

Abstract
The interaction of neutrophils (PMNs) and epithelial cells are requisite lines of communication during mucosal inflammatory responses. Consequences of such interactions often determine endpoint organ function, and for this reason, much interest has developed around defining the constituents of the tissue microenvironment of inflammatory lesions. Physiologic in vitro and in vivo models have aided in discovery of components that define the basic inflammatory machinery that mold the inflammatory tissue microenvironment. Here, we will review the recent literature related to the contribution of PMNs to molding of the tissue microenvironment, with an emphasis on the gastrointestinal (GI) tract. We focus on endogenous pathways for promoting tissue homeostasis and the molecular determinants of neutrophil-epithelial cell interactions during ongoing inflammation. These recent studies highlight the dynamic nature of these pathways and lend insight into the complexity of treating mucosal inflammation.
Keywords: metabolism, hypoxia-inducible factor, inflammation, nucleoside, nucleotidase, mucosa, colitis, epithelium
Introduction
The recruitment of neutrophils (polymorphonuclear leukocyte, PMN) to sites of tissue injury and infection has long been recognized as a hallmark of acute inflammation. It is increasingly appreciated that the presence of PMNs at sites of injury do not necessarily prove causation to tissue damage, and in fact, may provide clues to healing. The contribution(s) of PMN to the molding of the tissue microenvironment is an area of signifcant interest. These studies have shown that recruited PMN communicate with the surrounding parenchymal tissues in ways that fundamentally change the milieu to promote tissue restitution, wound healing and homeostasis. In this review, we will summarize recent literature related to the contribution of PMN the the tissue inflammatory microenvironment, with a particular focus on the GI mucosa.
The microenvironment of the intestinal mucosa
An especially unique feature of the GI microenvironment is the presence of large numbers of microbes on the lumenal surface. The mammalian GI tract harbors trillions of bacteria, where a finely balanced mutualism exists (1). Lined by epithelial cells, a primary function of the intestinal mucosa is to provide a selective barrier to the outside world. At these same surfaces exists the potential for infection by pathogenic organisms and the neccessity to control commensal microorganisms at homeostatic levels. In this regard, the tissue healing following injury occurs in conjunction with the constant flux of antigenic material and requires that the mucosal immune system appropriately dampen inflammatory and immunological reactions to harmless ingested antigens. The overlying epithelium plays an important role in coordinating both inflammation and wound healing. The epithelium lies juxtaposed to the mucosal immune system and lines the entire gastrointestinal tract. Covering a surface area of nearly 300 m2, the human adult intestinal epithelium consists of a monolayer of cells with intercellular tight junctions, a complex three dimensional structure and a thick mucous gel layer that provides a dynamic and regulated barrier to the flux of the luminal contents to the lamina propria (2, 3). It is widely understood that the gastrointestinal tract exists in a state of low-grade inflammation, resulting from the constant sampling and processing of luminal antigenic material during the development of oral tolerance and the priming of the mucosal immune system for rapid and effective responses to antigens or microbes that may penetrate the barrier (4).
The GI tract may be somewhat unique in the manner of handling PMN. For example, the depletion of circulating PMN using anti-Gr1 antibodies results in the exacerbation of symptoms in a number of different murine colitis models, strongly implicating PMN as a protective factor in ongoing inflammation (5). By contrast, the depletion of PMN in acute lung injury models appears to serve an anti-inflammatory role (6), although this idea has been revisited (7). Nonetheless, these results suggest fundamental differences in mechanisms of inflammation between various mucosal organs. Below, we discuss some potential mechanisms that may contribute to the unique mucosal niche established by infiltrating PMN.
Contribution of infiltrating PMN to the inflammatory microenvironment
The role of PMN as a first line of defense against invading pathogens has been long recognized as a primary function of PMN at sites of inflammation. PMN diapedesis, during which PMN migrate from the circulatory system to areas of tissue damage and inflammation, results in priming of PMN to express and release a number of factors that drive inflammation in the microenvironment. Activated PMN release multiple pro-inflammatory cytokines, including IL-1, IL-6, TGF-β and TNF-α (8). Chemokines such as CXCL8, CXCL1, CXCL9, CCL2, and CCL3, and other chemoattractants are also produced, resulting in further autocrine and paracrine activation of inflammatory pathways and recruitment of leukocytes (8). Lipid mediators, including leukotriene B4 and prostaglandin E2, as well as key enzymes in arachidonic acid metabolic pathways are also produced by activated neutrophils (9). Proteases such as PMN elastase, gelatinase, and collagenase are released during the process of PMN degranulation and can lead to further activation of cytokines and cause tissue damage (10). Further collateral tissue damage in the microenvironment may occur through the production of reactive oxygen species (ROS) by PMN. Thus, as PMN infiltrate inflamed tissues, they have the ability to cause further tissue injury as well as perpetuate inflammation through multiple mechanisms. While most studies indicate a protective role for PMN in colitis (5, 11), some reports suggest that PMN exacerbate inflammation (12). Seemingly paradoxical to their proinflammatory potential, the preponderance of evidence in the GI mucosa suggests that PMN promote the resolution of chronic inflammation. The remainder of this review will focus on mechanisms by which PMN create a microenvironment that dampens the inflammatory response and promotes tissue healing.
