

Abstract
The role of androgen receptor (AR) in the initiation and progression of prostate cancer (PCa) is well established. Competitive inhibition of the AR ligand-binding domain (LBD) has been the staple of antiandrogen therapies employed to combat the disease in recent years. However, their efficacy has often been limited by the emergence of resistance, mediated through point mutations, and receptor truncations. As a result, the prognosis for patients with malignant castrate resistant disease remains poor. The amino-terminal domain (NTD) of the AR has been shown to be critical for AR function. Its modular activation function (AF-1) is important for both gene regulation and participation in protein-protein interactions. However, due to the intrinsically disordered structure of the domain, its potential as a candidate for therapeutic intervention has been dismissed in the past. The recent emergence of the small molecule EPI-001 has provided evidence that AR-NTD can be targeted therapeutically, independent of the LBD. Targeting of AR-NTD has the potential to disrupt multiple intermolecular interactions between AR and its coregulatory binding partners, in addition to intramolecular cross-talk between the domains of the AR. Therapeutics targeting these protein-protein interactions or NTD directly should also have efficacy against emerging AR splice variants which may play a role in PCa progression. This review will discuss the role of intrinsic disorder in AR function and illustrate how emerging therapies might target NTD in PCa.
Keywords: androgen receptor, inhibitor, intrinsically disordered structure, N-terminal domain, prostate cancer, small molecules
INTRODUCTION
The discovery in 1941 by Dr. Charles Brenton Huggins that castration or androgen ablation had a beneficial effect in patients with prostate cancer (PCa) opened the door for the development of therapeutic androgen receptor (AR) antagonists in this disease.1 Cyproterone acetate was the first antiandrogen to be used clinically (1964), competitively antagonizing the AR and inhibiting gonadotropin secretion. This was quickly superseded by nonsteroidal antiandrogens such as hydroxyflutamide, nilutamide, and bicalutamide, which showed lower hepatotoxicity and higher selectivity for the AR.2
Despite advances, treatment of PCa has been limited by the development of resistance to antiandrogen therapy and progression to castrate resistant disease (CRPC). Specific point mutations in the ligand-binding domain (LBD) of AR have been identified in patient samples and immortalized PCa cell lines which confer resistance to all first-generation antiandrogens, in addition to second-generation antiandrogens enzalutamide and ARN-509.3,4 In addition, the presence of splice variants of the AR lacking the LBD in patients with advance CRPC may also contribute to the development of resistance.5,6 As a result, alternative targets have been proposed for novel PCa therapies which would inhibit AR action, including androgen synthesis inhibitors, such as abiraterone,7 molecules with the capacity to block the interaction between AR and its coregulatory proteins,8 compounds targeting the AR for degradation,9 and molecules which target other functional regions of AR such as DNA-binding domain (DBD) or intrinsically disordered amino-terminal domain (NTD)10,11 (Figure 1).
Figure 1.
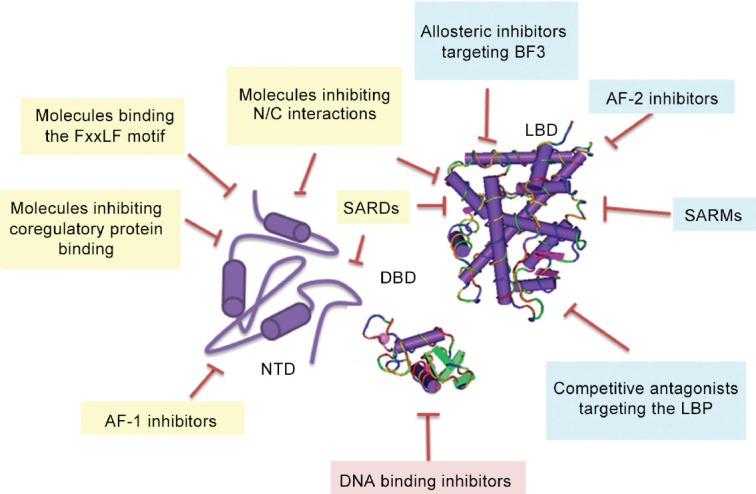
Proposed mechanisms for the inhibition of the AR and its splice variants in CRPC. The AR protein has a globular ligand (LBD) and DNA (DBD)-binding domains and a large intrinsically disordered amino-terminal domain (NTD). A number of therapies in clinical and preclinical development competitively target the AR-LBD. These include small molecules and peptides binding to the ligand-binding pocket (LBP) and the AF-2 and BF3 surfaces. Therapeutic targeting of the AR-DBD is also possible, but high sequence conservation between different members of the steroid hormone receptor family makes off-target effects more likely. Targeting of the AR-NTD with molecules which interact with FxxLF motif, inhibit protein-protein interactions or impair receptor transactivation would all be beneficial in downregulating AR in PCa. Combining or conjugating these therapies with chemicals able to degrade the androgen receptor would also be beneficial.
STRUCTURE AND FUNCTION OF THE AR
The AR (NR3C4) is a ligand-gated transcription factor and member of the steroid hormone receptor family.12 In normal physiology, the AR mediates responses to endogenous androgens such as testosterone and 5α-dihydrotestosterone (DHT), facilitating male sexual differentiation and sperm production, in addition to roles in metabolism, the nervous system, and promotion of skeletal muscle growth.13,14 The AR has also been shown to play a key role in driving the initiation and progression of PCa and as such has become a major therapeutic target in this disease.1,15 Pathological and protective roles for the AR have also been suggested in the development of breast, ovarian, and endometrial cancers.16,17
The human AR gene is located on the X chromosome and encodes eight exons which are transcribed and translated to a 110 kDa protein composed of four functional domains (Figure 2).18,19 LBD (amino acids 671–920) contains the ligand-binding pocket (LBP), site of binding for endogenous androgens and exogenous antiandrogens. The domain also contains activation function (AF)-2, an important surface for interaction between N- and C-termini of the receptor, facilitating cross-talk between receptor domains. Mutations within the LBD have been shown to alter both agonist and antagonist binding of AR, as well as the binding of receptor chaperone and coregulatory proteins, and approximately 45% of mutations in PCa have been mapped to this domain.3,4,20 Mutations linked to resistance to antiandrogens have been identified, including point mutation T877A which confers resistance to hydroxyflutamide in LNCaP PCa cell line and missense mutation F876L conferring resistance to second-generation antiandrogens enzalutamide and ARN-509.3 Resistance to these therapies is also conferred by receptor splice variants which lack the LBD (Figure 2).6 The AR gene mutations database contains a comprehensive overview of all the mutations identified in patients with AR associated disorders.20
Figure 2.
