could block ligand-dependent and ligand-independent AR transactivation and hence, be active in hormone-sensitive and castrate-resistant PCas. Nevertheless, the design of inhibitors targeting the AF-1 site is partially hampered by the lack of structural information for the NTD, which is highly flexible and intrinsically disordered (Figure 4c). However, little is known about the functional dynamics of the NTD in PCa cells due to a high degree of intrinsic disorder in this domain.134 Previous studies reported on helices in AR transcriptional activation, a178 LKDIL182 motif resident in the AF1a region of the TAU1 domain135,136 and a435 WHTLF439 motif in the TAU5 domain.137 Following a combinatorial approach that is the most applied in these cases71,138,139 to target the AR-NTD, a library of marine sponge extracts were screened in a transactivation assay.140 Several short chlorinated peptides from the sponge Dysidea sp. named Sintokamides were identified and no cytotoxicity was observed in a cell viability assay. Sintokamide A 15 was found to reduce PSA expression and block AR-NTD transactivation at 5 mg ml−1 in luciferase reporter gene assays and to inhibit proliferation of AR-positive LNCaP cells but not AR-negative PC3 cells. Another screen was more recently performed on extracts of the marine sponge Niphates digitalis leading to the discovery of niphatenones, a group of glycerol-ether lipids, as AR-NTD inhibitors at different disease stages.141
Important advances in the structure-function relationships of the AR and structural interactomic studies of AR splice variants in PCa evolution have provided a detailed knowledge of the AR axis in PCa. The limited treatment options for CRPC patients and the increasing appreciation of the role of AR in advanced PCa prompt the development of new approaches to target AR signaling. Different strategies to destabilize the AR, prevent its nuclear translocation, inhibit its binding to DNA or block co-activator recruitment are being explored. The latter, mediated by PPIs, offers the opportunity to develop selective noncompetitive AR antagonists that would overcome resistance to traditional anti-androgens and remain effective in advanced PCa. PPI inhibitors have been developed using a wide variety of approaches including structure-based drug design, screening of natural compound libraries, ligand-based peptidomimetics and different combinations of virtual library screening and experimental evaluation. While many compounds described need more comprehensive SAR studies and pharmacological optimization to progress into clinical evaluation, some including EPI-001 and oligobenzamides have already shown very encouraging results in vivo.142
In this scenario, an important study focused on the modulation of AR functions by nucleophosmin1 (NPM1/B23), a member of the histone chaperone family, that in turn contains an IDR. Further NPM1 resulted overexpressed in PCa.143 NPM1 is an abundant multifunctional protein which is present in high quantities in the granular region of nucleoli.144 It is capable of shuttling between the nucleus and cytoplasm and is involved in many cellular functions such as the regulation of ribosome biogenesis, chromatin remodeling, DNA replication, recombination, transcription repair, and the control of centrosome duplication.145 Notably, NPM1 has been identified as the most frequently mutated gene in acute myeloid leukemia (AML) patients, accounting for approximately 30% of cases.146 Besides its primary role as a therapeutic target in AML drug discovery programs, it represents an important model for multidomain proteins.25,39,147 AR and NPM1 interact in vitro and in vivo, and NPM1 is critical for androgen-dependent transcriptional activation in LNCaP cells as an anti-NPM, siRNA downregulates transcription of a transfected ARE-containing reporter promoter as well as expression of the endogenous androgen-responsive PSA gene. NPM1 is highly co-expressed with the AR in secretory epithelial cells of localized PCas and strongly binds to the DNA-binding domain (DBD)/hinge domain of AR in vitro and facilitates AR binding to its consensus ARE suggesting that other proteins are probably involved in this AR–NPM1 interaction. In addition, it has been shown that AR binds to full-length NPM1 as well as to both the N-terminal domain and to the CTD-containing IDRs.147 Deletion of these two regions abolished the interaction of NPM with AR indicating that the acidic/nuclear localization signal (Ac/NLS) domain of NPM1 is not sufficient for binding to AR. Altogether, these data indicate that AR and NPM1 interact in vitro through multiple domains and that other proteins seem to modulate this interaction in vivo and thus, even if IDRs of NPM1 are unable to directly bind to AR they seem to be crucial for the binding. Thus, the NPM1–AR interaction is linked to the regulation of gene expression by androgens during prostate carcinogenesis. Immunohistochemistry studies demonstrated that a strong and extensive staining for NPM1 was found in neoplastic prostate tissues while it is present at lower levels in the basal and luminal epithelial cells. Interestingly, AR also expresses more in the same sections of adenocarcinomas.148
CONCLUSIONS
In PPI networks disordered binding regions provide specific but transient interactions that enable IDPs to play central roles in important signaling pathways.19 IDPs are endowed with crucial abilities to interact with multiple protein partners without losing specificity or affinity. The main challenging goal in this field lies in elucidating the structural basis for promiscuous binding and the mechanisms that lead to specific responses to a particular cellular signal.149 The occurrence of intrinsic disorder in cancer-associated proteins strongly suggests that disorder needs to be seriously evaluated in the drug discovery process toward the development of novel therapeutic compounds. Unfortunately, this area has remained largely unexplored primarily due to the lack of effective screening tools. Structural and interactomic studies have helped to investigate pathways that are involved in prostate carcinogenesis and to unveil signaling events that are important for tumor progression. In addition to the ones that have been analyzed, many other PPIs involving IDPs have been selected as potential targets. For example, recently the Cancer/Testis Antigen (CTA), Prostate-associated Gene 4 (PAGE4) that is upregulated in PCa,150 and several other CTAs such as the MAGE proteins, represent novel therapeutic targets for CRPC for which there are currently no effective therapeutics.44,151
COMPETING FINANCIAL INTERESTS
The authors declared no conflict of interest.
