

Summary
Francisella tularensis in an intracellular bacterial pathogen that causes a potentially lethal disease called tularemia. Studies performed nearly 100 years ago revealed that neutrophil accumulation in infected tissues correlates directly with the extent of necrotic damage during F. tularensis infection. However, the dynamics and details of bacteria-neutrophil interactions have only recently been studied in detail. Herein we review current understanding regarding the mechanisms that recruit neutrophils to F. tularensis infected lungs, opsonization and phagocytosis, evasion and inhibition of neutrophil defense mechanisms, as well as the ability of F. tularensis to prolong neutrophil lifespan. In addition, we discuss distinctive features of the bacterium, including its ability to act at a distance to alter overall neutrophil responsiveness to exogenous stimuli, and the evidence which suggests that macrophages and neutrophils play distinct roles in tularemia pathogenesis, such that macrophages are major vehicles for intracellular growth and dissemination whereas neutrophils drive tissue destruction by dysregulation of the inflammatory response.
Keywords: apoptosis, phagocytosis, NADPH oxidase, tularemia, mitochondria, neutrophils, inflammation
Introduction
Neutrophils and innate host defense
Polymorphonuclear leukocytes (PMNs) are the most abundant leukocyte population in humans and are rapidly recruited from the bloodstream to sites of infection by chemotactic factors of host and/or microbial origin (1). In this locale PMNs phagocytose microbes and utilize a combination of NADPH oxidase-derived reactive oxygen species (ROS), cytotoxic granule components and antimicrobial peptides to generate a highly lethal intraphagosomal environment that is essential for efficient microbe killing and degradation (1, 2). The ability of neutrophils to mobilize their intracellular granules within seconds of microbe binding and a robust respiratory burst are features that distinguish neutrophils from macrophages, which modify phagosome composition slowly over the course of 30–60 min or more via sequential fusion with endosomal pathway organelles (3).
PMNs are also distinguished from macrophages and other leukocytes by their short lifespan. Thus, whereas tissue macrophages can survive for months and are self-renewing (4), neutrophils are inherently programmed to die by constitutive (spontaneous, physiological) apoptosis approximately 24 h after release into circulation, and at sites of infection PMN death is accelerated further by phagocytosis and oxidant production, a process called phagocytosis-induced cell death (PICD) (5–7). At the molecular level, PMN apoptosis is regulated not only by intracellular signaling, but also by global changes in gene expression that comprise an ‘apoptosis differentiation program’ (7–9). Tight spatial and temporal control of PMN apoptosis is critical for elimination of infection and resolution of the inflammatory response, thus, phagocytic and proinflammatory capacity are down-regulated, release of toxic cell component is prevented, and tissue damage is minimized (7, 10, 11). If this process is disrupted PMNs can develop a proinflammatory phenotype that promotes tissue necrosis and granuloma formation and sustains infection (11, 12). For this reason, defects in PMN turnover are indicative of an ineffective and dysregulated inflammatory response (13, 14). Consistent with this notion, recent data indicate that neutrophils can exhibit significant phenotypic plasticity in vivo and are also important immunoregulatory cells with the capacity to directly influence the function of other leukocytes including natural killer cells, dendritic cells, macrophages and lymphocytes (14–16).
Francisella tularensis and tularemia
Francisella tularensis is a small, hardy, Gram-negative, coccobacillus and the causative agent of tularemia. This disease was first described as a plague-like illness of ground squirrels in Tulare County, California in the early 20th century, and F. tularensis is now known to be distributed throughout the Northern Hemisphere in association with both terrestrial and acquatic environments (17–21). This pathogen infects more than 200 species of mammals including but not limited to rabbits, hares, coyotes, ground squirrels, beavers, rats, voles and other rodents, as well as ticks and other arthropod vectors (21–23). Ticks are a permanent reservoir that maintains F. tularensis in the environment (20), perhaps in conjunction with infection of resistant mammals such as horses, cattle, red foxes and wild boars (20, 21). Three subspecies of F. tularensis have been described that differ in geographic distribution and virulence (24–26). F. tularensis subspecies tularensis (type A) is the most virulent, accounts for nearly all lethal infections with this bacterium in humans, and is exclusive to North America. F. tularensis subspecies holarctica (type B) is less virulent and is found throughout the Northern Hemisphere, whereas F. tularensis subspecies mediasiatica exhibits low virulence, is found only in central Asia, and is rarely studied.
Humans are highly susceptible to tularemia, and individuals particularly at risk include hunters, butchers, farmers, landscapers, hikers, and other individuals with an increased probability of contact with infected animals, the carcasses or bodily fluids of these animals, or infected ticks. Several routes of infection and disease manifestations have been described (20, 27). Ulceroglandular tularemia is the most common, and ensues following the bite of an infected arthropod vector or bacterial contact with broken or abraded skin. A papule forms at the site of inoculation, which ulcerates concomitant with bacterial dissemination, massive enlargement of draining lymph nodes that may progress to suppuration, and spreading of the infection to the spleen, liver and lungs. A similar disease course without ulceration follows bacterial penetration of intact skin. Oropharyngeal tularemia is caused by consumption of contaminated water, food or undercooked meat, whereas oculoglandular tularemia is caused by self-inoculation of the eye with subsequent bacterial penetration of the conjunctiva. Pneumonic tularemia is by far the most severe form of this disease and can ensue following inhalation of as few as 10 type A organisms, and in this case extensive replication of bacteria in lung phagocytes is followed by dissemination to the liver and spleen. Mortality rates for pneumonic tularemia can be as high as 30–60% in the absence of appropriate antibiotic treatment, and the proximal cause of death is almost invariably bronchopneumonia (20, 28–30). In contrast, the overall mortality for all types of untreated tularemia is 5–15%, with convalescence of survivors lasting for 3–12 months and lymph node suppuration occurring as long as 22–24 months after infection (20). Regardless of infection route, tularemia is noted for very sudden onset of fever and extensive necrosis of infected tissues that directly parallels neutrophil density (20).
There has long been a biosafety concern regarding F. tularensis (31–33). Due to the bacterium’s extraordinarily low infectious dose and ability to be easily aerosolized, in addition to the high mortality rate of untreated pneumonic tularemia, the Center for Disease Control and Prevention has classified F. tularensis as a Tier 1 select agent. Moreover, the United States, Japan, and the former Soviet Union stockpiled F. tularensis for potential use as a bioweapon; however, these biowarfare programs were subsequently dissolved and were, in at least some cases, replaced with research programs aimed at counteracting the threat posed by F. tularensis and other Tier 1 pathogens (31, 33). Studies of type A and type B F. tularensis strains require biosafety level (BSL)-3 containment. However, an attenuated variant of F. tularensis subspecies holarctica was developed by the former Soviet Union. This live vaccine strain (LVS) can be used in BSL-2 and retains many features of fully virulent organisms in cell culture and mouse models, but is not licensed for use as a vaccine in the United States as little is understood about its mode of attenuation (32). In the past few years it has become apparent that F. tularensis infects several types of cells including endothelial cells, alveolar type (AT) II cells, macrophages, neutrophils, and hepatocytes, and manipulates multiple aspects of innate host defense (32, 34–37). A majority of studies have focused on F. tularensis infection of macrophages, as these cells appear to be major vehicles for bacterial growth and dissemination in vivo. Herein we summarize the results of recent studies by our laboratory and others that provide fundamental insight into the unique role that neutrophils play during infection with this complex and fascinating bacterial pathogen.
Bacterial virulence factors
F. tularensis has a small genome and many incomplete metabolic pathways, consistent with its fastidious growth requirements and adaptation to an intracellular lifestyle (32). Major virulence factors of this bacterium include lipopolysaccharide (LPS) and capsule that are essential for virulence and confer resistance to complement-mediated lysis, and several genes required for LPS O-antigen and capsule synthesis have recently been identified (32, 38–42). F. tularensis LPS also contributes directly to immune evasion via its atypical structure that does not interact efficiently with host endotoxin binding proteins, and for this reason does not elicit signaling via Toll-like receptor (TLR)4 or TLR2 (32, 43, 44). A capacity for TLR and IL-1 receptor signaling later in infection is also undermined by miR-155-mediated downregulation of MyD88 in macrophages (45). These findings underscore the stealth-like nature of F. tularensis and its ability to prevent induction of a potentially protective inflammatory response until very late stages of infection when bacterial load is overwhelming. At the same time, studies of capsule and O-antigen-deficient mutants suggest that these surface carbohydrates may also enhance bacterial structural integrity, favoring bacterial replication to high density and perhaps enhancing evasion of cytosolic host defense mechanisms (40, 46).
The Francisella pathogenicity island (FPI) is a 30 kb region of the genome that is present in two identical copies, each of which contain 19 genes organized in two large operons (47, 48). Nearly all genes in the FPI are essential for phagosome escape, intracellular growth, and in vivo virulence, including iglC, iglI and iglJ (48, 49). Although the specific functions of FPI genes remain elusive, this locus may encode a type VI-like secretion system (48). In contrast, F. tularensis lacks genes for type III, type IV, and type V secretion systems that are common in other bacterial pathogens and modulate host cell function via secretion of effector proteins (32). Three major regulatory factors, MglA, FevR and MigR, control expression of approximately 100 genes in F. tularensis, including those in the FPI, and are also essential for intracellular bacterial growth and virulence (48, 50–52). MigR and FevR are also critical for inhibition of oxidative host defense at the level of the phagocyte NADPH oxidase complex (discussed below), but in this case relevant downstream target genes appear to reside outside of the FPI (51, 53). Finally, the F. tularensis genome encodes three tolC homologues despite its lack of known toxins, which are some of the primary substrates for type I secretion (32, 54).
Neutrophils and tularemia pathogenesis
Insights from in vivo studies
Aerosol infection of nonhuman primates with virulent type B F. tularensis isolates in the 1970s confirmed and extended prior knowledge regarding the features of pneumonic tularemia and provided additional evidence to demonstrate that neutrophils play a role in host tissue destruction and disease progression (55–57). In particular, the data demonstrate that neutrophils are abundant in infected lungs and alveoli, and that bronchioles become progressive clogged with PMNs, bacteria and necrotic debris. Also present are granulomas that contain live neutrophils, bacteria and necrotic debris surrounded by epithelial syncytia. Similar data were obtained for infected rabbits, rats and mice, and ex vivo analyses of infected PMNs confirmed the presence of viable intracellular bacteria (58). Rhesus monkeys, mice, rabbits and many humans succumb to acute pneumonic infection with type A F. tularensis (20, 56, 59). These organisms replicate faster than type B strains, and progression to moribund status is characterized by profound accumulation of PMNs in the airway and extensive necrotic tissue damage such that death often ensues prior to the development of a potentially protective adaptive immune response (60–62). In agreement with the histopathology findings, detailed analyses by flow cytometry of lung cells following intranasal infection of mice demonstrated that alveolar macrophages are infected first and account for 70–90% of infected cells at 4–24 h post-infection with either the type A F. tularensis strain Schu S4 or LVS (63, 64). At later time points this pattern changes markedly, as neutrophils account for at least 50% of F. tularensis-infected cells by day three of infection, whereas infected AT II cells and dendritic cells were rare at all time points examined (63).
