

Abstract
The study of patients with inherited bleeding problems is a powerful approach in determining the function and regulation of important proteins in human platelets and their precursor, the megakaryocyte. The normal range of platelet counts in the bloodstream ranges from 150 000 to 400 000 platelets per microliter and is normally maintained within a narrow range for each individual. This requires a constant balance between thrombopoiesis, which is primarily controlled by the cytokine thrombopoietin (TPO), and platelet senescence and consumption. Thrombocytopenia can be defined as a platelet count of less than 150 000 per microliter and can be acquired or inherited. Heritable forms of thrombocytopenia are caused by mutations in genes involved in megakaryocyte differentiation, platelet production and platelet removal. In this review, we will discuss the main causative genes known for inherited thrombocytopenia and highlight their diverse functions and whether these give clues on the processes of platelet production, platelet function and platelet lifespan. Additionally, we will highlight the recent advances in novel genes identified for inherited thrombocytopenia and their suggested function.
Keywords: Inherited thrombocytopenia, megakaryocytes, platelets, gene mutations, bleeding
Introduction
Inherited thombocytopenias (ITs) are a heterogeneous group of disorders characterized by a sustained reduction in platelet count manifesting as a bleeding diathesis. Since the discovery of disease inheritance patterns in disorders such as Bernard Soulier Syndrome (BSS), genetic studies of thrombocytopenia have been a vital tool in determining megakaryocyte and platelet physiology [1]. As a result of parallel whole exome and whole genome sequencing over the past 5–10 years, we are discovering increasing numbers of novel genes with a critical role in platelet physiology. However, are any of these genes giving us clues to platelet lifespan? Is IT predominately caused by a defect in the process of platelet production or is there more to the story than we currently know?
To date, there are 31 genes suspected to cause 27 separate forms of inherited thrombocytopenia (Table I). The majority of patients with thrombocytopenia present secondary to syndromic disorders usually affecting the immune system or in cases of immune thrombocytopenic purpura (ITP). However, a number of cases present as a bleeding diathesis as a result of a reduced platelet count, sometimes alongside additional platelet dysfunction, indicating an effect solely on cells of the hematopoietic lineage. As a consequence of the varied phenotypic display and clinical presentation of inherited thrombocytopenias, there are several ways in which they are characterized. One such way, which has been favored following molecular characterization of the effect of mutations, is to group genes based upon their effect on megakaryocyte differentiation, platelet production and platelet function. Therefore, to understand whether a link to platelet lifespan can be established it is best to first consider all genes implicated in a reduced platelet count.
Table I.
Genetic causes of inherited thrombocytopenia, the encoded protein and their associated diseases. Grouped into their suggested role within megakaryopoiesis, platelet production or clearance/other.
| Area of mutational effect | Gene | Protein | Associated disease | References |
|---|---|---|---|---|
| Megakaryopoiesis | ANKRD26 | Ankyrin repeat domain 26 | ANKRD26-related thrombocytopenia | [25] |
| ETV6 | Transcription factor ETV6 | THC5 | [7] | |
| FLI1 | Friend leukaemia integration 1 transcription factor | Paris Trousseau type thrombocytopenia/Jacobsen (11q23 del) | [8] | |
| FYB | FYN-binding protein | Novel thrombocytopenia | [26] | |
| GATA1 | Erythroid transcription factor | GATA1 related disease (XLT and XLTT) | [9] | |
| GFI1B | Zinc finger protein Gfi-1b | Grey Platelet Syndrome + novel thrombocytopenia | [10] | |
| HOXA11 | Homeobox protein Hox-A11 | Amegakaryocytic thrombocytopenia with radio-ulnar synostosis | [74] | |
| MPL | Thrombopoietin receptor | Congentical amegakaryocytic thrombocytopenia | [12] | |
| NBEAL2 | Neurobeachin-like protein 2 | Grey Platelet Syndrome | [20] | |
| RBM8A | RNA-binding protein 8A | Thrombocytopenia with absent radii | [16] | |
| RUNX1 | Runt-related transcription factor | Familial platelet disorder and predisposition to AML | [11] | |
| THPO | Thrombopoietin | Mild novel thrombocytopenia (heterozygous) | [13] | |
| Platelet production | ACTN1 | Alpha-actinin-1 | Bleeding disorder, platelet-type 15 | [36] |
| CYCS | Cytochrome C | CYCS-related thrombocytopenia | [45] | |
| GP1BA | Platelet glycoprotein 1b alpha chain | Bernard–Soulier Syndrome + Platelet type von-Willebrand disease | [75] | |
| GP1BB | Platelet glycoprotein 1b beta chain | [76] | ||
| GP9 | Platelet glycoprotein IX | [77] | ||
| ITGA2B | Integrin alpha-Iib | ITGA2B/ITGB3-related thrombocytopenia | [78] | |
| ITGB3 | Integrin beta-3 | [79] | ||
| MKL1 | MKL/myocardin-like protein 1 | Thrombocytopenia with immunodeficiency | [31] | |
| MYH9 | Myosin 9 | MYH9 related disease | [80] | |
| PRKACG | cAMP-dependant protein kinase catalytic subunit gamma | Bleeding disorder, platelet-type 19 | [39] | |
| TUBB1 | Tubulin beta-1 chain | TUBB1-related macrothrombocytopenia | [29] | |
| WAS | Wiskott–Aldrich syndrome protein | Wiskott-Aldrich syndrome, X-linked thrombocytopenia | [81] | |
| Platelet clearance/other | ABCG5 | ABC transporter G family member 5 | Thromobocytopenia associated with sitosterolaemia | [57] |
| ABCG8 | ABC transporter G family member 8 | [57] | ||
|
ADAMTS13 GNE |
Disintegrin/metalloproteinase with thrombospondin motifs 13 Glucosamine (UDP-N-Acetyl)-2-Epimerase |
TTP, Upshaw–Schulman syndrome GNE myopathy with congenital thrombocytopenia |
[51] [65] |
|
| SLFN14 | Schalfen family member 14 | Novel thrombocytopenia | [62] | |
|
STIM1 vWF |
Stromal interaction molecule 1 Von Willebrand factor |
Stormorken Syndrome Von Willebrand disease type 2B |
[82] [83] |
Genes that effect megakaryocyte differentiation and maturation