AUTHOR CONTRIBUTIONS
DM and EN conceived the manuscript; DM, AR, AM, and SLM wrote the manuscript.
REFERENCES
- 1.Shariat SF, Scherr DS, Gupta A, Bianco FJ, Jr, Karakiewicz PI, et al. Emerging biomarkers for prostate cancer diagnosis, staging, and prognosis. Arch Esp Urol. 2011;64:681–94. [PubMed] [Google Scholar]
- 2.Albiges-Sauvin L, Levy A, Massard C, Fizazi K. [Prognosis and predictive factors in prostate cancer] Bull Cancer. 2009;96:439–49. doi: 10.1684/bdc.2009.0846. Article in French. [DOI] [PubMed] [Google Scholar]
- 3.Divrik RT, Turkeri L, Sahin AF, Akdogan B, Ates F, et al. Prediction of response to androgen deprivation therapy and castration resistance in primary metastatic prostate cancer. Urol Int. 2012;88:25–33. doi: 10.1159/000334539. [DOI] [PubMed] [Google Scholar]
- 4.Bluemn EG, Nelson PS. The androgen/androgen receptor axis in prostate cancer. Cur Opin Oncol. 2012;24:251–7. doi: 10.1097/CCO.0b013e32835105b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoffman-Censits J, Kelly WK. Enzalutamide: a novel antiandrogen for patients with castrate-resistant prostate cancer. Clin Cancer Res. 2013;19:1335–9. doi: 10.1158/1078-0432.CCR-12-2910. [DOI] [PubMed] [Google Scholar]
- 7.Reid P, Kantoff P, Oh W. Antiandrogens in prostate cancer. Invest New Drugs. 1999;17:271–84. doi: 10.1023/a:1006344807086. [DOI] [PubMed] [Google Scholar]
- 8.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 9.Parker C, Nilsson S, Heinrich D, Helle SI, O’sullivan JM, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro D, Tareen B. Current and emerging treatments in the management of castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2012;12:951–64. doi: 10.1586/era.12.59. [DOI] [PubMed] [Google Scholar]
- 11.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–33. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 12.Uversky VN. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 2011;43:1090–103. doi: 10.1016/j.biocel.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Schuster N, Dünker N, Krieglstein K. Transforming growth factor-β induced cell death in the developing chick retina is mediated via activation of c-jun N-terminal kinase and downregulation of the anti-apoptotic protein Bcl-XL. Neurosci Lett. 2002;330:239–42. doi: 10.1016/s0304-3940(02)00801-7. [DOI] [PubMed] [Google Scholar]
- 14.Uversky VN. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front Biosci (Landmark Ed) 2009;14:5188–238. doi: 10.2741/3594. [DOI] [PubMed] [Google Scholar]
- 15.Uversky VN. Under-folded proteins: conformational ensembles and their roles in protein folding, function, and pathogenesis. Biopolymers. 2013;99:870–87. doi: 10.1002/bip.22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uversky VN, Dave V, Iakoucheva LM, Malaney P, Metallo SJ, et al. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem Rev. 2014;114:6844–79. doi: 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–64. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uversky VN, Santambrogio C, Brocca S, Grandori R. Length-dependent compaction of intrinsically disordered proteins. FEBS Lett. 2012;586:70–3. doi: 10.1016/j.febslet.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 19.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 20.Dunker AK, Uversky VN. Drugs for ‘protein clouds’: targeting intrinsically disordered transcription factors. Curr Opin Pharmacol. 2010;10:782–8. doi: 10.1016/j.coph.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 21.Tompa P, Schad E, Tantos A, Kalmar L. Intrinsically disordered proteins: emerging interaction specialists. Curr Opin Struct Biol. 2015;35:49–59. doi: 10.1016/j.sbi.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chem Rev. 2014;114:6561–88. doi: 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- 23.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 24.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, et al. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marasco D, Scognamiglio PL. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int J Mol Sci. 2015;16:7394–412. doi: 10.3390/ijms16047394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 27.Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Protein disorder in the human diseasome: unfoldomics of human genetic diseases. BMC Genomics. 