Neutrophil recruitment into the lung
Mechanisms of neutrophil chemotaxis, including cell migration into the lung, have been studied extensively (65–68). Inhaled bacteria that are not removed by mucociliary clearance typically engage TLRs or other receptors on alveolar macrophages and AT II cells, leading to synthesis and secretion of neutrophil chemoattractants such as IL-8 (in humans) and KC (in mice), as well as GRO-α, MIP-2 and MCP-1. Other relevant factors include G-CSF, IL-1β, LIX/CXCL5, C5a, the eicosanoids LTB4 and PGE2, and proline-glycine-proline (PGP), a peptide that is generated by cleavage of collagen in the extracellular matrix by matrix metalloproteinase (MMP)-8 and MMP-9 (69). Microbial factors such as formyl peptides also stimulate PMN chemotaxis, and neutrophils themselves can contribute to this process by secretion of IL-8, IL-1β, LTB4, and/or MMP-9. As the signals mediating recruitment can influence PMN phenotype and function (70, 71), it is noteworthy that a specific subset of signals appears to control neutrophil migration into the lungs during tularemia. For example, it is established that relatively little IL-8 is secreted by macrophages, AT II cells or the vascular endothelium under these conditions, and neither IL-8 nor MCP-1 is essential for neutrophil recruitment (71, 72). Instead, F. tularensis upregulates MMP-9, thereby driving PMN accumulation via the collagen-MMP-9-PGP pathway (73). Notably, PGP can initiate and sustain tissue neutrophil accumulation in the absence of other stimuli and N-acetylation enhances its activity and stability (74), but whether PGP is modified in this manner during tularemia remains to be determined. Mechanisms that mediate PMN migration into other infected tissues or into the lung following F. tularensis inoculation into the skin have not been characterized. Nevertheless, it is noteworthy that neutrophils also play a major role in the pathology associated with chronic obstructive pulmonary disease (COPD), and in this case PMN accumulation in the lungs is also PGP-dependent (69).
Effects of local or systemic PMN depletion
The increased susceptibility to infection that characterizes persons with neutropenia or or inherited defects in neutrophil function underscores the critical role of PMNs in innate immunity and host defense (2, 75). Accordingly, neutrophil depletion strategies are often used to discern the role of this cell type in animal models of various infectious or inflammatory disorders. Two different antibodies have been developed for this purpose (76). RB6-8C5 was developed first, and because it efficiently depletes neutrophils in mouse models has been used in numerous studies. However, it was later revealed that this antibody binds to Ly6C into addition to Ly6G and, at a minimum, causes depletion of inflammatory monocytes, plasmacytoid dendritic cells, and some CD8+ T-cell populations in addition to neutrophils (77–79). As multiple cell types are depleted by RB6-8C5, the desired specificity for PMNs cannot be achieved. To address this limitation, the Ly6G-specific antibody 1A8 was developed, which appears to be PMN-specific and is currently the preferred reagent for studies of neutrophil depletion in mice (76, 79). Direct comparisons of RB6-8C5 and 1A8 have shown that some phenotypes previously attributed to PMN depletion are actually caused by loss of monocytes or other cell populations, necessitating reinterpretation of prior results (77, 78).
With regard to F. tularensis, no data for antibody1A8 are available. However, a 1994 study concluded that neutrophils are essential for innate defense against intradermal F. tularensis LVS, as RB6-8C5-treated mice succumbed to what would otherwise be a sublethal infection (80). Conversely, a subsequent study found that RB6-8C5 did not increase lethality following intranasal infection with type A F. tularensis Schu S4 (81). Differences in bacterial virulence, route of infection, or RB6-8C5 concentration may contribute to these discordant findings, and based on these data the role of neutrophils in tularemia cannot be discerned. For this reason, the study of Malik et al. is particularly important (73). These investigators used MMP-9 null mice to demonstrate that inhibition of PMN migration into the lungs significantly reduces tissue damage and is sufficient to enhance mouse survival following intranasal infection with either Schu S4 or LVS. These data therefore confirm the role of neutrophils in tissue destruction following F. tularensis infection, and also definitively demonstrate that neutrophils contribute to tularemia pathogenesis and disease progression rather than effective host defense. This conclusion is further supported by additional studies which show that hepatotoxicity and tularemia severity are significantly enhanced by conditions that induce neutrophilia (82, 83). Notably, neutrophils are also detrimental during infection with Mycobacterium tuberculosis, and in this case the beneficial effects of IFNγ are in part due to prevention of tissue PMN accumulation (15). IFNγ is also important for control of F. tularensis, but studies to date have focused on its role in upregulation of macrophage iNOS, and whether tissue neutrophil load is influenced, particularly in humans, is unknown (84, 85). Thus, additional studies that include analyses of PMN immunoregulatory properties are needed to determine the specific mechanisms of neutrophil-mediated disease exacerbation.
Opsonization and phagocytosis
Phagocytosis is a fundamental aspect of innate defense against infection that requires ligation of specific cell surface receptors and is driven by local actin polymerization. Some receptors that mediate phagocytosis bind directly to microbe surface molecules, whereas others bind indirectly to microbes that have been opsonized with complement components or IgG. Complement opsonization markedly enhances the efficiency of F. tularensis phagocytosis by neutrophils as well as macrophages, yet at the same time the extent of C3 fixation to the bacterial surface is low, and recent studies by us and others provide insight into the underlying molecular mechanisms (86–91). F. tularensis does not efficiently trigger the alternative pathway and does not engage mannose-binding lectin, the critical initiating factor of the lectin complement pathway (88, 89, 92). Rather, opsonization occurs via the classical pathway and is absolutely dependent on natural IgM that is present in normal, non-immune human serum at low levels and binds to LPS O-antigen and/or capsule on the bacterial surface (91, 93). Consistent with classical pathway activation, C1q and C3 are also essential, whereas C5 is not (90, 91). Nevertheless, C3b deposition on the bacterial surface is relatively inefficient, which together with Factor H binding is likely important for evasion of complement-mediated lysis (89–91). In keeping with this, F. tularensis capsule and O-antigen mutants are highly serum-sensitive, trigger robust complement activation via the alternative pathway, and are profoundly attenuated in vivo (40, 41, 90, 91, 94).
Receptor blocking studies indicate that phagocytosis of opsonized F. tularensis is mediated by complement receptor 1 (CR1, CD35) and CR3 (CD11b/CD18) on neutrophils, and by CR3, CR4 (CD11c/CD18), and/or scavenger receptor A on macrophages and dendritic cells (91, 95, 96). On the other hand, the mannose receptor and nucleolin mediate uptake of unopsonized F. tularensis by macrophages (88, 97), whereas the receptors used for uptake of unopsonized bacteria by PMNs are as yet unknown. It is established that mechanism of uptake influences microbe fate (98), and the mannose receptor and CR3 are generally considered ‘safe portals of entry’ that are exploited by many pathogens, including to F. tularensis, as they are not strongly coupled to host microbicidal mechanisms (88, 98, 99). For example, opsonins influence the magnitude of the respiratory burst (53, 92, 98, 100, 101), and a direct comparison of opsonized and unopsonized F. tularensis shows that CR3-mediated phagocytosis curtails TLR2-dependent proinflammatory signaling and NF-κB activation in human macrophages (99). In our hands, both opsonized and unopsonized F. tularensis grow to very high density in primary human macrophages in independent studies and direct side-by-side comparisons (40, 49), whereas other investigators report that opsonization with human serum diminishes bacterial growth in murine macrophages (102). What accounts for this discordance is unknown, but differences in bacterial growth conditions, inherent difference between these two macrophage types, effects of human complement on murine cells, or other aspects of experimental design should be considered.
Regulation of NADPH oxidase assembly and activation
The phagocyte NADPH oxidase complex is comprised of six subunits: two integral membrane proteins p22phox and gp91phox (Nox2) that form a heterodimer called flavocytochrome b558, and four soluble proteins p47phox, p67phox, p40phox, and the GTPase Rac2 (103, 104). This enzymatic complex catalyzes the conversion of molecular oxygen into superoxide anions, and its importance to innate host defense is exemplified by the fact that persons with inherited defects in genes that encode the subunits of this enzyme have chronic granulomatous disease (CGD) and suffer from repeated, life-threatening bacterial and fungal infections (105). As superoxide anions and other ROS can be toxic to host cells as well as microbes, NADPH oxidase activity is tightly controlled. In resting neutrophils the enzyme is unassembled and inactive with subunits segregated in distinct in distinct subcellular compartments (104). Flavocytochrome b558 is enriched in the membranes of specific granules (SG) and to a lesser extent is also found in gelatinase granules (GG), secretory vesicles (SV) and at the plasma membrane, whereas the other subunits are distributed throughout the cytosol. Soluble NADPH oxidase agonists (such as formyl peptides) trigger enzyme assembly at cell surface, whereas particulate agonists (such as bacteria and fungi) induce enzyme assembly on forming phagosomes. Regardless of the stimulus, a critical aspect of NADPH oxidase assembly and activation is membrane translocation of the soluble subunits, with subsequent docking on flavocytochrome b558 leading to formation of an active holoenzyme. Rac2 translocates to the membrane independently of the other subunits, whereas p47phox, p67phox, and p40phox translocate en bloc as a tripartite complex via a process that requires phosphorylation of several serine residues in p47phox by protein kinase C (PKC) (104).
The dynamics of NADPH oxidase assembly and activation on neutrophil phagosomes has been extensively studied, with significant insight resulting from the use of sensitive assays to quantify the kinetics of oxidant production together with synchronized phagocytosis and confocal microscopy to assess subunit localization and trafficking (106–108). The microscopy data demonstrate that NADPH oxidase assembly on forming phagosomes is extremely rapid and is apparent within 30 sec of particle binding (106). Both membrane and soluble enzyme components accumulate selectively on forming phagocytic cups and colocalize with F-actin and actin binding proteins such as coronin (106, 107). Restricting NADPH oxidase assembly and activation to phagosomes prevents host tissue damage and favors efficient microbe killing by concentrating oxidants in close proximity to the engulfed microbe. Measurements of PMN oxygen consumption using a Clark electrode and ROS production using sensitive luminescent probes confirms rapid activation of this enzyme during phagocytosis, and nitroblue tetrazolium (NBT) staining demonstrates directly that superoxide anions accumulate in the phagosome lumen and not the in extracellular space (107–109). Notably, superoxide anions are only weakly microbicidal and are subsequently converted into more toxic ROS. In neutrophils this is driven by myeloperoxidase (MPO), which is delivered into the phagosome lumen by fusion with azurophilic granules (AG), mediates dismutation of superoxide anions into hydrogen peroxide and also catalyzes conversion of hydrogen peroxide into highly lethal HOCl (1, 110, 111).
