

Abstract
A striking characteristic of cancer cells is their remarkable phenotypic plasticity, which is the ability to switch states or phenotypes in response to environmental fluctuations. Phenotypic changes such as a partial or complete epithelial to mesenchymal transition (EMT) that play important roles in their survival and proliferation, and development of resistance to therapeutic treatments, are widely believed to arise due to somatic mutations in the genome. However, there is a growing concern that such a deterministic view is not entirely consistent with multiple lines of evidence, which indicate that stochasticity may also play an important role in driving phenotypic plasticity. Here, we discuss how stochasticity in protein interaction networks (PINs) may play a key role in determining phenotypic plasticity in prostate cancer (PCa). Specifically, we point out that the key players driving transitions among different phenotypes (epithelial, mesenchymal, and hybrid epithelial/mesenchymal), including ZEB1, SNAI1, OVOL1, and OVOL2, are intrinsically disordered proteins (IDPs) and discuss how plasticity at the molecular level may contribute to stochasticity in phenotypic switching by rewiring PINs. We conclude by suggesting that targeting IDPs implicated in EMT in PCa may be a new strategy to gain additional insights and develop novel treatments for this disease, which is the most common form of cancer in adult men.
Keywords: epithelial to mesenchymal transition, intrinsically disordered proteins, prostate cancer, state-switching
INTRODUCTION
State or phenotypic switching is a fundamental physiological process, in which a cell/organism undergoes spontaneous, and potentially reversible, transitions between different phenotypes. Thus, phenotypic plasticity is a key feature of the development and normal function of cells within most multicellular organisms that enables the cell to respond to various intrinsic and external cues and stimuli in a concerted fashion, enabling them to “make” appropriate cellular decisions.1
Cancer cells not only retain this plasticity but also exploit it for opportunistic adaptation and progression of the disease from transformation to metastasis, which accounts for >90% of cancer-related deaths.2 Furthermore, phenotypic plasticity allows cancer to beget stem cell-like cells (the so-called cancer stem cells (CSCs) or Tumor Initiating cells) that are thought to be the main cause of therapeutic resistance and disease recurrence.3,4 CSCs that arise from somatic cells (nonstem cell cancer cells) are similar to normal stem cells in their ability to self-renew and to generate large populations of differentiated progeny. However, in contrast to the irreversible and hierarchical organization that is generally believed to be the mode of normal tissue homeostasis, phenotypic plasticity allows cancer cells to dynamically enter into and exit the stem-cell state.5 Thus, it is critical to understand the mechanisms underlying phenotypic plasticity in cancer and whether phenotypic changes resulting from this plasticity can be reversed so that ultimately, this knowledge can be used to develop new and effective cancer therapeutics.
Epithelial–mesenchymal transition (EMT) and its reverse mesenchymal–epithelial transition (MET) that appear to be closely associated with metastasis6 and the acquisition of stem-cell properties by cancer cells7 are the classical examples of phenotypic plasticity in biological systems. EMT, first discovered by Greenburg and Hay,8 is, in fact, a normal process in development and wound healing, in which epithelial cells undergo multiple biochemical changes that enable them to assume a mesenchymal phenotype which includes a loss of apico-basal polarity and cell-cell attachments to gain migratory capacity, invasiveness, and increased resistance to apoptosis.9,10
Based on the biological context in which they occur, three different types of EMTs have been identified with quite different functional consequences.10 Thus, while type I EMT is associated with implantation, embryo formation, and organ development, which are organized to generate diverse cell types that share common mesenchymal phenotypes, type II EMT is associated with wound healing, tissue regeneration, and organ fibrosis. In contrast, type III EMT occurs in cancer cells with genetic and epigenetic changes, specifically in genes regulating clonal outgrowth and the development of localized tumors. Such cells undergoing a type III EMT may invade and metastasize to distant locations leading to life-threatening manifestations of cancer progression. Furthermore, although the specific signals that induce type III EMTs are not fully understood, it is believed that cancer cells may pass through EMTs to differing extents. Thus, while some epithelial cells (E phenotype) may shed all vestiges of their epithelial origin to acquire a fully mesenchymal phenotype (M phenotype), others may display mesenchymal characteristics while still maintaining an epithelial identity (E/M phenotype).11 In other words, such cells display a hybrid phenotype that is hypothesized to be metastable12 or in our models, stabilized by OVOL expression. More importantly, cells with the hybrid phenotype have maximum cellular plasticity, possess heightened tumor-initiating properties,13,14 and are detected in several cancers including PCa, where they can migrate to distant locations via the bloodstream by moving collectively as a cluster of Circulating Tumor Cells (CTCs).15,16,17,18 Such CTC clusters have been reported to have up to 50× greater potential for metastasis than individual CTCs,19 making them the bad actors in metastasis.