2009;10(Suppl 1):S12. doi: 10.1186/1471-2164-10-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uversky VN. Wrecked regulation of intrinsically disordered proteins in diseases: pathogenicity of deregulated regulators. Front Mol Biosci. 2014;1:6. doi: 10.3389/fmolb.2014.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–6. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 30.Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–91. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–32. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 32.Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–8. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 33.Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–53. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Gasperini RJ, Klaver DW, Hou X, Aguilar MI, Small DH. Mechanisms of transthyretin aggregation and toxicity. Subcell Biochem. 2012;65:211–24. doi: 10.1007/978-94-007-5416-4_9. [DOI] [PubMed] [Google Scholar]
- 35.Moreau KL, King JA. Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med. 2012;18:273–82. doi: 10.1016/j.molmed.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker LC, LeVine H., 3rd Corruption and spread of pathogenic proteins in neurodegenerative diseases. J Biol Chem. 2012;287:33109–15. doi: 10.1074/jbc.R112.399378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuanalo-Contreras K, Mukherjee A, Soto C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int J Cell Biol 2013. 2013:638083. doi: 10.1155/2013/638083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 39.Di Natale C, Scognamiglio PL, Cascella R, Cecchi C, Russo A, et al. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions. FASEB J. 2015;29:3689–701. doi: 10.1096/fj.14-269522. [DOI] [PubMed] [Google Scholar]
- 40.Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, et al. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci U S A. 2008;105:5762–7. doi: 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mark WY, Liao JC, Lu Y, Ayed A, Laister R, et al. Characterization of segments from the central region of BRCA1: an intrinsically disordered scaffold for multiple protein-protein and protein-DNA interactions? J Mol Biol. 2005;345:275–87. doi: 10.1016/j.jmb.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 42.Uversky VN, Roman A, Oldfield CJ, Dunker AK. Protein intrinsic disorder and human papillomaviruses: increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J Proteome Res. 2006;5:1829–42. doi: 10.1021/pr0602388. [DOI] [PubMed] [Google Scholar]
- 43.Malaney P, Pathak RR, Xue B, Uversky VN, Dave V. Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci Rep. 2013;3:2035. doi: 10.1038/srep02035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem. 2011;112:3256–67. doi: 10.1002/jcb.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng ZP, Zhang X, Han P, Arora N, Anders RF, et al. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol. 2006;150:256–67. doi: 10.1016/j.molbiopara.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 46.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, et al. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–9. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Hegyi H, Buday L, Tompa P. Intrinsic structural disorder confers cellular viability on oncogenic fusion proteins. PLoS Comput Biol. 2009;5:e1000552. doi: 10.1371/journal.pcbi.1000552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonomi S, Gallo S, Catillo M, Pignataro D, Biamonti G, et al. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol. 2013;2013:962038. doi: 10.1155/2013/962038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ladomery M. Aberrant alternative splicing is another hallmark of cancer. Int J Cell Biol 2013. 2013:463786. doi: 10.1155/2013/463786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res. 2014;20:1135–45. doi: 10.1158/1078-0432.CCR-13-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng Z, Mizianty MJ, Xue B, Kurgan L, Uversky VN. More than just tails: intrinsic disorder in histone proteins. Mol Biosyst. 2012;8:1886–901. doi: 10.1039/c2mb25102g. [DOI] [PubMed] [Google Scholar]
- 52.Korneta I, Bujnicki JM. Intrinsic disorder in the human spliceosomal proteome. PLoS Comput Biol. 2012;8:e1002641. doi: 10.1371/journal.pcbi.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, et al. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci U S A. 2006;103:8390–5. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sette C. Alternative splicing programs in prostate cancer. Int J Cell Biol 2013. 2013:458727. doi: 10.1155/2013/458727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Y, Dobi A, Sreenath T, Cook C, Tadase AY, et al. Delineation of TMPRSS2-ERG splice variants in prostate cancer. Clin Cancer Res. 2008;14:4719–25. doi: 10.1158/1078-0432.CCR-08-0531. [DOI] [PubMed] [Google Scholar]