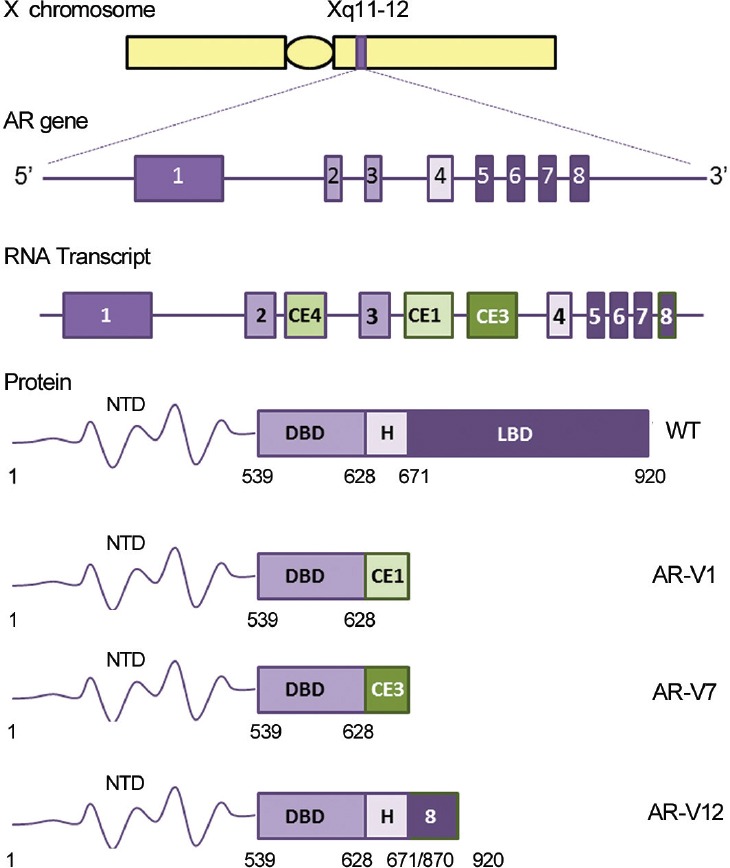
Structure of the AR gene, transcript, and protein. The AR gene is located on the X chromosome and encoded by eight exons which are transcribed and translated to form a 110 kDa protein with four functional domains: the LBD, hinge, DBD, and NTD. Alternative splicing of the RNA transcript can lead to the emergence of constitutively active AR splice variants lacking various portions of the AR C-terminal region. Expression of AR splice variants AR-V1, AR-V7, and AR-V12 have all been documented in both prostate cancer cell lines and patient samples. Number for the human AR is based on the NCBI reference sequence NM_000044.3.
Adjacent to the LBD is a flexible hinge region (amino acids 629–670), with a bipartite nuclear localization signal formed by the sequence RKLKKL (amino acids 629–634), which is a target for acetylation, ubiquitylation, and methylation.21 Crystallization of importin-α and AR shows binding via this motif, highlighting the importance of the hinge region in nuclear translocation.22 In addition, the RKLKKL motif, through forming part of the carboxyterminal extension (CTE) of the DNA-binding domain (DBD) and interacting with selective androgen response elements (AREs), provides a role for the hinge in DNA binding.23 Somatic point mutations R629Q and K630T identified in a patient with CRPC were shown to have increased transcriptional potency in transfected HeLa cells compared to WT AR.24 Deletion of amino acids 629–636 was also shown to increase N/C interaction, suggesting an inhibitory role for the hinge region in AR transactivation. The evidence is still growing to suggest that the hinge region may be a distinct functional unit of the receptor.24 However, its potential as a drug target remains unknown.
The AR-DBD (amino acids 539–628) is composed of two zinc finger regions required for receptor dimerization and binding DNA elements in the promoter and enhancer regions of androgen-responsive genes.18 While essential for the function of the receptor, this region is highly conserved within the steroid receptor family, making it a challenging target for therapeutic manipulation in PCa.
THE ROLE OF THE AR-NTD IN RECEPTOR FUNCTION
Previous work using AR deletion mutants has shown that the NTD (amino acids 1–538) is critical for the transactivation and function of the AR.25,26 The AR-AF-1 is composed of two units: the ligand-dependent TAU-1 (amino acids 101–307) and ligand-independent TAU-5 (amino acids 360–528). In addition, a FxxLF motif (amino acids 23–27) is important for facilitating N/C terminal interactions and coregulatory protein binding. Phosphorylation of serine residues within the NTD is implicated in receptor function, with specific effects on gene expression, coregulatory protein binding, cellular localization, and receptor degradation.27 A growing number of coregulatory proteins have been described binding to the NTD, of particular note are members of the p160 coactivator family (SRC-1 and -2), the histone acetyltransferase protein CBP and the general transcription factor TFIIF, which all bind to the AR-AF1 domain.28 Also, of note is the “transcriptional hub” protein MAGE-A11, which binds to the FxxLF motif in the AR-NTD, interacts with CBP (p300) and p160 proteins, and represents a potential tissue-selective AR transcriptional coactivator.29 The AR-NTD has also been shown to modulate binding affinity of the AR-DBD although this relies on covalent binding of the two domains.30 It has also been suggested that expression of the NTD may drive androgen independent nuclear localization and suppress nuclear export of the AR in CRPC.31
The AR-NTD plays a direct role in the pathogenesis of several diseases, with 30% of AR mutations identified in PCa mapped to the NTD.20 The polyglutamine (polyQ) repeat in the AR-NTD is crucial in SBMA pathogenesis but may also play a role in regulating folding and structure within the NTD. While hyperextension of this repeat results in SBMA, shorter repeat lengths have been associated with an increased risk of PCa. Analysis of the AR-NTD with an expanded (Q45) or deleted polyQ region using circular dichroism (CD) spectroscopy revealed that expansion of the region promoted α-helical structure while deletion resulted in a loss of α-helical content and movement of four tryptophan residues.32
Experimental and bioinformatic analyses reveal that the AR-NTD conforms to the idea of an intrinsically disordered protein (IDP) and exists as an ensemble of conformations with collapsed disordered structure.27,33 This large collection of interconverting conformations thus allows rapid and reversible alterations in domain structure in response to the cellular environment and coregulatory protein binding. The advantage of this IDP structure is the ability for flexible binding to multiple protein partners with distinct outcomes and has been proposed to mediate allosteric regulation of receptor function.27,34 Analyses by circular dichroism (CD), Fourier transform infrared (FTIR) spectroscopy, secondary structure prediction, and mutagenesis have revealed that different regions of the NTD contain more or less stable secondary structure, including four regions of α-helix within the AR-AF1 domain.27,34
SPLICE VARIANTS OF AR AND THEIR ROLE IN CRPC
Alternative splicing of the AR can lead to generation of constitutively active splice variants lacking varying portions of the LBD and hinge regions.5 Interest has grown rapidly in the role of splice variants in prostate cancer progression, from both mechanistic and therapeutic standpoints. Splice variants show altered interactions with coregulatory proteins and transcription factors, DNA, and drugs used in PCa treatment.5,6,35,36 The dimerization and interaction of splice variants with full-length AR and altered cellular localization have also been studied.37 The best characterized AR splice variants in the context of PCa are the ARv7 and ARv567 (ARv12) (Figure 2).