Oxygen consumption by PMN as a driver an hypoxic microenvironmnet
Recent studies have shown that a contributing factor to inflammatory resolution in the GI mucosa is the unique nature of oxygen distribution and metabolism. The intestinal mucosa, for example, exhibits a particularly unique oxygenation profile, experiencing profound fluctuations in blood flow and metabolism. These fluctuations result in a steep oxygen gradient from the lumen to the lamina propria in the human intestinal tract. Breathable air at sea level contains a partial O2 pressure (pO2) of ~145 mmHg (approximately 21% O2). Measurements of the healthy lung alveolus have revealed a pO2 of 100–110 mmHg (13). By contrast, the most luminal aspect of the healthy colon exists at a pO2 below 10 mmHg (14–16). Such differences reflect a combination of O2 sources, local metabolism and the anatomy of blood flow (17). The pO2 drops precipitously along the radial axis from the intestinal submucosa to the lumen, largely driven by the vast number of lumenal microbes (18). It is perhaps not surprising that the GI mucosa has evolved a number of adaptive features to cope with these profound metabolic conditions. Studies comparing functional responses between epithelial cells from different tissues have revealed that intestinal epithelia appear to be uniquely resistant to hypoxia and that even very low levels of O2 within the normal mucosa (so-called “physiologic hypoxia”) may be a regulatory adaptation mechanism to the steep oxygen gradient across the intestinal mucosa (19). A key discovery was the observation that epithelial cells of the distal gut basally regulate hypoxia-inducible factor (HIF) (15), a master regulator of oxygen homeostasis (20). Kelly et al, recently demonstrated that the low-O2 conditions of the distal GI tract that enable microbial short chain fatty acid (SCFA) production (in particular butyrate), promotes epithelial O2 consumption to the extent that HIF is stabilized and fundamentally contributes to the expression of some antimicrobial peptides (21) as well as maintenance of epithelial barrier function (22).
Metabolic demand for oxygen is prominently enhanced during inflammation. It was recently demonstrated by Campbell et al., (11) that during acute inflammatory disease, infiltrating neutrophils shape the tissue microenvironment in ways that significantly promote the stabilization of HIF. Guided by unbiased microarray analysis of epithelial cells following PMN transmigration, these studies identified a prominent induction of a cohort of HIF target genes. Utilizing PMN depletion strategies, HIF reporter mice and transgenic mice lacking a respiratory burst (Gp91phox−/−) in acute colitis models, these studies discovered that transmigrating neutrophils rapidly deplete the microenvironment of molecular oxygen in an NADPH-oxidase-dependent manner. As such, it was shown that transmigrating PMN “transcriptionally imprint” a molecular signature that reflects PMN-mediated induction of HIF-target genes onto the surrounding tissue. Notably, this molecular signature promotes effective HIF-dependent inflammatory resolution. Indeed, Gp91phox−/− mice developed highly amplified colitic responses compared to controls. These differences included exaggerated PMN infiltration, diminished inflammatory hypoxia and increased microbial invasion. Clinical corollaries to these findings have revealed that patients which lack a functional NADPH oxidase (i.e. chronic granulomatous disease) often present with an IBD-like syndrome (23). CGD-associated IBD results from a number of single nucleotide polymorphisms that impact the NADPH oxidase complex are IBD candidate genes that can result in a disease termed very early onset IBD (i.e. occurs in the first 6 years of life) (24, 25). This NADPH oxidase complex is responsible for the generation of reactive oxygen species (ROS) and used by innate immune cells (esp. PMN and macrophages) to kill invading pathogens. Interestingly, chronic granulomatous disease (CGD) patients exhibit congenital defects in genes coding the subunits comprising the neutrophil NADPH oxidase complex and approximately 40% of CGD patients develop this IBD-like syndrome (26). A recent retrospective clinical study examined all CGD patients screened at the National Institutes of Health between 1990 and 2010. The authors observed that CGD patients with colonic involvement, 74% had skip lesions (indicative of Crohn’s disease) and 93% presented with anorectal disease (commonly associated with ulcerative colitis). Thus the authors propose that CGD-inflammatory bowel disease is a distinct entity (27). Most CGD patients exhibit mutations in gp91phox (CYBB) and p47phox (NCF1), with fewer exhibiting defects in p22phox (CYBA) and p67phox (NCF2) (28). More recently identified is a rare mutation in a fifth subunit, p40phox (NCF4), involved in phagocytosis-induced superoxide production. The patient presented with CGD and a p40phox mutation exhibited intractable IBD, distinct from infectious complications observed with other mutations (29). Taken together, these findings suggest that CGD-associated IBD represents a separate form of IBD which could represent a failure to resolve acute intestinal and colonic inflammatory insults.