- 56.Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60:199–215. doi: 10.1111/j.1365-2559.2011.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Narayanan NK, Nargi D, Horton L, Reddy BS, Bosland MC, et al. Inflammatory processes of prostate tissue microenvironment drive rat prostate carcinogenesis: preventive effects of celecoxib. Prostate. 2009;69:133–41. doi: 10.1002/pros.20862. [DOI] [PubMed] [Google Scholar]
- 58.Fan Y, Mao R, Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4:176–85. doi: 10.1007/s13238-013-2084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alexander WS, Hilton DJ. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol. 2004;22:503–29. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 60.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–21. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 61.Nicholson SE, Hilton DJ. The SOCS proteins: a new family of negative regulators of signal transduction. J Leukoc Biol. 1998;63:665–8. doi: 10.1002/jlb.63.6.665. [DOI] [PubMed] [Google Scholar]
- 62.Hilton DJ, Richardson RT, Alexander WS, Viney EM, Willson TA, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A. 1998;95:114–9. doi: 10.1073/pnas.95.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Babon JJ, McManus EJ, Yao S, DeSouza DP, Mielke LA, et al. The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol Cell. 2006;22:205–16. doi: 10.1016/j.molcel.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 64.Elliott J, Johnston JA. SOCS: role in inflammation, allergy and homeostasis. Trends Immunol. 2004;25:434–40. doi: 10.1016/j.it.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Bergamin E, Wu J, Hubbard SR. Structural basis for phosphotyrosine recognition by suppressor of cytokine signaling-3. Structure. 2006;14:1285–92. doi: 10.1016/j.str.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 66.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20:469–76. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bullock AN, Debreczeni JE, Edwards AM, Sundstrom M, Knapp S. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci U S A. 2006;103:7637–42. doi: 10.1073/pnas.0601638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Babon JJ, Sabo JK, Soetopo A, Yao S, Bailey MF, et al. The SOCS box domain of SOCS3: structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J Mol Biol. 2008;381:928–40. doi: 10.1016/j.jmb.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Flowers LO, Subramaniam PS, Johnson HM. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene. 2005;24:2114–20. doi: 10.1038/sj.onc.1208437. [DOI] [PubMed] [Google Scholar]
- 70.Waiboci LW, Ahmed CM, Mujtaba MG, Flowers LO, Martin JP, et al. Both the suppressor of cytokine signaling 1 (SOCS-1) kinase inhibitory region and SOCS-1 mimetic bind to JAK2 autophosphorylation site: implications for the development of a SOCS-1 antagonist. J Immunol. 2007;178:5058–68. doi: 10.4049/jimmunol.178.8.5058. [DOI] [PubMed] [Google Scholar]
- 71.Doti N, Scognamiglio PL, Madonna S, Scarponi C, Ruvo M, et al. New mimetic peptides of the kinase-inhibitory region (KIR) of SOCS1 through focused peptide libraries. Biochem J. 2012;443:231–40. doi: 10.1042/BJ20111647. [DOI] [PubMed] [Google Scholar]
- 72.Madonna S, Scarponi C, Doti N, Carbone T, Cavani A, et al. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur J Immunol. 2013;43:1883–95. doi: 10.1002/eji.201343370. [DOI] [PubMed] [Google Scholar]
- 73.Feng ZP, Chandrashekaran IR, Low A, Speed TP, Nicholson SE, et al. The N-terminal domains of SOCS proteins: a conserved region in the disordered N-termini of SOCS4 and 5. Proteins. 2012;80:946–57. doi: 10.1002/prot.23252. [DOI] [PubMed] [Google Scholar]
- 74.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bellezza I, Neuwirt H, Nemes C, Cavarretta IT, Puhr M, et al. Suppressor of cytokine signaling-3 antagonizes cAMP effects on proliferation and apoptosis and is expressed in human prostate cancer. Am J Pathol. 2006;169:2199–208. doi: 10.2353/ajpath.2006.060171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Neuwirt H, Puhr M, Cavarretta IT, Mitterberger M, Hobisch A, et al. Suppressor of cytokine signalling-3 is up-regulated by androgen in prostate cancer cell lines and inhibits androgen-mediated proliferation and secretion. Endocr Relat Cancer. 2007;14:1007–19. doi: 10.1677/ERC-07-0172. [DOI] [PubMed] [Google Scholar]