In this Special Issue article, we first discuss phenotypic plasticity in PCa. We identify the key factors implicated in EMT/MET and highlight the fact that they are predicted to be intrinsically disordered proteins (IDPs). Next, we discuss the possibility that phenotypic switching could be analogous to a phase transition phenomenon. It is driven by “noise” resulting from the conformational dynamics of the IDPs and contributes to the rewiring of the cell's protein interaction network (PIN). Thus, contrary to conventional wisdom, phenotypic switching may occur due to events that are stochastic rather than merely deterministic in nature. We conclude by suggesting that targeting IDPs may be a new strategy to gain additional insights and develop novel treatments for PCa.
EMT IN PCA
In PCa, epithelial disease is initially seen within the prostate, but as the disease progresses, markers for the mesenchymal phenotype become more apparent.20 Mesenchymal cells attach more strongly to the extracellular matrix (ECM) and are able to act as path generators by secreting matrix metalloproteinases (MMPs).21 MMPs degrade the extracellular matrix (ECM) and disrupt cell-cell and cell-ECM interactions, thereby promoting migration and invasion.22 Prostatic mesenchymal cells migrate from the primary tumor site and invade the bloodstream to metastasize to distant locations with the most frequent involvement being bone (90%), lung (46%), liver (25%), pleura (21%), and the adrenals (13%).23 It is tacitly assumed that these mesenchymal cells then become dormant and after a prolonged period of dormancy, which can last for decades, an unknown trigger causes the reverse transition, MET.24,25 MET enables tumor growth and leads to a clinically significant metastasis. Metastatic lesions can then re-seed the primary tumor resulting in a vicious cycle. Thus, EMT and MET serve as critical processes for the initial invasion and migration, and acceleration of the later stages of metastatic colonization and growth, respectively, are ideal examples of phenotypic plasticity in PCa and underscore their clinical importance.
A change from an epithelial to a mesenchymal phenotype coincides with a large number of genetic, epigenetic, and structural changes within the cell (Figure 1). Mesenchymal cells become elongated, lose cell-cell adhesions, and become motile. The most well-known target in epithelial cells is E-cadherin (CDH1), a cell membrane protein that allows for cell-cell attachment via a Ca2+ -dependent binding with E-cadherin molecules on adjacent cells. In a normal prostate epithelial cell, E-cadherin is also connected internally with actin filaments.26 Changes in the cytoskeleton are most evident within the intermediate filaments, which changes from keratin to vimentin (VIM) providing the mesenchymal cells with their structure and contributing to motility.27,28
Figure 1.

EMT Progression. Epithelial (E) cells are well anchored to the basement membrane, connected to each other in sheets via E-cadherin junctions and express various markers, which may vary depending on the individual cancer cell. E cells can then transition into mesenchymal cells by first going through a series of intermediates (E/M) that are usually metastable. Mesenchymal (M) cells are fully motile and can exhibit various genes and properties including the ability to adapt to stresses including chemotherapy.
Notwithstanding the differences in the types of EMTs, the phenotypic switch from E to M is mostly regulated by a common set of genes and signaling pathways. Among the signaling pathways that are observed to play critical roles are the transforming growth factor beta (TGFβ), epidermal growth factor (EGF), hepatocyte growth factor (HGF), Notch, fibroblast growth factor (FGF), hedgehog, Wnt, and insulin-like growth factor (IGF) together with mechanical factors such as extracellular matrix (ECM) density changes which can weaken cell-cell adhesions.29 Signals from these pathways are often involved in modulating the activities of several transcription factors that form a gene regulatory circuit with positive and negative feedback, and auto-regulatory loops (Figure 2a), and act very early during EMT before epigenetic changes to DNA, and histones make the transition more stable.
Figure 2.

Modeling of prostate cancer transitions. (a) (left) miR-200, OVOL (Ovo-Like Zinc Finger 1/2), and ZEB (Zinc finger E-box-binding homeobox 1/2) circuit with solid lines indicating direct transcriptional activation (arrows) or inhibition (bar). The dashed line is a miRNA-mediated translational inhibition whereas the dotted line is an indirect inhibition. Within the red dotted lines is the circuit considered by Jia et al. The graphs in the middle and right depict the correlation between the levels of ZEB1, CDH1 (E-cadherin), and OVOL in NCI-60 cell lines. The CellMiner website was used to query the corresponding gene expression levels. (b) Levels of CDH1 and miR-200 in three phenotypes, as predicted by the model considered in (a). (c) Graph indicates how the levels of SNAIL (which includes SNAI1/2 [Snail Family Zinc Finger 1/2]) protein change the levels of ZEB mRNA per cell. The yellow area is where only the E/M (Epithelial/Mesenchymal hybrid) state exists. Corresponding in silico predicted FACS (Fluorescence-activated cell sorting) (top) and “Waddington landscape” figures (below) have been included. (d) The phase diagram has the SNAIL signal split into two external inputs, which can either activate ZEB or repress miR-200. (b and d) Modified from Jia et al. 2015, while (a) is an extension of the model with the addition of E-cadherin and a detailed method provided in the supplemental information. (c) Modified from Jolly et al.