Oxidant production peaks 15–20 min after initiation of phagocytosis and then wanes slowly, returning to baseline after about 90 min (107, 108). Mechanisms that regulate termination of the respiratory burst are incompletely defined, but this correlates with enzyme disassembly, Rac deactivation, and changes in p47phox phosphorylation (107, 112). In addition, oxidants can directly damage the enzyme, and for this reason exchange of ‘fresh’ subunits may be required to sustain catalytic activity (112, 113). Additional insight as been gained by studies of neutrophils from persons with CGD. For example, studies of neutrophils lacking gp91phox revealed that signals required for membrane translocation of the soluble enzyme components are intact in the absence of flavocytochrome b558 (106), whereas an absence of p40phox has no apparent effect on NADPH oxidase activation at the plasma membrane, and instead plays a selective and essential role in sustaining NADPH oxidase assembly on maturing phagosomes (114).
Multiple mechanisms of NADPH oxidase inhibition
Inhibition of enzyme assembly
As noted above, phagocytosis is normally coupled to rapid and robust NADPH oxidase activation, and toxic oxidants kill most bacteria within the first 60 min of infection (2, 8, 107). A study published in 1975 was the first to suggest that neutrophil activation is impaired by infection with F. tularensis Schu S4 or LVS (115). Specifically, Proctor et al., demonstrated that in marked contrast to Salmonella typhi, Escherichia coli and latex beads assayed under identical conditions, F. tularensis that had been opsonized with normal human serum did not activate rhesus monkey neutrophils as indicated by measurements of hexose monophosphate shunt activity. We confirmed and extended these findings to show that F. tularensis opsonized with normal human serum does not activate human neutrophils for production of ROS even at very high multiplicities of infection (100). This phenotype is specific for live bacteria, as it is ablated by formalin fixation or periodate treatment, and is shared by virulent type A and type B strains of F. tularensis as well as LVS, indicating a conserved mechanism of virulence (53, 100). Confocal microscopy studies revealed a profound defect in NADPH oxidase assembly on F. tularensis-containing phagosomes, as indicated by exclusion of gp91phox and p22phox as well as p47phox and p67phox, and as a result, fewer than 5% of these compartments contained superoxide anions as indicated by NBT staining (53, 100). Although the absence of flavocytochrome b558 on F. tularensis compartments deprives the soluble subunits of their docking site and is therefore sufficient to account for the enzyme assembly defect, in vivo radiolabeling experiments revealed that the soluble subunits were also directly affected, as indicated by a profound defect in phosphorylation of p47phox that also extended to other PKC substrates (53). Thus, the data demonstrate that F. tularensis acts at multiple points to prevent NADPH oxidase assembly. Mechanisms that account for phagosome exclusion of flavocytochrome b558 have not been precisely defined, but this correlates with a general defect in phagosome-granule fusion that is apparent by transmission electron microscopy (TEM) (100) and is the subject of ongoing studies in our laboratory. Additional studies of the effects of F. tularensis on PKC signaling and p47phox phosphorylation are also warranted.
Effects of other pathogens
Microorganisms that survive in neutrophils must evade or withstand toxic oxidants. Consistent with this, other pathogens that manipulate NADPH oxidase assembly as part of their virulence strategies have also been identified. For example, Anaplasma phagocytophilum specifically blocks phagosome acquisition of flavocytochrome b558 and Rac (116, 117). In contrast, phagosomes containing Opa-negative Neisseria gonorrhoeae accumulate both gp91phox and p22phox, yet exhibit a partial defect in recruitment of p47phox and p67phox that is associated with reduced phosphorylation of p47phox (118). Coxiella burnetti also inhibits membrane translocation of p47phox and p67phox in PMNs by an unknown mechanism (119). Unlike pathogens that act at various points to inhibit NADPH oxidase assembly, a distinctly different strategy is used by Helicobacter pylori (108). In this case, live, unopsonized bacteria are avidly phagocytosed by PMNs and trigger a robust respiratory burst, yet at the same time NADPH oxidase targeting is altered, leading to enzyme accumulation at the cell surface rather than bacterial phagosomes. In this manner H. pylori evades intracellular killing and enhances host tissue damage via release of toxic oxidants into the extracellular milieu.
Post-assembly NADPH oxidase activity defects
An additional unexpected and distinctive feature of F. tularensis is its ability to significantly inhibit neutrophil activation by heterologous stimuli. Specifically, we were surprised to discover that within 10 min of infection with F. tularensis, the ability of neutrophils be activated by phorbol myristate acetate (PMA) was reduced by 75% (53, 100). As PMA is a diacylglycerol analog that directly activates PKC, we also tested soluble and particulate stimuli that trigger NADPH oxidase activation via ligation of surface receptors, including formyl peptides, opsonized zymosan (OpZ), Staphylococcus aureus, and H. pylori, and obtained similar results (53, 100). Notably, global perturbation of neutrophil activation is dose- and time- dependent, specific for live bacteria, and can also be achieved if F. tularensis is added along with or shortly after control stimuli (53, 100, 120). NBT staining confirmed a defect in superoxide production, and the significance of this effect is indicated by a 60% reduction in the ability of PMNs to kill S. aureus even though other aspects of cell function, such as phagocytosis, are unchanged (53).
Chemiluminescence assays provided insight into the dynamics of this response (53, 100). In particular, the data show that initiation of the respiratory burst occurs normally for all heterologous stimuli tested, as lag times and initial rates of oxidant production elicited by each agonist were indistinguishable from their respective controls, but shortly thereafter chemiluminescence declines sharply, and both peak and total oxidant production are profoundly diminished. These data strongly suggest that F. tularensis acts distal to enzyme assembly to curtail the magnitude and duration of the respiratory burst. We predicted that F. tularensis may accelerate enzyme disassembly as means to curtail ROS production, perhaps via effects on p47phox, as continuous phosphorylation of this protein is required to maintain superoxide production (112, 121). However, these predictions were incorrect as infection did not alter the extent of in vivo p47phox phosphorylation at 15 and 60 min after stimulation with OpZ or PMA, and detailed microscopy studies of OpZ phagosomes using antibodies to all six enzyme components demonstrated definitively that enzyme disassembly was not enhanced (53). Indeed, we obtained the opposite result, as NADPH oxidase accumulation at the membrane was significantly prolonged. Thus, whereas 80% of OpZ phagosomes in control PMNs shed p40phox, p47phox, p67phox and Rac2 by 60 min after uptake, these subunits, as well as gp91phox and p22phox, remained enriched on OpZ compartments during F. tularensis co-infection (53).
Taken together, the data indicate that global disruption of neutrophil activation is mediated by a post-assembly mechanism that is characterized by prolonged accumulation at the membrane of dysfunctional NADPH oxidase complexes. Notably, a similar if not identical mechanism undermines PMN activation by F. tularensis that have been opsonized with anti-LVS immune serum, indicating an ability of this pathogen to diminish the efficacy of a potentially protective adaptive immune response (53). How oxidase activity is disrupted remains to be determined. Potential mechanisms could potentially include effects on a conserved signaling intermediate, impaired capacity to renew enzyme complexes by exchange of fresh and spent subunits, site-specific changes in p47phox phosphorylation at the membrane or in the cytosol, altered phosphorylation of other subunits, disruption of membrane microdomains or protein-protein interactions within the assembled complex that are required to sustain electron flow, or loss of NADPH or FAD. Inactivation of Rac2 is less likely, as the antibody we used for microscopy is specific for the active form of this GTPase. It is also possible that the residual amount of NADPH oxidase activity that remains under these conditions is sufficient to prevent disassembly of the complex. Effects of F. tularensis on the phagocyte NADPH oxidase are summarized in Figure 1.
Fig. 1. Inhibition of NADPH oxidase assembly and activity by F. tularensis.
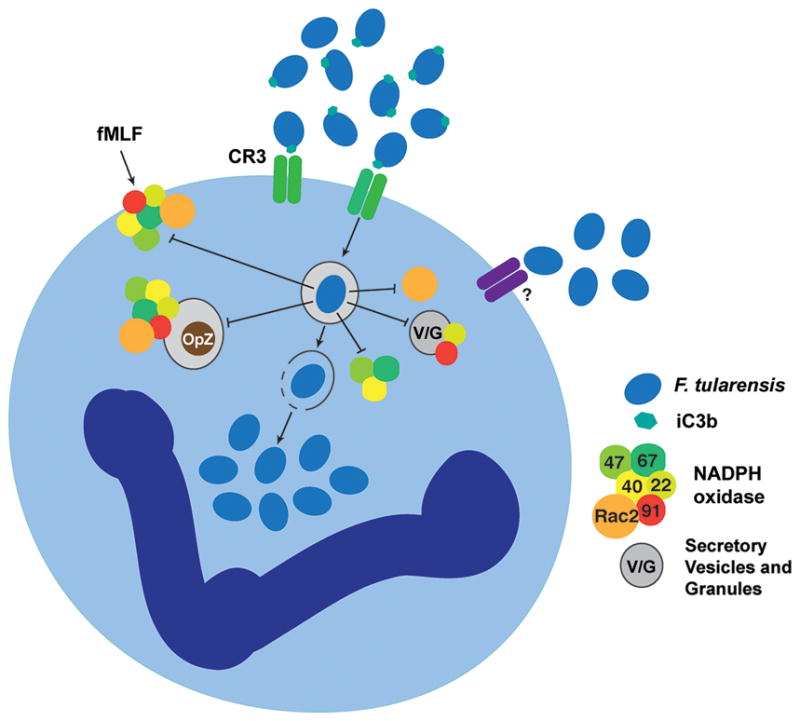
F. tularensis opsonized with iC3b binds CR3 on neutrophils, whereas unopsonized bacteria bind a receptor that is not yet defined. NADPH oxidase assembly at the F. tularensis phagosome is blocked, and this is followed by disruption of the phagosome membrane and bacterial growth in the cytosol. In addition, F. tularensis also acts by a post-assembly mechanism to inhibit NADPH oxidase activity triggered by heterologous soluble and particulate stimuli such as formyl-methionine-leucine-phenylalanine (fMLF) and opsonized zymosan particles (OpZ). V/G includes secretory vesicles, gelatinase granules and specific granules (which together comprise the intracellular stores of flavocytochrome b558 in resting neutrophils).