Like all blood cells, megakaryocytes are derived from hematopoietic stem cells (HSCs) via progressively differentiated progenitor cells [2]. This is a process that is underpinned by the synthesis and synergistic effect of the cytokine thrombopoietin (TPO) through its receptor c-Mpl [3]. However, the commitment to differentiate and commit along the myelo-erythroid lineage is also highly regulated by transcription factors. One such transcription factor is GATA1, which is critically regulated transcriptionally [4]. Additional transcription factors such as Runt-related transcription factor 1, encoded by the gene RUNX1, ETV6 and FLI-1 in tandem with the transcriptional repressor GFI1B, ensure maturation of the megakaryocytes by binding critical promoter regions in crucial megakaryocyte-expressed genes [5, 6]. With the exception of Friend of Gata-1 (FOG1), the co-factor to GATA1, variants in the aforementioned hematopoietic transcription factors have previously been shown to cause inherited thrombocytopenia [7–11]. In addition, variants have also been observed in the TPO receptor gene MPL, as well as in its ligand, encoded by the gene THPO in one large Micronesian family with affected individuals displaying idiopathic aplastic anemia and mild thrombocytopenia [12, 13]. As the crucial role of TPO and transcription factors in megakaryocyte production is widely known, it is unsurprising that variants within these genes are commonly found within patients with inherited thrombocytopenia. However, clinical presentation does vary (most likely as a result of secondary effects of transcription factors and chemokines), but, all mutations are consistent in their ability to reduce proliferation of CD34+ cells by altering transcription profiles of genes crucial in megakaryocyte development [14].
In addition to the aforementioned genes involved specifically in the commitment to the proliferation towards megakaryocytes, a number of other genes have putative roles in differentiation and maturation. These include HOXA11, RBM8A, ANKRD26 and NBEAL2.
Similar to FLI-1, ETV6 and AML-1, HOXA11 is a DNA-binding protein, which is expressed, like many other homeodomain box proteins, in human cord blood and hematopoietic precursor cells [15]. The specific molecular mechanism behind how variants in HOXA11 cause amegakaryocytic thrombocytopenia with radio-ulnar synostosis syndrome has yet to be established. However, it has been suggested that a point mutation in the third helix of the homeobox domain can lead to a reduction in CD61 expression in staurosporine-induced K562 cells [15].
RBM8A, the gene encoding the exon junction splicing complex component Y14, is interesting both in the genetic complexity of the disease and in its proposed molecular mechanism. Thrombocytopenia with absent radii (TAR) syndrome is known currently as a result of the co-inheritance of a variably sized deletion of the region surrounding 1q21.1, including RBM8A, alongside one of two relatively high-frequency SNPs within the regulatory region of RBM8A [16]. The clinical outcome is severe but heterogeneous and seems to be determined by a number of factors, including variable gene expression and possible epigenetic modifiers. Little is known on the specific cellular effect of the compound hypomorphic effect of mutations within RBM8A, but a reduction in megakaryocyte progenitors is observed and is thought to arise as a result of aberrant JAK2 signaling downstream of TPO [17, 18].
The production of intracellular granules, during megakaryocyte maturation, from the budding of small vesicles containing cargo from trans-Golgi network is crucial for the further differentiation of platelets [19]. Platelet α-granules play a crucial role in platelet function and amplification of activation during hemostasis. A number of ITs, including GATA1-related thrombocytopenia (RT) and GFI1B-RT, show reduced numbers of platelet α-granules. However, a complete absence of platelet α-granules is observed in patients with Grey Platelet Syndrome (GPS), a condition caused by genetic variations within NBEAL2 [20]. NBEAL2 localizes to the endoplasmic reticulum in platelets, where it is likely to function in membrane trafficking; however, the role of NBEAL2 in GPS and the subsequent reduction in platelet count is still largely unknown. Nbeal2−/- mice do recapitulate the phenotype of GPS, including an absence of platelet α-granules as well as delayed megakaryocyte maturation, decreased survival and decreased ploidy in cultured megakaryocytes [21]. Interestingly, however, a lack of Nbeal2 in mice does not affect initial α-granule formation, packaging or transport to the budding platelets within megakaryocytes [22]. Both GPS mice and patients do develop premature myelofibrosis within the bone marrow, which may be as a result of spontaneous granule release or “leaking” from the megakaryocytes [22–24].
Another gene where variants have been shown to cause a defect in megakaryocyte maturation resulting in reduced platelet α-granules is ANKRD26. Like RBM8A, variants affect gene expression and occur in the 5’-UTR of the gene, specifically in a short stretch of nucleotides from c.-134G to c.-113A [25]. In affected individuals, megakaryocytes are small, with hypolobulated nuclei as a result of dysmegakaryopoiesis.
One of the most recently discovered genes known to cause inherited thrombocytopenia is FYB. Although the effect of the mutation has yet to be determined, a role in megakaryopoiesis has been suggested due to a reduced percentage of mature megakaryocytes in the bone marrow [26].
It seems then that the progression of HSCs through the megakaryocytic lineage to achieve mature megakaryocytes is a prime pathway to be disrupted by genetic mutation. Indeed, most variants not only involve altered expression or downstream signaling of the chemokine TPO, but many also affect additional signaling pathways, leading to a wide range of secondary abnormalities.
Genes that affect pro-platelet formation and release
Following megakaryopoiesis, the next phase in platelet production is pro-platelet formation and release of platelets from pro-platelet tips [27]. As the expression profile alters to accommodate new functional processes of the megakaryocyte, so do the genes in which mutations mediate thrombocytopenia.
