

Summary
One of the most fascinating and exciting periods in my scientific career entailed dissecting the symbiotic relationship between two membrane transporters, the NADPH oxidase complex and voltage gated proton channels (HV1). By the time I entered this field, there had already been substantial progress toward understanding NADPH oxidase, but HV1 were known only to a tiny handful of cognoscenti around the world. Having identified the first proton currents in mammalian cells in 1991, I needed to find a clear function for these molecules if the work was to become fundable. The then-recent discoveries of Henderson, Chappell, and colleagues in 1987–1988 that led them to hypothesize interactions of both molecules during the respiratory burst of phagocytes provided an excellent opportunity. In a nutshell, both transporters function by moving electrical charge across the membrane: NADPH oxidase moves electrons and HV1 moves protons. The consequences of electrogenic NADPH oxidase activity on both membrane potential and pH strongly self-limit this enzyme. Fortunately, both consequences specifically activate HV1, and HV1 activity counteracts both consequences, a kind of yin-yang relationship. Notwithstanding a decade starting in 1995 when many believed the opposite, these are two separate molecules that function independently despite their being functionally interdependent in phagocytes. The relationship between NADPH oxidase and HV1 has become a paradigm that somewhat surprisingly has now extended well beyond the phagocyte NADPH oxidase -- an industrial strength producer of reactive oxygen species (ROS) -- to myriad other cells that produce orders of magnitude less ROS for signaling purposes. These cells with their seven NADPH oxidase (NOX) isoforms provide a vast realm of mechanistic obscurity that will occupy future studies for years to come.
Keywords: Eosinophils, Monocytes/Macrophages, Neutrophils, Phagocytosis, HVCN1, proton channel
Introduction
The topic of this review was essentially conceived in a seminal series of three papers published by Lydia Henderson, Brian Chappell, and Owen Jones at the University of Bristol in 1987–1988 (1–3). They discovered that NADPH oxidase in human neutrophils is electrogenic and that its activity is accompanied by electrogenic H+ efflux. They proposed that this H+ efflux serves to compensate for the charge separation resulting from NADPH oxidase extruding electrons and leaving protons behind in the cytoplasm, and was therefore necessary for sustained activity. Further, they proposed that the molecule responsible for the H+ efflux was a voltage-gated proton channel (HV1), the first voltage-clamp studies of which had been reported in snail neurons just five years earlier by Roger Thomas and Bob Meech, also in Bristol. Finally, because electron efflux was mediated by NADPH oxidase and H+ efflux by a separate molecule, the proton channel, a second proposed function of HV1 was to stabilize pHi by eliminating the protons left behind in the cell when electrons leave. These discoveries and their proposed explanations have all proven to be perfectly correct.
The decades since this primordial era have filled in many details of mechanisms, structures, functions, and unexpected intricacies. There have been some missteps and digressions along the way that might have seemed annoying or counterproductive at the time, but these did serve to reinforce and strengthen the eventual prevailing narrative. The present assigned task is to review the contributions of the author’s lab within the historical context of the field, and to speculate where future progress will occur. This is a perilous assignment for someone with a tendency toward autohagiography, but I will try to exercise equanimity!
The Transport Molecules: NADPH Oxidase
Electron flux via NADPH oxidase (NOX)
Phagocytes produce reactive oxygen species (ROS) primarily to attack invading bacteria, fungi, and protozoa. The entity responsible for this ROS production is NADPH oxidase, a multi-component enzyme complex that is not assembled in the cell until it is needed. NADPH oxidase is now called NOX, and it comes in seven varieties, NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2 (4). This nomenclature is arbitrary and imprecise, because the first discovered and by far the best known NOX is NOX2, the phagocyte NADPH oxidase. In addition, NOX specifically refers to the electron-transporting component of the enzyme (gp91phox in phagocytes (5, 6)), but in common parlance, the entire enzyme complex is often called NOX or NOX2.
The phagocyte NADPH oxidase enzyme complex (7) is formed from two membrane proteins: gp91phox (now rebranded as NOX2; the superscript phox refers to “phagocyte oxidase”) and p22phox that exist in membranes as a preformed heterodimer and four cytosolic proteins: p47phox, p67phox, p40phox that likely form a heterotrimer (8–12), and a small GTPase: Rac1 or Rac2 (Fig. 1). After assembly of the oxidase complex, a conformational change is thought to result in activation of the enzyme (13–16). Much has been learned about the importance and molecular composition of the enzyme from hereditary chronic granulomatous disease (17), CGD, in which a mutation in one of the components alters or eliminates function. Two-thirds of CGD cases exhibit mutations of gp91phox, one-fourth p47phox, 6% p22phox, 6% p67phox (18), and a single case has been shown to involve p40phox (19). ROS production by NADPH oxidase is impaired or abolished in CGD (20), bactericidal function is compromised (21), and patients present with chronic recurrent bacterial and fungal infections; without treatment, most die in childhood (12, 18). The existence of CGD directly demonstrates the importance of NADPH oxidase in innate immunity. In fact, CGD was first described as a “fatal granulomatous disease of childhood” (17, 22).
Fig. 1. Proton and electron fluxes into the phagosome.

The tan double line represents the phagocyte plasma membrane engulfing a bacterium into a nascent phagosome, which will seal off from the extracellular space and become internalized. The components of NADPH oxidase assemble in the phagosome membrane to produce an active complex, shown schematically. This enzyme removes two electrons from NADPH inside the cell and shuttles them across the membrane via an electron transport chain through gp91phox (Fig. 2) to reduce two O2 molecules sequentially to superoxide anion, O2•- (255, 256). O2•- rapidly dismutates into H2O2, most of which is converted by myeloperoxidase (MPO) (210) into hypochlorous acid (household bleach, HOCl) (42). Proton channels in the phagosome membrane (133–135, 217, 257) are activated by membrane depolarization and a drop in pHi, both resulting from the electrogenic activity of NADPH oxidase. NADPH consumed in this reaction is regenerated continuously by the hexose monophosphate shunt (HMS). [Adapted from (258)].
Fig. 1 depicts the stoichiometry of several key reactions (23–25) in the phagocyte respiratory burst. The term “respiratory burst” refers to the rapid generation of O2•- by NADPH oxidase during phagocytosis or stimulation by chemotactic peptides or other agents, and is a misnomer because the up to 100-fold increase in O2 consumption (26) is not really a “burst of extra respiration” (27), but rather the O2 serves as a substrate for O2•- production (28, 29). NADPH oxidase extracts two electrons from NADPH which traverse the membrane through gp91phox to reduce two molecules of O2 to superoxide anion, O2•-. For each NADPH oxidized, one proton is left behind. The product, NADP+ then enters the hexose monophosphate shunt (HMS) to regenerate NADPH (24, 29–31), in the process producing a second proton. This reaction occurs continuously, because otherwise the 50–100 µM free NADPH in a neutrophil would be depleted entirely within <100 msec (32). The importance of the HMS for ROS production is shown by the CGD-like symptoms of patients with glucose-6-phosphate dehydrogenase deficiency (33, 34). The HMS also generates one CO2 for every two NADP+ which could potentially form carbonic acid and release another proton. In sum, at least two protons are generated intracellularly for each NADPH consumed. Inside the phagosome, two O2•- dismutate spontaneously or through MPO-catalyzed reactions (35, 36) to form hydrogen peroxide, H2O2, a reaction that consumes two H+ and also produces O2. About half of the H2O2 is converted by myeloperoxidase (MPO) into the more reactive HOCl (37–42). HOCl has a pKa 7.53 (43) so that much exists in the undissociated, neutral, membrane-permeable form. Some enters the cytoplasm, exacerbating the acidification (44), but most of the HOCl kills engulfed microorganisms (36).
Is the NADPH oxidase complex activated by arachidonic acid or PKC or both?
A large number of stimuli lead to activation of NADPH oxidase, including opsonized microorganisms, chemotactic peptides, and an assortment of artificial agents (45). Depending on the stimulus, there is a lag of seconds to minutes between stimulus and response, presumably due to the multiple intervening biochemical processes that must occur (32). There are various signaling pathways, but most converge upon protein kinase C (PKC), which phosphorylates several cytoplasmic oxidase components, especially p47phox, leading to assembly of the complex (13, 46–49). The complex needs to be continually renewed or else deactivation occurs rapidly (32). A potent and highly reliable agonist for experimental purposes is the phorbol ester PMA (phorbol myristate acetate), whose best-known function is to activate PKC.