Bacterial factors required for disruption of oxidative host defense
Studies by us and others have begun to define the bacterial factors that are required for NADPH oxidase inhibition. FevR and MigR are major regulatory factors in F. tularensis that control expression of nearly 100 genes and are essential for many aspects of virulence including blockade of phagosome maturation, intracellular growth and in vivo virulence (48, 50–52). We used site-directed mutagenesis to disrupt fevR and migR in Schu S4 and LVS and demonstrated that these virulence regulators are essential for inhibition of the respiratory burst triggered by F. tularensis uptake as well as exposure to heterologous stimuli (51, 53). Although the relevant downstream target genes within the FevR and MigR regulons remain elusive, additional mutagenesis studies excluded a role for FPI genes iglI and iglJ (53). Based on these data it is attractive to predict that the factors required for evasion of oxidative host defense are distinct from those that mediate phagosome escape, and as such may be encoded by genes that reside outside the FPI. A critical but indirect role has also been demonstrated for carA, carB, and pyrB, which are required for bacterial pyrimidine biosynthesis (101).
On the other hand, the contribution of F. tularensis acid phosphatase, AcpA, to NADPH oxidase inhibition is controversial due to conflicting results. This enzyme is periplasmic and secreted, and in purified or recombinant form exhibits strong, nonspecific phosphomonoesterase activity at pH 6.0 that can inhibit activation of porcine neutrophils by an unknown mechanism (122, 123). Thus, it was proposed that AcpA secreted by F. tularensis may gain access to the cytosol and undermine phosphorylation of NADPH oxidase subunits as a means to inhibit enzyme assembly and activation. However, AcpA secretion, which is first apparent 30 min after synchronized phagocytosis (124), is too slow to prevent oxidase assembly on forming phagosomes, and AcpA activity is very low at cytosolic pH (122). Moreover, disruption of acpA in Schu S4 ablated bacterial acid phosphatase activity, yet in our hands had no effect on the ability of F. tularensis to curtail oxidant production at its own phagosome or elsewhere in infected PMNs (53). AcpA mutants generated by two other groups also exhibited no apparent in vitro or in vivo virulence defects (125, 126). In contrast, Mohapatra et al., reported that acpA mutants were unable to prevent p47phox translocation to phagosomes or ROS production (127). What accounts for these discordant findings is unknown, particularly as flavocytochrome b558 targeting was not analyzed in the latter study, and basal phosphorylation of p47phox in uninfected neutrophils, which should be absent, was very high.
Several factors that allow F. tularensis to resist endogenous and/or exogenous oxidative stress and contribute to intracellular survival have also been identified. These include a glutamate transporter (GadC) (128), superoxide dismutase (SodB) (129), and catalase (KatG) (130). Type A F. tularensis strains also maintain lower intrabacterial iron stores than type B isolates (131), which may account at least in part for the fact that type A organisms are more resistant to killing by direct exposure to HOCl and hydrogen peroxide than LVS (131, 132). It is also worth noting that bacterial growth conditions influence the extent of NADPH oxidase inhibition and capacity to resist oxidative stress. Specifically, we find that NADPH oxidase inhibition is maximized when F. tularensis is grown in brain heart infusion or on cysteine heart agar supplemented with sheep blood, conditions that have been shown to mimic the phenotype of bacteria that are actively replicating in host cells and confer optimal inhibition of host immune and inflammatory responses (133, 134). Conversely, we find that the capacity of bacteria grown in Mueller Hinton broth to prevent oxidant production is diminished, as is their ability to prevent macrophage proinflammatory cytokine production (133, 134).
Phagosome maturation, phagosome escape and replication in the cytosol
The intracellular lifecycle of F. tularensis was defined by studies of human and murine macrophages (135–138), which demonstrate that phagosomes containing the bacterium transiently acquire early endosome antigen 1 (EEA1), and then accumulate late endosome membrane protein (lamp)-1 and CD63, yet exclude the vacuolar proton ATPase and lysosomal enzymes such as cathepsin D and are only moderately acidified (pH 6.7). These data indicate that F. tularensis avoids trafficking to lysosomes and arrests phagosome maturation at a late endosome-like stage. Shortly thereafter F. tularensis escapes from its phagosome into the cytosol and replicates to very high density (135–138). How the phagosome membrane is breached is unclear, but multiple breaks in the membrane are apparent by TEM (135–138), and both phagosome egress and intracellular growth, as well as in vivo virulence, are absolutely dependent on MigR and FevR, as well as genes in the FPI, including iglI and iglJ, that appear to encode components of a type VI-like secretion system (47, 49–52, 139). These same general features also characterize the F. tularensis lifecycle in epithelial cells (140), and there is no evidence to suggest that F. tularensis can replicate inside phagosomes or other vesicular compartments in mammalian cells.
Less is known about the F. tularensis phagosome in neutrophils. Phagosome maturation in this cell type is defined by rapid fusion with GG, SG and AG (141). The fact that nearly all F. tularensis phagosomes are devoid of flavocytochrome b558 suggests a defect in phagosome-GG and phagosome-SG fusion (53, 100), and preliminary analyses of AG markers suggests that this aspect of phagosome maturation may also be impaired. TEM studies demonstrate that during the first few hours of infection F. tularensis occupies close-fitting phagosomes that do not appear to fuse with surrounding granules (100). Approximately 4 h after uptake the phagosome membrane is breached, bacteria are released into the cytosol and begin to replicate in this locale (100, 142). Thus, F. tularensis is one of few pathogens that can grow in neutrophils as well as mononuclear phagocytes (8, 36, 100, 143, 144), and a light microscopy image of bacteria replicating in PMN cytosol is shown in Figure 2. Of note, a subset of ingested bacteria fail to reach the cytosol and are eventually degraded, which coincides with phagosome-granule fusion and is first apparent by TEM at 9 h after uptake (100). Taken together, these data suggest that F. tularensis actively prevents phagosome maturation for several hours after entry into neutrophils. However, the capacity to prevent phagosome-granule fusion eventually wanes, thereby making bacteria that fail to reach the cytosol sensitive to intracellular killing. Additional studies are needed to define the composition of the early F. tularensis phagosome in detail, and to determine how disruption of phagosome-granule fusion is achieved.
Fig. 2. F. tularensis replicates in PMN cytosol.
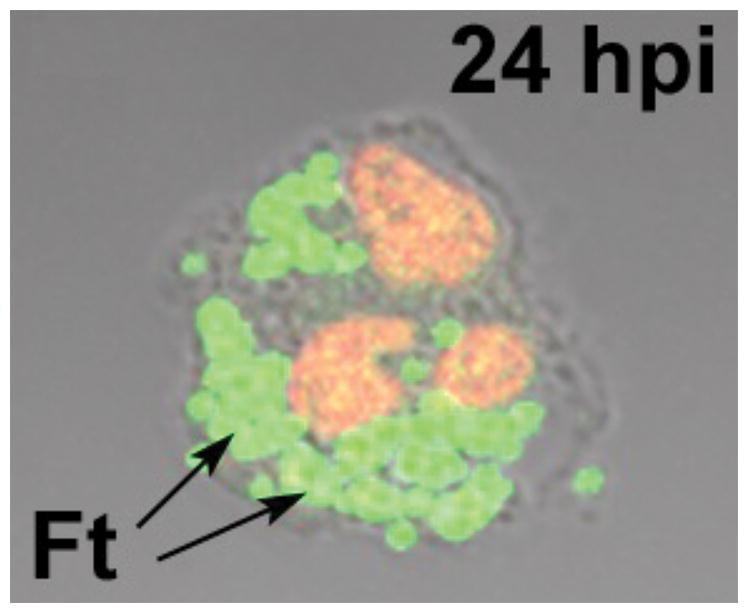
The light microcopy image shows a human neutrophil at 24 h post-infection (hpi) with F. tularensis (Ft). DNA in the PMN nucleus was stained red using TOPRO-3. The arrow indicates clusters of green fluorescent protein-expressing bacteria replicating in the cytosol.
Inhibition of neutrophil apoptosis
As critical effector cells of the innate immune system neutrophils are rapidly recruited to sites of infection, and under normal circumstances phagocytosed microbes are efficiently killed and degraded. This, in turn, accelerates PMN apoptosis via the PICD pathway and promotes clearance of dying cells by macrophages. During this process neutrophil proinflammatory capacity is downregulated, release of cytotoxic cell content is prevented, and macrophages are reprogrammed to a pro-resolving phenotype that is required for restoration of tissue homeostasis (1, 6, 8, 145). Conversely, defects in PMN turnover and clearance are hallmarks of an ineffective and dysregulated inflammatory response that favors PMN progression to secondary necrosis and markedly enhances tissue destruction, granuloma formation, and disease progression, particularly in the lungs (12, 13, 146–148). Summarized here are recent data which demonstrate that F. tularensis strains Schu S4 and LVS inhibit PMN apoptosis and prolong cell lifespan, findings that demonstrate additional mechanisms of innate immune evasion by this pathogen, and at the same time begin to define molecular molecular mechanisms to account for the profound neutrophil accumulation and necrotic tissue damage that is characteristic of pneumonic tularemia (20, 36, 142).
There are distinct biochemical and morphological changes that occur in all cells undergoing apoptosis that include externalization of plasma membrane phosphatidylserine (PS), membrane blebbing, nuclear condensation, and DNA fragmentation. These processes are significantly diminished and delayed in human neutrophils by F. tularensis, and activation of caspase-8, and caspase-9, and caspase-3 is impaired (36). Inhibition of apoptosis is dose-dependent and specific for live bacteria, and the vast majority of neutrophils remain viable until at least 48 h post-infection. Importantly, our data also show that F. tularensis not only fails to induce PICD, but also extends cell lifespan relative to aged, uninfected, controls, indicating that constitutive neutrophil apoptosis is impaired (36).
Mitochondria play an important role in regulation of PMN viability via control of the intrinsic apoptosis pathway, which mediates both constitutive and phagocytosis-induced PMN death. In healthy cells mitochondria are intact, whereas disruption of the outer mitochondrial membrane (OMM) is a very early event that may irreversibly commit cells to death (6, 146, 149). Loss of OMM integrity dissipates membrane potential and releases pro-apoptotic inner membrane space (IMS) proteins, including cytochrome c, into the cytosol. Liberated cytochrome c, in turn, initiates caspase-9 activation via apoptosome formation. At the molecular level OMM disruption is mediated by the pro-apoptotic protein BAX, which translocates to mitochondria from the cytosol, oligomerizes, and via its activation by tBID, inserts into the OMM, generating pores that mediate IMS protein release (as illustrated in Figure 3). Our data demonstrate that mitochondrial membrane potential and organelle integrity are sustained by LVS, BAX translocation to mitochondria is delayed, and conversion of BID to tBID is curtailed (150). Thus, mitochondrial stabilization is one mechanism used by F. tularensis to extend PMN lifespan, and this is due to downregulation of BAX mRNA and protein as well as defects in BID processing (142, 150).