MODEL FORMULATION
The model for the circuit shown in Figure 2a extends the framework presented in a study by Jia et al.31 by incorporating E-cadherin as an additional component and the mutual inhibition between ZEB1 and E-cadherin (representing the direct transcriptional inhibition of E-cadherin by ZEB1 and the sequestration of beta-catenin on the membrane by E-cadherin, thus restricting the activation of Wnt/beta-catenin pathway that can transcriptionally activate ZEB).32 The equation for ZEB mRNA presented in the study by Jia et al.31 was modified to incorporate the term representing the inhibition by E-cadherin (E).
Hs– (E, λE,mZ) denotes the shifted Hill function denoting inhibition of ZEB by E-cadherin as described above. HS– (E, λE,mZ) = H–(E) + λE,mZ (1 – H–(E)) where H–(E) is the negative Hill function defined by  and λE,mZ denotes the fold-change in the production rate of ZEB1 by E-cad.
and λE,mZ denotes the fold-change in the production rate of ZEB1 by E-cad.
The parameters used here are: λE,mZ = 0.8, nE,mZ = 2 and  molecules.
molecules.
The equation for E-cadherin is given by:
where, gE and kE denote the production and degradation rates for E-cad, respectively. The term HS– (Z, λZ,E) represents the shifted Hill function that represents the inhibition of E-cad by ZEB1. ZEB1 can bind to two E-boxes in E-cad promoter region,32 and the half-life of E-cadherin is about 5–10 h.20,21 Correspondingly, the parameters used are: gE = 5000 molecules, KE molecules h−1 λZ,E = 0.1, nZ,E = 2 and 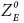 = 100 000 molecules. All the other parameters have been mentioned in the study by Jia et al.31
= 100 000 molecules. All the other parameters have been mentioned in the study by Jia et al.31
THE KEY DRIVERS OF EMT IN PCA
Biochemical evidence suggests that among the transcription factors constituting the EMT gene regulatory circuit in PCa, ZEB1 appears to play a crucial role by acting as a trigger for EMT when it is upregulated. More specifically, ZEB1 represses the expression of epithelial marker genes such as E-cadherin (CDH1) while activating genes that characterize a mesenchymal phenotype such as Vimentin (VIM).30 ZEB (ZEB1/2) expression itself is positively regulated by the transcription factor SNAIL (SNAI1/2). Further, ZEB directly inhibits and is directly inhibited by the transcription factors OVOL1/2 (OVOL) and microRNA-200c (miR-200c), forming mutually inhibitory feedback loops. In addition, OVOL can inhibit its own transcription,31 and E-cadherin can indirectly inhibit ZEB32 (Figure 2a). Downregulation of ZEB results in the reversal of EMT, which is known as an MET. Thus, while the mesenchymal (M) phenotype is characterized by elevated expression of ZEB and SNAIL and downregulation of miR-200c, CDH1, and OVOL, the opposite is true for an epithelial (E) phenotype, as also observed in the NCI-60 panel of cell lines that have been divided into E, E/M, and M phenotype based on CDH1/VIM ratio33,34 (Figure 2a).
Mechanism-based mathematical modeling of EMT by Lu et al.35 has elucidated that miR-200/ZEB can act as a three-way switch enabling transitions among the three phenotypes – E, hybrid E/M, and M – each corresponding to distinct levels of miR-200, ZEB1, and CDH1 (Figure 2b). Incorporating OVOL in the framework, Jia et al.31 have uncovered a crucial role for OVOL in modulating the phenotypic plasticity of PCa cells.31 The effect of OVOL can be visualized in terms of a bifurcation diagram of ZEB mRNA levels (Y-axis), when the miR-200/ZEB/OVOL/E-cadherin circuit is driven by varying SNAIL protein levels (X-axis) (Figure 2c). This diagram demonstrates how different levels of SNAIL enable transitions among different phenotypes; for example, at low SNAIL levels, cells can adopt either an E or a hybrid E/M phenotype, and therefore, cells with the same genetic background can be expected to resolve into two subpopulations (E and E/M) in a FACS experiment (green-shaded region in Figure 2c). Similarly, at higher levels of SNAIL, cells can adopt either a hybrid E/M or M phenotype (orange-shaded region in Figure 2c).
Furthermore, cells in one phenotype can stochastically undergo transition to adopt the other one. These rates of transition depend on how stable or robust a particular phenotype is to network perturbations, and the relative stability can be expressed via the canonical “epigenetic landscape,” a powerful metaphor in developmental biology introduced by Waddington to describe cell fate specification36 (Figure 2c). In this metaphorical representation, cells, represented by balls, roll downhill through a landscape of bifurcating valleys. Each new valley represents a possible cell fate and the ridges between the valleys maintain the cell fate once it has been chosen; the deeper the valley, the more stable the phenotype. Thus, to undergo transition from one state (phenotype) to another, cells with distinct phenotypes are separated by an energy barrier and would have to jump out of one valley into another valley, most likely the one adjacent. Thus, an E to M transition is much less likely than an E to E/M transition for low SNAIL levels. When SNAIL levels are low, the valley associated with M is very shallow; consequently, cells that undergo an EMT (i.e., transition to M) are unlikely to stay because the energy to undergo transition back into E/M is very low and thus cells cannot maintain a full-blown M phenotype, rather switch to being hybrid E/M.