Much has been learned from study of NADPH oxidase in “cell-free systems” in which the enzyme is assembled de novo from its component parts (15). An important discovery arising from these studies was that in addition to the protein components and the substrates NADPH and O2, it is necessary to include an anionic amphiphile (50–54), such as the physiological unsaturated fatty acid, arachidonic acid (AA), the physiological combination of phosphatidic acid and diacylglycerol (55–57), or an unphysiological detergent SDS (sodium dodecyl sulfate) (58, 59). Indeed, it was well known that arachidonic acid and other unsaturated fatty acids could activate NADPH oxidase very effectively in intact leukocytes (60–63). Saturated fatty acids are less effective or ineffective and are inhibited by Ca2+ (61, 63–65). Furthermore, AA is released by most, but not all activators of NADPH oxidase (62, 66). One interpretation is that arachidonic acid acts as a “second messenger” to activate NADPH oxidase (62, 67). This hypothesis was supported by the observation that certain inhibitors of phospholipase A2 (PLA2) prevented activation of NADPH oxidase (62), even by PMA (68). However, more specific cPLA2α inhibitors [see Introduction of (69)] do not inhibit ROS production (70) and AA can itself activate PKC (67, 71). In addition, electron current was activated normally in cPLA2α knockout mouse neutrophils (69).
AA appears capable of acting as a phosphomimetic to activate NADPH oxidase. PKC phosphorylation of p47phox removes auto-inhibition, allowing the molecule to interact with the membrane-bound components of the complex (13). The conformational changes in induced by phosphorylation of p47phox also occur when AA or SDS are applied at sufficiently high concentrations (16, 72, 73). AA and other anionic amphiphiles also have effects on cytochrome b558 (the heterodimeric complex of gp91phox and p22phox) (14, 15, 74–77). The requirement for anionic amphiphile can be eliminated by enriching the membrane with anionic phospholipids (78) or by prenylation of Rac1 (79).
In intact cells, AA can activate PKC, which then activates the oxidase (80). This possibility was demonstrated directly in human eosinophils in which the electron current elicited by AA was completely abolished by addition of PKC inhibitors (69). Our general conclusion is that the respiratory burst can be activated by multiple pathways, but physiological pathways mainly involve PKC.
Is enhanced gating due to AA or phosphorylation?
The phenomenology of “enhanced gating” of proton channels during the respiratory burst is discussed in more detail below. Here we simply ask the parallel question whether HV1 is “activated” by AA or by PKC (or both). Analogous suggestive studies had supported AA as the final activator of HV1, as was explicitly proposed by Henderson and Chappell (81). Again, use of specific cPLA2α inhibitors failed to prevent enhanced gating of HV1, and the responses of neutrophils from cPLA2α knock-out mice to PMA or fMLF were indistinguishable from those of WT mice (69). The most startling, and to me most convincing discovery occurred after an irksome manuscript referee demanded a large number of new experiments. We grudgingly obliged but were pleasantly surprised when one experiment turned out very differently than we had expected. The surprising result was that the PKC inhibitors GFX (GF109203X) and staurosporine reversed both the activation of NADPH oxidase (i.e., the electron current went away) and the enhanced gating of HV1 resulting from stimulation by AA (69)! Evidently, AA produces much of its effects on both NADPH oxidase and HV1 in phagocytes by activating PKC (69). This was especially surprising because AA was already known to have profound direct “pharmacological” effects on HV1, being one of the few molecules in the universe that alters proton currents even in whole-cell configuration (82–86)! Intriguingly, the potency sequence for fatty acids amplifying HV1 currents (85) roughly parallels that for activating NADPH oxidase (62, 63, 65, 67, 87).
One of the classical indicators of enhanced gating in phagocytes is a slowing of tail currents, which reflect the rate channels close upon repolarization (88–93), but AA increases H+ current without producing this effect, accelerating τtail under conditions in which PKC is not activated (85, 94, 95). A dual pathway was suggested to explain the differential effects of AA and phosphorylation on deactivation (HV1 tail current) kinetics (69):
The “direct” pathway produces larger H+ currents, faster channel opening and closing, and a 15–20 mV hyperpolarizing shift of the gH-V relationship, and occurs in whole-cell configuration (82–86) or inside-out patch configurations (95). The indirect pathway produces the classical “enhanced gating mode” response that reflects activation of PKC (69) and consequent phosphorylation of the channel (*HV1) at Thr29 (96), and is sensitive to PKC inhibition. PMA-induced enhanced gating produces larger currents and faster opening, but also includes profound slowing of τtail and a -40 mV shift of the gH-V relationship. Both direct and indirect effects of AA occur in perforated-patch configuration (69, 94), and likely also in intact cells. In intact cells, AA liberated by cPLA2 may augment H+ current by direct effects (86, 95), in addition to producing phosphorylation-mediated effects. HV1 closing (τtail) is slower not only in PMA-stimulated phagocytes, but also after physiological agonists like fMLF (69), suggesting that phosphorylation rather than AA is the main cause of physiological enhanced gating. Similarly, leukotriene B4 stimulation of human eosinophils appears to slow tail currents (97). Spontaneously enhanced gating, due presumably to adherence, is inhibited by the PKC inhibitor, staurosporine (69). Enhanced gating produced by lipopolysaccharide in dendritic cells requires PKC, and is prevented by GFX (98). All things considered, it is likely that AA introduced exogenously or released by cPLA2 (95) produces both direct and indirect effects in intact phagocytes, (69), but the main stimulus for enhanced gating of hHV1 is phosphorylation.
History of the demonstration of electron current
As mentioned above, in 1987 and 1988, Henderson, Chappell, and Jones published their landmark studies (1–3) showing that NADPH oxidase is electrogenic and proposing that voltage-gated proton channels exist in phagocytes and are required for sustained NADPH oxidase activity to compensate both charge and pH. They further proposed that AA was the physiological activator of HV1 (81). Eager to demonstrate a biological function for HV1, we obtained human neutrophils, usually our own, and confirmed that they did have proton channels with properties consistent with their proposed functions (82). From that moment onwards, every paper and talk included the hypothetical role of proton channels in compensating for the electrogenic activity of NADPH oxidase. But it never occurred to me to measure the electron current directly! Krause and colleagues were clever enough to attempt this, and they reported electron currents in human eosinophils (99). We worked very hard to reproduce these results in neutrophils. Only later did we realize that because NADPH oxidase activity is several times lower in neutrophils than in eosinophils (100–107), the electron currents in neutrophils are quite small indeed (Table 1), making their detection challenging! The cellular levels of various NADPH oxidase components appear to be similar in human eosinophils and neutrophils (107, 108), leading several authors to ascribe the more vigorous respiratory burst measured in human eosinophils mainly to the tendency of the oxidase to assemble predominantly in the plasma membrane in eosinophils, but intracellularly in neutrophils (105, 108). The rationale is that neutrophils mainly engulf small pathogens, whereas eosinophils (109) are more ambitious and attack helminthes many times their own size (110) by generating extracellular ROS (105), in the process inflicting tissue damage (111) and thereby being generally pro-inflammatory (112).
Table 1.