Fig. 3. Mechanisms that contribute to PMN apoptosis inhibition during F. tularensis infection.

The extrinsic, phagocytosis-induced, and intrinsic pathways are shown. F. tularensis inhibits extrinsic pathway activation triggered by Fas crosslinking, whereas blockade of NADPH oxidase activity prevents cathepsin D-mediated caspase-8 activation during PICD. At the same time, Bax translocation to mitochondria and tBID-mediated Bax activation are impaired. Sustained mitochondrial integrity prevents intrinsic pathway activation triggered by intermembrane space (IMS) protein release and apoptosome formation. Calpastatin also contributes to caspase-9 and caspase-3 inhibition by preventing calpain-mediated degradation of XIAP. Effects of F. tularensis on expression of genes that contribute to apoptosis regulation is also depicted. Please see the text for additional details.
Under normal circumstances phagocytosis accelerates PMN death in a dose-dependent manner (5, 8, 9), and the established hallmarks of PICD are upregulation of BAX and oxidant-driven activation of caspase-8 (8, 12, 151–154). Under these conditions, increased BAX abundance favors mitochondrial permeabilization and intrinsic pathway activation, whereas the extrinsic pathway is activated by mechanisms that are independent of surface death receptor ligation, and in this case caspase-8 is activated by cathepsin D that is released into the cytosol from AG that have been damaged by NADPH oxidase-derived ROS (153, 154). Similarly, the ability of Pseudomonas aeruginosa to trigger very rapid PMN apoptosis is cathepsin D-dependent, and during this process oxidants and bacterial pyocyanin act in concert to induce AG damage (155). Thus, the ability of F. tularensis to downregulation of BAX and prevent NADPH oxidase-driven AG damage likely accounts for the fact that PICD is not induced, and in keeping with this caspase-8 activation is nearly ablated (36).
During both constitutive and phagocytosis-induced PMN death apoptosis is controlled by global changes in gene expression that extend beyond upregulation of BAX and together comprise an ‘apoptosis differentiation program’ (7–9). We have shown that F. tularensis LVS alters the PMN transcriptome during the first 24 h of infection, significantly influencing the expression of nearly 3,500 PMN genes, including 365 distinct genes linked to apoptosis and survival (142). Among these are several genes that antagonize the extrinsic pathway including CFLAR (which encodes FLIP), SOD2, TNFAIP3 (which encodes A20), and TNFRSF10. NF-κB pro-survival signaling is also important for PMN viability, and we find that NF-κB transcription factors (NFKB1, NFKB2, and RELA) as well as several anti-apoptotic target genes are rapidly induced. Among these, GADD45β is of interest as it can counteract FAS-stimulated cell death (156). Also upregulated are BIRC4 and BIRC3, which encode the caspase-inhibitors XIAP and cIAP-2, respectively. XIAP binds directly to caspase-9 and caspase-3 and is the most potent caspase inhibitor in PMN cytosol (151). During apoptosis XIAP is degraded by calpain. However, F. tularensis prevents XIAP degradation by upregulating CAST, which encodes the endogenous calpain inhibitor, calpastatin (142, 150). The effects of F. tularensis on PMN apoptosis pathways are summarized in Figure 3.
It is also worth noting that F. tularensis has atypical effects on some PMN survival factors including Mcl-1 and proliferating cell nuclear antigen (PCNA) (142, 150, 152). Mcl-1 is a short-lived protein that binds directly to BAX and prevents its ability to form pores in the OMM (146, 152). Continued synthesis of Mcl-1 is required for PMN survival, and up-regulation of MCL1 contributes to the ability of GM-CSF and pathogens such as A. phagocytophilum to extend neutrophil lifespan (146, 152). In contrast, MCL1 expression is slightly but significantly reduced by F. tularensis, but despite this Mcl-1 protein levels are sustained by an unknown mechanism (142, 150). PCNA is a scaffolding protein that resides in the nucleus of most cell types but is exclusively cytosolic in mature human neutrophils (152, 157). In this locale PCNA binds to procaspase-9 and procaspase-3 and impairs their activation. As PCNA is upregulated by G-CSF and disappears during apoptosis, it has been suggested that PCNA may also be essential for neutrophil survival (152, 157). However, PCNA expression is progressively downregulated beginning 6 h after infection with F. tularensis, and PCNA protein disappears rapidly and selectively from infected neutrophils by 18 h after infection, despite sustained cell viability (142, 150). What accounts for this is uncertain, but loss of PCNA correlates directly with F. tularensis-mediated induction of p21Waf1 (142), and peptides derived from p21Waf1 can displace PCNA from procaspase-9, leading to its degradation by the proteasome (152, 157). These data demonstrate that PCNA is not required for neutrophil survival in the context of F. tularensis infection, and as such underscore the ability of bacterial pathogens to inform studies of fundamental biological processes, including apoptosis.
Manipulation of neutrophil function and phenotype does not require direct infection
F. tularensis can markedly delay PMN apoptosis at very low doses and when only a subset of cells in the population are infected (36). Separation of cells and bacteria using a Transwell provided direct evidence that LVS can act at a distance to influence PMN function, and suggested a role for secreted or shed factors in apoptosis inhibition (36). In addition, Moreland et al., demonstrated that human neutrophils will traverse polarized monolayers of human pulmonary microvascular endothelial cells in response to LVS, and in this process acquire an altered phenotype that impairs their ability to produce ROS or secrete elastase when exposed to OpZ or other stimuli (71). Moreover, although endothelial cells were infected with LVS under the conditions of this study, the PMNs were not. Based on these data, we propose a model in which F. tularensis acts at a distance to influence neutrophil function and phenotype as these cells enter infected tissues at early stages of infection, and that manipulation of neutrophil function is subsequently reinforced by mechanisms linked to phagocytosis and intracellular growth. These data are consistent with the notion that macrophages and PMNs play distinct roles in tularemia; with avid replication in macrophages promoting F. tularensis dissemination, and manipulation of neutrophil activation state and lifespan driving tissue necrosis and granuloma formation. As apoptosis inhibition and phagocytosis can be uncoupled, the data also distinguish F. tularensis from obligate intracellular pathogens, such as A. phagocytophilum and Chlamydia pneumoniae, that must delay PMN apoptosis to sustain viability of their replicative niche (146).
Potential for therapeutic intervention
Challenges in treatment of pneumonic tularemia patients include the nonspecific nature of the presenting symptoms and the potentially short interval between the onset of symptom and severe illness or death. For this reason there is interest in the development of adjunctive treatment strategies that can be used in combination with antibiotics to enhance control of this potentially lethal infection (158). Cyclin-dependent kinases (CDKs) control the growth of most cell types and are essential for PMN survival (152). R-roscovitine is a CDK inhibitor and apoptosis-inducing agent that is currently in clinical trials and was developed for the treatment of cancer (158). Recent studies have shown that R-roscovitine also induces apoptosis in neutrophils, and in this manner reverses the pro-survival effects of GM-CSF, promotes resolution of inflammatory disorders that are characterized by aberrant PMN accumulation including carrageenan-induced pleurisy, bleomycin-induced lung injury and arthritis, (159, 160), and also synergizes with antibiotics for treatment of pneumococcal meningitis (161). We confirmed the ability of R-roscovitine to induce human neutrophil apoptosis, yet also found that its efficacy is undermined by F. tularensis LVS, which correlates directly with LVS-mediated upregulation of CDK2 and CDK7 (142, 150). Whether other PMN-targeted strategies may be beneficial for treatment of tularemia is unknown, but another candidate of interest is Roflumilast, which has shown promise in pilot studies of persons with COPD and blocks PMN migration into the lungs via direct effects on the MMP-9/PGP pathway (162).
Summary and conclusions
F. tularensis is a facultative intracellular pathogen capable of causing severe disease at low infectious doses. The pathogenicity of this organism is dependent on its ability to evade innate immune defenses and infect a number of cell types, including phagocytes and epithelial cells. In the past few years our understanding of neutrophil-F. tularensis interactions has increased dramatically. Neutrophils are recruited to the F. tularensis-infected lung by an MMP-9 and PGP-dependent pathway (73). LPS and capsule play multiple roles in virulence, conferring serum resistance and also mediating a specific mechanism of complement opsonization that promotes infection of macrophages as well as neutrophils, but does not elicit TLR4-mediated proinflammatory responses (32, 43, 91, 99). A fundamental characteristic of F. tularensis is its ability to disrupt neutrophil host defense mechanisms, including NAPDH oxidase-derived ROS production, and this is achieved not merely by controlling the composition of its own phagosome, but also by rapidly altering global cell responsiveness (53, 71, 100), and it is attractive to predict that additional studies of post-assembly NADPH oxidase inhibition during F. tularensis infection may provide fundamental insight into mechanisms that control termination of the respiratory burst. Early evasion of oxidative defense mechanisms and phagosome-granule fusion is followed by phagosome escape and bacterial replication in PMN cytosol, as well as initiation of profound changes in gene expression that inhibit apoptosis and markedly extend PMN lifespan (36, 100, 142, 150). Our understanding of the bacterial factors that control this complex mechanism of virulence are only beginning to be defined. Finally, F. tularensis also reduces the efficacy of the apoptosis-inducing drug R-roscovitine, and infected neutrophils may therefore provide a model system for future studies of CDKs and their role in PMN survival (150).
Since the earliest studies of F. tularensis in the 1920s it has been apparent that extensive neutrophil accumulation and necrotic damage are defining features of tissues infected with this pathogen (20). Although our understanding of F. tularensis-PMN interactions has increased significantly, many important questions remain unanswered. Major challenges include identification and characterization of the virulence factors that curtail apoptosis and disrupt neutrophil defense mechanisms, both before and after phagocytosis. As the inflammatory response is aberrantly regulated during tularemia, studies of neutrophil immunomodulatory properties and interactions with other leukocytes are also warranted.
Acknowledgments
This work was supported in part by a Department of Veteran’s Affairs Merit Review Grant 5 I01 BX002108 and funds from the NIH (NIAID P01 AI044642) awarded to L.-A.H. Allen. L.C.K. is supported by an NIH/NIAID predoctoral fellowship via T32 AI007511.