As megakaryocytes reach their mature state, they undergo fundamental changes to be able to release platelets into circulation via bone marrow sinusoids. These changes are underpinned by cytoskeletal rearrangements and subsequent cellular signaling. Causative mutations of IT are actually most commonly found to functionally disrupt these processes.
The fundamental action leading to the release of platelets is the production of proplatelet extensions from a demarcated membrane system. Once in the vascular niche, mature megakaryocytes produce protrusions by microtubule sliding [28]. β1-Tubulin is the main isoform within human megakaryocytes and mutations within the gene encoding TUBB1 are known to cause an autosomal dominant form of inherited thrombocytopenia [29].
Like β1- tubulin, F-actin is present throughout proplatelets and allows proplatelet branching. These actin polymerization processes are promoted by nucleation factors such as WASp, encoded by WAS, which is selectively expressed in hematopoietic cells [30]. Mutations in WAS generally follow a genotype–phenotype correlation, where variants which abolish WASp expression or result in the expression of a truncated protein are associated with Wiskott–Aldrich Syndrome (WAS). Those causing decreased levels of WASp result in X-linked thrombocytopenia (XLT). Missense variants are most common in XLT and those occurring in the first three coding exons and the correlating EVH1/WH1 domain tend to cause the mild X-linked thrombocytopenia, which is not associated with the immunodeficiency observed in WAS.
Actin polymerization and the production of F-actin has recently been shown to be affected in a patient with a homozygous nonsense mutation within the gene encoding megakaryoblastic leukemia 1; MKL1 [31]. In addition to severe immunodeficiency, the patient suffers from mild-to-moderate thrombocytopenia (platelet counts 50–150 × 109/l). The cause of this is thought to be a loss of filamentous actin production due to a reduction in globular actin (G-actin) coupled with a reduction in megakaryocyte migration, which phenocopies mice deficient for murine Mkl1 [32, 33].
Myosins, in particular, IIA and IIB, are also involved in proplatelet formation by interacting with actin filaments generating contractile forces. One myosin, the heavy chain of nonmuscle myosin IIA (NMMHC-IIA) encoded by the gene MYH9, is thought to play a slightly different role as mutations actually increase proplatelet formation [34]. Mutations causing MYH9-related disease therefore cause thrombocytopenia through the most proximal part of platelet formation, where a disruption in the Rho-Rho kinase-myosin IIA pathway leads to a lack of reactivation of NMMHC-IIA. This is a process that is required for the budding of preplatelets and subsequent separation into platelets; therefore, mutations in MYH9 cause a loss of induced fragmentation of preplatelets promoting the formation of a reduced number of large platelets [35].
Cross linking of actin filaments is required for their binding to the actin cytoskeleton. This is mediated in structures known as actin-binding domains (ABDs), which contain α-actinin. One of the four isoforms, which is expressed in megakaryocytes, is ACTN1. To date, 13 pathogenic missense variants have been found within the encoding gene ACTN1; all cause a mild dominantly inherited thrombocytopenia where megakaryocyte tips are reduced in number, increased in size and exogenous expression of the mutations inhibits actin filament assembly [36].
Filamins, via an N-terminal ABD, anchor the filaments to the cell membrane. Variations in the most abundant filamin in platelets, FLNA, are known to cause periventricular nodular heteropia as well as an isolated thrombocytopenia [37]. Some patient megakaryocytes from cultured peripheral CD34+ blood cells show frayed structures and signs of cytoplasmic degradation favoring abnormal distribution into incorrectly packaged platelet-like fragments. However, a population of platelets negative for FLNA was observed, indicating it may not be necessary in proplatelet formation, therefore, playing a noncrucial role [37].
There may remain an important role for FLNA in platelet biogenesis as its proteolysis is protected by phosphorylation at S2152 by cAMP-dependent protein kinase A, which is similar to the protection observed in GP1bβ [38]. The kinase consists of two catalytic subunits, the y-isoform of which is encoded by PRKACG. To date, only one variant has been found within PRKACG, which results in rapid degradation of FLNA and a reduction in the percentage of proplatelet-bearing megakaryocytes [39].
Complementary to the physical formation of proplatelets is the relay of extracellular signals to the cytoskeleton via membrane-bound receptors. Two main receptors are affected by mutations in inherited thrombocytopenia. The first is the receptor for Von-Willebrand factor (VWF), GP1b-IX-V. Consisting of four subunits, GP1bα, GP1bβ, GPIX and GPV, activation of the receptor via interaction of VWF with GP1bα relays signals to aid in proplatelet formation. Variants in the encoding genes, GP1BA, GP1BB and GP9, are known to cause both monoallelic and biallelic forms of BSS and lead to a reduction in proplatelet-forming megakaryocytes in culture [40, 41].
The second receptor known to be affected in the spectrum of inherited thrombocytopenias is the integrin GPIIb-IIIa, the receptor for fibrinogen. Encoded by the genes ITGA2B and ITGB3, mutations are associated with Glanzmann thrombasthenia (GT). GT often causes bleeding with no alteration in platelet count; however, gain of function variants exists that lead to a thrombocytopenia, suggesting a role for the interaction of fibrinogen for platelet biogenesis [42]. Recently, it has been suggested that cytoskeleton rearrangement can be impaired, arresting actin turnover at the stage of polymerization, by permanent triggering of the aIIbβ3-mediated outside-in signaling [43]. Murine megakaryocytes transduced with an ITGB3 variant (c.del647-683) generated proplatelets with a reduced number of large tips and barbell-proplatelets, suggesting impaired cytoskeletal remodeling as the cause of thrombocytopenia in patients with GT.