Although detected at low levels in hormone-naive samples, transcripts of ARv4, ARv7, and ARv567 have been found to be enriched in prostate epithelium of patients with prostate cancer and in CRPC bone metastasis where they were associated with increased nuclear AR, aberrant cell cycle regulation, and reduced overall survival.35,36 More recently, expression ARv7 was detected in circulating tumor cells of patients treated with antiandrogen enzalutamide (12 of 31) or Cyp17A1 inhibitor abiraterone (6 of 31), and expression was correlated with therapy-resistance.38 The identification of splice variants in clinical prostate cancer samples and particularly their enrichment in metastasis not only suggests a mechanism for the progression of CRPC in the absence of androgens but also highlights the need for therapies with the capacity to target these variants which lack the LBD. The altered transcriptional profile of splice variants suggests that they act in different pathways to the full-length receptor to promote PCa progression.39 Although there are conflicting reports on whether expression of the full-length receptor is also required for their action, targeting the NTD of the receptor would be beneficial in either circumstance.
The ARv567 lacks exons 5, 6, and 7 (Figure 2) and is capable of increasing the expression of full-length AR when exogenously expressed in LNCaP cells and conferring resistance to castration in a xenograft model. This variant is capable of both binding and stabilizing AR protein, increasing the full-length receptor's sensitivity to hormone, and also functioning as a dominant constitutively active splice variant with an alternative regulation of gene expression.35
The ARv7 variant consists of exons 1, 2, and 3 plus an untranslated region caused by alternative splicing and inclusion of exon CE3 in the RNA transcript (Figure 2). ARv7 has been shown to differentially regulate FOXA1 sensitive genes in relevant PCa cell lines. Krause et al.40 generated LNCaP cell lines coexpressing doxycycline inducible ARv7 and an AR-NTD-DBD construct. ARv7 was found to be less effective at inducing expression of TMPRSS2 and could not induce hormone-regulated genes RASSF3 and EXTL2 which require FOXA1 for AR-mediated induction. However, EDN2 and ETS2 were notably upregulated by ARv7 and AR-NTD-DBD. Knockdown of FOXA1 by siRNA had no effect on ARv7 protein expression or induction of EDN2 and ETS2 but attenuated R1881-mediated expression of RASSF3 and eliminated hormone driven repression of EDN2.40 Similarly, EDN2 and ETS were upregulated in VCaP cells transfected with ARv7 and in the 22Rv1 cell line endogenously expressing ARv7. The upregulation of a unique set of target genes by ARv7, independent of the hormone regulated full-length AR, may constitute a growth and/or survival advantage to cells expressing this variant in PCa.40
In addition, similarity of results obtained using ARv7 and AR-NTD-DBD variants, which differ only in the hinge region sequence, indicates that the transcriptional activity of specific variants is driven by the loss of the LBD and may not differ between variants. As a result, overall levels of splice variant might be a sufficient indicator of the biology of the tumor.40
Although often examined for their independent effects on gene regulation, AR splice variants may also modify the action of full-length AR. Cao et al.37 showed that ARv7 and ARv567 were both capable of facilitating the nuclear localization of full-length AR in the absence of androgens. Perhaps more importantly the translocation of both variants was unaffected by enzalutamide in the presence or absence of hormone, and the presence of splice variants also prevented the cytoplasmic sequestration of full-length AR by enzalutamide in the presence and absence of R1881. Using ChIP analysis, they identified that in the 22Rv1 cell line, ARv7 and full-length AR are capable of cooccupying the PSA promoter in androgen independent conditions in a codependent manner. However, in the presence of androgen, ARv7 attenuates full-length AR transactivation and reduces the response of PSA and TMPRSS2 to hormone. Together with ARv7 knockdown studies in vitro and in xenograft models, these data suggest a mechanism of enzalutamide resistance whereby ARv7 reduces the response of cells to hormone-dependent signaling and growth.37
In addition to targeted anti-androgen therapy, cytotoxic taxane chemotherapy is also approved by the FDA for treatment in CRPC. Cabazitaxel and docetaxel are approved for first- and second-line chemotherapies and are the only class of chemotherapeutics to prolong survival in castrate-resistant disease. Taxanes have also been shown to inhibit ligand-induced AR nuclear translocation and subsequently downstream signaling in CRPC circulating tumor cells.41 Interestingly, taxane sensitivity may be modified by the expression of AR splice variants. As these variants lack varying regions of the LBD and hinge regions, they may utilize alternative mechanisms of nuclear translocation to the full-length receptor. In their 2014 study, using a microtubule cosedimentation assay, Thadani-Mulero et al. identified that association of the AR with the microtubule cytoskeleton was mediated by the C-terminal domain of the receptor, with contributions from the LBD, hinge region, and DBD.42 When examining the clinically relevant ARv567 and ARv7 splice variants, tagged with GFP and transfected into the M12 PCa cell line, they found that nuclear accumulation of ARv567 and wild-type AR was impaired following docetaxel treatment, but there was no effect on ARv7 translocation. In addition, docetaxel treatment impaired FKBP51 transcription induced by the ARv567 variant, but not ARv7. Similarly, in xenograft tumors of LuCAP86.2 cells expressing ARv567, docetaxel treatment impaired tumor growth and nuclear localization, a result not seen in LuCAP23.1 xenograft models (expressing WT AR and ARv7).42
Further investigations suggest that unlike AR and ARv567, ARv7 does not utilize dynein-dependent transport for nuclear localization. ARv7 is truncated at aa 644, lacking a C-terminal portion of the hinge region which has been proposed to be important in the association with dynein protein. In contrast, ARv567 has a fully constituted hinge-region, lacking exons 5, 6, and 7 of the LBD.42 This evidence conflicts with the conclusions of Krause et al.40 and suggests that splice variants which differ in the hinge region sequence may have alternative mechanisms of action.