Fine tuning ROS generation and dismutation at sites of injury or infection is likely a critical pivotal juncture in influencing either resolution of acute inflammation or progression towards a chronic inflammatory state. Aside from phagocyte oxidases, other enzymes in this family are expressed throughout the body and respond to inflammatory stimuli to generate ROS, including Nox1, Nox3, Nox4, Nox5, Duox1 and Duox2 (30). Both Nox1 and Duox2 are expressed at high levels within the colonic epithelium. Nox1 is expressed cytosolically, conversely Duox2 is localized to the apical epithelial membrane (31), as such, Nox1 is more likely involved in intracellular signaling, whereas Duox2 likely elicits extracellular ROS release. Proinflammatory stimuli such as IFN-γ (32) and LPS challenge (33) lead to up-regulation of Nox1, whereas Duox2 has been shown to be increased in a cohort of Crohn’s disease patients (34), intestinal staining was negative in the epithelial compartment. Moreover, murine Duox2 has been implicated in restricting Helicobacter felis colonization by secretion of H2O2 into the gastric lumen(35). In a recent study the authors suggest that mucosal oxidases perform in a sequential fashion during inflammation, prior to recruitment of Nox2-expressing phagocytes. Here the authors examined the influence of epithelial Nox1/Duox2 on the virulence of Campylobacter jejuni and determined that epithelial-derived ROS (36). Furthermore, novel mutations in both Nox1 and Duox2 have been identified and implicated in very early onset IBD (37). Studies by Jones et. al., have implicated a protective role for epithelial-expressed NOX1 generating ROS in response to symbiotic microbes, including Lactobacilli species (38). They subsequently demonstrated that this epithelial cytoprotection is most likely due to xenobiotic-induced regulation of the Nrf2 pathway (39). Most recently they have demonstrated that wounded epithelia rely on Nox1 expression to facilitate wound healing, which is significantly induced by mucosally-associated microbes, including Akkermansia mucinophilia (40). Taken together these studies implicate mucosally-expressed oxidases in mediating homeostasis and highlight the intimate association between microbiota, epithelium and underlying immune system. Moreover, they emphasize the active role attributed to the epithelium in responding to environmental conditions and coordinating adaptive changes in both the mucosally-associated microbiota and immune response.
Significant evidence indicates that the large amounts of localized oxygen consumption associated with acute inflammation signals epithelial restitution and inflammatory resolution through the stabilization of HIF (41). Once stabilized, HIF triggers the transcription of a cohort of genes that enable intestinal epithelial cells to resolve defective barrier function (19, 42–44). Originally studies using microarray analysis of intestinal epithelial cells subjected to low O2 revealed influences on barrier-related genes (45) that have now been validated in a number animal models of intestinal inflammation (15, 46–50) and in human intestinal tissues (11, 51–53). The functional proteins encoded by these HIF targets include those that localize to the most luminal aspect of polarized epithelia that contribute fundamentally to effective barrier function, including tight junction proteins (e.g. claudin-1(54)), mucins (55), mucin modifiers (e.g. intestinal trefoil factor (19), antimicrobials (21), xenobiotic clearance (42) and nucleotide signaling/metabolism (e.g. ecto-5′-nucleotidase)(44, 45)).
It is also notable that one of the more prominent epithelial genes induced by PMN transmigration was cyclooxygenase-2 (COX-2) (11). COX-2 contributes fundamentally to both inflammation and resolution (56, 57). During epithelial-PMN interactions, pro-resolving lipid mediators (e.g. lipoxin and resolvins) are amplified by transcellular biosynthesis through the interactions of two or more cell types, each contributing an enzymatic product; in this case epithelial cell COX-2 generates 18-HEPE from dietary omega-3 fatty acids and PMN-expressed 5-LO to generate resolvins (58). These locally generated lipid mediators bind to and activate surface expressed receptors (e.g. ChemR23), which in turn has been shown to upregulate a number of antimicrobial peptides in the mucosa (59).
HIF activity has also been shown to directly impact PMN function. In original studies by Cramer (60) and Peyssonnaux (61), these authors provided an essential role for HIF-1 in innate immunity of myeloid phagocytes, particularly related to bacterial killing. Utilizing conditional deletion of Hif1a in myeloid populations (lysM-Cre) revealed profoundly decreased bactericidal capacity and a failure to restrict systemic sepsis when myeloid phagocytes lacked functional Hif1a.
It is important to note that the majority of these studies on neutrophil and epithelial oxidases and the roles of HIF have utilized animal models, thus extending conclusions to humans should be done cautiously. All things considered, these studies have provided new insight into how PMN commuinicate with tissues during active inflammation. The described “transcriptional imprinting“ response of select molecular pathways, notably HIF, provides mucosal memory of trafficked PMN that elicits functional responses that may be central to tissue homeostasis. While PMN accumulation as crypt abscesses has long served as a hallmark of mucosal inflammation, these findings reveal that PMN accumulation, coinciding with the surrounding tissue response, is likely a neccesity for appropriate wound healing.
Neutrophils in the tumour microenvironment
Inflammation has long been recognized as an important feature of cancer development and progression. The DNA damaging and pro-mutagenic conditions that exist during chronic inflammation have long been suspected to initiate and promote tumor development. As such, significant evidence exists that neutrophils play an important role in tumor promotion (62). This topic was recently reviewed in detail by Liang and Ferrara (63), but some relevant points are noteworthy here.