Electron Currents in Mammalian Phagocytes and Other Cells
| Cell | Species | Stimulus | °C | pHo | pHi | Ie (-pA) | -pA/pF | Reference |
|---|---|---|---|---|---|---|---|---|
| Neutrophil | Human | PMA | 20 | 7.0 | 7.0 | 2.3 | - | (91) |
| Neutrophil | Mouse | PMA | 22.5 | 7.0 | 7.0 | 2.6 | - | (69) |
| Neutrophil | Mouse | fMLF | 22.5 | 7.0 | 7.0 | 3.3a | - | (69) |
| Neutrophil | Mouse | PMA | 23 | 7.0 | 7.0 | 1.2 | - | (44) |
| Neutrophil | Mouse | PMA | 22.5 | 7.0 | 7.0 | 2.37 | - | (130) |
| Eosinophil | Human | none | 23.5 | 7.1 | 7.6 | - | 6.26b | (99) |
| Eosinophil | Human | PMA | 21 | 7.0 | 7.0 | 6.0 | - | (89) |
| Eosinophil | Human | AA | 21 | 7.0 | 7.0 | 7.4 | - | (94) |
| Eosinophil | Human | PMA | 31.1 | 7.0 | 7.0 | 19.8 | - | (140) |
| Eosinophil | Human | PMA | 33.9 | 7.0 | 7.0 | 30.5 | - | (140) |
| Monocyte | Human | PMA | 25 | 7.0 | 7.0 | 8.3 | - | (92) |
| Osteoclast | Mouse | PMA | 22 | 7.3 | 7.3 | 8.4 | - | (266) |
| COSphox | Monkey | AA | 21 | 7.0 | 7.0 | 0.26 | - | (198) |
| Dendritic | Mouse | LPS | RTc | 7.1 | 7.6 | - | 0.16 | (98) |
Mean electron current in 7 of 12 cells tested. Cells that failed to respond to fMLF did subsequently respond to PMA.
The pipette solution included 8 mM NADPH as substrate.
RT is room temperature.
For this table, Ie,max is simply the largest electron current reported or depicted in each study. If no mean value was provided, a value extracted from a figure is given. Some authors normalize the currents according to the size of the cell, whose membrane surface area is indicated by the capacity (in pF); granulocytes typically have a capacity of 2–3 pF (82, 83, 99, 211, 267). Cells are freshly isolated or in primary culture, except the COSphox cells are the COS-7 cell line with the four required NADPH oxidase components transfected in. When a temperature range is reported, the mean value is given.
We could not see any effect of PMA on neutrophils in whole-cell recordings. Therefore we used the “perforated-patch” approach in order to preserve intracellular constituents – conventional whole-cell methods rapidly dialyze diffusible cytoplasmic contents into the pipette (113). Including a pore-forming molecule like amphotericin in the pipette makes the membrane patch permeable to small ions, but not larger molecules, enabling electrical access to the entire cell membrane while preserving other cytoplasmic constituents and second messenger pathways (114–116). The electron currents in neutrophils were only 2–3 pA and they turned on slowly after stimulation of the cell with PMA, requiring 3 min to reach half their final value (91). The NADPH oxidase inhibitor DPI (diphenylene iodonium (117)) also works slowly, so the whole phenomenon comprised a very slow increase and then decrease of inward current. We had to see this happen in at least a dozen cells before we began to believe that it was real. Fig. 2 illustrates the time course of electron current turn-on and turn-off in human eosinophils, which have much greater NADPH oxidase activity than neutrophils, and thus larger electron currents (Table 1). Even in eosinophils, it is evident that an electrically “tight” cell (the nonspecific “leak” current was typically <2 pA) with a very stable baseline is required. To give some perspective, the name “gigaohm-seal” patch-clamp technique refers to a seal resistance between the pipette tip and membrane of at least 1 GΩ (1 gigaohm = 109 ohms); to have a 2-pA leak current at -60 mV requires a 30 GΩ seal. It was useful to record the entire experiment on a chart recorder, in addition to the usual patch-clamper’s “oscilloscope” or computer monitor equivalent which displays only a brief section of current data (typically one pulse) at a time. With a greatly compressed timebase, small excursions of the electron current from baseline appear more convincing (Fig. 2).
Fig. 2. Activation of electron currents in three human eosinophils by AA and PMA.

The membrane was held by voltage-clamp at -60 mV throughout. The current is interrupted when test pulses were applied. At the arrows, 5 µM AA, 60 nM PMA, or 6 µM DPI were introduced by complete bath change. Electron current is inward (a downward deflection relative to baseline) because the direction of current flow is defined as the direction positive charge moves. Electrons that leave the cell carry negative charge out, which by definition is an inward current. Electron current was activated spontaneously in the cell in A at the start of the record, likely by adherence to the glass chamber, as confirmed when the inward current was inhibited by DPI. [From (94)]
NADPH oxidase is electrogenic because it functions by moving electrons across the membrane along a redox chain (Fig. 3). This process must be sensitive to membrane potential, and from other electrogenic transporters, we knew that the voltage dependence might be highly non-linear (118, 119). For these measurements, we used human eosinophils, because their electron currents are larger (Table 1). We applied voltage ramps, which we had found to be very useful for studying single channel currents. After repeating the ramp many times, one may accumulate and average sections of currents with one channel open or sections with all channels closed and subtract one from the other. The software was written by Gary Yellen (then at Harvard), who gave it to us when I was in Mike Cahalan’s lab at the University of California at Irvine, in the elder days when scientists actually helped each other instead of competing viciously! To measure electron currents, we applied repeated voltage ramps, added PMA, and waited for the electron current to turn on. Later, addition of DPI gradually inhibited the oxidase and eliminated the electron current, confirming its reality. In principle one could subtract “before PMA” from “after PMA” curves, but we found the most reliable method was to subtract the current in DPI from that at the peak response. It is not necessary to wait until the response is completely inhibited, which may take minutes (Fig. 2) because the shape of the difference curve was the same whether inhibition was partial or complete. This was a useful control, because it confirmed that the voltage dependence was genuine and invariant.
Fig. 3. Electron transport chain in NADPH oxidase.

This enzyme removes two electrons (e1 and e2) from NADPH inside the cell and shuttles them across the membrane via an electron transport chain whose chemical midpoint potentials are shown that includes flavine adenine dinucleotide (FAD) and two hemes, sequentially reducing two O2 molecules to superoxide anion, O2•- (255, 256). Both electrons pass through the same two hemes, but at different times and with different energetics. The first electron follows the lower path, the second the upper path. All components of this reaction including the binding sites for NADPH and O2 are contained within gp91phox, the catalytic subunit of NADPH oxidase. [Figure originally created by Andrew R. Cross, reprinted in (256)]
The voltage dependence that we found (Fig. 4) was interesting in three respects. First, it showed that depolarization to +200 mV abolished the electron current completely. Although something like this was in general expected, it is still an amazing thing to realize that an enzyme can be turned off simply by changing the voltage across the membrane in which it resides. We toyed with the idea of trying to interpret this voltage, perhaps in terms of the midpoint potentials of the electron transport chain (Fig. 3), but then realized that these are measured under standard conditions, which certainly did not apply to our measurements. We also considered whether there ought to be a “reversal potential” beyond which outward current might be observed. Measurement of the reversal potential is de rigueur when studying a new ion channel and its value is predictable from the Nernst potentials of the permeant ions present. But in thinking about what we were actually measuring, namely electron flux across the membrane culminating in the reduction of O2 to O2•-, it seemed clear that the final step must be irreversible.
Fig. 4. The voltage dependence of electron currents in human eosinophils shows that NADPH oxidase cannot work at high voltages.