SNAIL activity may also differ depending on the cofactors and how it is posttranslationally modified, and therefore, can be thought of as two separate signals: one repressing miR-200c and one activating ZEB1 (Figure 2d). The resulting phase diagram demonstrates the existence of combinations of different distinct phenotypes. In fact, all the three may co-exist as a function of SNAIL's differential repressing or activating activities.31,35 The validation of this prediction by mathematical modeling is seen in recent experiments showing that cell lines belonging to multiple cancers can harbor multiple subpopulations corresponding to E, E/M, and M phenotypes.13,37
The extent of epithelial or mesenchymal characteristic of a PCa cell is reflected by E-cadherin (CDH1) expression in the cell. The transcription factors to which E-cadherin expression correlates most strongly are OVOL1, OVOL2, and GRHL2 in a panel of 877 different cancer cell lines representing 36 tumor types including PCa.38,39 Among all genes, GRHL2 is the only one in the top 10 (Supplementary Table 1 (176.8KB, tif) ); however, OVOL1/2 are #41 and #24, respectively (data not shown), and are hypothesized to act interchangeably. On the other hand, there is a strong negative correlation between ZEB1 expression and E-cadherin levels (Supplementary Table 2 (176.8KB, tif) shows the top 10 negatively correlated genes) while SNAI1 is only very weakly correlated with E-cadherin (Pearson − 0.18, Spearman − 0.14) (data not shown). These correlations corroborate the modeling results that the ZEB1/miR-200c feedback loop forms the “decision-making switch” for EMT/MET, while the SNAIL/miR-34 feedback loop by itself behaves as an integrator, restricting aberrant activation of EMT.27 As shown in Figure 2d, at low SNAIL levels, the E phenotype is dominant and at high levels, the M phenotype is dominant. However, in the intermediate regime of SNAIL, preceding a threshold value for the transition to an M phenotype defines the E/M hybrid phenotype. Together, the modeling of this dynamical system underscores that phenotypic plasticity in the context of EMT/MET is not dependent on a linear upregulation of transcription but rather a phase transition through a bifurcation, indicative of a network rewiring process. The equations that were used in formulating the mathematical model are described in the Model Formulation section.
List of genes that are highly co-expressed with CDH1 according to Pearson correlation coefficient. The Spearman correlation coefficient is also indicated on the table but was not used in ranking. The cBioPortal website was used to query 877 tumor samples from the Cancer Cell Line Encyclopedia
List of genes that are inversely expressed with CDH1 according to Pearson correlation coefficient. The Spearman correlation coefficient is also indicated on the table but was not used in ranking. The cBioPortal website was used to query 877 tumor samples from the Cancer Cell Line Encyclopedia
IDPS AND EMT
A hallmark of the factors implicated in phenotypic switching whether in cancer or in normal cells is that they are IDPs. Thus, for example, the products of most oncogenes such as Jun, Fos, Myc,40,41 the Yamanaka factors, namely, OCT3/4, SOX2, Myc, NANOG, and KLF4 that induce reprograming of pluripotent stem (iPS) cells,42 and >90% of the Cancer/Testis Antigens43 several of which are implicated in EMT44,45 are predicted, and in many cases, they have been experimentally verified to be IDPs.46,47,48,49 Consistent with these observations, we demonstrate here that the key factors implicated in EMT/MET, namely, OVOL1/2, ZEB1, and SNAI1 are also strongly predicted to be IDPs (Figure 3).
Figure 3.

Disorder prediction for OVOL1/2 (Ovo-Like Zinc Finger 1/2), ZEB1 (Zinc finger E-box-binding homeobox 1), and SNAI1 (Snail Family Zinc Finger 1). The MetaPrDOS metaserver was used to predict the disorder. MetaPrDOS predicts the disorder tendency of each residue using support vector machines from the prediction results of the seven independent predictors. The method has been evaluated by using the Critical Assessment of Techniques for Protein Structure Prediction 7 (CASP7) targets to avoid an overestimation due to the inclusion of proteins used in the training set of some component predictors. As a result, the meta-approach achieves higher prediction accuracy than all methods participating in CASP7. Amino acid numbers are indicated on the X-axis and the disorder likelihood on the Y-axis with a red line indicating the 0.5 (50%) level. (a) OVOL1 (b) OVOL2 (c) SNAI1 (d) ZEB1.