While PMN are typically thought of as short-lived immune cells, their recruitment to tissues and exposure to micronevironmental factors such as GM-CSF (64) or hypoxia (65) prolong PMN lifespan and regulate their function. The tumor microenvironment provides a plethora of factors that recruit PMN and support the activity of PMN function (Figure 1). PMN are initially recruited to tumors through the local generation of several chemokines, but the primary signal appears to be CXCL8 (human IL-8, murine KC) (66). The reservoir for these CD11b+ Ly6Ghi cells appears to be the spleen (67). It remains unclear exactly what signaling cascade elicits tumor-derived IL-8/KC and whether this signaling is tumor-specific. Nonetheless, early in tumor development, these tumor-associated neutrophils (TAN) migrate to the periphery of the tumor, where they remain adherent to the tumor stroma (68). As tumors grow and develop, TANs appear to penetrate and associate among the tumor cells, even deep into the tumor bed. Some studies have shown that while at the tumor site, some TANs may phenotypically differentiate. For example, analogous to the macrophage M1 and M2 dichotomy, TANs appear to develop a more protumorigenic (N2) phenotype in untreated tumors, driven largely by the presence of tumor-derived TGF-β (69). Blocking TGF- β with neutralizing antibodies convert TANs to a more antitumor (N1) phenotype (70), termed N1 TANs that produce higher levels of mediators such as nitric oxide, hydrogen peroxide, and TNF-α and appear to be tumor cytotoxic (71). It is also interesting to note that the depletion of peripheral neutrophils with anti-Gr-1 antibodies resulted in decreased expression of tumor pro-angioenic factors (e.g. VEGF, MMP9, CXCR4) and inhibited overall tumor growth (70). It is notable that the majority of pro-angiogenic factors are direct HIF target genes (72) and from this perspective, it is intriguing to speculate that PMN oxygen consumption/HIF stabilization within the tissue microenvironment (see above), may function to amplify the production of pro-tumorigenic factors within this niche to enhance tumor growth and/or tumor metastasis. Considering the disparate actions of M1 versus M2 polarized macrophages in tumor biology compared with inflammation, it is tempting to speculate on the presence of distinctly polarized PMN in inflammatory conditions. The first report to characterize different subsets of PMN based on resistance or sensitivity to methicillin-resistant Staphylococcus aureus (Figure 1) (73). The authors identified distinct subsets of PMN with differential surface antigen expression, macrophage activation potential and cytokine/chemokine production. Aside from PMN that polarize in situ, recent findings indicate that subpopulations of PMN may preexist in circulation and respond to disparate chemoattractive stimuli. Massena et al recently demonstrated a CD49d+CXCR4highVEGFR1high population of PMN that specifically migrate to sites of hypoxia and promote angiogenesis (74). CD49d is a differentiation marker downregulated on mature PMN. Given the relatively low percentage of circulating PMN that exhibit this phenotype, it is unclear if this population represents an immature PMN population or a distinct VEGF-responsive PMN that responds selectively to hypoxic injury (75). On the polar end of the maturation spectrum, immature PMN progenitors have been characterized to adopt a hybrid PMN/dendritic cell phenotype when exposed to GM-CSF, capable of both antimicrobial activities and professional antigen presentation (76). Furthermore, this novel subset of PMN has been identified in vivo, demonstrated to accelerate clearance of Escherichia coli strains and present antigen to CD4 T cells (77). Placed in context, consideration for divergent PMN functions and polarities should not be disregarded in future investigations.
FIGURE 1. Neutrophil polarization in response to microenvironmental reprogramming.
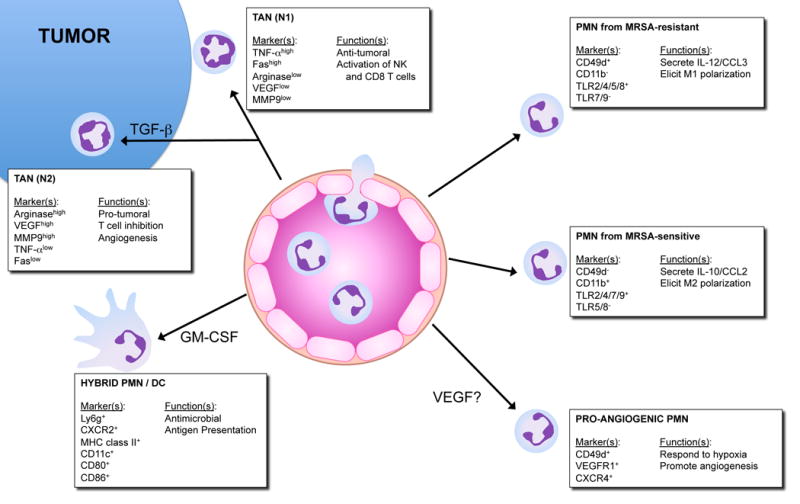
PMN respond to environmental cues to elicit differential polarization and function. Counter-clockwise from the top: 1) TAN-N1 migrate to tumor periphery, their nuclei exhibit a “hypersegmented” appearance and they mediate anti-tumoral activities. 2) TAN-N2 respond to tumor-derived TGF-β, their nuclei appear “circular” and they elicit pro-tumoral role. 3) Hybrid PMN/DC represent an immature PMN precursor exposed to GM-CSF and adopt a hybrid antimicrobial and antigen presenting phenotype. 4) Pro-angiogenic PMN may be derived from longer-lived mature PMN and respond specifically to sites of ischemia/hypoxia. 5) PMN isolated from MRSA-sensitive hosts secrete anti-inflammatory IL-10 and promote alternative activation in macrophages. 6) PMN isolated from MRSA-resistant hosts secrete IL-12 and influence classical activation in macrophages.
TAN: tumor associated neutrophil; DC: dendritic cell; MRSA: methicillin-resistant Staphylococcus aureus.