References
- 1.Kennedy A, DeLeo F. Neutrophil apoptosis and the resolution of infection. Immunol Res. 2009;43:25–61. doi: 10.1007/s12026-008-8049-6. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 3.Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Kleer I, Willems F, Lambrecht B, Goriely S. Ontogeny of myeloid cells. Front Immunol. 2014;5:423. doi: 10.3389/fimmu.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;156:3986–3992. [PubMed] [Google Scholar]
- 6.Kobayashi SD, DeLeo FR. Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med. 2009;1:309–333. doi: 10.1002/wsbm.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi SD, et al. An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J Leukoc Biol. 2003;73:315–322. doi: 10.1189/jlb.1002481. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi SD, et al. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci, USA. 2003;100:10948–10953. doi: 10.1073/pnas.1833375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi SD, Voyich JM, Buhl CL, Stahl RM, DeLeo FR. Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: Cell fate is regulated at the level of gene expression. Proc Natl Acad Sci U S A. 2002;99:6901–6906. doi: 10.1073/pnas.092148299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun. 2010;2:216–227. doi: 10.1159/000284367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kobayashi SD, Voyich JM, Braughton KR, DeLeo FR. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. 2003;170:3357–3368. doi: 10.4049/jimmunol.170.6.3357. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi SD, et al. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol. 2004;172:636–643. doi: 10.4049/jimmunol.172.1.636. [DOI] [PubMed] [Google Scholar]
- 13.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 14.Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519–531. doi: 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- 15.Jaillon S, Galdiero MR, Del Prete D, Cassatella MA, Garlanda C, Mantovani A. Neutrophils in innate and adaptive immunity. Semin Immunopathol. 2013;35:377–394. doi: 10.1007/s00281-013-0374-8. [DOI] [PubMed] [Google Scholar]
- 16.Pillay J, et al. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J Clin Invest. 2012;122:327–336. doi: 10.1172/JCI57990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoy GW, Chapin CW. A plague-like disease or rodents. Public Health Bull. 1911;43:53–71. [Google Scholar]
- 18.McCoy GW, Chapin CW. Bacterium tularense, the cause of a plaguelike disease of rodents. Public Health Bull. 1912;53:17–23. [Google Scholar]
- 19.Wherry WB, Lamb BH. Infection of man with Bacterium tularense. J Infect Dis. 1914;15:331–340. doi: 10.1093/infdis/189.7.1321. [DOI] [PubMed] [Google Scholar]
- 20.Francis E. History of tularaemia. In: Simon CE, editor. The Damar Lectures, 1927–1928 of the School of Hygiene and Public Health, Johns Hopkins University. Batimore, MD: Williams and Wilkins; 1928. pp. 411–432. [Google Scholar]
- 21.Hestvik G, et al. The status of tularemia in Europe in a one-health context: a review. Epidemiol Infect. 2015;143:2137–2160. doi: 10.1017/S0950268814002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morner T. The ecology of tularaemia. Rev Sci Tech. 1992;11:1123–1130. [PubMed] [Google Scholar]
- 23.Francis E. Tularemia. JAMA. 1925;84:1243–1250. [Google Scholar]
- 24.Olsufiev NG, Emelyanova OS, Dunaeva TN. Comparative study of strains of B. tularense in the old and new world and their taxomony. J Hyg Epidemiol Microbiol Immunol. 1959;3:138–149. [PubMed] [Google Scholar]
- 25.Sandstrom G, Sjosted A, Forsman M, Pavlovich NV, Mishankin BN. Characterization and classification of strans of Francisella tularensis isolated in the central Asian focus of the Soviet Union and Japan. J Clin Microbiol. 1992;30:172–175. doi: 10.1128/jcm.30.1.172-175.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keim P, Johansson A, Wagner DM. Molecular Epidemiology, Evolution, and Ecology of Francisella. Ann NY Acad Sci. 2007;1105:30–66. doi: 10.1196/annals.1409.011. [DOI] [PubMed] [Google Scholar]
- 27.Sjostedt A. Tularemia: History, Epidemiology, Pathogen Physiology, and Clinical Manifestations. Ann NY Acad Sci. 2007;1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- 28.Saslaw S, Eigelsbach HT, Wilson HE, Prior JA, Carhart S. Tularemia vaccine study. I. Intracutaneous challenge. Arch Intern Med. 1961;107:689–701. doi: 10.1001/archinte.1961.03620050055006. [DOI] [PubMed] [Google Scholar]
- 29.Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. Tularemia vaccine study. II. Respiratory challenge. Arch Intern Med. 1961;107:702–714. doi: 10.1001/archinte.1961.03620050068007. [DOI] [PubMed] [Google Scholar]
- 30.McCrumb FR. Aerosol infection of man with Pasturella tularensis. Bacteriol Rev. 1961;25:262–267. doi: 10.1128/br.25.3.262-267.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dennis DT, et al. Tularemia as a biological weapon: medical and public health management. JAMA. 2001;285:2763–2773. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 32.McLendon MK, Apicella M, Allen L-AH. Francisella tularensis: taxomony, genetics and immunopathogenesis of a potenial agent of biowarfare. Annu Rev Microbiol. 2006;60:167–185. doi: 10.1146/annurev.micro.60.080805.142126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oyston PCF, Sjostedt A, Titball RW. Tularemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 34.Anthony LD, Burke RD, Nano FE. Growth of Francisella spp. in rodent macrophages. Infect Immun. 1991;59:3291–3296. doi: 10.1128/iai.59.9.3291-3296.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindemann SR, McLendon MK, Apicella MA, Jones BD. An In Vitro Model System Used To Study Adherence and Invasion of Francisella tularensis Live Vaccine Strain in Nonphagocytic Cells. Infect Immun. 2007;75:3178–3182. doi: 10.1128/IAI.01811-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz JT, Barker JH, Kaufman J, Fayram DC, McCracken JM, Allen L-AH. Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J Immmunol. 2012;188:3351–3363. doi: 10.4049/jimmunol.1102863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faron M, Fletcher JR, Rasmussen JA, Apicella MA, Jones BD. Interactions of Francisella tularensis with alveolar type II epithelial cells and the murine respiratory epithelium. PLoS ONE. 2015;10:e0127458. doi: 10.1371/journal.pone.0127458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandstrom G, Lofgren S, Tarnvik A. A capsule-deficient mutant of Francisella tularensis LVS exhibits enhanced sensitivity to killing by serum but diminished sensitivity to killing by polymorphonuclear leukocytes. Infect Immun. 1988;56:1194–1202. doi: 10.1128/iai.56.5.1194-1202.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Apicella MA, et al. Identification, characterization and immunogenicity of an O-antigen capsular polysaccharide of Francisella tularensis. PLoS ONE. 2010;5:e11060. doi: 10.1371/journal.pone.0011060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindemann SR, et al. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect Immun. 2011;79:581–594. doi: 10.1128/IAI.00863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen JA, et al. Francisella tularensis Schu S4 Lipopolysaccharide Core Sugar and O-Antigen Mutants Are Attenuated in a Mouse Model of Tularemia. Infect Immun. 2014;82:1523–1539. doi: 10.1128/IAI.01640-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rasmussen JA, Fletcher JR, Long ME, Allen LA, Jones BD. Characterization of Francisella tularensis Schu S4 mutants identified from a transposon library screened for O-antigen and capsule deficiencies. Front Microbiol. 2015;6:338. doi: 10.3389/fmicb.2015.00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barker JH, Weiss J, Apicella MA, Nauseef WM. Basis for the Failure of Francisella tularensis Lipopolysaccharide To Prime Human Polymorphonuclear Leukocytes. Infect Immun. 2006;74:3277–3284. doi: 10.1128/IAI.02011-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Teghanemt A, Zhang D, Levis EN, Weiss JP, Gioannini TL. Molecular Basis of Reduced Potency of Underacylated Endotoxins. J Immmunol. 2005;175:4669–4676. doi: 10.4049/jimmunol.175.7.4669. [DOI] [PubMed] [Google Scholar]
- 45.Bandyopadhyay S, Long ME, Allen L-AH. Differential Expression of microRNAs in Francisella tularensis-Infected Human Macrophages: miR-155-Dependent Downregulation of MyD88 Inhibits the Inflammatory Response. PLoS One. 2014;9:e109525. doi: 10.1371/journal.pone.0109525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Case ED, et al. The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell Microbiol. 2014;16:862–877. doi: 10.1111/cmi.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nano FE, et al. A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol. 2004;186:6430–6436. doi: 10.1128/JB.186.19.6430-6436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broms JE, Sjostedt A, Lavander M. The Role of the Francisella Tularensis Pathogenicity Island in Type VI Secretion, Intracellular Survival, and Modulation of Host Cell Signaling. Front Microbiol. 2010;1:136. doi: 10.3389/fmicb.2010.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Long ME, Lindemann SR, Rasmussen JA, Jones BD, Allen L-AH. Disruption of Francisella tularensis Schu S4 iglI, iglJ, and pdpC Genes Results in Attenuation for Growth in Human Macrophages and In vivo Virulence in Mice and Reveals a Unique Phenotype for pdpC. Infect Immun. 2013;81:850–861. doi: 10.1128/IAI.00822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brotcke A, Monack DM. Identification of fevR, a Novel Regulator of Virulence Gene Expression in Francisella novicida. Infect Immun. 2008;76:3473–3480. doi: 10.1128/IAI.00430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buchan BW, McCaffrey RL, Lindemann SR, Allen L-AH, Jones BD. Identification of migR, a regulatory element of the Francisella tularensis Live Vaccine Strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect Immun. 2009;77:2517–2529. doi: 10.1128/IAI.00229-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meibom KL, Barel M, Charbit A. Loops and networks in control of Francisella tularensis virulence. Future Microbiol. 2009;4:713–729. doi: 10.2217/fmb.09.37. [DOI] [PubMed] [Google Scholar]
- 53.McCaffrey RL, et al. Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. J Leukoc Biol. 2010;88:791–805. doi: 10.1189/jlb.1209811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gil H, et al. Deletion of TolC orthologues in Francisella tularensis identified roles in drug resistance and virulence. Proc Natl Acad Sci U S A. 2006;103:12897–12902. doi: 10.1073/pnas.0602582103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tulis JJ, Eigelsbach HT, Kerpsack RW. Host-parasite relationship in Monkeys administered live tularemia vaccine. Am J Pathol. 1970;58:329–336. [PMC free article] [PubMed] [Google Scholar]
- 56.Schricker RL, Eigelsbach HT, Mitten JO, Hall WC. Pathogenesis of tularemia in monkeys aerogenically exposed to Francisella tularensis 425. Infect Immun. 1972;5:734–744. doi: 10.1128/iai.5.5.734-744.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall WC, Kovatch RM, Schricker RL. Tularemic pneumonia: pathogenesis of the aerosol-induced disease in monkeys. J Pathol. 1973;110:193–201. doi: 10.1002/path.1711100302. [DOI] [PubMed] [Google Scholar]