IDPs are proteins that lack a three-dimensional structure; however, many IDPs can undergo transition from disorder to order upon interacting with a target50,51 or in response to posttranslational modifications such as phosphorylation.52 With multiple conformational states and rapid conformational dynamics, they engage in a myriad of often “promiscuous” interactions. These stochastic interactions between IDPs and their partners result in noise, defined as conformational noise,53 which is an inherent characteristic of IDP interactions. Recent progress has revealed that many biological processes are driven by probabilistic events, underscoring the importance of “noise” in biological systems.54 While current research on biological noise has focused on low gene copy numbers as the predominant source of noise, noise arising from stochastic protein interactions has not been fully appreciated. Just as transcriptional noise plays an important role in probabilistic differentiation and adaptation, noise inherent in protein interactions may underlie the activation of latent pathways and cellular transformation. Mahmoudabadi et al. have hypothesized that noise due to stochasticity in protein interaction networks (PINs) contributed by the conformational dynamics of IDPs may play a critical role in phenotypic/state switching.53 Consistent with this argument, there are now numerous examples of remodeling of the IDP conformational ensemble in response to binding and/or posttranslational modifications. For example, while Forman-Kay and co-workers52 found that phosphorylation can induce folding of an IDP, He et al.55 found that phosphorylation of PAGE4 can remodel the ensemble to populate a different conformation, both of which have large functional consequences. Together, these observations tend to suggest that contrary to the prevailing wisdom that phenotype specification is highly deterministic, stochasticity may be a confounding factor in specifying the cell fate. This thinking may also help explain how a given cell can reversibly switch phenotypes as seen in EMT and MET or for that matter, a drug-sensitive cell from developing resistance and switching back to drug sensitivity.56 Indeed, such stochasticity in phenotypic switching is also thought to underlie cellular differentiation,54 generation of induced pluripotent stem cells (iPS cells),57 tumor heterogeneity,58,59 and emergence of cancer stem cells from nonstem cancer cells.60 Implicit in this stochastic model, the PIN configuration contains information that specifies the cell's phenotype.
Pursuant to the theoretical framework proposed by Mahmoudabadi et al. Kulkarni et al. suggested that state switching may be analogous to a phase transition phenomenon where noise from the stochastic interactions initiated by the IDPs in response to a specific input allows the system to sample the network interaction space to rewire PINs and activate the previously masked options, resulting in a transition from one state to another,61 for example, from phenotype A which represents E to phenotype B which represents M. As depicted in Figure 4, phenotype A is characterized by a specific configuration of its PIN. When subjected to perturbations, the levels of certain IDPs such as ZEB1 and SNAI1 are upregulated and promote promiscuous interactions to rewire the PIN. If the new configurations of the rewired PIN remain within the threshold characteristic of phenotype A, the E phenotype is retained notwithstanding minor fluctuations in the network topology. However, if the search unmasks the latent PIN configurations that cross the threshold, the cell transitions to phenotype B, i.e., M or even E/M. Each cell has the same probability of switching to phenotype B, and once the perturbation exceeds a threshold (vertical line), the majority of cells in the population will be in phenotype B. Of note, depending on the network topology, lowering the perturbation (e.g., repressing ZEB/SNAI1 expression by OVOL/miR-200) can result in the PIN again rewiring itself to the E (default) network configuration, thereby reversing the phenotypic switch (M to E). In this stochastic model, each cell has an equal probability to undergo a specific phenotypic transition in response to the given input.
Figure 4.
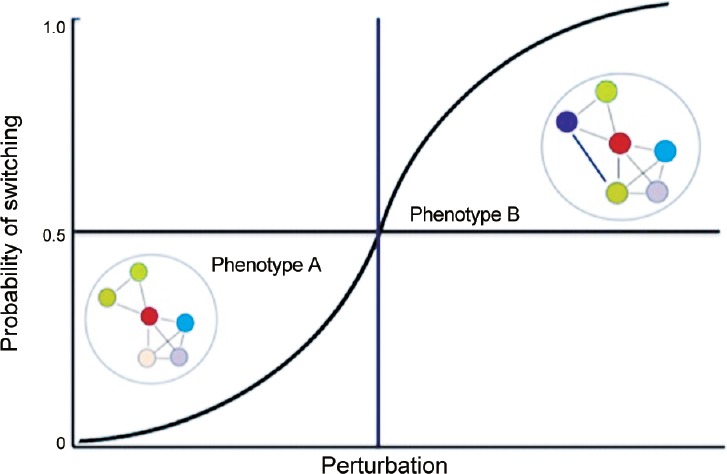
Schematic representation of phenotypic switching driven by noise in protein interaction networks. Phenotype A represents a normal cell that is characterized by a specific configuration of its protein interaction network (PIN). When subjected to perturbations, the levels of certain intrinsically disordered proteins (IDPs) increase and promote promiscuous interactions to rewire the PIN. If the new configurations of the rewired PIN remain within the threshold characteristic of phenotype A, this phenotype is retained notwithstanding minor fluctuations in the network topology. However, if the search unmasks latent PIN configurations that cross the threshold, the cell transitions to a cancer cell represented by phenotype B. Each cell has the same probability of switching to phenotype B, and once the perturbation exceeds a threshold (vertical line), the majority of cells in the population will be in phenotype B.