THERAPEUTIC TARGETING OF IDPS
IDPs are particularly enriched in signaling pathways where they offer flexible binding to multiple protein partners with rapid and selective outcomes. The ability to target these proteins therapeutically has been the goal in several diseases, including the targeting of BRCA1 in breast cancer and tau in Alzheimer's disease. However, the lack of secondary or tertiary structure in IDPs limits the capability of structural drug discovery techniques, such as in silico molecular modelling.
So far, the identification of compounds with the capacity to target the AR-NTD has involved screening of marine sponge extracts.10,43,44 Research is now shifting to include both high throughput screening of compounds with activity at the AR, alongside the design of peptides antagonists against the AR-AF-1 region and selective androgen receptor downregulators (SARDs). The following sections discuss the latest compounds with the ability to inhibit the AR, their mechanism of action, and their relevance to the inhibition of the AR and its splice variants via the NTD.
SMALL MOLECULES AND NATURAL PRODUCT INHIBITORS OF THE AR-NTD
Chlorinated peptide sinkotamide A, small molecules EPI-001 and glycerol ether naphetenone B (Figure 3) associating with the AR-NTD inhibit receptor transactivation and function. Sintokamides A–E are bioactive chlorinated peptides isolated from the sponge Dysidea sp. although a microbial origin has been suggested (cyanobacteria).44 In PSA-luciferase reporter gene assays, Sintokamide A (Figure 3a) inhibited hormone-dependent AR-induced reporter gene activity. To examine AR-NTD transactivation, LNCaP was transfected with an AR NTD-Gal4DBD fusion protein, stimulated with forskolin, and activity was measured with the Gal4-luciferase reporter. Sintokamide A inhibited forskolin-induced transactivation of the AR-NTD, and additionally inhibited proliferation in LNCaP cells, although did not inhibit proliferation of the PC-3 AR-negative cell line, suggesting that AR expression is required for efficacy.
Figure 3.
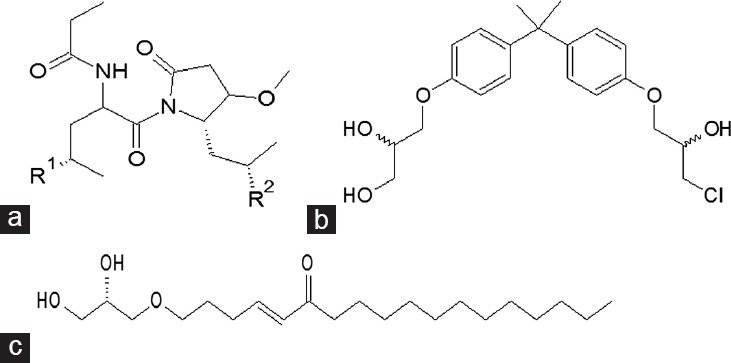
Small molecule compounds identified from marine library screening with inhibitory activity at the AR-NTD. (a) Sinkotamides are chlorinated peptides isolated from marine sponge Dysidea sp. (b) EPI-001 is a bisphenol A derivative. (c) Glycerol ether Naphetenone B from the marine sponge Niphates digitalis.
EPI-001 is a bisphenol A diglycidyl ether derivative10 (Figure 3b). Bisphenol A has previously been described as an androgen disrupter.45,46,47 However, the ability of EPI-001 to bind the AF-1 region of the AR-NTD and inhibit receptor function provided an excellent proof-of-concept for driving investigations into other small molecules with the capacity to inhibit the AR in this fashion. EPI-001 (and its trans isomer EPI-002) has been shown to inhibit forskolin-induced AR-NTD transactivation, inhibit proliferation of cell lines expressing AR, selectively block receptor-protein interactions and recruitment of the AR to DNA response elements, and has an additive effect on androgen-responsive reporter gene activity when used in combination with low concentrations of antiandrogen bicalutamide.10,48 Steady-state fluorescence experiments with the AR-AF1 region have shown that EPI-001 requires some α-helical structure in this region to bind and this can be disrupted using urea.48
The activity of EPI-001 compounds has been attributed to the presence of the chlorohydrin group. Using a biotinylated version of EPI-002, Myung et al.48 showed directly that EPI binds AR covalently in LNCaP cells. In addition, compounds lacking the chlorohydrin group did not have any effect on the weight of androgen sensitive organs in vivo, in contrast to the reduction seen following EPI-002 treatment. Both EPI-001 and EPI-002 are capable of reducing tumor volume in xenograft mouse models of PCa.10,48 An analogue of the EPI compounds is currently in phase I/II trials (NCT02606123), which represents the first AR-NTD inhibitor to be tested in patients with metastatic castrate-resistant prostate cancer.
More recently, Brand and colleagues found that 50 μmol l−1 EPI-001 resulted in a 90% inhibition of AR-mediated luciferase reporter activity.49 With the use of AR NTD-Gal4 DBD deletion constructs, EPI-001 was shown to inhibit the transcriptional activity of both TAU1 and TAU5. EPI-001 also inhibited AR mRNA and protein expression in LNCaP, C4-2, LAPC4, and 22Rv1 cells, including the expression of known endogenous splice variants. The reduction in mRNA was due to inhibition of AR transcription rather than increased mRNA degradation.49 Interestingly, protein and mRNA expression were not affected by EPI-001 in CWR-R1 cells. Growth suppression was observed in both AR positive and negative PCa cell lines, with the former notably sensitive to low concentrations, below 25 μmol l−1, of EPI-001.49 Supporting the argument that EPI-001 can selectively target the AR. However, it is worth noting that this study by Dehm and coworkers also found evidence for EPI-001 acting as a “selective PPARγ modulator” at higher concentrations and a role for this nuclear receptor in the inhibition of androgen-regulated target genes.5 Furthermore, this study suggested that EPI-001 could act as a general alkylating agent, however at neutral pH, this effect was very modest. In contrast, the original in vivo work from Sadar and co-workers found no evidence for general toxicology in mice injected with EPI-001.44 The outcomes from the ongoing clinical trials, on efficacy and tolerance, are therefore awaiting with considerable interest.