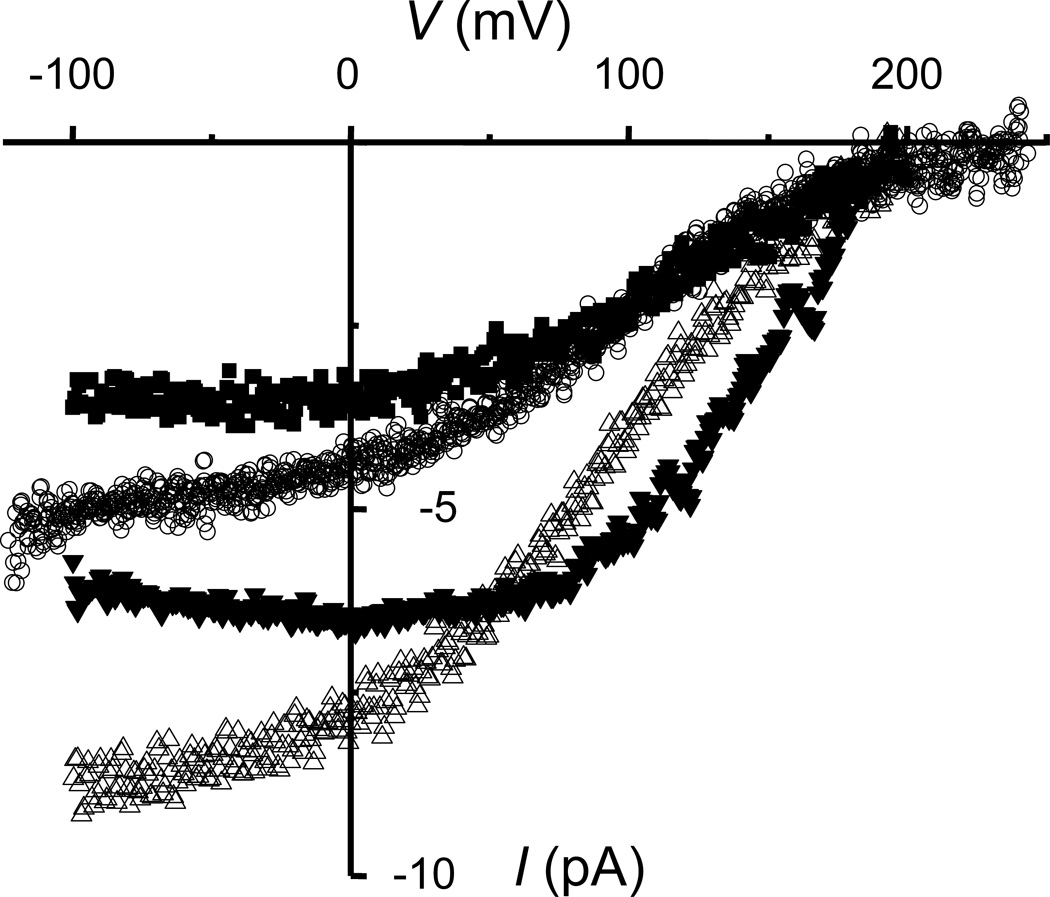
The electron currents in four human eosinophils measured over a range of membrane potentials (V) are shown. The electron current amplitude is directly proportional to NADPH oxidase activity, because each electron translocated produces one O2•- molecule. Voltage ramps were applied and the current after DPI addition was subtracted from the current before to obtain net electron current. The electrogenic nature of NADPH oxidase inherently causes depolarization that without compensation would terminate its own function within 10 msec in a human eosinophil (45). [From: (120)]
The second intriguing aspect of the electron current-voltage curve (Fig. 4) was that between +50 and +200 mV it is strongly voltage dependent. This means that the movement of electrons across the membrane in this voltage range is rate-limiting. Because the heme-1 to heme-2 transfer is uphill energetically (Fig. 3), and because the hemes are located within the membrane perpendicular to its surface (6), we proposed this step to be rate limiting in this voltage range (120). In previous studies using cell-free reconstitution systems (in which the membrane potential is almost certainly 0 mV), the rate-limiting step had been considered to be electron transfer from NADPH to FAD (121) or to heme (122). Petheő and Demaurex (123) showed that the curvature of the Ie-V curve (Fig. 4) is somewhat less pronounced at higher-than physiological NADPH concentrations, suggesting that the flat part of the curve at negative voltages may reflect limitation due to substrate availability.
The third feature is that the electron current is nearly constant over the entire voltage range encountered by a living neutrophil, from the resting membrane potential of roughly -60 to -80 mV [numerous estimates are discussed on p. 541 of (45)] up the voltage attained at the peak of the respiratory burst, which is roughly +50 mV (124–126). This extensive region of weak voltage dependence is teleologically useful, because it means the NADPH oxidase is free to depolarize the membrane by 100 mV or so before it begins to seriously limit its own activity. The job of HV1 is to short-circuit the depolarization at that point, because beyond +50 mV enzyme activity is drastically reduced (Fig. 4). Numerous studies have confirmed that inhibiting HV1 with Zn2+ or genetic KO (knock-out) of HV1 increases the depolarization that occurs during the respiratory burst (1, 88, 126–130), consequently attenuating ROS production (2, 120, 129–138). Finally, a quantitative electrophysiological model of NADPH oxidase and HV1 in phagocytes (42), to be discussed below, reproduces this and other observed electrophysiological behavior in great detail (Fig. 7 and Fig. 10, below).
Fig. 7. Minuscule NADPH oxidase activity suffices to depolarize the membrane rapidly.

The membrane depolarization response of human neutrophils stimulated with PMA or fMLP (now called fMLF, formyl Met-Leu-Phe, a chemotactic peptide) reported by Rada et al. (126) is highly non-linear. Movement of a small number of charges across the membrane produces rapid depolarization toward the Nernst potential for H+. A quantitative model of the electrical responses of human neutrophils including HV1 and NADPH oxidase predicts precisely this kind of response (dashed line). The depolarization is strongly blunted as HV1 open. [Figure from (42)]
Fig. 10. Depolarization during the respiratory burst in model or reality is greatly exacerbated by inhibiting hHV1 with Zn2+.

The model of the neutrophil respiratory burst, described in Fig. 7, here predicts depolarization of the membrane potential, Vm, during a PMA-stimulated respiratory burst. There is rapid depolarization to just beyond 0 mV, then slower depolarization. The dashed and solid lines show the predicted membrane potential response with (‘PMA’) or without enhanced gating. Enhanced gating enables hHV1 to compensate for electron efflux more efficiently, requiring less depolarization, and consequently, less self-inhibition. The Zn2+ curve shows the extreme depolarization resulting from inhibiting HV1. The BK curve shows that a single open BK channel would clamp the membrane potential near the Nernst potential for K+, in contrast to the 100 mV depolarization observed in real human neutrophils (124–126). The inset shows actual data recorded by G. L. Petheő and N. Demaurex in a membrane patch from a human eosinophil with activated NADPH oxidase. The observed depolarization is massively increased by Zn2+. NBT is nitroblue tetrazolium, which reacts with O2•-. [From (42)]
When the electron current is just starting to turn on, one always has nagging doubts that perhaps the cell is just becoming leaky – electron currents have no time-dependence, and one current is indistinguishable from another. In contrast, most voltage-gated ion channels turn on during a voltage pulse with a highly distinctive and immediately recognizable time course (their signature). In order to maximize the information available, when we apply a stimulus, we always give test pulses that activate proton current (Fig. 5, right panel), so we can see HV1 properties changing as it enters the “enhanced gating mode” (a kind of Dr. Jekyll and Mr. Hyde transformation that massively increases the proton flux under any given conditions) that will be described later. The first change is usually a progressive increase in the H+ current amplitude together with faster activation (smaller τact). The electron current typically turns on later and more slowly (89). The onset of enhanced gating mode behavior was a strong indicator that a cell was becoming “activated” and that any inward current that appeared was likely electron current. Nonphysiological changes in the holding current – i.e., current at the negative “holding potential” that simulates the resting membrane potential – commonly do occur over time during experiments, and apparent electron current might simply be an increase in the “leak” current. However, a crucial difference is that leak current is nonspecific and reverses near 0 mV, whereas electron current is inward all the way up to +200 mV. This means that the electron current at a depolarizing test potential (for example +60 mV in Fig. 5) will be inward even though the proton current is outward. As a result, the current just at the start of the test pulse, before the proton channels begin to open, drops lower when real electron current turns on, due to superposition of inward and outward currents, as can be seen in Fig. 5 by comparing current “a” at rest and “b” after PMA. If the -10 pA of inward electron current had been leak, the current at the start of the pulse would have been more outward (higher). This indication of inward current at a large positive voltage unambiguously identifies electron current. An independent and rigorous test of the reality of electron current is to add DPI or GFX and see it disappear.
Fig. 5. The time course of the appearance of electron current and enhanced gating of proton current.