Purine nucleotides and neutrophils in the microenvironment
Neutrophils are an important source of extracellular nucleotides found within the microenvironment of inflammatory lesions (78). A number of studies have revealed that PMN serve as a prominent reservoir for adenosine precursors that are made available for metabolism to adenosine (79). During inflammation, PMN actively release ATP, ADP and AMP (80). Release of ATP occurs in an activation-dependent manner through a mechanism involving connexin 43 (Cx43) hemichannels expressed on the surface of PMN (80). Other studies have shown that human PMNs release ATP along the leading edge of their cell surface as a mechanism to amplify chemotactic signals and direct cell orientation by feedback through P2Y2 nucleotide receptors (81).
It is now well documented that the phosphohydrolysis of ATP and AMP by surface apyrases (e.g. CD39) and ecto-5′-nucleotidase (CD73), respectively, represents the major pathway for accumulation of extracellular adenosine (44, 45, 82). Once liberated in the extracellular space, adenosine is either recycled (e.g. through dipyridamole-sensitive carriers) or interacts with cell surface Ado receptors (83). Presently, four subtypes of G protein-coupled Ado receptors exist, designated AA1R, AA2AR, AA2BR or AA3R and are classified according to utilization of pertussis toxin-sensitive (A1 and A3) or insensitive (A2A and A2B) pathways (83). Recent work has specifically implicated the AA2BR in mucosal healing responses, wherein activation of this receptor elicits potent inhibition of inflammatory signaling cascades mediated by NF-κB (84). Work in various cell types has shown that adenosine inhibits NF-κB activation through a number of distinct mechanisms. First, adenosine signaling results in elevation of intracellular cyclic adenosine monophosphate (cAMP) and activation of protein kinase A (PKA), blocking IκB phosphorylation and thus inhibiting NF-κB activation(85). In addition, adenosine signaling leads to inhibition of phosphorylation and degradation of IκBα, resulting in attenuated NF-κB activation due to increased SUMO-1 modification of IκBα (86). Lastly, NF-κB activity is also secondarily inhibited by a reduction in tumor necrosis factor (TNF)-α, which leads to a decrease in nuclear translocation of active NF-κB without influencing IκB phosphorylation or degradation(87).
Platelets also provide a rich source of extracellular ATP during mucosal inflammation. Platelets release nucleotides at high concentration upon activation by collagen or ADP through dense granule release (88). It was shown that interactions between platelets and PMN provide important signals for the resolution of intestinal inflammation and fluid transport via in an adenosine-dependent manner (89). Indeed, these studies showed that platelets migrate across intestinal epithelia in a PMN-dependent manner. Furthermore, platelet-PMN comigration was observed in intestinal tissue derived from human patients with IBD. The translocated platelet-PMN clusters were found to release large quantities of ATP, which was sequentially metabolized to adenosine via a 2-step enzymatic reaction involving CD73 and CD39-like molecules expressed on IEC(90). These studies identified a mechanism involving adenosine-mediated activation of electrogenic chloride secretion, with concomitant water movement into the intestinal lumen (91). This physiologic response has been demonstrated to serve as a protective flushing mechanism for mucosal-associated bacteria(92).
Given its long history, it is somewhat surprising how little is known about the molecular mechanisms of adenosine responses. The signal transduction through the various adenosine receptors (ARs) has been well characterized, however, less is known about the post-receptor events. One potentially important mechanism has revealed that adenosine stabilizes HIF (93) and inhibits NF-κB (84, 94) through actions on Cullin neddylation pathways. Neddylation is a form of post-translational protein modification, whereby Nedd8 (neutral precursor expressed, developmentally downregulated) moieties are added to proteins(95). Cullin proteins are key components of the ubiquitin ligase complexes, regulating protein ubiquitination and proteasomal processing. Neddylation of cullins is essential for functional ubiquitin ligase complexes, similarly, deneddylation of cullins acts to inhibit ubiquitination(95). Original studies implicated commensal bacterial inhibition of NF-κB through Cullin-1 (Cul-1) deneddylation (96). Bacterial fermentation products (e.g. butyrate) can increase intracellular ROS and lead to impaired neddylation of Cullins thus influencing NFκB signaling, implicating an important role for the commensal intestinal flora for the regulation of inflammatory processes(97). An analogous signaling complex exists upstream of HIF-α proteins, which is comprised of Cul-2, Elongin B/C and the von Hippel-Lindau protein (pVHL). Similar to the Cul-1-IκB-NF-κB axis, neddylation of Cul-2 is required for the ubiquitination of hydroxylated HIF-1α in normoxic conditions. HIF-α subunits are hydroxylated by prolyl hydroxylases (PHDs) to target the HIF subunits for degradation. In the absence of oxygen, the HIF-α subunits are not targeted for degradation and therefore translocate into the nucleus to dimerize with constitutively expressed HIF-1β subunits, turning on targeting genes(78).