- 77.Singh N, Hussain S, Bharadwaj M, Kakkar N, Singh SK, et al. Overexpression of signal transducer and activator of transcription (STAT-3 and STAT-5) transcription factors and alteration of suppressor of cytokine signaling (SOCS-1) protein in prostate cancer. J Recept Signal Transduct Res. 2012;32:321–7. doi: 10.3109/10799893.2012.733885. [DOI] [PubMed] [Google Scholar]
- 78.Neuwirt H, Puhr M, Santer FR, Susani M, Doppler W, et al. Suppressor of cytokine signaling (SOCS)-1 is expressed in human prostate cancer and exerts growth-inhibitory function through down-regulation of cyclins and cyclin-dependent kinases. Am J Pathol. 2009;174:1921–30. doi: 10.2353/ajpath.2009.080751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Puhr M, Santer FR, Neuwirt H, Susani M, Nemeth JA, et al. Down-regulation of suppressor of cytokine signaling-3 causes prostate cancer cell death through activation of the extrinsic and intrinsic apoptosis pathways. Cancer Res. 2009;69:7375–84. doi: 10.1158/0008-5472.CAN-09-0806. [DOI] [PubMed] [Google Scholar]
- 80.Puhr M, Santer FR, Neuwirt H, Marcias G, Hobisch A, et al. SOCS-3 antagonises the proliferative and migratory effects of fibroblast growth factor-2 in prostate cancer by inhibition of p44/p42 MAPK signalling. Endocr Relat Cancer. 2010;17:525–38. doi: 10.1677/ERC-10-0007. [DOI] [PubMed] [Google Scholar]
- 81.Yokogami K, Yamashita S, Takeshima H. Hypoxia-induced decreases in SOCS3 increase STAT3 activation and upregulate VEGF gene expression. Brain Tumor Pathol. 2013;30:135–43. doi: 10.1007/s10014-012-0122-0. [DOI] [PubMed] [Google Scholar]
- 82.Vegran F, Berger H, Ghiringhelli F, Apetoh L. Socs3 induction by PPARgamma restrains cancer-promoting inflammation. Cell Cycle. 2013;12:2157–8. doi: 10.4161/cc.25370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fu W, Yao J, Huang Y, Li Q, Li W, et al. LXR agonist regulates the carcinogenesis of PCa via the SOCS3 pathway. Cell Physiol Biochem. 2014;33:195–204. doi: 10.1159/000356662. [DOI] [PubMed] [Google Scholar]
- 84.Horndasch M, Culig Z. SOCS-3 antagonizes pro-apoptotic effects of TRAIL and resveratrol in prostate cancer cells. Prostate. 2011;71:1357–66. doi: 10.1002/pros.21353. [DOI] [PubMed] [Google Scholar]
- 85.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: endogenous danger signaling. Mol Med. 2008;14:476–84. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–13. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 87.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr Opin Genet Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 88.Czura CJ, Wang H, Tracey KJ. Dual roles for HMGB1: DNA binding and cytokine. J Endotoxin Res. 2001;7:315–21. doi: 10.1177/09680519010070041401. [DOI] [PubMed] [Google Scholar]
- 89.Gebhardt C, Riehl A, Durchdewald M, Nemeth J, Furstenberger G, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205:275–85. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gnanasekar M, Kalyanasundaram R, Zheng G, Chen A, Bosland MC, et al. HMGB1: a promising therapeutic target for prostate cancer. Prostate Cancer 2013. 2013:157103. doi: 10.1155/2013/157103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parrish W, Ulloa L. High-mobility group box-1 isoforms as potential therapeutic targets in sepsis. Methods Mol Biol. 2007;361:145–62. doi: 10.1385/1-59745-208-4:145. [DOI] [PubMed] [Google Scholar]
- 92.Li T, Gui Y, Yuan T, Liao G, Bian C, et al. Overexpression of high mobility group box1 with poor prognosis in patients after radical prostatectomy. BJU Int. 2012;110:E1125–30. doi: 10.1111/j.1464-410X.2012.11277.x. [DOI] [PubMed] [Google Scholar]
- 93.Shetty AV, Thirugnanam S, Dakshinamoorthy G, Samykutty A, Zheng G, et al. 18alpha-glycyrrhetinic acid targets prostate cancer cells by down-regulating inflammation-related genes. Int J Oncol. 2011;39:635–40. doi: 10.3892/ijo.2011.1061. [DOI] [PubMed] [Google Scholar]
- 94.Kuniyasu H, Chihara Y, Kondo H, Ohmori H, Ukai R. Amphoterin induction in prostatic stromal cells by androgen deprivation is associated with metastatic prostate cancer. Oncol Rep. 2003;10:1863–8. [PubMed] [Google Scholar]