A human eosinophil studied in the perforated-patch configuration was stimulated with PMA and then treated with 3 µM GFX. The spikes in the upper left record are proton currents during steps to +60 mV applied every 30 s from the holding potential, -60 mV. The lower record shows the same current at -60 mV at higher gain, with the currents during the test pulses blanked, revealing the time course of inward electron current. On the right are proton currents during selected pulses to +60 mV with an expanded time-base, recorded in this experiment at the times indicated in the left panel by lowercase letters. Note that both enhanced proton current and electron currents were reversed by the PKC inhibitor GFX; evidence that both are regulated by PKC. [From: (69)]
The ability to detect electron current in a single cell makes it possible to measure properties of NADPH oxidase in new ways, using each cell as its own control. For example, Fig. 6A shows the strong effects of pH, changed symmetrically, on electron current amplitude. Fig. 6B illustrates the agreement between the mean normalized electron current amplitude plotted as a function of pH (blue stars) and previous measurements of pH effects on the rate of superoxide anion production by NADPH oxidase from human neutrophils. NADPH oxidase has a pH optimum near neutral, and is strongly inhibited by pH deviations in either direction. With the patch-clamp we could also vary pH asymmetrically, which showed clearly that the pH effects were due to pHi not pHo (139).
Fig. 6. The pH dependence of NADPH oxidase activity measured biochemically or electrophysiologically is similar.

(A) Electron current was elicited by PMA stimulation of a human eosinophil in perforated-patch configuration. The pH was changed symmetrically using symmetrical 50 mM NH4+ to equilibrate pH across the membrane (91, 259). (B) The relative amplitude of the electron current in many cells is plotted as blue stars with SEM bars (n = 4–38). Also plotted using different symbols are biochemical measurements of O2•- production by human neutrophils in cell-free systems (52, 260–265), in which pH is presumably symmetrical. [A and blue stars in B from (139)]
It was straightforward to vary the temperature during an experiment, which revealed surprisingly strong temperature dependence of electron current, which is evident in Table 1, with Q10 4.2 (140).
Directly-recorded electron currents were recently reported for another heme-containing membrane protein, SDR2 (141). To date, no reports exist of electron currents in other NOXes, although they presumably exist.
The scale of the phagocyte respiratory burst
The rate and quantity of O2•- production by NADPH oxidase during the respiratory burst is truly prodigious. Expressed in electrical terms, the electron current in a human neutrophil would depolarize the membrane by 100 mV in just 91 msec (45), if uncompensated. Given the strong inhibition of NADPH oxidase by depolarization (Fig. 4), the enzyme would thereby shut itself off in a fraction of a second. Because each electron that exits the cell leaves a H+ inside, the same electron flux would, in effect, acidify the cytoplasm, but much more slowly due to the high buffering capacity of cytoplasm. This temporal disparity between the two main consequences of NOX activity caused us to suspect that the main purpose of HV1 during the respiratory burst was charge compensation, as discussed below. However, inhibiting HV1 or knocking it out genetically results in failure of pHi recovery from a spike of acidification that occurs during phagocytosis (44), demonstrating that pHi maintenance by HV1 is also necessary. With HV1 eliminated, pHi in phagocytosing human neutrophils drops to levels that directly inhibit NADPH oxidase (139), completing the circle.
The turnover rate of the electron transport chain in NADPH oxidase is 300–330 electrons s−1 (142, 143). Multiplying this by the elementary charge (1.6 × 10−19 C) gives a single-NOX2 electron current of 50 aA (1 Coulomb = 1 ampere for 1 second; 1 attoampere = 10−18 amperes). The single HV1 channel conductance extrapolated to physiological pH and body temperature is 78 fS (144) which at +58 mV, the level of depolarization achieved in intact neutrophils (124–126), and assuming ΔpH = 0.2 (pHo 7.4, pHi 7.2), would result in a single-channel H+ current of 5.4 fA (1 femtoampere = 10−15 amperes), corresponding to the translocation of 34,000 H+ s−1. Thus, one proton channel can fully compensate for the electron flux through 100 NADPH oxidase molecules. Rough calculations indicate that there are 20 NADPH oxidase complexes per HV1 in human neutrophils (45).
It is difficult to appreciate how few electrons need to cross the membrane to produce distinct depolarization. The extrusion just 3 amoles (1 attomole = 10−18 moles), or 2 × 106, of uncompensated electrons will depolarize the phagocyte membrane by 100 mV. This represents <0.01% of the 3 × 1010 electrons translocated by a human eosinophil during the respiratory burst (42). One consequence is that a number of careful studies published between 1978 and 1983 concluded that membrane depolarization was the trigger for the respiratory burst, because measurable depolarization preceded detectable superoxide production (145–151). Of course, thanks to the incisive studies of Henderson, Chappell, and colleagues (1–3), we now know that the opposite is the case – depolarization is a direct consequence of the electrogenic activity of NADPH oxidase. The number of electrons needed to change the membrane potential is simply much smaller than the number required to produce detectable quantities of ROS.
Another manifestation of this phenomenon is illustrated in Fig. 7. Erzsébet Ligeti’s group in Budapest measured membrane depolarization in human neutrophils upon NADPH oxidase activation, varying enzyme activity over a wide range by using several concentrations of the inhibitor DPI. They found that a tiny level of enzyme activity sufficed to produce a large depolarization (126). Ricardo Murphy produced a quantitative model of the phagocyte respiratory burst, in which every parameter was measured in actual cells (42). This model (“model” in Fig. 7), which includes only proton and electron currents, predicts precisely this extraordinary sensitivity of membrane potential to small levels of electron current (i.e., NADPH oxidase activity). In addition, the model predicts a biphasic response, precisely like the data. The membrane “rapidly” depolarizes, but then the response flattens out. Depolarization up to 0 mV is facile, because no ion channels are open in this range. The input resistance of human eosinophils, measured in perforated-patch configuration with K+-containing solutions is 53 GΩ between -60 and 0 mV (42). Above 0 mV, HV1 channels begin to open and this dampens any further depolarization, which is, after all, one of the primary functions of HV1 in phagocytes!
The Transport Molecules: the Voltage Gated Proton Channel (HV1)
The molecule and gene
In 1972, John Woodland “Woody” Hastings became the first human being to conceive the idea of a voltage gated proton channel. Assimilating a variety of extant data, he proposed that such a molecule could trigger the bioluminescent flash in certain dinoflagellates (152). In 2003, 31 years later, I wrote what I believed to be a comprehensive, if not exhaustive, 105-page review with 1120 references on voltage-gated proton channels and related molecules. After its publication I received a very polite note from Woody Hastings, in which he said that I might be interested to learn that he had considered the idea of such a channel well before proton currents were discovered in snail neurons. “Many years ago I proposed that they [proton channels] are involved in the triggering of the flashing of dinoflagellates, and the evidence continues to mount in support of that.” Needless to say, I was chagrined, but immediately invited him to give a seminar, which he did. We discussed his ideas and I submitted a series of grant applications to address this idea, without success until years later during Obama’s ARRA economic stimulus. A small NSF grant supported a fabulous expedition to the wilds of the Texas coast to hunt the wily dinoflagellate, Noctiluca scintillans. To make a long story shorter, after a 4 decade hiatus, Woody’s hypothesis was finally strongly supported by the identification of a bona fide voltage gated proton channel gene in a nonbioluminescent dinoflagellate in Hastings’ final publication (153). More recently, we have confirmed another HV1 in a bioluminescent dinoflagellate species, Lingulodinium polyedrum (154).
In 1982, a decade after Hastings’ proposal, Roger Thomas and Bob Meech, without knowledge of this concept (155), independently discovered voltage-gated proton currents in Helix aspersa snail neurons (156). Byerly, Meech, and Moody carried out a thorough electrophysiological study (157) that defined the main properties of voltage gated proton channels in Lymnaea stagnalis in 1984, which was recapitulated in Helix pomatia by Doroshenko et al. (158). Also in 1984 Barish and Baud (working near Moody and Byerly at UCLA) reported proton currents in salamander eggs (159, 160), but then the field became dormant.