- 58.Dunaeva TN, Shlygina KN. Phagocytic activity of the neutrophils in tularemia in animals with varying infective sensitivity. Zh Mikrobiol Epidomiol Immunobiol. 1975;10:22–26. [PubMed] [Google Scholar]
- 59.Eigelsbach HT, Tulis JJ, McGavran MH, White JD. Live tularemia vaccine I. Host parastie relationship in monkeys vaccinated intracutaneously or aerogenically. J Bacteriol. 1962;84:1020–1027. doi: 10.1128/jb.84.5.1020-1027.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Conlan JW, Chen W, Shen H, Webb A, KuoLee R. Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb Pathog. 2003;34:239. doi: 10.1016/s0882-4010(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 61.Lamps LW, Havens JM, Sjostedt A, Page DL, Scott MA. Histologic and molecular diagnosis of tularemia: a potential bioterrorism agent endemic to North America. Mod Pathol. 2004;17:489–495. doi: 10.1038/modpathol.3800087. [DOI] [PubMed] [Google Scholar]
- 62.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active Suppression of the Pulmonary Immune Response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 63.Hall JD, et al. Infected-Host-Cell Repertoire and Cellular Response in the Lung following Inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roberts LM, et al. Identification of early interactions between Francisella and the host. Infect Immun. 2014;82:2504–2510. doi: 10.1128/IAI.01654-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil Recruitment to the Lungs during Bacterial Pneumonia. Infect Immun. 2009;77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. Mechanisms of Neutrophil Accumulation in the Lungs Against Bacteria. Am J Respir Cell Mol Biol. 2010;43:5–16. doi: 10.1165/rcmb.2009-0047TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sadik CD, Kim ND, Iwakura Y, Luster AD. Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcγR signaling. Proc Natl Acad Sci U S A. 2012;109:E3177–E3185. doi: 10.1073/pnas.1213797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fullerton JN, O’Brien AJ, Gilroy DW. Lipid mediators in immune dysfunction after severe inflammation. Trends Immunol. 2013 doi: 10.1016/j.it.2013.10.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weathington NM, et al. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 70.Moreland JG, Bailey G, Nauseef WM, Weiss JP. Organism-Specific Neutrophil-Endothelial Cell Interactions in Response to Escherichia coli, Streptococcus pneumoniae, and Staphylococcus aureus. J Immunol. 2004;172:426–432. doi: 10.4049/jimmunol.172.1.426. [DOI] [PubMed] [Google Scholar]
- 71.Moreland JG, Hook JS, Bailey G, Ulland T, Nauseef WM. Francisella tularensis directly interacts with the endothelium and recruits neutrophils with a blunted inflammatory phenotype. Am J Physiol Lung Cell Mol Physiol. 2009;296:L1076–L1084. doi: 10.1152/ajplung.90332.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gentry M, et al. Role of Primary Human Alveolar Epithelial Cells in Host Defense against Francisella tularensis Infection. Infect Immun. 2007;75:3969–3978. doi: 10.1128/IAI.00157-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malik M, et al. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J Immunol. 2007;178:1013–1020. doi: 10.4049/jimmunol.178.2.1013. [DOI] [PubMed] [Google Scholar]
- 74.Snelgrove RJ, et al. A Critical Role for LTA4H in Limiting Chronic Pulmonary Neutrophilic Inflammation. Science. 2010;330:90–94. doi: 10.1126/science.1190594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andrews T, Sullivan KE. Infections in Patients with Inherited Defects in Phagocytic Function. Clin Micrbiol Rev. 2003;16:597–621. doi: 10.1128/CMR.16.4.597-621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 77.Dunay IR, Fuchs A, Sibley LD. Inflammatory Monocytes but Not Neutrophils Are Necessary To Control Infection with Toxoplasma gondii in Mice. Infect Immun. 2010;78:1564–1570. doi: 10.1128/IAI.00472-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wojtasiak M, et al. Depletion of Gr-1+, but not Ly6G+, immune cells exacerbates virus replication and disease in an intranasal model of herpes simplex virus type 1 infection. J Gen Virol. 2010;91:2158–2166. doi: 10.1099/vir.0.021915-0. [DOI] [PubMed] [Google Scholar]
- 79.Carr KD, Sieve AN, Indramohan M, Break TJ, Lee S, Berg RE. Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur J Immunol. 2011;41:2666–2676. doi: 10.1002/eji.201041363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sjostedt A, Conlan JW, North RJ. Neutrophils are critical for host defense against primary infection with the facultative intracellular bacterium Francisella tularensis in mice and participate in defense against reinfection. Infect Immun. 1994;62:2779–2783. doi: 10.1128/iai.62.7.2779-2783.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.KuoLee R, Harris G, Conlan JW, Chen W. Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect. 2011;13:447–456. doi: 10.1016/j.micinf.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 82.Bosio CM, Elkins KL. Susceptibility to Secondary Francisella tularensis Live Vaccine Strain Infection in B-Cell-Deficient Mice Is Associated with Neutrophilia but Not with Defects in Specific T-Cell-Mediated Immunity. Infect Immun. 2001;69:194–203. doi: 10.1128/IAI.69.1.194-203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Melillo AA, Foreman O, Elkins KL. IL-12Rβ2 is critical for survival of primary Francisella tularensis LVS infection. J Leukoc Biol. 2013;93:657–667. doi: 10.1189/jlb.1012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lindgren H, Stenman L, Tarnvik A, Sjostedt A. The contribution of reactive nitrogen and oxygen species to the killing of Francisella tularensis LVS by murine macrophages. Microbes Infect. 2005;7:467–475. doi: 10.1016/j.micinf.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 85.Lindgren H, Stenmark S, Chen W, Tarnvik A, Sjostedt A. Distinct Roles of Reactive Nitrogen and Oxygen Species To Control Infection with the Facultative Intracellular Bacterium Francisella tularensis. Infect Immun. 2004;72:7172–7182. doi: 10.1128/IAI.72.12.7172-7182.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lofgren S, Tarnvik A, Carlsson J. Influence of complement on the chemiluminescent response of human leukocytes to immune complex. Infect Immun. 1980;29:335–341. doi: 10.1128/iai.29.2.335-341.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lofgren S, Tarnvik A, Carlsson J. Demonstration of opsonizing antibodies to Francisella tularensis by leukocyte chemiluminescence. Infect Immun. 1980;29:329–334. doi: 10.1128/iai.29.2.329-334.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schulert GS, Allen L-AH. Differential infection of mononuclear phagoctyes by Francisella tularensis: role of the macrophage mannose receptor. J Leukoc Biol. 2006;80:563–571. doi: 10.1189/jlb.0306219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ben Nasr A, Klimpel GR. Subversion of complement activation at the bacterial surface promotes serum resistance and opsonophagocytosis of Francisella tularensis. J Leukoc Biol. 2008;84:77–85. doi: 10.1189/jlb.0807526. [DOI] [PubMed] [Google Scholar]
- 90.Clay CD, Soni S, Gunn JS, Schlesinger LS. Evasion of complement-mediated Lysis and complement C3 deposition are regulated by Francisella tularensis lipopolysaccharide O-antigen. J Immunol. 2008;181:5568–5578. doi: 10.4049/jimmunol.181.8.5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwartz JT, Barker JH, Long ME, Kaufman J, McCracken J, Allen L-AH. Natural IgM Mediates Complement-Dependent Uptake of Francisella tularensis by Human Neutrophils via Complement Receptors 1 and 3 in Nonimmune Serum. J Immunol. 2012;189:3064–3077. doi: 10.4049/jimmunol.1200816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barker JH, McCaffrey RL, Baman NK, Allen LA, Weiss JP, Nauseef WM. The role of complement opsonization in interactions between F. tularensis subsp. novicida and human neutrophils. Microbes Infect. 2009;11:762–769. doi: 10.1016/j.micinf.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sorokin VM, Pavlovich NV, Prozorova LA. Francisella tularensis resistance to bactericidal action of normal human serum. FEMS Immunol Med Microbiol. 1996;13:249–252. doi: 10.1111/j.1574-695X.1996.tb00246.x. [DOI] [PubMed] [Google Scholar]
- 94.Raynaud C, et al. Role of the wbt locus of Francisella tularensis in lipopolysaccharide O-antigen biogenesis and pathogenicity. Infect Immun. 2007;75:536–541. doi: 10.1128/IAI.01429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pierini LM. Uptake of serum-opsonized Francisella tularensis by macrophages can be mediated by class A scavenger receptors. Cell Microbiol. 2006;8:1361–1370. doi: 10.1111/j.1462-5822.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 96.Ben Nasr A, Haithcoat J, Masterson JE, Gunn JS, Eaves-Pyles T, Klimpel GR. Critical role for serum opsonins and complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in phagocytosis of Francisella tularensis by human dendritic cells (DC): uptake of Francisella leads to activation of immature DC and intracellular survival of the bacteria. J Leukoc Biol. 2006;80:774–786. doi: 10.1189/jlb.1205755. [DOI] [PubMed] [Google Scholar]
- 97.Barel M, Hovanessian AG, Meibom K, Briand JP, Dupuis M, Charbit A. A novel receptor-ligand pathway for entry of Francisella tularensis into monocyte-like THP-1 cells: interaction between surface nucleolin and bacterial elongation factor Tu. BMC Microbiol. 2008;8:145. doi: 10.1186/1471-2180-8-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 99.Dai S, Rajaram MVS, Curry HM, Leander R, Schlesinger LS. Fine Tuning Inflammation at the Front Door: Macrophage Complement Receptor 3-mediates Phagocytosis and Immune Suppression for Francisella tularensis. PLoS Pathog. 2013;9:e1003114. doi: 10.1371/journal.ppat.1003114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McCaffrey RL, Allen L-AH. Pivotal Advance: Francisella tularensis evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol. 2006;80:1224–1230. doi: 10.1189/jlb.0406287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schulert GS, et al. Francisella tularensis Genes Required for Inhibition of the Neutrophil Respiratory Burst and Intramacrophage Growth Identified by Random Transposon Mutagenesis of Strain LVS. Infect Immun. 2009;77:1324–1336. doi: 10.1128/IAI.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Geier H, Celli J. Phagocytic receptors dictate phagosomal escape and intracellular proliferation of Francisella tularensis. Infect Immun. 2011;79:2204–2214. doi: 10.1128/IAI.01382-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 104.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 105.Curnutte JT. Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol. 1993;67:S2–S15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- 106.Allen L-AH, DeLeo FR, Gallois A, Toyoshima S, Suzuki K, Nauseef WM. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood. 1999;93:3521–3530. [PubMed] [Google Scholar]
- 107.DeLeo FR, Allen L-AH, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163:6732–6740. [PubMed] [Google Scholar]
- 108.Allen L-AH, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol. 2005;174:3658–3667. doi: 10.4049/jimmunol.174.6.3658. [DOI] [PubMed] [Google Scholar]