- 95.Lotze MT, Tracey KJ. High-mobility group box1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 96.Stott K, Watson M, Bostock MJ, Mortensen SA, Travers A, et al. Structural insights into the mechanism of negative regulation of single-box high mobility group proteins by the acidic tail domain. J Biol Chem. 2014;289:29817–26. doi: 10.1074/jbc.M114.591115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knapp S, Muller S, Digilio G, Bonaldi T, Bianchi ME, et al. The long acidic tail of high mobility group box1 (HMGB1) protein forms an extended and flexible structure that interacts with specific residues within and between the HMG boxes. Biochemistry. 2004;43:11992–7. doi: 10.1021/bi049364k. [DOI] [PubMed] [Google Scholar]
- 98.Stott K, Watson M, Howe FS, Grossmann JG, Thomas JO. Tail-mediated collapse of HMGB1 is dynamic and occurs via differential binding of the acidic tail to the A and B domains. J Mol Biol. 2010;403:706–22. doi: 10.1016/j.jmb.2010.07.045. [DOI] [PubMed] [Google Scholar]
- 99.Sahu D, Debnath P, Takayama Y, Iwahara J. Redox properties of the A-domain of the HMGB1 protein. FEBS Lett. 2008;582:3973–8. doi: 10.1016/j.febslet.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–12. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 101.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Snider L, Thirlwell H, Miller JR, Moon RT, Groudine M, et al. Inhibition of Tcf3 binding by I-mfa domain proteins. Mol Cell Biol. 2001;21:1866–73. doi: 10.1128/MCB.21.5.1866-1873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Furstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol. 2002;3:298–302. doi: 10.1016/s1470-2045(02)00731-3. [DOI] [PubMed] [Google Scholar]
- 104.Wu J, Yu E. Insulin-like growth factor receptor-1 (IGF-IR) as a target for prostate cancer therapy. Cancer Metastasis Rev. 2014;33:607–17. doi: 10.1007/s10555-013-9482-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sutherland BW, Knoblaugh SE, Kaplan-Lefko PJ, Wang F, Holzenberger M, et al. Conditional deletion of insulin-like growth factor-I receptor in prostate epithelium. Cancer Res. 2008;68:3495–504. doi: 10.1158/0008-5472.CAN-07-6531. [DOI] [PubMed] [Google Scholar]
- 106.Heidegger I, Ofer P, Doppler W, Rotter V, Klocker H, et al. Diverse functions of IGF/insulin signaling in malignant and noncancerous prostate cells: proliferation in cancer cells and differentiation in noncancerous cells. Endocrinology. 2012;153:4633–43. doi: 10.1210/en.2012-1348. [DOI] [PubMed] [Google Scholar]
- 107.Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, et al. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR) Cancer Res. 2001;61:6276–80. [PubMed] [Google Scholar]
- 108.Salatino M, Schillaci R, Proietti CJ, Carnevale R, Frahm I, et al. Inhibition of in vivo breast cancer growth by antisense oligodeoxynucleotides to type I insulin-like growth factor receptor mRNA involves inactivation of ErbBs, PI-3K/Akt and p42/p44 MAPK signaling pathways but not modulation of progesterone receptor activity. Oncogene. 2004;23:5161–74. doi: 10.1038/sj.onc.1207659. [DOI] [PubMed] [Google Scholar]
- 109.Heidegger I, Pircher A, Klocker H, Massoner P. Targeting the insulin-like growth factor network in cancer therapy. Cancer Biol Ther. 2011;11:701–7. doi: 10.4161/cbt.11.8.14689. [DOI] [PubMed] [Google Scholar]
- 110.Alberobello AT, D’Esposito V, Marasco D, Doti N, Ruvo M, et al. Selective disruption of insulin-like growth factor-1 (IGF-1) signaling via phosphoinositide-dependent kinase-1 prevents the protective effect of IGF-1 on human cancer cell death. J Biol Chem. 2010;285:6563–72. doi: 10.1074/jbc.M109.097410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heidegger I, Massoner P, Sampson N, Klocker H. The insulin-like growth factor (IGF) axis as an anticancer target in prostate cancer. Cancer Lett. 2015;367:113–21. doi: 10.1016/j.canlet.2015.07.026. [DOI] [PubMed] [Google Scholar]
- 112.Sato A, Nishimura S, Ohkubo T, Kyogoku Y, Koyama S, et al. 1H-NMR assignment and secondary structure of human insulin-like growth factor-I (IGF-I) in solution. J Biochem. 1992;111:529–36. doi: 10.1093/oxfordjournals.jbchem.a123791. [DOI] [PubMed] [Google Scholar]
- 113.Torres AM, Forbes BE, Aplin SE, Wallace JC, Francis GL, et al. Solution structure of human insulin-like growth factor II. Relationship to receptor and binding protein interactions. J Mol Biol. 1995;248:385–401. doi: 10.1016/s0022-2836(95)80058-1. [DOI] [PubMed] [Google Scholar]