Martin et al.50 also demonstrated a reduction in cell viability of 22Rv1 PCa cells (expressing AR with duplication of exon 3 and ARv7) in response to docetaxel treatment, which was enhanced by treatment with EPI-002. Both EPI and docetaxel monotherapy and combination therapies also enhanced cytotoxicity in the LNCaP95 cell line expressing full-length AR (with T877A mutation) and ARv7. This suggests a synergistic effect and highlights the benefit of targeting the NTD alongside microtubule-mediated AR translocation. 22Rv1 xenograft models responded to treatment with docetaxel, and cotreatment with docetaxel and EPI-002, but not to EPI-002 alone. However, no change in cellular localization of AR could be seen with docetaxel treatment, suggesting the response to taxane therapy was not mediated by AR. Combination therapy did decrease AR-mediated transcription in reporter gene assays. However, effects of docetaxel and EPI-002 monotherapy and combination therapy were not sustained when expression of mRNA for gene targets PSA/KLK3 (WT AR) and UGT2B17 (ARv7) were examined.50
Niphatenone B is a glycerol ether from the marine sponge Niphates digitalis43 (Figure 3c). The compound showed inhibitory activity against the AR in reporter gene assays and inhibited androgen driven proliferation of LNCaP, but not cells lacking the AR. Niphatenone B blocks AR N/C interactions (Figure 1) and expression of AR-regulated genes. However, it also covalently binds both the AR and glucocorticoid receptor AF-1 regions.51 The reported roles of the glucocorticoid receptor in PCa are conflicting, acting as a tumor suppressor in some circumstances, and promoting PCa progression in others. However, in CRPC cell lines lacking AR expression, the GR is capable of driving AR-responsive gene expression and cell proliferation, and this suggests that cotargeting of both the AR and glucocorticoid receptor might be beneficial in some patients.52
TARGETING PROTEIN-PROTEIN INTERACTIONS IN THE AR-NTD
Upregulated expression of the AR in both wildtype and mutated forms has been extensively documented in advanced PCa. However, the upregulation of coactivator proteins has also been reported and could be an important mediator of sustained AR activity in low androgen conditions. Both Steroid Receptor Coactivator 1 (SRC1) and Transcriptional Intermediary Factor 2 (TIF2, also called SRC2) have been shown to be overexpressed in CRPC15 and altered expression and localization of AR corepressors in prostate cancer has also been described.
Brooke et al.8 have described engineered repressors of the AR - short peptides consisting of an interaction motif fused to repression domains from AR corepressors (Figure 4). These repressors contain an FxxLF α-helix and as such competed for binding to the AR-LBD AF-2. Colocalization of the AR and engineered repressors was shown by fusion of MAD7–35-AR1–54 with GFP, which was nuclear in the presence of hormone. Immunoprecipitation with α-GFP also resulted in ligand-dependent pull down of AR.8 In reporter gene assays, repression domains alone had no effect on AR activity. In isolation, the FxxLF peptide alone reduced reporter gene activity by 34%. However, the fused engineered repressors inhibited receptor transactivation up to 86%, in both transiently and stably transfected cells. Some inhibition of GR, PR, and ER activity was seen, albeit at lower levels than the AR, suggesting some selectivity for the AR. Independent mutation of either the FxxLF motif or repression domain both reduced inhibitory action of the engineered repressors. The engineered repressors were also able to inhibit SRC-1 enhanced AR activity, which is often seen in CRPC and were effective against the AR with the T877A or H874Y mutations associated with antiandrogen resistance. LNCaP proliferation and colony formation were also reduced in the presence of engineered repressors.8 It would be interesting to see if a similar approach could be employed to target the AR-AF-1 region using peptides complementary to known protein-protein interactions.
Figure 4.
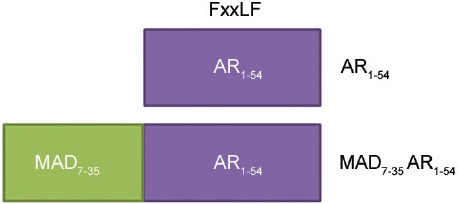
Engineered repressors of the AR. Potential peptide repressors of the AR containing the FxxLF motif of the AR-NTD would be predicted to compete for binding to the AF-2 surface on the LBD. This competition would disrupt the AR-N/C interaction and/or coregulatory proteins binding to AF2.
In a similar study, Ravindranathan et al. described small molecule peptidomimetic D2 which mimics the NR-box LxxLL motif and disrupts the interaction between the AR and coregulatory proteins expressing LxxLL.53 D2 was capable of blocking hormone driven AR transactivation in LNCaP and LAPC4 cells, as well as transcription of AR-regulated genes in C4-2 and CWR22Rv1 cells, and reduced receptor-dependent xenograft tumor growth. However, the peptidomimetic was not capable of blocking all LxxLL-containing coregulatory protein interactions.53 Again, the interaction in this study occurs at the AR-LBD and it will be interesting to pursue the development of peptidomimetics which mimic sequences common to AR-NTD-targeting coregulatory proteins.
Interestingly, Quayle et al.54 showed that stable expression of the AR-NTD (AR1–588) inhibited the transactivation of full-length AR in LNCaP cells. Endogenous PSA expression was also reduced. In xenograft models created with the stably transfected LNCaP cells, tumor incidence and size were reduced compared to those created with LNCaP lacking the AR-NTD decoy molecule. Serum PSA was also reduced, along with a delay in progression to castrate resistance. The same results were achieved when the AR-NTD decoy molecule was delivered to established tumors. Ex vivo analysis suggested that expression of the AR-NTD decoy reduced proliferation and increased apoptosis within the tumour.54
SELECTIVE ANDROGEN RECEPTOR DOWNREGULATOR (SARDS)
Fulvestrant is an ER downregulator which was approved by the FDA for the treatment of advanced breast cancer in 2002. In 2011, Bradbury et al.55 suggested that similar specific downregulation or degradation of the AR might prove useful in the treatment of CRPC (Figure 1). Chemical manipulation of nortestosterone and testosterone resulted in the generation of compounds with the ability to downregulate AR as shown by decreased nuclear expression of an AR-GFP fusion protein in LNCaP. The efficacy of these compounds was confirmed in vivo using the Hershberger assay, and subsequently, 100 000 related compounds were synthesized and measured for affinity at the rat AR-LBD by fluorescence polarization. Rounds of chemical optimization and analysis in pharmacokinetic and functional studies led to the identification of AZD3514 (Figure 5a), a small molecule AR downregulator.56
Figure 5.