In 1991, while exploring new research areas for a grant renewal, I decided to study the effects of pH changes on chloride currents in rat alveolar epithelial cells. This seemed a logical approach, because one of my mentors, Otto F. Hutter of Glasgow University, along with Anne Warner, had famously demonstrated dramatic effects of high and low pH on chloride currents in frog skeletal muscle (161, 162). When I applied low external pH (pHo) the outward Cl− currents turned off during sustained depolarization (Fig. 8), but on repolarization there was a prominent inward tail current whose amplitude grew larger with prepulse duration, wildly inconsistent with the turn-off kinetics of the outward Cl− current. Given that the solutions contained mostly impermeant ions, the only possible explanation was a proton conductance, which had been discussed several years earlier in a journal club by Mark Estacion in the Cahalan lab. I was overjoyed at discovering the first proton currents in mammalian cells (163) and felt certain that NIH funding was assured. However, despite their “high-risk, high-reward” claims, the NIH categorically refused to support the only lab in the world studying this novel ion channel. Fortunately, the American Heart Association kept my tiny lab afloat until a few other groups entered the field, thereby establishing that I was not a crackpot.
Fig. 8. The first evidence for voltage gated proton channels in mammalian cells.

Currents in a rat alveolar epithelial cell during depolarizing pulses of various lengths are superimposed. Note that the outward current decays over time, but the inward tail current seen upon repolarization becomes progressively larger after longer pulses. The decaying outward current is Cl− current, but the inward current is H+ tail current that was activated, with a characteristically slow time course, by depolarization. No H+ current can be seen during the pulse to +80 mV, because pHi was 7.4 and pHo was 6.0, hence EH was +82 mV, very near the test voltage and thus providing little driving force. Unpublished data by the author.
During the next decade, we enjoyed working in a sparsely populated field, where the absence of competition allowed us to take the time to do each study at our own pace, to our own satisfaction. We systematically explored the behavior of HV1 over a range of pHo and pHi, learning that the strong shifts in the position of the gH-V relationship, which can be encapsulated in Vthreshold (i.e., the voltage at which the channel turns on) that had been noted by Byerly et al. (1984) could be described quantitatively as a strict dependence on the pH gradient, ΔpH, defined as pHo-pHi (164). This unique control mechanism, in which pHo and pHi are equally effective in shifting the gH-V relationship by 40 mV/Unit change in pH, results in the channel opening only when there is an outward electrochemical gradient for protons. The HV1 channel thus opens only when doing so will result in H+ leaving the cell, making it an ideal acid extrusion device. We studied the channel with heavy water and observed deuterium currents with intriguing properties, suggesting that the channel was not a simple water-filled pore like other ion channels (165). Varying the buffer concentration revealed that deprotonation of buffer was not a rate-determining step in permeation, and that highly buffered solutions (i.e., 100 mM buffer!) were necessary to control pHi (166). Using inside-out patches of membrane we found that lowering pHi as a powerful activator of HV1; lowering pHi by 1 unit accelerated channel opening 5-fold (167). We found that both the conductance (Q10 > 2) and the kinetics of gating (channel opening and closing, Q10 6–9) had stronger temperature dependence than almost any other ion channel (168). These and other unique properties of HV1 were recently ascribed to stabilization of the closed channel by cation-π interactions between tryptophan and arginine in the S4 helix (169). Its perfect proton selectivity led to the concept that HV1 was an ideal biological pH meter (170, 171); that measuring the reversal potential was a better indicator of the true pH than was the nominal pH of the solutions.
During this halcyon period, we discovered that the potent inhibition of HV1 by polyvalent cations, particularly Zn2+, was strongly inhibited at low pH (172). In retrospect it was surprising that this discovery was so belated; for 17 years everyone (ourselves included) had used divalent cations to inhibit HV1, and everyone had varied pH to show that the channel was proton selective. When we saw how weak Zn2+ was at low pH we thought at first that we had made a 100-fold error in diluting our 1 M ZnCl2 stock solution; at pHo 5, Zn2+ was 100 times less effective than at pHo 6 (Fig. 9)! But the result was real. By comparing the predictions of several models of competitive binding, we found that the competition between H+ and Zn2+ at different pH could be explained only if the Zn2+ binding site included two or more titratable groups with a pKa near that of histidine (172). Very simple 2- or 3-site models (Fig. 9 legend) could fit all the data simultaneously and required only one simple assumption: when Zn2+ is bound to the closed channel, it cannot open. When the human HVCN1 gene was identified seven years later its product, hHV1 had two His that were essential for Zn2+ binding (173), and the recent crystal structure of the mouse HV1 (thought to be in a closed state) actually has a Zn2+ bound at a site comprised mainly of these two His (174). Few things are more satisfying than making a prediction based only on electrophysiological data that is later verified by the gene and then the structure!
Fig. 9. The competition between H+ and Zn2+ can be explained if Zn2+ binds at a bidentate site comprising two groups with a p Ka 6.3 (e.g two histidines).

The mean relative slowing of the time constant for the turn-on of H+ current (τact) during a pulse, measured at pHo ranging 5–8 is plotted, with 1.0 being no effect. The curves are all drawn with one equation (Eq. A6 of (172)) using three fixed parameters (inset): pKM = the Zn2+ binding constant, pKa = H+ affinity, and a = a cooperativity factor. The model assumes that when Zn2+ is bound to its receptor on the H+ channel, the channel cannot open. The opening rate (1/τact) will be slowed by the factor (1-PZn), where PZn is the probability that the receptor is occupied by Zn2+. The ratio of τact in the presence of ZnCl2 to that in its absence will be simply (1-PZn)−1. Given these assumptions, τact is slowed by a factor of 2 at the KM of Zn2+. [From (172)]
A final fundamental measurement from this era revealed that single HV1 channel currents are so tiny that they can just barely be detected directly under the most favorable conditions, including teraohm (1012 Ω) seals. Our meticulous colleague, Richard A. Levis, one of the pioneers of patch-clamp circuit design, insisted on checking the data himself to be sure that we really had attained such a high resistance with the gigaohm (a mere 109 Ω!) seal technique. Under these extraordinary conditions, we could just detect ∼10 fA current steps that with a good imagination resembled single-channel currents (144), the smallest ever recorded directly. The unitary current amplitude can be determined more reliably by using current fluctuation (noise) analysis, and it is very small indeed (144). The data assimilated for this “noise” paper were collected over more than a decade. When we started an experiment by forming a seal between the pipette and cell membrane, before rupturing the patch to go to “whole-cell” configuration, we would first check for high resistance, stability, and absence of channels other than HV1, and if the patch met these criteria, we would abandon whatever experiment had been planned and instead do noise measurements. Taking all the time necessary to do things thoroughly and correctly was possible while there was little competition. All good things must come to an end and with the discovery of proton channel genes in 2006 (173, 175), the field immediately mushroomed. All facetiousness aside, this development has in fact turned out well, because people approach problems in different ways, and progress in the structure-function realm has been quite rapid, although I suspect that competition increases the error rate. From nearly total obscurity, the proton channel field has blossomed, and the GenBank now informs us that a huge variety of species have HV1 (176).
Several features emerge from Table 2, which lists only mammalian cells in primary culture (not cell lines) that have been found to have proton currents. Species differences in expression levels can be pronounced. In humans, T lymphocyte currents are tiny, 100 times smaller than in B cells, but murine T cells have large H+ currents. The large H+ currents in human sperm dwarf the almost nonexistent currents in mouse sperm. All leukocytes have proton currents, and these are generally large. H+ currents are smaller in human neutrophils than human eosinophils, and tiny in human myotubes. Human eosinophils have 10-fold more HV1 protein than human neutrophils (108). The seductive argument that the respiratory burst is larger in eosinophils than in neutrophils because of their greater ROS production does not account for basophils, which have large H+ currents but no respiratory burst. On the other hand, every leukocyte that exhibits a respiratory burst has HV1 (Table 1 is a subset of Table 2), and it has been observed that in every cell available for comparison, the magnitude of the H+ conductance is at least 10 times greater than adequate to compensate fully for the respiratory burst (177). In cells lacking a respiratory burst, HV1 obviously must perform other functions.
Table 2.