CONCLUSIONS
In this perspective article, we have briefly summarized new thinking regarding PCa by underscoring the transient nature of the phenotypic plasticity of cancer cells and how plasticity at the molecular level (remodeling of the IDP conformational ensemble) may contribute to it. Thus, it follows from the foregoing that IDPs represent nodal points of phenotypic plasticity in cancer including PCa, and thus, targeting them pharmacologically may represent a new strategy to develop novel therapeutics for PCa. Indeed, at present, one of the most popular therapeutic targets in PCa is the androgen receptor (AR), an IDP. In fact, AR is known to interact with more than 160 different62 proteins and a large region of the N-terminal half of the molecule (first ~500 amino acids) that includes its powerful transactivation domain is significantly intrinsically disordered.63,64 Ironically, however, every AR blockade that is in use in the clinic is directed against its highly structured ligand-binding domain. While initially being very effective in treating PCa, patients typically end up developing drug resistance and therefore, there is a dire need for new therapies. Recent attempts to target the AR disordered region have yielded small molecule inhibitors that attenuate the growth of castrate-resistant PCa xenografts.65
Other researchers working on c-Myc, another important player in PCa, which is also an IDP, have identified small molecules that disrupt its dimerization with Max.66,67,68 While there can be no guarantee that newer drugs targeting intrinsically disordered regions or proteins will not result in drug resistance, it is important to understand that PINs adapt a scale-free architecture that follows a power law distribution, making them more resilient to perturbations. Therefore, malfunction of critical hubs in the network can incapacitate the network.69 Thus, identifying IDPs that occupy such critical hubs and can rewire the cancer cell's PIN to exploit its plasticity70 provides an opportunity for developing new and effective therapeutics. Of note, major efforts to map the Arabidopsis interactome71,72 has helped researchers show that certain plant pathogens actually target hub proteins to control the host's cellular machinery,73 lending credence to our argument.
GLOSSARY OF TERMS
Bifurcation: A splitting of one thing into two. Bifurcation theory is the mathematical study of changes in the qualitative or topological structure of a given family; for example, a family of differential equations. In dynamical systems theory, the term refers to the splitting of steady states or fixed points. A bifurcation occurs when a small smooth change made to the parameter values (the bifurcation parameters) of a system causes a sudden ‘qualitative’ or topological change in its behaviour.
Bifurcation diagram: in dynamical systems, a bifurcation diagram shows the values visited or approached asymptotically (fixed points, periodic orbits, or chaotic attractors) of a system as a function of a bifurcation parameter in the system.
Conformational dynamics: Transitions of ensemble conformations of intrinsically disordered proteins that lack a rigid 3D structure.
Conformational noise: Defined as stochastic interactions between intrinsically disordered proteins and their partners.
Dynamical systems: In mathematics, a dynamical system is a system in which a function describes the time dependence of a point in a geometrical space. For example, the mathematical models that describes the swinging of a clock pendulum.
Epigenetic landscape: A metaphor introduced by Conrad Waddington where a ball, representing a stem cell, rolling across a rugged landscape with hills and valleys symbolizes development through time. The valleys continue to bifurcate and eventually the cell lands in one of many terminal sub-valleys at the bottom of the hill that represent terminally differentiated states where it is held permanently by high valley walls. In Waddington's terminology, the steeper the walls and the narrower the valleys, the more ‘canalized’ the cell fate. However, in response to environmental perturbations, the cell can be pushed from one developmental pathway to another and genetic assimilation acts as an evolutionary process to heighten the ridges of the landscape. Consequently, over time, increasingly greater perturbations are needed to shift the ball from one developmental trajectory to another.
Genetic (or gene) regulatory circuits: Functional clusters of genes that impact each other's expression through inducible transcription factors and cis-regulatory elements.
Phase diagram: A type of chart used to show conditions at which thermodynamically distinct phases occur and coexist at equilibrium.
Power law: A relationship between two quantities such that one is proportional to a fixed power of the other.
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing financial interests.
Supplementary information is linked to the online version of the paper on the Asian Journal of Andrology website.