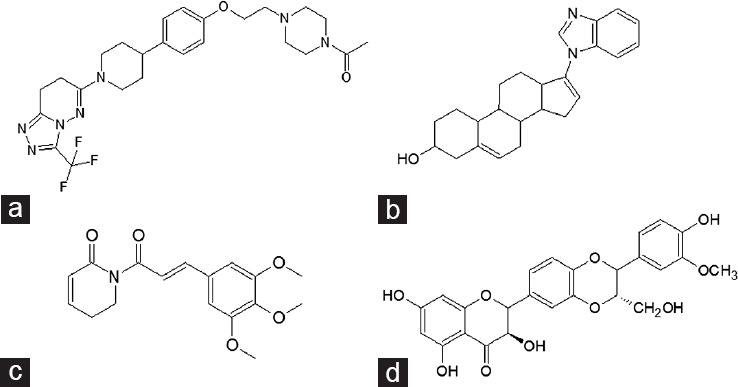
Selective AR downregulators (SARDs) (a) AZD3514. (b) Galaterone is a nonsteroidal antiandrogen and CYP17A1 inhibitor. (c) Piperlongumine is a naturally occurring alkaloid derived from Piper longum. (d) Isosylibin B, derived from milk thistle downregulates AR expression in human prostate cancer cells.
AZD3514 was found to inhibit proliferation of AR positive LAPC4 and LNCaP cells, without affecting growth of the AR-negative DU145 cell line and downregulated receptor protein expression in the presence and absence of DHT over 24 h, and also inhibited nuclear localization in response to hormone.57 Using mass spectrometry, Loddick et al.57 measured the rate of AR synthesis and degradation (Hsp90 inhibitor geldanamycin was used as a positive control for AR degradation) and observed reduced levels of an AR peptide suggesting a decrease in AR synthesis, but no effect on degradation. This was confirmed in an AR turnover assay utilizing pulse-chase labelling. Despite this, AZD3514 reduced tumor growth in Copenhagen rats bearing Dunning R3327H prostate tumors, as well as reducing AR expression within the tumor, specifically nuclear AR. This suggests that targeting both AR degradation and AR synthesis might be viable in the treatment of PCa. Two phase I trials of AZD3514 were completed in 2015.58 Although significant antitumor activity was observed, the drug was not well tolerated with significant side effects including nausea and vomiting. While AZD3514 may not be viable in the clinic, it provides a valuable proof-of concept for a new class of SARDs.
Galeterone (Figure 5b) inhibits CYP17 and antagonizes the AR. It reduces AR expression by increasing degradation and can reduce both full-length AR and ARv7 expression.7,59 Galeterone is also active against the T877A and F876L mutant ARs. Phase I and II clinical trials have been undertaken with galeterone, showing that the drug is well tolerated and has measurable effects on PSA levels. In ARMOR1 (phase I), there was a 30% or higher decrease in PSA in 49% patients; with 22.4% patients showing >50% decline in PSA. In ARMOR2 (phase II), 72.2% of patients demonstrated a 30% or greater decline in PSA; in 54.5% of patients, this decline exceeded 50%. All the participants had progressive disease despite antiandrogen therapy. PSA decline was also observed in patients deemed to have elevated levels of AR splice variant expression.60 As a result, the AMOR3-SV trial is now recruiting, where patients with expression of ARv7 in circulating tumor cells will be randomized to enzalutamide or galeterone treatment.
Piperlongumine (Figure 5c), a naturally occurring alkaloid of the long pepper (Piper longum), has also been shown to induce rapid depletion of AR in PCa cell lines. Golovine et al.61 showed that piperlongumine depleted AR protein expression in, and proliferation of, LNCaP and receptor transfected PC-3 cell lines at concentrations <5 μmol l−1. The compound was also capable of depleting mutant receptor lacking LBD (ARΔLBD) and inhibited transactivation of the full-length AR. However, there was no attempt by the authors to show direct binding of piperlongumine to the AR-NTD/DBD. Instead, they suggest that the compound accelerated degradation of the receptor via the ubiquitin-proteasome pathway.61 It would be interesting to investigate the mechanism of action of piperlongumine further, particularly with regard to NTD binding or interaction with AR coregulatory proteins. Other natural products have also been suggested to enhance degradation of the AR, including Isosilybin B (Figure 5d).62,63
THE FUTURE OF ANTIANDROGEN THERAPY
While pharmaceutical companies continue to improve on LBD targeted antiandrogen therapy, the mechanisms of resistance that drive CRPC suggest that a fundamental change in the type of therapy offered is required. A number of options for the targeted inhibition of the AR are outlined in Figure 1. The requirement for the NTD in AR function suggests that despite its intrinsically disordered nature, it is an ideal candidate for AR inhibition. Lack of significant sequence homology between members of the steroid hormone receptor family within the NTD also suggests that targeted this domain will be more selective for the AR, yielding fewer side effects. These inhibitors would also be effective against the AR splice variants which may play a role in driving CRPC in low androgen conditions.
The possibility for dual targeting of the receptor, by direct inhibition of the NTD and tagging of the AR for degradation are both appealing. Linking small molecules or peptides which target the AR for degradation or chemotherapies is a further alternative to inhibition of the AR-LBD alone. Recently, Gustafson and colleagues linked RU59063 to a hydrophobic adamantyl group via a short PEG linker to create SARD279 and SARD033.9 The two SARDs were able to specifically downregulate AR protein levels and were effective in inhibiting proliferation in LNCaP at least as well as enzalutamide, and antagonized R1881 induced gene expression. Significantly, efficacy was retained in cells expressing the F876L mutation conferring enzalutamide resistance. Other examples of targeted AR degradation molecules include proteolysis targeting chimeric molecules (PROTACs) which link a target moiety to a recognition element for ubiquitin-protein ligase,17 and inhibitors of apoptosis proteins (IAPs) which can induce proteasomal degradation by tagging proteins with ubiquitin chains.64 Conjugating targeting moieties to metallo-based cytotoxic agents such as cisplatin have also been proposed.65 It would be interesting to see if these approaches could be employed with a small molecule or peptide targeting the AR-NTD. Alternatively, targeting of protein-protein interactions between the AR-NTD and coactivator proteins may also provide new ways to downregulate AR activity.
Targeting AR domains or surfaces distinct from the LBP offers both significant challenges and potential for developing “selective androgen receptor modulators” and novel inhibitors to overcome drug resistance. In this respect, obtaining further insight into the folding and function of the intrinsically disordered AR-NTD will be of fundamental importance.
COMPETING INTEREST
The authors declared that they have no competing financial interests.
AUTHOR CONTRIBUTIONS
AEM and IJM contributed to the writing and editing of this review.
ACKNOWLEDGMENTS
AEM was supported by a BBSRC-CASE studentship award. Research in the IJM laboratory is currently supported by the Chief Scientist's Office of the Scottish Government and the charity Friends of Anchor.