Proton Currents in Mammalian Phagocytes and Other Primary Tissue Cells
| Cell | Species | °C | pHo | pHi |
IH,max (pA) |
IH,max (pA/pF) |
Reference |
|---|---|---|---|---|---|---|---|
| Neutrophil | Human | 20.5 | 7.4 | 7.0 | 10 | - | (82) |
| Neutrophil | Human | 20 | 7.0 | 7.0 | 22a | - | (91) |
| Neutrophil | Human | RT | 7.4 | 7.3 | 107a | - | (211) |
| Neutrophil | Mouse | RT | 7.4 | 7.3 | 63a | - | (211) |
| Neutrophil | Mouse | 22.5 | 7.0 | 7.0 | 70 | - | (69) |
| Neutrophil | Mouse | 24 | 7.0 | 6.0 | - | 336 | (134) |
| Neutrophil | Mouse | RT | 7.5 | 6.5 | 58.6 | - | (135) |
| Neutrophil | Mouse | RT | 7.4 | 6.6 | - | 43 | (268) |
| Eosinophil | Human | 22.5 | 7.0 | 7.0 | 193 | - | (83) |
| Eosinophil | Human | 20 | 7.5 | 5.7 | - | 338 | (84) |
| Eosinophil | Human | 20.5 | 7.0 | 7.0 | 115a | - | (89) |
| Basophil | Human | 21 | 7.0 | 5.5 | 102 | - | (267) |
| Basophil | Human | 22.5 | 7.0 | 7.0 | 106a | - | (203) |
| Mast cell | Mouse | 32 | 7.3 | 7.3 | 12 | 2.4 | (184) |
| Macrophage | Mouse | 22 | 7.5 | 6.5 | - | 13.2 | (180) |
| Macrophage | Mouse | 22 | 7.5 | 6.8 | - | 11.1 | (85) |
| Macrophage | Rat | RT | 6.5 | 6.5 | 150 | - | (269) |
| Osteoclast | Rabbit | 22 | 7.5 | 6.0 | 267 | 6.7 | (185) |
| Osteoclast | Mouse | 22 | 7.3 | 7.4 | - | 3.4 | (266) |
| Monocyte | Human | 22 | 7.5 | 6.0 | 209 | 3.45 | (183) |
| Monocyte | Human | 21.5 | 7.0 | 7.0 | 175* | - | (92) |
| Microglia | Mouse | 21.5 | 7.4 | 5.8 | 275 | - | (270) |
| Microgliab | Mouse | 35 | 7.4 | 7.3 | 239 | 6.3 | (271) |
| Microgliab | Mouse | RT | 7.2 | 5.5 | 428 | 12.6 | (138) |
| Microglia | Mouse | 21.5 | 7.5 | 6.0 | 1488 | 52.2 | (272) |
| Microglia | Mouse | 21.5 | 7.5 | 6.0 | 1016 | - | (273) |
| Microglia | Rat | RT | 7.2 | 5.5 | 23 | 1.0 | (138) |
| Microglia | Rat | 23 | 7.3 | 6.8 | - | 5.4 | (274) |
| Microglia | Human | RT | 7.5 | 5.5 | 1200 | - | (138) |
| Dendritic cell | Mouse | RTc | 7.0 | 7.0 | - | 29.6 | (98) |
| T lymphocyte | Mouse | 24 | 8.0 | 6.0 | 294 | - | (137) |
| T lymphocyte | Human | 21.5 | 7.5 | 6.0 | 1.46 | 0.9 | (275) |
| B lymphocyte | Human | 21.5 | 7.5 | 6.0 | 170 | 94.7 | (275) |
| B lymphocyte CLL | Human | 21 | 7.0 | 7.0 | 135 | - | (224) |
| Fibroblast | Human | 24 | 7.4 | 6.2 | - | 1.1 | (276) |
| Alveolar epithelium | Rat | 20 | 7.0 | 5.5 | 334 | 27.3 | (163) |
| Alveolar epithelium | Rat | 20 | 7.0 | 6.5 | - | 17 | (164) |
| Sperm | Human | 24 | 7.4 | 5.5 | - | 104.3 | (277) |
| Sperm | Mouse | 24 | 7.4 | 5.5 | - | 3.6 | (277) |
| Chondrocyte | Cow | 21 | 7.0 | 6.0 | - | 7.2 | (278) |
| Myotube | Human | 21 | 7.33 | 7.0 | 19 | 0.1 | (279) |
Currents in cells stimulated with PMA (enhanced gating mode).
These microglia were in brain slices.
RT is room temperature.
For this table, IH,max is simply the largest current reported or depicted in each study. Some authors normalize the currents according to the size of the cell, whose membrane surface area is indicated by the capacity (in pF). Comparisons under different conditions are imprecise because proton currents are exquisitely sensitive to temperature (168, 280) and pH, and measurements were made at different voltages. When multiple pH were studied, the measurement nearest to physiological was selected. All cells are freshly isolated or in primary culture. When a temperature range is reported, the median value is given.
Fine, but what good are they?
What never changes is the need for funding, and credible relevance of proton channels to human health was needed. There were huge proton currents in rat type II alveolar epithelial cells, but it was not immediately clear why alveolar epithelial cells needed proton channels. The possibility that HV1 contributed to facilitated diffusion of CO2 across the blood-gas barrier in the lung was considered, but seemed improbable due to the lack of carbonic anhydrase in the alveolar subphase fluid (178). Consequently, we developed a renewed interest in the studies of Henderson and colleagues that led them to postulate the existence of voltage gated proton channels in human neutrophils (1–3, 81). We decided to test their hypothesis by voltage-clamping human neutrophils. There were small but unequivocal proton currents, whose properties were consistent with those required for sustaining NADPH oxidase activity (82). The same year, other labs reported proton currents in human cell lines (179, 180). Indirect evidence that demonstrated functional proton channels in human neutrophils came from Sergio Grinstein (181), including an exciting report that the activation of a conductive H+ efflux was defective in CGD neutrophils (182). Evidently, either the activation of H+ efflux was contingent on the assembly of NADPH oxidase, or the oxidase itself acted as a proton channel. Grinstein immediately ruled out the latter idea by patch-clamping monocytes from CGD patients that lacked functional NADPH oxidase, revealing proton currents of identical size and properties to those in normal neutrophils (183).
Despite the confirmation that proton channels were present in an increasing variety of cells and could be shown to affect pHi recovery (184–189), skepticism remained. Because HV1 channels turned on at such positive voltages (e.g., at +20 mV at symmetrical pH, pHo = pHi (164)), skeptics said they would never be activated in intact cells, most of which have negative resting membrane potentials. The BK potassium channel suffered the same aspersions for many years, but by now has been shown to play various roles in human cells. Two papers convincingly debunked this argument for HV1. Using human neutrophils loaded with voltage sensing fluorescent dyes, Geiszt et al. (124) and Jankowski and Grinstein (125) demonstrated that during the respiratory burst stimulated by either PMA or the chemotactic peptide fMLF, the membrane potential depolarizes massively, to +50 or +60 mV, and remains there for minutes. This showed that in their best-studied functional role, proton channels would certainly be activated just when they were needed most. The idea is that as NADPH oxidase-induced depolarization progressed, proton channels would continue to open until they permitted enough H+ efflux to perfectly balance the electron efflux, preventing further depolarization. Supporting this idea, divalent cations at concentrations that inhibit HV1 (1, 88, 126–128) and genetic removal of HV1 (129, 130) result in greater depolarization during the respiratory burst. Fig. 10 shows that the model predicts depolarization during the respiratory burst that is greatly exaggerated when HV1 is inhibited by Zn2+ and that both phenomena are observed in real phagocyte membranes (Fig. 10 inset).
Remarkably, this model further predicts that introducing other ion channels would disrupt the delicate balance between these two molecules (42). For example, a single open BK channel (‘BK’ trace in Fig. 10) would hyperpolarize the membrane potential toward EK, the Nernst potential for K+, which clearly does not happen; the respiratory burst produces depolarization (1, 88, 124–130, 145–147, 149–151, 190–192).
Will the real proton channel please stand up?