One gene whose involvement in platelet production currently, through the intrinsic apoptosis pathway, is controversial is Cytochrome C (CYCS). CYCS initiates apoptosis through the activation of caspase 9 and subsequently caspase 3, a dysregulation of which was originally suggested as a feature of platelet production [44, 45]. To date, two variants (Y49H [p.48 in original publication] and G42S [p.41 in original publication]) have been observed in the apoptosis-linked gene CYCS [45, 46]. Indeed, electron microscopy of patient bone marrow shows intramedullary naked megakaryocyte nuclei, indicative of dysregulated megakaryopoiesis and premature platelet release. Furthermore, mouse lung fibroblasts transduced with CycsY48H and CycsG41S show increased apoptotic activity in response to staurosporine. It was therefore first believed that apoptosis had a fundamental role in the timing of pro-platelet release, hypothesizing that inhibition of actin polymerization, a process required for the formation of proplatelets, activates the apoptotic pathway [47]. However, platelet production can proceed regardless of both the intrinsic and extrinsic apoptotic pathways, suggesting there is more to the role of apoptosis in megakaryopoiesis than currently meets the eye [48].
Genes with a molecular function outside of platelet production
It is clear and obvious that the fundamental processes governing the production of platelets are easily affected by deleterious mutations leading to thrombocytopenia. This is summarized in Figure 1. However, disease affecting megakaryocytes and the formation of platelets do not cover all of the genes currently known to cause thrombocytopenia. These alternative genes do not play role in megakaryopoiesis/thrombopoiesis. Their molecular causes of disease are unique or not currently known and it is best to consider them individually and separate from the aforementioned genes.
Figure 1.
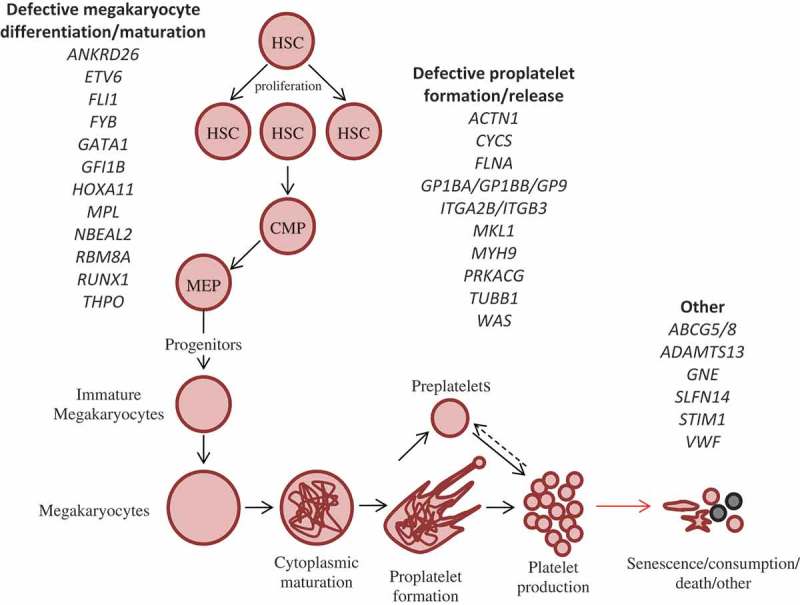
Megakaryopoiesis, platelet production and other causes of IT. Differentiation from HSCs to platelets proceeds by a number of intermediate cell types, which leads to the formation of megakaryocytes which fragment via proplatelet formation to produce mature platelets. This process is driven by a number of genes encoded a number of transcription factors and proteins. Defects in these genes have been shown to give rise to thrombocytopenia by mostly affecting the relevant stage of platelet production they are labeled under. Variants that do not play a role in platelet production by megakaryopoiesis are included in the third subgroup entitled platelet removal, death and other. HSC: Hematopoietic stem cell, CMP: Common myeloid progenitor, MEP: Megakaryocyte-erythroid progenitor.
VWF is particularly well known due to its importance in hemostasis, illustrated with the functional deficiency of vWF known as von Willebrand disease (VWD), a well-characterized and common collection of bleeding disorders. One particular type of VWD is known as type 2B, which refers to gain-of-function mutations that increase the affinity of vWF for GP1b on the platelet surface. The net effect of this is the presence of giant platelets due to continuous interactions between vWF and GP1bα disrupting megakaryopoiesis [49]. More interestingly, though, large multimers of vWF associated with platelets are also observed in VWD type 2B patients [50]. In vWF knock-in mice, these specific complexes are taken up efficiently by macrophages in the liver and spleen, indicating an increased clearance of platelets from the circulation.
One interesting gene with a mechanism of pathogenesis for thrombocytopenia is ADAMTS13. Variants in the metalloproteinase ADAMTS13 are the genetic cause of Upshaw–Schulman syndrome, alternatively known as congenital thrombotic thrombocytopenic purpura (TTP) [51]. TTP is a life-threatening systemic illness characterized by hemolytic anemia, neurological symptoms, renal dysfunction as well as thrombocytopenia. Normally in-circulation ADAMTS13 is the von Willebrand factor-cleaving protease. However, loss-of-function mutations within ADAMTS13 and subsequent TTP result in vWF not being cleaved at the Tyr 842-Met 843 peptide bond resulting in the accumulation of large vWF multimers similar to a gain-of-function mutations in vWF[52]. Unlike VWD type 2B, the reduction in platelet count observed in TTP may be due to platelet aggregation under sheer stress because of an inability to cleave vWF [53]. This has the propensity to explain vWF–platelet aggregation in the arterioles and capillaries, a phenotype characteristic of TTP.
An increased clearance of platelets is also present in patients with activating mutations of STIM1 within Stormorken syndrome and more recently York syndrome [54, 55]. Platelets in these patients are circulating in a preactivated state due to a constitutively active store operated Ca(2+) release-activated Ca(2+) (CRAC) channel and increase in Ca2+ entry. The net effect of which causes a reduction in the number of circulating platelets, but not due to an early ageing of platelets but an increased clearance of pre-activated platelets within the spleen, which is recapitulated in Stim1Sax/+ mice [56].