REFERENCES
- 1.Denmeade SR, Isaacs JT. A history of prostate cancer treatment. Nat Rev Cancer. 2002;2:389–96. doi: 10.1038/nrc801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haendler B, Cleve A. Recent developments in antiandrogens and selective androgen receptor modulators. Mol Cell Endocrinol. 2012;352:79–91. doi: 10.1016/j.mce.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 4.McDonald S, Brive L, Agus DB, Scher HI, Ely KR. Ligand responsiveness in human prostate cancer: structural analysis of mutant androgen receptors from LNCaP and CWR22 tumors. Cancer Res. 2000;60:2317–22. [PubMed] [Google Scholar]
- 5.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo Z, Yang X, Sun F, Jiang R, Linn DE, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem. 2005;48:2972–84. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]
- 8.Brooke GN, Powell SM, Lavery DN, Waxman J, Buluwela L, et al. Engineered repressors are potent inhibitors of androgen receptor activity. Oncotarget. 2014;5:959–69. doi: 10.18632/oncotarget.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gustafson JL, Neklesa TK, Cox CS, Roth AG, Buckley DL, et al. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew Chem Int Ed Engl. 2015;54:9659–62. doi: 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 11.Dalal K, Roshan-Moniri M, Sharma A, Li H, Ban F, et al. Selectively targeting the DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289:26417–29. doi: 10.1074/jbc.M114.553818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu NZ, Wardell SE, Burnstein KL, Defranco D, Fuller PJ, et al. International union of pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol Rev. 2006;58:782–97. doi: 10.1124/pr.58.4.9. [DOI] [PubMed] [Google Scholar]
- 13.Davison SL, Bell R. Androgen physiology. Semin Reprod Med. 2006;24:71–7. doi: 10.1055/s-2006-939565. [DOI] [PubMed] [Google Scholar]
- 14.Sinnesael M, Claessens F, Laurent M, Dubois V, Boonen S, et al. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: evidence from targeted AR disruption in mouse osteocytes. J Bone Miner Res. 2012;27:2535–43. doi: 10.1002/jbmr.1713. [DOI] [PubMed] [Google Scholar]
- 15.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–9. [PubMed] [Google Scholar]
- 16.Gibson DA, Simitsidellis I, Collins F, Saunders PT. Evidence of androgen action in endometrial and ovarian cancers. Endocr Relat Cancer. 2014;21:T203–18. doi: 10.1530/ERC-13-0551. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–11. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brinkmann AO, Faber PW, van Rooij HC, Kuiper GG, Ris C, et al. The human androgen receptor: domain structure, genomic organization and regulation of expression. J Steroid Biochem. 1989;34:307–10. doi: 10.1016/0022-4731(89)90098-8. [DOI] [PubMed] [Google Scholar]
- 19.Trapman J, Klaassen P, Kuiper GG, van der Korput JA, Faber PW, et al. Cloning, structure and expression of a cDNA encoding the human androgen receptor. Biochem Biophys Res Commun. 1988;153:241–8. doi: 10.1016/s0006-291x(88)81214-2. [DOI] [PubMed] [Google Scholar]
- 20.Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. The androgen receptor gene mutations database: 2012 update. Hum Mutat. 2012;33:887–94. doi: 10.1002/humu.22046. [DOI] [PubMed] [Google Scholar]
- 21.Clinckemalie L, Vanderschueren D, Boonen S, Claessens F. The hinge region in androgen receptor control. Mol Cell Endocrinol. 2012;358:1–8. doi: 10.1016/j.mce.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 22.Cutress ML, Whitaker HC, Mills IG, Stewart M, Neal DE. Structural basis for the nuclear import of the human androgen receptor. J Cell Sci. 2008;121:957–68. doi: 10.1242/jcs.022103. [DOI] [PubMed] [Google Scholar]
- 23.Helsen C, Dubois V, Verfaillie A, Young J, Trekels M, et al. Evidence for DNA-binding domain – Ligand-binding domain communications in the androgen receptor. Mol Cell Biol. 2012;32:3033–43. doi: 10.1128/MCB.00151-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haelens A, Tanner T, Denayer S, Callewaert L, Claessens F. The hinge region regulates DNA binding, nuclear translocation, and transactivation of the androgen receptor. Cancer Res. 2007;67:4514–23. doi: 10.1158/0008-5472.CAN-06-1701. [DOI] [PubMed] [Google Scholar]
- 25.Simental JA, Sar M, Lane MV, French FS, Wilson EM. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Biol Chem. 1991;266:510–8. [PubMed] [Google Scholar]
- 26.Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–6. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- 27.Kumar R, McEwan IJ. Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation. Endocr Rev. 2012;33:271–99. doi: 10.1210/er.2011-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lavery DN, McEwan IJ. Functional characterization of the native NH2-terminal transactivation domain of the human androgen receptor: binding kinetics for interactions with TFIIF and SRC-1a. Biochemistry. 2008;47:3352–9. doi: 10.1021/bi702220p. [DOI] [PubMed] [Google Scholar]
- 29.Wilson EM. Primate-specific multi-functional androgen receptor coregulator and proto-oncogene melanoma antigen-A11 (MAGE-A11) In: McEwan IJ, Kumar R, editors. Nuclear Receptors: From Structure to the Clinic. Switzerland: Springer; 2015. p. 137. [Google Scholar]
- 30.Brodie J, McEwan IJ. Intra-domain communication between the N-terminal and DNA-binding domains of the androgen receptor: modulation of androgen response element DNA binding. J Mol Endocrinol. 2005;34:603–15. doi: 10.1677/jme.1.01723. [DOI] [PubMed] [Google Scholar]
- 31.Dar JA, Masoodi KZ, Eisermann K, Isharwal S, Ai J, et al. The N-terminal domain of the androgen receptor drives its nuclear localization in castration-resistant prostate cancer cells. J Steroid Biochem Mol Biol. 2014;143:473–80. doi: 10.1016/j.jsbmb.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies P, Watt K, Kelly SM, Clark C, Price NC, et al. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J Mol Endocrinol. 2008;41:301–14. doi: 10.1677/JME-08-0042. [DOI] [PubMed] [Google Scholar]
- 33.Lavery DN, McEwan IJ. Structural characterization of the native NH2-terminal transactivation domain of the human androgen receptor: a collapsed disordered conformation underlies structural plasticity and protein-induced folding. Biochemistry. 2008;47:3360–9. doi: 10.1021/bi702221e. [DOI] [PubMed] [Google Scholar]
- 34.McEwan IJ. Intrinsic disorder in the androgen receptor: identification, characterisation and drugability. Mol Biosyst. 2012;8:82–90. doi: 10.1039/c1mb05249g. [DOI] [PubMed] [Google Scholar]