- 114.Kuang Z, Yao S, McNeil KA, Forbes BE, Wallace JC, et al. Insulin-like growth factor-I (IGF-I): solution properties and NMR chemical shift assignments near physiological pH. Growth Horm IGF Res. 2009;19:226–31. doi: 10.1016/j.ghir.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 115.Cooke RM, Harvey TS, Campbell ID. Solution structure of human insulin-like growth factor 1: a nuclear magnetic resonance and restrained molecular dynamics study. Biochemistry. 1991;30:5484–91. doi: 10.1021/bi00236a022. [DOI] [PubMed] [Google Scholar]
- 116.Hiort O. The differential role of androgens in early human sex development. BMC Med. 2013;11:152. doi: 10.1186/1741-7015-11-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schmidt LJ, Tindall DJ. Androgen receptor: past, present and future. Curr Drug Targets. 2013;14:401–7. doi: 10.2174/1389450111314040002. [DOI] [PubMed] [Google Scholar]
- 118.Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer cell. 2009;16:458–62. doi: 10.1016/j.ccr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 119.Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9:155–70. doi: 10.1677/erc.0.0090155. [DOI] [PubMed] [Google Scholar]
- 120.Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. J Natl Cancer Inst. 2001;93:1687–97. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- 121.Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, et al. FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol Cell Biol. 2010;30:1243–53. doi: 10.1128/MCB.01891-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Centenera MM, Harris JM, Tilley WD, Butler LM. The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol Endocrinol. 2008;22:2373–82. doi: 10.1210/me.2008-0017. [DOI] [PubMed] [Google Scholar]
- 123.Perner S, Cronauer MV, Schrader AJ, Klocker H, Culig Z, et al. Adaptive responses of androgen receptor signaling in castration-resistant prostate cancer. Oncotarget. 2015;6:35542–55. doi: 10.18632/oncotarget.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McEwan IJ. Intrinsic disorder in the androgen receptor: identification, characterisation and drugability. Mol Biosyst. 2012;8:82–90. doi: 10.1039/c1mb05249g. [DOI] [PubMed] [Google Scholar]
- 125.Needham M, Raines S, McPheat J, Stacey C, Ellston J, et al. Differential interaction of steroid hormone receptors with LXXLL motifs in SRC-1a depends on residues flanking the motif. J Steroid Biochem Mol Biol. 2000;72:35–46. doi: 10.1016/s0960-0760(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 126.van de Wijngaart DJ, van Royen ME, Hersmus R, Pike AC, Houtsmuller AB, et al. Novel FXXFF and FXXMF motifs in androgen receptor cofactors mediate high affinity and specific interactions with the ligand-binding domain. J Biol Chem. 2006;281:19407–16. doi: 10.1074/jbc.M602567200. [DOI] [PubMed] [Google Scholar]
- 127.van Royen ME, Cunha SM, Brink MC, Mattern KA, Nigg AL, et al. Compartmentalization of androgen receptor protein-protein interactions in living cells. J Cell Biol. 2007;177:63–72. doi: 10.1083/jcb.200609178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lallous N, Dalal K, Cherkasov A, Rennie PS. Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int J Mol Sci. 2013;14:12496–519. doi: 10.3390/ijms140612496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sadar MD. Small molecule inhibitors targeting the “achilles’ heel” of androgen receptor activity. Cancer Res. 2011;71:1208–13. doi: 10.1158/0008-5472.CAN_10-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer. 2011;18:R183–96. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lavery DN, McEwan IJ. Structure and function of steroid receptor AF1 transactivation domains: induction of active conformations. Biochem J. 2005;391:449–64. doi: 10.1042/BJ20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Quayle SN, Mawji NR, Wang J, Sadar MD. Androgen receptor decoy molecules block the growth of prostate cancer. Proc Natl Acad Sci U S A. 2007;104:1331–6. doi: 10.1073/pnas.0606718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.McEwan IJ. Molecular mechanisms of androgen receptor-mediated gene regulation: structure-function analysis of the AF-1 domain. Endocr Relat Cancer. 2004;11:281–93. doi: 10.1677/erc.0.0110281. [DOI] [PubMed] [Google Scholar]