One interesting case of thrombocytopenia is as a secondary symptom with Sitosterolemia. Sitosterolemia is a rare, autosomal recessive disease caused by mutations in two adjacent ATP-binding cassette transport genes; ABCG5 and ABCG8 [57]. Hematological abnormalities, including thrombocytopenia, are a symptom of this complex disorder which results in decreased excretion and therefore accumulation of dietary sterols. ABCG5 and ABCG8, which normally act as a heterodimeric efflux pump, are not present on blood cells or platelets. The effects of mutations are therefore considered to be because of the relative toxicity they possess in high levels to cellular membranes [58, 59]. Interestingly, in ABCG5/ABCG8 knock-out mice, which are fed on high-plant-sterol diet, platelets are noted to be hyper-activatable and show impaired function [60].
One of the most recently discovered genes known to cause inherited thrombocytopenia is SLFN14. SLFN14 has recently been identified as an endoribonuclease, functioning to destroy mRNA, which cannot be correctly translated, and causes degradation of ribosomal subunits [61]. Three consecutive heterozygous missense mutations were identified in three unrelated families predicted to encode substitutions K218E, K219N and V220D [62]. Alongside the reduced number of platelets, they also show platelet dysfunction and an increased immature platelet fraction, a noninvasive measure indicative of increased platelet clearance. However, the exact mechanism through which mutations in SLFN14 mediate thrombocytopenia and aberrant platelet function is unknown.
There is similar lack of knowledge for the molecular effect of one of the most recently reported causative genes of IT, GNE, Mutations within the gene encoding Glucosamine (UDP-N-Acetyl)-2-Epimerase/N-Acetylmannosamine kinase (GNE) are most noted as the molecular cause of Sialuria (OMIM#269921) and Hereditary Inclusion Body Myopathy (HIBM; OMIM#600737) [63, 64]. Recently, however, two related individuals presented with GNE myopathy associated with congenital thrombocytopenia [65]. Both patients suffered from a severe reduction in platelet count; however, the hematological effect of mutations within GNE has yet to be suggested.
Clearly, it is not solely platelet production that is affected by deleterious mutation. Lifespan can be inadvertently altered by pre-activation, the production of aggregated multimers and by toxicity. However, with platelet lifespan so intrinsically controlled by levels of pro-apoptotic proteins versus pro-survival proteins, why is it that no variants have thus far been discovered in this specific pathway? For us to understand this further, we need to consider how platelet lifespan is controlled and how it could theoretically be disturbed.
In platelets, it was long suggested that the Bcl-2 family of pro-survival proteins was required to suppress the pro-apoptotic signals of BAK and BAX [66]. It was Mason et al. who first established the link of degradation of Bcl-XL (encoded in humans by BCL2L1) to platelet death [67]. This subsequent loss of the restrained pro-apoptotic signal was therefore suggested as a molecular clock for platelet lifespan, an idea which was pioneered by the group of B. Kile. [67]. Megakaryocyte-specific deletion of Bcl-xL in mice does indeed profoundly reduce the platelet count to 2% of that observed in wild-type mice [68]. Additionally, platelet survival is markedly increased in Bak and Bax knock out and megakaryocyte-specific depleted mice [68].
So in a process so highly controlled by expression and protein levels, why are there not any variants in Bcl-xL or Bak/Bax that are known to cause thrombocytopenia?
There are two possible explanations. One is that loss-of-function variants in BCL2L1 are lethal. While Bcl-XL is involved in preventing mature megakaryocytes from apoptosis it is not essential alone for their growth and development, whereas presence of either Bcl-XL or Mcl-1, another member of the pro survival Bcl-2 family, is required for organism viability [69]. However, Bcl-XL’s functions are not limited solely to platelet lifespan and it follows a wide expression pattern both in adult and in embryonic tissue [70, 71]. Interestingly, Jak2, which is thought to positively regulate Bcl-XL expression in megakaryoblastic cells, is associated with decreased survival when selectively deleted in mouse hematopoietic cells [69, 72]. Therefore, at this moment in time, it is difficult to rule out the possibility that an organism-specific effect may be occurring within humans where genetic variations in BCL2L1 are simply not viable.
The other point to consider is the possibility that we haven’t determined a molecular cause of disease within BCL2L1 or any other genes involved in platelet lifespan simply due to their rarity. The most recent large-scale targeted genomics study into inherited thrombocytopenia revealed that only 50% of patients have a defined genetic etiology of disease [73]. With the progression of next-generation sequencing and the introduction of large-throughput whole exome and whole genome sequencing, new variants are being discovered in these previously undiagnosed patients [7, 62]. Inherited thrombocytopenias though, as a spectrum of disorders, are still fundamentally underdiagnosed. Yet, as our knowledge increases, we learn more about the effect of deleterious variants gaining insights into new mechanisms within platelet production, function and lifespan.
Acknowledgments
The work in the author’s laboratories is supported by the British Heart Foundation (PG/13/36/30275; FS/13/70/30521) and BJ is supported by an MRC Doctoral Training Grant (DTG) in Biomedical and Life Sciences. We thank Dr Steve Thomas for critical and constructive comments on our manuscript and Professor Ben Kile for his guidance regarding platelet lifespan.
Declaration of interest
The authors report no declarations of interest.