- 35.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS One. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao B, Qi Y, Zhang G, Xu D, Zhan Y, et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget. 2014;5:1646–56. doi: 10.18632/oncotarget.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krause WC, Shafi AA, Nakka M, Weigel NL. Androgen receptor and its splice variant, AR-V7, differentially regulate FOXA1 sensitive genes in LNCaP prostate cancer cells. Int J Biochem Cell Biol. 2014;54:49–59. doi: 10.1016/j.biocel.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, et al. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res. 2011;71:6019–29. doi: 10.1158/0008-5472.CAN-11-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, et al. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. 2014;74:2270–82. doi: 10.1158/0008-5472.CAN-13-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meimetis LG, Williams DE, Mawji NR, Banuelos CA, Lal AA, et al. Niphatenones, glycerol ethers from the sponge Niphates digitalis block androgen receptor transcriptional activity in prostate cancer cells: structure elucidation, synthesis, and biological activity. J Med Chem. 2012;55:503–14. doi: 10.1021/jm2014056. [DOI] [PubMed] [Google Scholar]
- 44.Sadar MD, Williams DE, Mawji NR, Patrick BO, Wikanta T, et al. Sintokamides A to E, chlorinated peptides from the sponge Dysidea sp. that inhibit transactivation of the N-terminus of the androgen receptor in prostate cancer cells. Org Lett. 2008;10:4947–50. doi: 10.1021/ol802021w. [DOI] [PubMed] [Google Scholar]
- 45.Takeuchi T, Tsutsumi O. Serum bisphenol a concentrations showed gender differences, possibly linked to androgen levels. Biochem Biophys Res Commun. 2002;291:76–8. doi: 10.1006/bbrc.2002.6407. [DOI] [PubMed] [Google Scholar]
- 46.Takeuchi T, Tsutsumi O, Ikezuki Y, Takai Y, Taketani Y. Positive relationship between androgen and the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr J. 2004;51:165–9. doi: 10.1507/endocrj.51.165. [DOI] [PubMed] [Google Scholar]
- 47.Ye L, Zhao B, Hu G, Chu Y, Ge RS. Inhibition of human and rat testicular steroidogenic enzyme activities by bisphenol A. Toxicol Lett. 2011;207:137–42. doi: 10.1016/j.toxlet.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 48.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–60. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brand LJ, Olson ME, Ravindranathan P, Guo H, Kempema AM, et al. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget. 2015;6:3811–24. doi: 10.18632/oncotarget.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin SK, Banuelos CA, Sadar MD, Kyprianou N. N-terminal targeting of androgen receptor variant enhances response of castration resistant prostate cancer to taxane chemotherapy. Mol Oncol. 2015;9:628–39. doi: 10.1016/j.molonc.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banuelos CA, Lal A, Tien AH, Shah N, Yang YC, et al. Characterization of niphatenones that inhibit androgen receptor N-terminal domain. PLoS One. 2014;9:e107991. doi: 10.1371/journal.pone.0107991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–22. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, et al. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4:1923. doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- 54.Quayle SN, Mawji NR, Wang J, Sadar MD. Androgen receptor decoy molecules block the growth of prostate cancer. Proc Natl Acad Sci U S A. 2007;104:1331–6. doi: 10.1073/pnas.0606718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bradbury RH, Hales NJ, Rabow AA, Walker GE, Acton DG, et al. Small-molecule androgen receptor downregulators as an approach to treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2011;21:5442–55. doi: 10.1016/j.bmcl.2011.06.122. [DOI] [PubMed] [Google Scholar]
- 56.Bradbury RH, Acton DG, Broadbent NL, Brooks AN, Carr GR, et al. Discovery of AZD3514, a small-molecule androgen receptor downregulator for treatment of advanced prostate cancer. Bioorg Med Chem Lett. 2013;23:1945–58. doi: 10.1016/j.bmcl.2013.02.056. [DOI] [PubMed] [Google Scholar]
- 57.Loddick SA, Ross SJ, Thomason AG, Robinson DM, Walker GE, et al. AZD3514: a small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol Cancer Ther. 2013;12:1715–27. doi: 10.1158/1535-7163.MCT-12-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Omlin A, Jones RJ, van der Noll R, Satoh T, Niwakawa M, et al. AZD3514, an oral selective androgen receptor down-regulator in patients with castration-resistant prostate cancer – Results of two parallel first-in-human phase I studies. Invest New Drugs. 2015;33:679–90. doi: 10.1007/s10637-015-0235-5. [DOI] [PubMed] [Google Scholar]
- 59.Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015;6:27440–60. doi: 10.18632/oncotarget.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Montgomery B, Eisenberger MA, Rettig MB, Chu F, Pili R, et al. Androgen receptor modulation optimized for response (ARMOR) phase I and II studies: galeterone for the treatment of castration-resistant prostate cancer. Clin Cancer Res. 2015;22:1356–63. doi: 10.1158/1078-0432.CCR-15-1432. [DOI] [PubMed] [Google Scholar]
- 61.Golovine KV, Makhov PB, Teper E, Kutikov A, Canter D, et al. Piperlongumine induces rapid depletion of the androgen receptor in human prostate cancer cells. Prostate. 2013;73:23–30. doi: 10.1002/pros.22535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deep G, Raina K, Singh RP, Oberlies NH, Kroll DJ, et al. Isosilibinin inhibits advanced human prostate cancer growth in athymic nude mice: comparison with silymarin and silibinin. Int J Cancer. 2008;123:2750–8. doi: 10.1002/ijc.23879. [DOI] [PubMed] [Google Scholar]
- 63.Deep G, Oberlies NH, Kroll DJ, Agarwal R. Isosilybin B causes androgen receptor degradation in human prostate carcinoma cells via PI3K-Akt-Mdm2-mediated pathway. Oncogene. 2008;27:3986–98. doi: 10.1038/onc.2008.45. [DOI] [PubMed] [Google Scholar]
- 64.Itoh Y, Ishikawa M, Kitaguchi R, Sato S, Naito M, et al. Development of target protein-selective degradation inducer for protein knockdown. Bioorg Med Chem. 2011;19:3229–41. doi: 10.1016/j.bmc.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 65.Huxley M, Sanchez-Cano C, Browning MJ, Navarro-Ranninger C, Quiroga AG, et al. An androgenic steroid delivery vector that imparts activity to a non-conventional platinum (II) metallo-drug. Dalton Trans. 2010;39:11353–64. doi: 10.1039/c0dt00838a. [DOI] [PubMed] [Google Scholar]