Recently, pharmacological inhibition of Cullin neddylation has been employed using a small molecular inhibitor, MLN4924. Interestingly, MLN4924 is a structural analog of adenosine monophosphate (AMP) and functions by inhibiting the Nedd8-activating enzyme, thereby inhibiting the neddylation of Cullin proteins (98). The protective role of HIF and the contribution of the neddylation pathway were confirmed in a murine DSS-colitis model. Colitic mice exposed to MLN4924 displayed an overall diminished inflammation, reflected as decreased weight loss, decreased colonic shortening and decreased histologic injury (99). Identification of the Nedd8-specific protease DEN1/SENP8 has provided new insight into this emerging field. DEN1 contains dual isopeptidase activity capable of directly deneddylating Cullin targets (100, 101). Knockdown of DEN-1 in vascular endothelial and intestinal epithelial cells using lentiviral shRNA approaches revealed decreased Cul-1 and Cul-2 neddylation and significantly abrogated barrier dysfunction in vitro (93, 94).
Therapeutic considerations of PMN and the tissue microenvironment
Given the central role of HIF in mucosal inflammation, substantial efforts have been made in the development of pharmacologic compounds that stabilize HIF and enhance the expression of HIF target genes(102). In most instances, the pharmacologic approach to achieve HIF stabilization in normoxia has involved the inhibition of HIF prolyl-hydroxylases (PHDs), originally described as products of genes related to C. elegans egl-9(103) and later cloned in mammals as three isoforms of PHDs (PHD1, PHD2 and PHD3) that hydroxylate HIF-α and promote degradation through VHL and proteasome-dependent pathways(103, 104). Targeting the catalytic domain of PHDs was initially achieved by a screen of molecules that interfere with critical cofactors such as the 2-oxoglutarate mimicry (e.g. dimethyloxalylglycine, DMOG)(105). The original studies using PHD inhibition in mucosal inflammation demonstrated a protective role for HIF stabilizers in different models of intestinal inflammation. The use of DMOG for the treatment of intestinal inflammation in chemically-induced colitis proved highly effective in abrogating mucosal inflammation, at least in part through blocking epithelial apoptosis (46). A parallel study published at the same time utilized a HIF stabilizer (FG-4497) that was based on a screen to identify erythropoietin enhancers. Similar to DMOG, FG-4497 blocks the active site of PHDs(105). In both cases, HIF stabilization was associated with the abrogation of mucosal inflammation, most prominently related to enhanced barrier function (46, 49). More HIF-1 selective stabilizers, such as AKB-4924, have shown significant protection in colitis models as well as anti-microbial activity in stimulating the killing of the pathogens (11, 106, 107). Results from these studies in pre-clinical models indicate that IBD may be one of the more promising applications for PHD inhibitor-based therapies.
Conclusion
The migration of PMN into sites of injury and infection represents one of the most significant changes to the tissue microenvironment. In the mucosa, restitution of the epithelial barrier defines a critical determinant of the healing response. Recent studies investigating changes within the microenvironment of acute inflammation have revealed important new signaling cascades initiated by activated PMN. Of particular recent interest is the shift in tissue oxygenation toward hypoxia and its relevance to HIF-target genes that are strongly associated with promoting barrier function and altering tissue bioenergetics. Studies in animal models have demonstrated an overall protective and anti-inflammatory influence of prolyl hydroxylase inhibition (i.e. HIF stabilization), identifying the mucosa as a strong candidate for targeted HIF-based therapy.
Acknowledgments
This work was supported by National Institutes of Health Grants DK103639, DK50189, DK104713 and DK95491 and by funding from the Crohn’s and Colitis Foundation of America.
Footnotes
The authors declare no financial interests in any of the work submitted here.
Disclosure statement: The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as influencing the objectivity of this review.
References
- 1.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 4.Koch S, Nusrat A. The life and death of epithelia during inflammation: lessons learned from the gut. Annu Rev Pathol. 2012;7:35–60. doi: 10.1146/annurev-pathol-011811-120905. [DOI] [PubMed] [Google Scholar]
- 5.Kuhl AA, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 6.Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol. 2009;40:519–535. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Persson C, Uller L. Transepithelial exit of leucocytes: inflicting, reflecting or resolving airway inflammation? Thorax. 2010;65:1111–1115. doi: 10.1136/thx.2009.133363. [DOI] [PubMed] [Google Scholar]
- 8.Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. 2014;5:1–7. doi: 10.3389/fimmu.2014.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5:354–366. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 11.Campbell EL, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40:66–77. doi: 10.1016/j.immuni.2013.1011.1020. Epub 2014 Jan 1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Natsui M, et al. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J Gastroenterol Hepatol. 1997;12:801–808. doi: 10.1111/j.1440-1746.1997.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 13.Schaible B, Schaffer K, Taylor CT. Hypoxia, innate immunity and infection in the lung. Respir Physiol Neurobiol. 2010;174:235–243. doi: 10.1016/j.resp.2010.08.006. doi: 210.1016/j.resp.2010.1008.1006. Epub 2010 Aug 1013. [DOI] [PubMed] [Google Scholar]
- 14.Albenberg L, et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology. 2014;18:1055–1063. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karhausen J, Ibla JC, Colgan SP. Implications of hypoxia on mucosal barrier function. Cell Mol Biol. 2003;49:77–87. [PubMed] [Google Scholar]