REFERENCES
- 1.Balazsi G, van Oudenaarden A, Collins JJ. Cellular decision making and biological noise: from microbes to mammals. Cell. 2011;144:910–25. doi: 10.1016/j.cell.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 3.van der Horst G, Bos L, van der Pluijm G. Epithelial plasticity, cancer stem cells, and the tumor-supportive stroma in bladder carcinoma. Mol Cancer Res. 2012;10:995–1009. doi: 10.1158/1541-7786.MCR-12-0274. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg RA. New York: Garland Science; 2007. The Biology of Cancer. [Google Scholar]
- 5.Scheel C, Weinberg RA. Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer. 2011;129:2310–4. doi: 10.1002/ijc.26311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mooney SM, Parsana P, Hernandez JR, Liu X, Verdone JE, et al. The presence of androgen receptor elements regulates ZEB1 expression in the absence of androgen receptor. J Cell Biochem. 2015;116:115–23. doi: 10.1002/jcb.24948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 8.Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982;95:333–9. doi: 10.1083/jcb.95.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, et al. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 2016 doi: 10.18632/oncotarget.8166. doi: 10.18632/oncotarget.8166. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klymkowsky MW, Savagner P. Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol. 2009;174:1588–93. doi: 10.2353/ajpath.2009.080545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grosse-Wilde A, Fouquier d’Herouel A, McIntosh E, Ertaylan G, Skupin A, et al. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS one. 2015;10:e0126522. doi: 10.1371/journal.pone.0126522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jolly MK, Huang B, Lu M, Mani SA, Levine H, et al. Towards elucidating the connection between epithelial-mesenchymal transitions and stemness. J R Soc Interface. 2014;11:20140962. doi: 10.1098/rsif.2014.0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–4. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, et al. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer. 2011;105:1338–41. doi: 10.1038/bjc.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou JM, Krebs M, Ward T, Sloane R, Priest L, et al. Circulating tumor cells as a window on metastasis biology in lung cancer. Am J Pathol. 2011;178:989–96. doi: 10.1016/j.ajpath.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marin-Aguilera M, Codony-Servat J, Reig O, Lozano JJ, Fernandez PL, et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol Cancer Ther. 2014;13:1270–84. doi: 10.1158/1535-7163.MCT-13-0775. [DOI] [PubMed] [Google Scholar]
- 21.Mooney SM, Rajagopalan K, Williams BH, Zeng Y, Christudass CS, et al. Creatine kinase brain overexpression protects colorectal cells from various metabolic and non-metabolic stresses. J Cell Biochem. 2011;112:1066–75. doi: 10.1002/jcb.23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Festuccia C, Angelucci A, Gravina GL, Villanova I, Teti A, et al. Osteoblast-derived TGF-beta1 modulates matrix degrading protease expression and activity in prostate cancer cells. Int J Cancer. 2000;85:407–15. [PubMed] [Google Scholar]
- 23.Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, et al. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–83. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 24.Morrissey C, Vessella RL, Lange PH, Lam HM. The biology and clinical implications of prostate cancer dormancy and metastasis. J Mol Med (Berl) 2016;94:259–65. doi: 10.1007/s00109-015-1353-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang KR, Mooney SM, Zarif JC, Coffey DS, Taichman RS, et al. Niche inheritance: a cooperative pathway to enhance cancer cell fitness through ecosystem engineering. J Cell Biochem. 2014;115:1478–85. doi: 10.1002/jcb.24813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inge LJ, Rajasekaran SA, Wolle D, Barwe SP, Ryazantsev S, et al. alpha-Catenin overrides Src-dependent activation of beta-catenin oncogenic signaling. Mol Cancer Ther. 2008;7:1386–97. doi: 10.1158/1535-7163.MCT-07-2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JJ, Yin B, Christudass CS, Terada N, Rajagopalan K, et al. Acquisition of paclitaxel resistance is associated with a more aggressive and invasive phenotype in prostate cancer. J Cell Biochem. 2013;114:1286–93. doi: 10.1002/jcb.24464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget. 2015;6:130–43. doi: 10.18632/oncotarget.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolly MK, Boareto M, Huang B, Jia D, Lu M, et al. Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol. 2015;5:155. doi: 10.3389/fonc.2015.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gemmill RM, Roche J, Potiron VA, Nasarre P, Mitas M, et al. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011;300:66–78. doi: 10.1016/j.canlet.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jia D, Jolly MK, Boareto M, Parsana P, Mooney SM, et al. OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget. 2015;6:15436–48. doi: 10.18632/oncotarget.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–66. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 33.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, et al. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72:3499–511. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu M, Jolly MK, Levine H, Onuchic JN, Ben-Jacob E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc Natl Acad Sci U S A. 2013;110:18144–9. doi: 10.1073/pnas.1318192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davila-Velderrain J, Martinez-Garcia JC, Alvarez-Buylla ER. Modeling the epigenetic attractors landscape: toward a post-genomic mechanistic understanding of development. Front Genet. 2015;6:160. doi: 10.3389/fgene.2015.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andriani F, Bertolini G, Facchinetti F, Baldoli E, Moro M, et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol Oncol. 2016;10:253–71. doi: 10.1016/j.molonc.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 41.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–46. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 42.Xue B, Oldfield CJ, Van YY, Dunker AK, Uversky VN. Protein intrinsic disorder and induced pluripotent stem cells. Mol Biosyst. 2012;8:134–50. doi: 10.1039/c1mb05163f. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem. 2011;112:3256–67. doi: 10.1002/jcb.23252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang P, Huo Z, Liao H, Zhou Q. Cancer/testis antigens trigger epithelial-mesenchymal transition and genesis of cancer stem-like cells. Curr Pharm Des. 2015;21:1292–300. doi: 10.2174/1381612821666141211154707. [DOI] [PubMed] [Google Scholar]