References
- Bernard J, SOULIER J. Sur une nouvelle variete de dystrphie thrombocytaire-hemorragipare congenitale. Sem Hop Paris. 1948;24:3217–3223. [PubMed] [Google Scholar]
- Ogawa M. Differentiation and proliferation of hematopoietic stem cells. Blood. 1993;81(11):2844–2853. [PubMed] [Google Scholar]
- Kaushansky K, Lok S, Holly RD, Broudy VC, Lin N, Bailey MC. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369(6481):568–571. doi: 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- Arinobu Y, Mizuno S, Chong Y, Shigematsu H, Iino T, Iwasaki H. Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell. 2007;1(4):416–427. doi: 10.1016/j.stem.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Tijssen MR, Ghevaert C. Transcription factors in late megakaryopoiesis and related platelet disorders. J Thrombosis Haemostasis: JTH. 2013;11(4):593–604. doi: 10.1111/jth.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A, Kramer DJ, Qin J, Ye D, Behlich AS, Mordecai S. Distinct, strict requirements for Gfi-1b in adult bone marrow red cell and platelet generation. J Exp Med. 2014;211(5):909–927. doi: 10.1084/jem.20131065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015;47(2):180–185. doi: 10.1038/ng.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunnacliffe A, Jones C, Le Paslier D, Todd R, Cherif D, Birdsall M. Localization of Jacobsen syndrome breakpoints on a 40-Mb physical map of distal chromosome 11q. Genome Res. 1999;9(1):44–52. [PMC free article] [PubMed] [Google Scholar]
- Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3):266–270. doi: 10.1038/73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson WS, Morel-Kopp MC, Chen Q, Liang HP, Bromhead CJ, Wright S. GFI1B mutation causes a bleeding disorder with abnormal platelet function. J Thrombosis Haemostasis: JTH. 2013;11(11):2039–2047. doi: 10.1111/jth.12368. [DOI] [PubMed] [Google Scholar]
- Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- Ihara K, Ishii E, Eguchi M, Takada H, Suminoe A, Good RA. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc Natl Acad Sci U S A. 1999;96(6):3132–3136. doi: 10.1073/pnas.96.6.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasouki MJ, Rafi SK, Olm-Shipman AJ, Wilson NR, Abhyankar S, Ganter B. Exome sequencing reveals a thrombopoietin ligand mutation in a Micronesian family with autosomal recessive aplastic anemia. Blood. 2013;122(20):3440–3449. doi: 10.1182/blood-2012-12-473538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songdej N, Rao AK. Hematopoietic transcription factor mutations and inherited platelet dysfunction. F1000prime Rep. 2015;7:66. doi: 10.12703/P7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvat-Switzer RD, Thompson AA. HOXA11 mutation in amegakaryocytic thrombocytopenia with radio-ulnar synostosis syndrome inhibits megakaryocytic differentiation in vitro. Blood Cells Mol Diseases. 2006;37(1):55–63. doi: 10.1016/j.bcmd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44(4) doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homans AC, Cohen JL, Mazur EM. Defective megakaryocytopoiesis in the syndrome of thrombocytopenia with absent radii. Br J Haematol. 1988;70(2):205–210. doi: 10.1111/j.1365-2141.1988.tb02465.x. [DOI] [PubMed] [Google Scholar]
- Fiedler J, Strauss G, Wannack M, Schwiebert S, Seidel K, Henning K. Two patterns of thrombopoietin signaling suggest no coupling between platelet production and thrombopoietin reactivity in thrombocytopenia-absent radii syndrome. Haematologica. 2012;97(1):73–81. doi: 10.3324/haematol.2011.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer EM, Harrison P, Savidge GF, Wilbourn B, Debili N, Vainchenker W. Uncoordinated expression of alpha-granule proteins in human megakaryocytes. Prog Clin Biol Res. 1990;356:131–142. [PubMed] [Google Scholar]
- Gunay-Aygun M, Falik-Zaccai TC, Vilboux T, Zivony-Elboum Y, Gumruk F, Cetin M. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet alpha-granules. Nat Genet. 2011;43(8):732–734. doi: 10.1038/ng.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahr WH, Lo RW, Li L, Pluthero FG, Christensen H, Ni R. Abnormal megakaryocyte development and platelet function in Nbeal2(-/-) mice. Blood. 2013;122(19):3349–3358. doi: 10.1182/blood-2013-04-499491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero JA, Bennett C, van der Weyden L, McKinney H, Chin M, Nurden P. Gray platelet syndrome: proinflammatory megakaryocytes and alpha-granule loss cause myelofibrosis and confer metastasis resistance in mice. Blood. 2014;124(24):3624–3635. doi: 10.1182/blood-2014-04-566760. [DOI] [PubMed] [Google Scholar]
- Larocca LM, Heller PG, Podda G, Pujol-Moix N, Glembotsky AC, Pecci A. Megakaryocytic emperipolesis and platelet function abnormalities in five patients with gray platelet syndrome. Platelets. 2015;26(8):751–757. doi: 10.3109/09537104.2014.994093. [DOI] [PubMed] [Google Scholar]
- Nurden AT, Nurden P. The gray platelet syndrome: clinical spectrum of the disease. Blood Rev. 2007;21(1):21–36. doi: 10.1016/j.blre.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Noris P, Perrotta S, Seri M, Pecci A, Gnan C, Loffredo G. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117(24):6673–6680. doi: 10.1182/blood-2011-02-336537. [DOI] [PubMed] [Google Scholar]
- Levin C, Koren A, Pretorius E, Rosenberg N, Shenkman B, Hauschner H. Deleterious mutation in the FYB gene is associated with congenital autosomal recessive small-platelet thrombocytopenia. J Thrombosis Haemostasis: JTH. 2015;13(7):1285–1292. doi: 10.1111/jth.12966. [DOI] [PubMed] [Google Scholar]
- Machlus KR, Italiano JE., Jr. The incredible journey: From megakaryocyte development to platelet formation. J Cell Biol. 2013;201(6):785–796. doi: 10.1083/jcb.201304054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender M, Thon JN, Ehrlicher AJ, Wu S, Mazutis L, Deschmann E. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood. 2015;125(5):860–868. doi: 10.1182/blood-2014-09-600858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima S, Kobayashi R, Itoh TJ, Hamaguchi M, Saito H. Mutation of the beta1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood. 2009;113(2):458–461. doi: 10.1182/blood-2008-06-162610. [DOI] [PubMed] [Google Scholar]
- Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann New York Acad Sci. 2013;1285:26–43. doi: 10.1111/nyas.12049. [DOI] [PubMed] [Google Scholar]
- Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood. 2015;126(13):1527–1235. doi: 10.1182/blood-2014-12-611012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles L, Bluteau D, Boukour S, Chang Y, Zhang Y, Robert T. MAL/SRF complex is involved in platelet formation and megakaryocyte migration by regulating MYL9 (MLC2) and MMP9. Blood. 2009;114(19):4221–4232. doi: 10.1182/blood-2009-03-209932. [DOI] [PubMed] [Google Scholar]
- Cheng EC, Luo Q, Bruscia EM, Renda MJ, Troy JA, Massaro SA. Role for MKL1 in megakaryocytic maturation. Blood. 2009;113(12):2826–2834. doi: 10.1182/blood-2008-09-180596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Naveiras O, Balduini A, Mammoto A, Conti MA, Adelstein RS. The May-Hegglin anomaly gene MYH9 is a negative regulator of platelet biogenesis modulated by the Rho-ROCK pathway. Blood. 2007;110(1):171–179. doi: 10.1182/blood-2007-02-071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinler KR, Shin JW, Lambert MP, Discher DE. Myosin-II repression favors pre/proplatelets but shear activation generates platelets and fails in macrothrombocytopenia. Blood. 2015;125(3):525–533. doi: 10.1182/blood-2014-05-576462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima S, Okuno Y, Yoshida K, Shiraishi Y, Sanada M, Muramatsu H. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Human Genet. 2013;92(3):431–438. doi: 10.1016/j.ajhg.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurden P, Debili N, Coupry I, Bryckaert M, Youlyouz-Marfak I, Sole G. Thrombocytopenia resulting from mutations in filamin A can be expressed as an isolated syndrome. Blood. 2011;118(22):5928–5937. doi: 10.1182/blood-2011-07-365601. [DOI] [PubMed] [Google Scholar]
- Jay D, Garcia EJ, Lara JE, Medina MA. de la Luz Ibarra M. Determination of a cAMP-dependent protein kinase phosphorylation site in the C-terminal region of human endothelial actin-binding protein. Arch Biochem Biophys. 2000;377(1):80–84. doi: 10.1006/abbi.2000.1762. [DOI] [PubMed] [Google Scholar]
- Manchev VT, Hilpert M, Berrou E, Elaib Z, Aouba A, Boukour S. A new form of macrothrombocytopenia induced by a germ-line mutation in the PRKACG gene. Blood. 2014;124(16):2554–2563. doi: 10.1182/blood-2014-01-551820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduini A, Malara A, Pecci A, Badalucco S, Bozzi V, Pallotta I. Proplatelet formation in heterozygous Bernard-Soulier syndrome type Bolzano. J Thrombosis Haemostasis: JTH. 2009;7(3):478–484. doi: 10.1111/j.1538-7836.2008.03255.x. [DOI] [PubMed] [Google Scholar]
- Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat. 2014;35(9):1033–1045. doi: 10.1002/humu.22607. [DOI] [PubMed] [Google Scholar]
- Kunishima S, Kashiwagi H, Otsu M, Takayama N, Eto K, Onodera M. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the alphaIIbbeta3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood. 2011;117(20):5479–5484. doi: 10.1182/blood-2010-12-323691. [DOI] [PubMed] [Google Scholar]
- Bury L, Falcinelli E, Chiasserini D, Springer TA, Italiano JE, Jr., Gresele P. Haematologica. 2015. Cytoskeletal perturbation leads to platelet dysfunction and thrombocytopenia in Glanzmann variants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Wang X. Cytochrome C-mediated apoptosis. Ann Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- Morison IM, Cramer Borde EM, Cheesman EJ, Cheong PL, Holyoake AJ, Fichelson S. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat Genet. 2008;40(4):387–389. doi: 10.1038/ng.103. [DOI] [PubMed] [Google Scholar]
- De Rocco D, Cerqua C, Goffrini P, Russo G, Pastore A, Meloni F. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim Biophys Acta. 2014;1842(2):269–274. doi: 10.1016/j.bbadis.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Avanzi MP, Izak M, Oluwadara OE, Mitchell WB. Actin inhibition increases megakaryocyte proplatelet formation through an apoptosis-dependent mechanism. PloS One. 2015;10(4):e0125057. doi: 10.1371/journal.pone.0125057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson EC, Burnett DL, Lebois M, Debrincat MA, White MJ, Henley KJ. Platelet production proceeds independently of the intrinsic and extrinsic apoptosis pathways. Nat Commun. 2014;5:3455. doi: 10.1038/ncomms4455. [DOI] [PubMed] [Google Scholar]
- Nurden P, Debili N, Vainchenker W, Bobe R, Bredoux R, Corvazier E. Impaired megakaryocytopoiesis in type 2B von Willebrand disease with severe thrombocytopenia. Blood. 2006;108(8):2587–2595. doi: 10.1182/blood-2006-03-009449. [DOI] [PubMed] [Google Scholar]
- Casari C, Du V, Wu YP, Kauskot A, de Groot PG, Christophe OD. Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2B-associated thrombocytopenia. Blood. 2013;122(16):2893–2902. doi: 10.1182/blood-2013-03-493312. [DOI] [PubMed] [Google Scholar]
- Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–494. doi: 10.1038/35097008. [DOI] [PubMed] [Google Scholar]
- Moake JL, McPherson PD. Abnormalities of von Willebrand factor multimers in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. Am J Med. 1989;87(3N):9N–15N. [PubMed] [Google Scholar]