- 17.Zheng L, Kelly CJ, Colgan SP. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol. 2015;309:C350–360. doi: 10.1152/ajpcell.00191.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kelly CJ, et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe. 2015;17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuta GT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semenza GL. Oxygen sensing, homeostasis, and disease. NEJM. 2011;365:537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 21.Kelly CJ, et al. Fundamental role for HIF-1alpha in constitutive expression of human beta defensin-1. Mucosal Immunol. 2013;6:1110–1118. doi: 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly CJ, et al. Host-microbe crosstalk between short-chain fatty acids and intestinal epithelial HIF provides a new mechanism to augment tissue barrier function. Cell Host Microbe Cell Host Microbe. 2015 doi: 10.1016/j.chom.2015.03.005. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang JS, et al. Chronic granulomatous disease caused by a deficiency in p47(phox) mimicking Crohn’s disease. Clin Gastroenterol Hepatol. 2004;2:690–695. doi: 10.1016/s1542-3565(04)00292-7. [DOI] [PubMed] [Google Scholar]
- 24.McGovern DP, Kugathasan S, Cho JH. Genetics of Inflammatory Bowel Diseases. Gastroenterology. 2015;149:1163–1176. doi: 10.1053/j.gastro.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muise AM, et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 2012;61:1028–1035. doi: 10.1136/gutjnl-2011-300078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Werlin SL, Chusid MJ, Caya J, Oechler HW. Colitis in chronic granulomatous disease. Gastroenterology. 1982;82:328–331. [PubMed] [Google Scholar]
- 27.Khangura SK, et al. Gastrointestinal Features of Chronic Granulomatous Disease Found During Endoscopy. Clin Gastroenterol Hepatol. 2016;14:395–402 e395. doi: 10.1016/j.cgh.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kannengiesser C, et al. Molecular epidemiology of chronic granulomatous disease in a series of 80 kindreds: identification of 31 novel mutations. Human mutation. 2008;29:E132–149. doi: 10.1002/humu.20820. [DOI] [PubMed] [Google Scholar]
- 29.Matute JD, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geiszt M. NADPH oxidases: new kids on the block. Cardiovascular research. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contributions to microbiology. 2008;15:164–187. doi: 10.1159/000136357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuwano Y, et al. Interferon-gamma activates transcription of NADPH oxidase 1 gene and upregulates production of superoxide anion by human large intestinal epithelial cells. American journal of physiology Cell physiology. 2006;290:C433–443. doi: 10.1152/ajpcell.00135.2005. [DOI] [PubMed] [Google Scholar]
- 33.O’Leary DP, et al. TLR-4 signalling accelerates colon cancer cell adhesion via NF-kappaB mediated transcriptional up-regulation of Nox-1. PLoS One. 2012;7:e44176. doi: 10.1371/journal.pone.0044176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csillag C, et al. Expression of the genes dual oxidase 2, lipocalin 2 and regenerating islet-derived 1 alpha in Crohn’s disease. Scandinavian journal of gastroenterology. 2007;42:454–463. doi: 10.1080/00365520600976266. [DOI] [PubMed] [Google Scholar]
- 35.Grasberger H, El-Zaatari M, Dang DT, Merchant JL. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology. 2013;145:1045–1054. doi: 10.1053/j.gastro.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Corcionivoschi N, et al. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe. 2012;12:47–59. doi: 10.1016/j.chom.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayes P, et al. Defects in NADPH Oxidase Genes and in Very Early Onset Inflammatory Bowel Disease. Cellular and molecular gastroenterology and hepatology. 2015;1:489–502. doi: 10.1016/j.jcmgh.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones RM, et al. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 2013;32:3017–3028. doi: 10.1038/emboj.2013.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones RM, et al. Lactobacilli Modulate Epithelial Cytoprotection through the Nrf2 Pathway. Cell Rep. 2015;12:1217–1225. doi: 10.1016/j.celrep.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alam A, et al. The microenvironment of injured murine gut elicits a local pro-restitutive microbiota. Nature Micro. 2016;1 doi: 10.1038/nmicrobiol.2015.21. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer research. 2002;62:3387–3394. [PubMed] [Google Scholar]
- 43.Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. Faseb J. 2002;16:583–585. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 44.Eltzschig HK, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Ex Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Synnestvedt K, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cummins EP, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Han IO, Kim HS, Kim HC, Joe EH, Kim WK. Synergistic expression of inducible nitric oxide synthase by phorbol ester and interferon-gamma is mediated through NF-kappaB and ERK in microglial cells. J Neurosci Res. 2003;73:659–669. doi: 10.1002/jnr.10706. [DOI] [PubMed] [Google Scholar]
- 48.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 49.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shah YM, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–2048. 2048 e2031–2033. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giatromanolaki A, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. Journal of clinical pathology. 2003;56:209–213. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mariani F, et al. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279:221–229. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 53.Matthijsen RA, Derikx JP, Kuipers D, van Dam RM, Dejong CH, Buurman WA. Enterocyte shedding and epithelial lining repair following ischemia of the human small intestine attenuate inflammation. PLoS One. 2009;4:e7045. doi: 10.1371/journal.pone.0007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saeedi BJ, et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell. 2015;26:2252–2262. doi: 10.1091/mbc.E14-07-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–1627. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 56.Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cycloxygenase may have anti-inflammatory properties. Nat Med. 1999;5:698–701. doi: 10.1038/9550. [DOI] [PubMed] [Google Scholar]
- 57.Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res. 2011;107:1170–1184. doi: 10.1161/CIRCRESAHA.110.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell EL, Serhan CN, Colgan SP. Antimicrobial aspects of inflammatory resolution in the mucosa: a role for proresolving mediators. J Immunol. 2011;187:3475–3481. doi: 10.4049/jimmunol.1100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cramer T, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peyssonnaux C, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagerling C, Werb Z. Neutrophils: Critical components in experimental animal models of cancer. Semin Immunol. 2016 doi: 10.1016/j.smim.2016.02.003. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang W, Ferrara N. The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis. Cancer Immunol Res. 2016;4:83–91. doi: 10.1158/2326-6066.CIR-15-0313. [DOI] [PubMed] [Google Scholar]