- 45.Grelet S, Andries V, Polette M, Gilles C, Staes K, et al. The human NANOS3 gene contributes to lung tumour invasion by inducing epithelial-mesenchymal transition. J Pathol. 2015;237:25–37. doi: 10.1002/path.4549. [DOI] [PubMed] [Google Scholar]
- 46.Zeng Y, He Y, Yang F, Mooney SM, Getzenberg RH, et al. The cancer/testis antigen prostate-associated gene 4 (PAGE4) is a highly intrinsically disordered protein. J Biol Chem. 2011;286:13985–94. doi: 10.1074/jbc.M110.210765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajagopalan K, Qiu R, Mooney SM, Rao S, Shiraishi T, et al. The Stress-response protein prostate-associated gene 4, interacts with c-Jun and potentiates its transactivation. Biochim Biophys Acta. 2014;1842:154–63. doi: 10.1016/j.bbadis.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hellman M, Tossavainen H, Rappu P, Heino J, Permi P. Characterization of intrinsically disordered prostate associated gene (PAGE5) at single residue resolution by NMR spectroscopy. PLoS One. 2011;6:e26633. doi: 10.1371/journal.pone.0026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gjerstorff MF, Rosner HI, Pedersen CB, Greve KB, Schmidt S, et al. GAGE cancer-germline antigens are recruited to the nuclear envelope by germ cell-less (GCL) PLoS One. 2012;7:e45819. doi: 10.1371/journal.pone.0045819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–64. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–9. doi: 10.1038/nature13999. [DOI] [PubMed] [Google Scholar]
- 53.Mahmoudabadi G, Rajagopalan K, Getzenberg RH, Hannenhalli S, Rangarajan G, et al. Intrinsically disordered proteins and conformational noise: implications in cancer. Cell Cycle. 2013;12:26–31. doi: 10.4161/cc.23178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eldar A, Elowitz MB. Functional roles for noise in genetic circuits. Nature. 2010;467:167–73. doi: 10.1038/nature09326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He Y, Chen Y, Mooney SM, Rajagopalan K, Bhargava A, et al. Phosphorylation-induced conformational ensemble switching in an intrinsically disordered cancer/testis antigen. J Biol Chem. 2015;290:25090–102. doi: 10.1074/jbc.M115.658583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamanaka S. Elite and stochastic models for induced pluripotent stem cell generation. Nature. 2009;460:49–52. doi: 10.1038/nature08180. [DOI] [PubMed] [Google Scholar]
- 58.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–94. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Terada N, Shiraishi T, Zeng Y, Aw-Yong KM, Mooney SM, et al. Correlation of Sprouty1 and Jagged1 with aggressive prostate cancer cells with different sensitivities to androgen deprivation. J Cell Biochem. 2014;115:1505–15. doi: 10.1002/jcb.24805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–44. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 61.Kulkarni P, Shiraishi T, Kulkarni RV. Cancer: tilting at windmills? Mol Cancer. 2013;12:108. doi: 10.1186/1476-4598-12-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocrine Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 63.Reid J, Kelly SM, Watt K, Price NC, McEwan IJ. Conformational analysis of the androgen receptor amino-terminal domain involved in transactivation. Influence of structure-stabilizing solutes and protein-protein interactions. J Biol Chem. 2002;277:20079–86. doi: 10.1074/jbc.M201003200. [DOI] [PubMed] [Google Scholar]
- 64.Reid J, Betney R, Watt K, McEwan IJ. The androgen receptor transactivation domain: the interplay between protein conformation and protein-protein interactions. Biochem Soc Trans. 2003;31:1042–6. doi: 10.1042/bst0311042. [DOI] [PubMed] [Google Scholar]
- 65.Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–60. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc. 2009;131:7390–401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 67.Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, et al. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther. 2010;335:715–27. doi: 10.1124/jpet.110.170555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berg T. Small-molecule modulators of c-Myc/Max and Max/Max interactions. Curr Top Microbiol Immunol. 2011;348:139–49. doi: 10.1007/82_2010_90. [DOI] [PubMed] [Google Scholar]
- 69.Barabasi AL, Albert R. Emergence of scaling in random networks. Science. 1999;286:509–12. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 70.Mooney SM, Rajagopalan K, Rangarajan G, Kulkarni P. Cancer/testis antigens and obligate participation in multiple hallmarks of cancer: an update. Asian J Androl. 2016 doi: 10.4103/1008-682X.174858. doi: 10.4103/1008-682X.174858. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mukhtar MS, Carvunis AR, Dreze M, Epple P, Steinbrenner J, et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science. 2011;333:596–601. doi: 10.1126/science.1203659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Muers M. Systems biology: plant networks. Nat Rev Genet. 2011;12:586. doi: 10.1038/nrg3058. [DOI] [PubMed] [Google Scholar]
- 73.Landry CR. Cell biology. A cellular roadmap for the plant kingdom. Science. 2011;333:532–3. doi: 10.1126/science.1209753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of genes that are highly co-expressed with CDH1 according to Pearson correlation coefficient. The Spearman correlation coefficient is also indicated on the table but was not used in ranking. The cBioPortal website was used to query 877 tumor samples from the Cancer Cell Line Encyclopedia
List of genes that are inversely expressed with CDH1 according to Pearson correlation coefficient. The Spearman correlation coefficient is also indicated on the table but was not used in ranking. The cBioPortal website was used to query 877 tumor samples from the Cancer Cell Line Encyclopedia