- Tsai HM. Pathophysiology of thrombotic thrombocytopenic purpura. Int J Hematol. 2010;91(1):1–19. doi: 10.1007/s12185-009-0476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesin V, Wiley G, Kousi M, Ong EC, Lehmann T, Nicholl DJ. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci USA. 2014;111(11):4197–4202. doi: 10.1073/pnas.1312520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markello T, Chen D, Kwan JY, Horkayne-Szakaly I, Morrison A, Simakova O. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol Genet Metabol. 2015;114(3):474–482. doi: 10.1016/j.ymgme.2014.12.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse J, Braun A, Varga-Szabo D, Beyersdorf N, Schneider B, Zeitlmann L. An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J Clinic Invest. 2007;117(11):3540–3550. doi: 10.1172/JCI32312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science (New York, NY) 2000;290(5497):1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- Matsuo M. ATP-binding cassette proteins involved in glucose and lipid homeostasis. Biosci Biotechnol Biochem. 2010;74(5):899–907. doi: 10.1271/bbb.90921. [DOI] [PubMed] [Google Scholar]
- Su Y, Wang Z, Yang H, Cao L, Liu F, Bai X. Clinical and molecular genetic analysis of a family with sitosterolemia and co-existing erythrocyte and platelet abnormalities. Haematologica. 2006;91(10):1392–1395. [PubMed] [Google Scholar]
- Kanaji T, Kanaji S, Montgomery RR, Patel SB, Newman PJ. Platelet hyperreactivity explains the bleeding abnormality and macrothrombocytopenia in a murine model of sitosterolemia. Blood. 2013;122(15):2732–2742. doi: 10.1182/blood-2013-06-510461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisareva VP, Muslimov IA, Tcherepanov A, Pisarev AV. Characterization of novel ribosome-associated endoribonuclease SLFN14 from rabbit reticulocytes. Biochemistry. 2015;54(21):3286–3301. doi: 10.1021/acs.biochem.5b00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher SJ, Johnson B, Lowe GC, Bem D, Drake S, Lordkipanidze M. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clinic Invest. 2015;125(9):3600–3605. doi: 10.1172/JCI80347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seppala R, Lehto VP, Gahl WA. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am J Human Genet. 1999;64(6):1563–1569. doi: 10.1086/302411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–87. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- Izumi R, Niihori T, Suzuki N, Sasahara Y, Rikiishi T, Nishiyama A. GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromusc Disorders: NMD. 2014;24(12):1068–1072. doi: 10.1016/j.nmd.2014.07.008. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Claudio E, Rucker EB, 3rd, Riedlinger G, Broussard C, Schwartzberg PL. Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Dev (Cambridge, England) 2000;127(22):4949–4958. doi: 10.1242/dev.127.22.4949. [DOI] [PubMed] [Google Scholar]
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med. 2011;208(10):2017–2031. doi: 10.1084/jem.20110750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Hikita H, Kawaguchi T, Shigekawa M, Shimizu S, Hayashi Y. Mcl-1 and Bcl-xL regulate Bak/Bax-dependent apoptosis of the megakaryocytic lineage at multistages. Cell Death Different. 2012;19(11):1856–1869. doi: 10.1038/cdd.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels J, Kepp O, Senovilla L, Lissa D, Castedo M, Kroemer G. Functions of BCL-X L at the interface between cell death and metabolism. Int J Cell Biol. 2013;2013:705294. doi: 10.1155/2013/705294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB. Bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Dev (Cambridge, England) 1994;120(10):3033–3042. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- Grisouard J, Hao-Shen H, Dirnhofer S, Wagner KU, Skoda RC. Selective deletion of Jak2 in adult mouse hematopoietic cells leads to lethal anemia and thrombocytopenia. Haematologica. 2014;99(4):e52–e54. doi: 10.3324/haematol.2013.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduini CL, Pecci A, Noris P. Inherited thrombocytopenias: the evolving spectrum. Hamostaseologie. 2012;32(4):259–270. doi: 10.5482/ha12050001. [DOI] [PubMed] [Google Scholar]
- Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet. 2000;26(4):397–398. doi: 10.1038/82511. [DOI] [PubMed] [Google Scholar]
- Ware J, Russell SR, Vicente V, Scharf RE, Tomer A, McMillan R. Nonsense mutation in the glycoprotein Ib alpha coding sequence associated with Bernard-Soulier syndrome. Proc Natl Acad Sci USA. 1990;87(5):2026–2030. doi: 10.1073/pnas.87.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budarf ML, Konkle BA, Ludlow LB, Michaud D, Li M, Yamashiro DJ. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. Hum Mol Genet. 1995;4(4):763–766. doi: 10.1093/hmg/4.4.763. [DOI] [PubMed] [Google Scholar]
- Wright SD, Michaelides K, Johnson DJ, West NC, Tuddenham EG. Double heterozygosity for mutations in the platelet glycoprotein IX gene in three siblings with Bernard-Soulier syndrome. Blood. 1993;81(9):2339–2347. [PubMed] [Google Scholar]
- Burk CD, Newman PJ, Lyman S, Gill J, Coller BS, Poncz M. A deletion in the gene for glycoprotein IIb associated with Glanzmann’s thrombasthenia. J Clinic Invest. 1991;87(1):270–276. doi: 10.1172/JCI114982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus JC, O’Toole TE, Plow EF, Glass A, Frelinger AL, 3rd, Ginsberg MH. A beta 3 integrin mutation abolishes ligand binding and alters divalent cation-dependent conformation. Sci (New York, NY) 1990;249(4971):915–918. doi: 10.1126/science.2392682. [DOI] [PubMed] [Google Scholar]
- Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo Nigro C. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The. May-Heggllin/Fechtner Syndrome Consortium. Nat Genet. 2000;26(1):103–105. doi: 10.1038/79063. [DOI] [PubMed] [Google Scholar]
- Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78(4):635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. New England J Med. 2009;360(19):1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney KA, Nichols WC, Bruck ME, Bahou WF, Shapiro AD, Bowie EJ. The molecular defect in type IIB von Willebrand disease. Identification of four potential missense mutations within the putative GpIb binding domain. J Clinic Invest. 1991;87(4):1227–1233. doi: 10.1172/JCI115123. [DOI] [PMC free article] [PubMed] [Google Scholar]