- 64.Lee A, Whyte MK, Haslett C. Inhibition of apoptosis and prolongation of neutrophil functional longevity by inflammatory mediators. J Leukoc Biol. 1993;54:283–288. [PubMed] [Google Scholar]
- 65.Hannah S, et al. Hypoxia prolongs neutrophil survival in vitro. FEBS Lett. 1995;372:233–237. doi: 10.1016/0014-5793(95)00986-j. [DOI] [PubMed] [Google Scholar]
- 66.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–6741. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 67.Cortez-Retamozo V, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109:2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim J, Bae JS. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediators Inflamm. 2016 doi: 10.1155/2016/6058147. In press:Epub 2016 Feb 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fridlender ZG, et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One. 2012;7:e31524. doi: 10.1371/journal.pone.0031524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120:1151–1164. doi: 10.1172/JCI37223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71:2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 72.LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. 2016;18:356–365. doi: 10.1038/ncb3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN, Suzuki F. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity. 2004;21:215–226. doi: 10.1016/j.immuni.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 74.Massena S, et al. Identification and characterization of VEGF-A-responsive neutrophils expressing CD49d, VEGFR1, and CXCR4 in mice and humans. Blood. 2015;126:2016–2026. doi: 10.1182/blood-2015-03-631572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Campbell EL. Hypoxia-recruited angiogenic neutrophils. Blood. 2015;126:1972–1973. doi: 10.1182/blood-2015-09-666578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matsushima H, et al. Neutrophil differentiation into a unique hybrid population exhibiting dual phenotype and functionality of neutrophils and dendritic cells. Blood. 2013;121:1677–1689. doi: 10.1182/blood-2012-07-445189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Geng S, et al. Emergence, origin, and function of neutrophil-dendritic cell hybrids in experimentally induced inflammatory lesions in mice. Blood. 2013;121:1690–1700. doi: 10.1182/blood-2012-07-445197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153–175. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eltzschig HK, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 81.Chen Y, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 82.Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. 1997;81:154–164. doi: 10.1161/01.res.81.2.154. [DOI] [PubMed] [Google Scholar]
- 83.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annual review of pharmacology and toxicology. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 84.Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–711. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hong G, Zhang B, Harbrecht BG. Cyclic AMP inhibits IL-1beta plus IFNgamma-induced NF-kappaB translocation in hepatocytes by a PKA independent mechanism. J. Surg Res. 2010;159:565–571. doi: 10.1016/j.jss.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu Q, Li J, Khoury J, Colgan SP, Ibla JC. Adenosine signaling mediates SUMO-1 modification of IkappaBalpha during hypoxia and reoxygenation. J Biol Chem. 2009;284:13686–13695. doi: 10.1074/jbc.M809275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene. 2003;22:1206–1218. doi: 10.1038/sj.onc.1206184. [DOI] [PubMed] [Google Scholar]
- 88.Gordon JL, Extracellular ATP. effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weissmuller T, et al. PMNs facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-NTPDases. J Clin Invest. 2008;118:3682–3692. doi: 10.1172/JCI35874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73) Purinergic Signal. 2006;2:351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Madara JL, et al. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J Clin Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Keely S, et al. Activated fluid transport regulates bacterial-epithelial interactions and significantly shifts the murine colonic microbiome. Gut Microbes. 2012;3:250–260. doi: 10.4161/gmic.20529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Curtis VF, et al. Stabilization of HIF through inhibition of Cullin-2 neddylation is protective in mucosal inflammatory responses. FASEB J. 2015;29:208–215. doi: 10.1096/fj.14-259663. doi: 210.1096/fj.1014-259663. Epub 252014 Oct 259617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ehrentraut SF, et al. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J Immunol. 2013;190:392–400. doi: 10.4049/jimmunol.1202041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Curtis VF, Ehrentraut SF, Colgan SP. Actions of adenosine on cullin neddylation: implications for inflammatory responses. Comput Struct Biotechnol J. 2015;13:273–276. doi: 10.1016/j.csbj.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neish AS, et al. Prokaryotic regulation of epithelial responses by inhibition of IkappaB- alpha ubiquitination. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 97.Kumar A, et al. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. Embo J. 2007;26:4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soucy TA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 99.Cowley BP, Smith ME. Modulatin of E-cadherin expression and morphological phenotype in the intravascular component of adenocarcinomas. Int J Cancer. 1995;60:325–329. doi: 10.1002/ijc.2910600308. [DOI] [PubMed] [Google Scholar]
- 100.Mendoza HM, et al. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 101.Wu K, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–28891. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 102.Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014;13:852–869. doi: 10.1038/nrd4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Epstein AC, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 104.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 105.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]
- 106.Keely S, et al. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol. 2014;7:114–123. doi: 10.1038/mi.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Okumura CY, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med. 2012;28:1079–1089. doi: 10.1007/s00109-012-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]