- 109.Dahlgren C, Karlsson A. Respiratory burst in human neutrophils. J Immmunol Methods. 1999;232:3–14. doi: 10.1016/s0022-1759(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 110.Kettle AJ, Anderson RF, Hampton MB, Winterbourn CC. Reactions of superoxide with myeloperoxidase. Biochemistry. 2007;46:4888–4897. doi: 10.1021/bi602587k. [DOI] [PubMed] [Google Scholar]
- 111.Kettle AJ, Maroz A, Woodroffe G, Winterbourn CC, Anderson RF. Spectral and kinetic evidence for reaction of superoxide with compound I of myeloperoxidase. Free Radic Biol Med. 2011;51:2190–2194. doi: 10.1016/j.freeradbiomed.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 112.Decoursey TE, Ligeti E. Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci. 2005;62:2173–2193. doi: 10.1007/s00018-005-5177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jandl RC, Andre-Schwartz J, Borges-DuBois L, Kipnes RS, McMurrich BJ, Babior BM. Termination of the respiratory burst in human neutrophils. J Clin Invest. 1978;61:1176–1185. doi: 10.1172/JCI109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Matute JD, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Proctor RA, White JD, Ayala E, Canonico PG. Phagocytosis of Francisella tularensis by rhesus monkey peripheral leukocytes. Infect Immun. 1975;11:146–152. doi: 10.1128/iai.11.1.146-151.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ijdo JW, Mueller AC. Neutrophil NADPH oxidase is reduced at the Anaplasma phagocytophilum phagosome. Infect Immun. 2004;72:5392–5401. doi: 10.1128/IAI.72.9.5392-5401.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Carlyon JA, Abdel-Latif D, Pypaert M, Lacy P, Fikrig E. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect Immun. 2004;72:4772–4783. doi: 10.1128/IAI.72.8.4772-4783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Smirnov A, Daily KP, Criss AK. Assembly of NADPH Oxidase in Human Neutrophils Is Modulated by the Opacity-Associated Protein Expression State of Neisseria gonorrhoeae. Infect Immun. 2014;82:1036–1044. doi: 10.1128/IAI.00881-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Siemsen DW, Kirpotina LN, Jutila MA, Quinn MT. Inhibition of the human neutrophil NADPH oxidase by Coxiella burnetii. Microbes Infect. 2009;11:671–679. doi: 10.1016/j.micinf.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Allen L-AH, McCaffrey RL. To activate or not to activate: distinct strategies used by Helicobacter pylori and Francisella tularensis to modulate the NADPH oxidase and survive in human neutrophils. Immunol Rev. 2007;219:103–117. doi: 10.1111/j.1600-065X.2007.00544.x. [DOI] [PubMed] [Google Scholar]
- 121.Heyworth PG, Badwey JA. Continuous phosphorylation of both the 47 and the 49 kDa proteins occurs during superoxide production by neutrophils. Biochem Biopys Acta. 1990;1052:299–305. doi: 10.1016/0167-4889(90)90225-3. [DOI] [PubMed] [Google Scholar]
- 122.Reilly TJ, Baron GS, Nano FE, Kuhlenschmidt MS. Characterization and sequencing of a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. J Biol Chem. 1996;271:10973–10983. doi: 10.1074/jbc.271.18.10973. [DOI] [PubMed] [Google Scholar]
- 123.Reilly TJ, Felts RL, Henzl MT, Calcutt MJ, Tanner JJ. Characterization of recombinant Francisella tularensis acid phosphatase A. Protein Expr Purif. 2006;45:132. doi: 10.1016/j.pep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 124.Dai S, Mohapatra NP, Schlesinger LS, Gunn JS. The Acid Phosphatase AcpA Is Secreted In Vitro and in Macrophages by Francisella spp. Infect Immun. 2012;80:1088–1097. doi: 10.1128/IAI.06245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Child R, Wehrly TD, Rockx-Brouwer D, Dorward DW, Celli J. Acid Phosphatases Do Not Contribute to the Pathogenesis of Type A Francisella tularensis. Infect Immun. 2010;78:59–67. doi: 10.1128/IAI.00965-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Baron GS, Reilly TJ, Nano FE. The respiratory burst-inhibiting acid phosphatase AcpA is not essential for the intramacrophage growth or virulence of Francisella novicida. FEMS Microbiol Lett. 1999;176:85–90. doi: 10.1111/j.1574-6968.1999.tb13646.x. [DOI] [PubMed] [Google Scholar]
- 127.Mohapatra NP, et al. Francisella Acid Phosphatases Inactivate the NADPH Oxidase in Human Phagocytes. J Immmunol. 2010;184:5141–5150. doi: 10.4049/jimmunol.0903413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ramond E, et al. Glutamate Utilization Couples Oxidative Stress Defense and the Tricarboxylic Acid Cycle in Francisella Phagosomal Escape. PLoS Pathog. 2014;10:e1003893. doi: 10.1371/journal.ppat.1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bakshi CS, et al. Superoxide Dismutase B Gene (sodB)-Deficient Mutants of Francisella tularensis Demonstrate Hypersensitivity to Oxidative Stress and Attenuated Virulence. J Bacteriol. 2006;188:6443–6448. doi: 10.1128/JB.00266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Avi-Dor Y, Yaniv H. The activity of catalase in Pasturella Tularensis. J Bacteriol. 1952;63:751–757. doi: 10.1128/jb.63.6.751-757.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lindgren H, Honn M, Salomonsson E, Kuoppa K, Forsberg Å, Sjöstedt A. Iron Content Differs between Francisella tularensis Subspecies tularensis and Subspecies holarctica Strains and Correlates to Their Susceptibility to H2O2-Induced Killing. Infect Immun. 2011;79:1218–1224. doi: 10.1128/IAI.01116-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lofgren S, Tarnvik A, Thore M, Carlsson J. A wild and an attenuated strain of Francisella tularensis differ in susceptibility to hypochlorous acid: a possible explanation of their different handling by polymorphonuclear leukocytes. Infect Immun. 1984;43:730–734. doi: 10.1128/iai.43.2.730-734.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hazlett KRO, et al. Adaptation of Francisella tularensis to the Mammalian Environment Is Governed by Cues Which Can Be Mimicked In Vitro. Infect Immun. 2008;76:4479–4488. doi: 10.1128/IAI.00610-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zarrella TM, et al. Host-Adaptation of Francisella tularensis Alters the Bacterium’s Surface-Carbohydrates to Hinder Effectors of Innate and Adaptive Immunity. PLoS ONE. 2011;6:e22335. doi: 10.1371/journal.pone.0022335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Clemens DL, Lee BY, Horwitz MA. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun. 2004;72:3204–3217. doi: 10.1128/IAI.72.6.3204-3217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Clemens DL, Lee B-Y, Horwitz MA. Francisella tularensis Phagosomal Escape Does Not Require Acidification of the Phagosome. Infect Immun. 2009;77:1757–1773. doi: 10.1128/IAI.01485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. From the Cover: Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjostedt A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun. 2003;71:5940–5950. doi: 10.1128/IAI.71.10.5940-5950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Clemens DL, Ge P, Lee BY, Horwitz MA, Zhou ZH. Atomic structure of T6SS reveals interlaced array essential to function. Cell. 2015;160:940–951. doi: 10.1016/j.cell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Craven RR, Hall JD, Fuller JR, Taft-Benz S, Kawula TH. Francisella tularensis Invasion of Lung Epithelial Cells. Infect Immun. 2008;76:2833–2842. doi: 10.1128/IAI.00043-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol. 2011;90:271–284. doi: 10.1189/jlb.0810457. [DOI] [PubMed] [Google Scholar]
- 142.Schwartz JT, et al. Francisella tualrensis alters human neutrophil gene expression: insights into the molecular basis of delayed neutrophil apoptosis. J Innate Immun. 2013;5:5124–5136. doi: 10.1159/000342430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Weinrauch Y, Drujan D, Shapiro SD, Weiss J, Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. 2002;417:91–94. doi: 10.1038/417091a. [DOI] [PubMed] [Google Scholar]
- 144.Mandic-Mulec I, Weiss J, Zychlinsky A. Shigella flexneri is trapped in polymorphonuclear leukocyte vacuoles and efficiently killed. Infect Immun. 1997;65:110–115. doi: 10.1128/iai.65.1.110-115.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Savill JS, Wuyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McCracken JM, Allen L-AH. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death. 2014;7:15–23. doi: 10.4137/JCD.S11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Resp Crit Care Med. 1999;160:S5–S11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 148.Fialkow L, et al. Neutrophil apoptosis: a marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit Care. 2006;10:R155. doi: 10.1186/cc5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11:143–153. doi: 10.1038/sj.cdd.4401320. [DOI] [PubMed] [Google Scholar]
- 150.McCracken JM, Kinkead LC, McCaffrey RL, Allen L-AH. Francisella tularensis modulates a dstinct set of regulatory factors and sustains mitochondrial integrity to impair human neutrophil apoptosis. J Innate Immun. 2016;8 doi: 10.1159/000443882. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell Death Differ. 2011;18:1457–1469. doi: 10.1038/cdd.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Witko-Sarsat V, Pederzoli-Ribeil M, Hirsh E, Sozzani S, Cassatella MA. Regulating neutrophil apoptosis: new players enter the game. Trends Immunol. 2011;32:117–124. doi: 10.1016/j.it.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 153.Conus S, et al. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med. 2008;205:685–698. doi: 10.1084/jem.20072152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Conus S, Pop C, Snipas SJ, Salvesen GS, Simon HU. Cathepsin D primes caspase-8 activation by multiple intra-chain proteolysis. J Biol Chem. 2012;287:21142–21151. doi: 10.1074/jbc.M111.306399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Prince LR, et al. Subversion of a Lysosomal Pathway Regulating Neutrophil Apoptosis by a Major Bacterial Toxin, Pyocyanin. J Immunol. 2008;180:3502–3511. doi: 10.4049/jimmunol.180.5.3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hoffman B, Liebermann DA. Gadd45 modulation of intrinsic and extrinsic stress responses in myeloid cells. J Cell Physiol. 2009;218:26–31. doi: 10.1002/jcp.21582. [DOI] [PubMed] [Google Scholar]
- 157.Witko-Sarsat V, et al. Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J Exp Med. 2010;207:2631–2645. doi: 10.1084/jem.20092241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Allen L-AH. Neutrophils: potential therapeutic targets in tularemia? Front Cell Infect Microbiol. 2013;3:109. doi: 10.3389/fcimb.2013.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rossi AG, et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 160.Leitch AE, Haslett C, Rossi AG. Cyclin-dependent kinase inhibitor drugs as potenital novel anti-inflammatory and pro-resolution agents. Br J Pharmacol. 2009;158:1004–1016. doi: 10.1111/j.1476-5381.2009.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Koedel U, et al. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental Pneumococcal meningitis. PLoS Pathog. 2009;5:e1000461. doi: 10.1371/journal.ppat.1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wells JM, et al. A Randomized, Placebo-controlled Trial of Roflumilast. Effect on Proline-Glycine-Proline and Neutrophilic Inflammation in Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med. 2015;192:934–942. doi: 10.1164/rccm.201503-0543OC. [DOI] [PMC free article] [PubMed] [Google Scholar]