- 134.Hilser VJ, Thompson EB. Structural dynamics, intrinsic disorder, and allostery in nuclear receptors as transcription factors. J Biol Chem. 2011;286:39675–82. doi: 10.1074/jbc.R111.278929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Claessens F, Denayer S, Van Tilborgh N, Kerkhofs S, Helsen C, et al. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008;6:e008. doi: 10.1621/nrs.06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dubbink HJ, Hersmus R, Verma CS, van der Korput HA, Berrevoets CA, et al. Distinct recognition modes of FXXLF and LXXLL motifs by the androgen receptor. Mol Endocrinol. 2004;18:2132–50. doi: 10.1210/me.2003-0375. [DOI] [PubMed] [Google Scholar]
- 137.Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res. 2007;67:10067–77. doi: 10.1158/0008-5472.CAN-07-1267. [DOI] [PubMed] [Google Scholar]
- 138.Marasco D, Perretta G, Sabatella M, Ruvo M. Past and future perspectives of synthetic peptide libraries. Curr Protein Pept Sci. 2008;9:447–67. doi: 10.2174/138920308785915209. [DOI] [PubMed] [Google Scholar]
- 139.Scognamiglio PL, Doti N, Grieco P, Pedone C, Ruvo M, et al. Discovery of small peptide antagonists of PED/PEA15-D4alpha interaction from simplified combinatorial libraries. Chem Biol Drug Des. 2011;77:319–27. doi: 10.1111/j.1747-0285.2011.01094.x. [DOI] [PubMed] [Google Scholar]
- 140.Sadar MD, Williams DE, Mawji NR, Patrick BO, Wikanta T, et al. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org Lett. 2008;10:4947–50. doi: 10.1021/ol802021w. [DOI] [PubMed] [Google Scholar]
- 141.Meimetis LG, Williams DE, Mawji NR, Banuelos CA, Lal AA, et al. Niphatenones, glycerol ethers from the sponge Niphates digitalis block androgen receptor transcriptional activity in prostate cancer cells: structure elucidation, synthesis, and biological activity. J Med Chem. 2012;55:503–14. doi: 10.1021/jm2014056. [DOI] [PubMed] [Google Scholar]
- 142.Biron E, Bedard F. Recent progress in the development of protein-protein interaction inhibitors targeting androgen receptor-coactivator binding in prostate cancer. J Steroid Biochem Mol Biol. 2015;151 doi: 10.1016/j.jsbmb.2015.07.006. pii: S0960-0760(15)30017-0. Doi: 10.1016/j.jsbmb. 2015.07.006. In Press. [DOI] [PubMed] [Google Scholar]
- 143.Teiten MH, Gaigneaux A, Chateauvieux S, Billing AM, Planchon S, et al. Identification of differentially expressed proteins in curcumin-treated prostate cancer cell lines. OMICS. 2012;16:289–300. doi: 10.1089/omi.2011.0136. [DOI] [PubMed] [Google Scholar]
- 144.Kang YJ, Olson MO, Jones C, Busch H. Nucleolar phosphoproteins of normal rat liver and Novikoff hepatoma ascites cells. Cancer Res. 1975;35:1470–5. [PubMed] [Google Scholar]
- 145.Okuwaki M, Iwamatsu A, Tsujimoto M, Nagata K. Identification of nucleophosmin/B23, an acidic nucleolar protein, as a stimulatory factor for in vitro replication of adenovirus DNA complexed with viral basic core proteins. J Mol Biol. 2001;311:41–55. doi: 10.1006/jmbi.2001.4812. [DOI] [PubMed] [Google Scholar]
- 146.Balusu R, Fiskus W, Rao R, Chong DG, Nalluri S, et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood. 2011;118:3096–106. doi: 10.1182/blood-2010-09-309674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Scognamiglio PL, Di Natale C, Leone M, Poletto M, Vitagliano L, et al. G-quadruplex DNA recognition by nucleophosmin: new insights from protein dissection. Biochim Biophys Acta. 2014;1840:2050–9. doi: 10.1016/j.bbagen.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 148.Leotoing L, Meunier L, Manin M, Mauduit C, Decaussin M, et al. Influence of nucleophosmin/B23 on DNA binding and transcriptional activity of the androgen receptor in prostate cancer cell. Oncogene. 2008;27:2858–67. doi: 10.1038/sj.onc.1210942. [DOI] [PubMed] [Google Scholar]
- 149.Gsponer J, Babu MM. The rules of disorder or why disorder rules. Prog Biophys Mol Biol. 2009;99:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 150.Zeng Y, He YN, Yang F, Mooney SM, Getzenberg RH, et al. The cancer/testis antigen prostate-associated gene 4 (PAGE4) is a highly intrinsically disordered protein. J Biol Chem. 2011;286:13985–94. doi: 10.1074/jbc.M110.210765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suyama T, Shiraishi T, Zeng Y, Yu W, Parekh N, et al. Expression of cancer/testis antigens in prostate cancer is associated with disease progression. Prostate. 2010;70:1778–87. doi: 10.1002/pros.21214. [DOI] [PMC free article] [PubMed] [Google Scholar]