Despite having made what turned out to be brilliant and completely accurate predictions about the roles of proton channels in compensating charge and pH for an active NADPH oxidase (1–3), Henderson and Chappell noted in 1992 that arachidonic acid (AA) activated both O2•- production and the associated H+ efflux, speculating that “the H+ channel is a component of the NADPH oxidase complex” (81). The year 1995 produced a bombshell when Henderson and colleagues reported that the gp91phox component of NADPH oxidase was in fact the phagocyte proton channel (193). I immediately invited Lydia Henderson to visit my lab so we could witness this discovery ourselves, and get in on the ground floor of molecular and genetic studies. Six frustrating weeks of recording from control and gp91phox-transfected CHO cells produced nothing but a weak paper showing that CHO cells have small intrinsic proton currents (194). We never saw the large proton currents in transfected cells that Lydia had seen in her lab. There are always unlimited tedious explanations for things that do not work in science, so although we had become skeptical, we could not draw strong conclusions. It was not immediately clear how to test the idea further. A second visit by Lydia in September, 2000 produced identical non-results, so we abandoned the gp91phox transfected cells, and switched to exploring effects of AA on human eosinophils. It was great working with an AA expert who could tell if a newly-purchased bottle was off simply by the smell. With her supervision, we detected effects of AA at 1–10 µM concentrations (Fig. 2) (94), an order of magnitude lower than the 20–100 µM we had reported previously (82). Presumably, despite all precautions we had taken, most of the AA had been oxidized in our earlier work. We found that the effects of AA on proton currents were reminiscent of those of PMA, but with some distinct differences (94).
How could the “gp91phox is a proton channel” idea have been resurrected, after the CGD results of Grinstein and colleagues had so clearly disproved it? This was accomplished by a genuine and significant discovery gone awry. In 1999, Bánfi and a distinguished group of scientists in Geneva reported that whereas unstimulated human eosinophils have voltage gated proton channels like those in other cells, under conditions in which NADPH oxidase was active, a different type of H+ channel appeared (88). It turned on faster, at more negative voltages, and even appeared to be more sensitive to inhibition by Zn2+. Because it was seen only when NADPH oxidase was active, they concluded (logically enough) that it was either a part of, or closely related to, the active NADPH oxidase complex.
Improving on perfection: The enhanced gating mode
Meanwhile, we adapted the “perforated-patch” approach to study phagocytes stimulated to undergo a respiratory burst. After a decade of studying what seemed to be the most inert ion channel in the universe, we were astounded by the results. We had previously tested numerous agents in search of a channel blocker or any agonist that might trigger some kind of physiological response, but found very little. The perforated-patch method preserves intracellular contents (114–116), which diffuse out of the cell and into the pipette when studied with the standard whole-cell patch-clamp configuration (113). The response of phagocyte proton currents to PMA stimulation in perforated-patch configuration was magnificent (91). Changes occurred with a credible time course, they progressed over several minutes, and their magnitude was spectacular (Figs. 5, 11, 12). Other channels might exhibit a 5-mV shift in response to a physiological stimulus, or a 30% change in kinetics, but the proton channel kinetics – both opening and closing - changed 4–6-fold, opening becoming faster and closing becoming slower; the H+ current doubled or tripled in size; and the gH-V relationship shifted a massive -40 mV – so far that inward currents could often be elicited! This constellation of effects was eventually dubbed “enhanced gating mode” behavior (45), and has been seen with remarkable consistency in a variety of cells (Table 1 in (93)). It also was unquestionably the same phenomenon that had been reported by Bánfi and colleagues. They had proposed that enhanced gating reflected the appearance of a new, separate channel (namely a component of NOX). We argued that there is one type of proton channel, that it is converted to enhanced gating mode by PMA (presumably by phosphorylation), and that there was no need to postulate a second type of proton channel (89–91). There was no evidence of two populations of channels – the kinetics were single exponential, and the gH-V relationship could be fitted by a single Boltzmann distribution that simply shifted negatively as the PMA effects set in. There was no correlation between the amplitude of proton and electron currents, as should exist if they were mediated by the same molecule. These were somewhat indirect arguments, and in retrospect, not surprisingly, they did not convert many skeptics.
Fig. 11. Evidence that gp91phox is not a proton channel: convincing, but quirky.

Test pulses to +60 mV in a PLB cell with gp91phox (A) and one without (B), after addition of PMA. The H+ current amplitude increases to the same extent in both, showing that this response does not require gp91phox. Mysteriously, however, the slowing of τtail (the current decay upon repolarization after the pulse) evident in PLB91 in A does not occur in PLBKO. (C) The magnitude of PMA responses of the four parameters that define enhanced gating are compared in two cells lacking gp91phox (X-CGD are neutrophils from a patient with CGD; PLBKO are PLB-985 cells with gp91phox knocked out by gene targeting) and in three cells with intact gp91phox (PLB91 are PLBKO that had gp91phox restored; PMN are human neutrophils, EOS are human eosinophils). The relative change in the first three parameters (PMA/control) is referenced to the dashed line which indicates no response. Both τact and Vthreshold responded but the changes were larger with gp91phox. The presence of gp91phox was necessary for the τtail slowing, but not for the increase in proton current amplitude, IH. [From (90)]
Fig. 12. Final unequivocal evidence that gp91phox could not possibly be a proton channel.

(A) Phagocytes from HVCN1 deficient (KO) mice have no proton current. Currents for pulses to +100 mV before (left) and after (right) 150 nM PMA reflect time-independent leak. (B) Electron current elicited by PMA in the same cell at +20 mV, showing inhibition by 20 µM DPI, an NADPH oxidase inhibitor. (C) PMA response of proton currents in a WT mouse cell during pulses to +80 mV applied at 1 min intervals before and after PMA. (D) Electron current elicited by PMA at -40 mV in the same cell. That identical electron current is activated in cells lacking HVCN1 and detectable proton current shows that all components of NADPH oxidase, including the catalytic subunit gp91phox, are present and functional. [From (44)]
One of the arguments for two types of H+ channels was that the novel channel associated with NAPDH oxidase activity was apparently 20 times more sensitive to inhibition by Zn2+ (88). This measurement had been done by using different test pulses to activate the two putative populations of channels, a logical approach because they appeared to be low- and high-threshold channels. However, the mechanism by which Zn2+ inhibits H+ current by is not simple block, in which the current is scaled down proportionally at all voltages, but is mainly the result of shifting the gH-V relationship positively (172), reminiscent of the effects of many divalent cations on other voltage-gated ion channels (195). We used quantitative modeling to show that this mechanism (Zn2+ shifting the gH-V relationship positively) will produce precisely what had been observed, namely that currents during a smaller test pulse will appear to be inhibited more strongly, and to the extent reported (89). Still the field remained unconvinced.
Having twice failed to reproduce the gp91phox heterologous expression result, we had become quite skeptical, and being further intrigued by the discovery of a putative novel H+ channel (88), we approached Mary Dinauer who had developed a leukocyte cell line (PLB-985) in which gp91phox was stably knocked out and another with it knocked out and then reinserted (196). This seemed the most direct way to test whether gp91phox really was a proton channel or not. The PLB cells had nice, big proton currents, and they responded well to PMA in perforated-patch studies. The PLBKO cells (lacking gp91phox) had proton currents identical to those in control PLB cells, demonstrating once again that in resting cells, the presence or absence of gp91phox is entirely irrelevant (90). The responses to PMA in perforated-patch studies were convincing to us, but not quite as clear-cut as we had hoped (Fig. 11). The PMA-stimulated increase in H+ current amplitude was identical in cells with or without gp91phox which shows that gp91phox is not a proton channel. Normally, one assumes that the size of the current is directly proportional to the number of channels activated. To maintain the “two channels” idea in the face of this evidence requires believing that somehow the gp91phox-derived channels displace ordinary H+ channels, changing their properties but not the total current amplitude. However, some of the effects of PMA were clearly different in cells that lacked gp91phox. There was no change in τtail, reinforcing the idea that this effect is coupled to the induction of electron current (89, 91). In addition, the negative shift of the gH-V relationship was much smaller in PLBKO or CGD cells, less than half as large as in normal PLB cells, neutrophils, or eosinophils. Needless to say, skeptics remained.
We next attacked the problem from the opposite direction with another cell line from the Dinauer lab. The COS-7 (monkey kidney) cell line is used widely as a gene expression system. Dinauer’s group had stably transfected all of the essential NADPH oxidase subunits to produce COSphox cells, which were capable of ROS production, proving that all the key NADPH oxidase components, including gp91phox, were present and functional (197). These cells had no detectable proton currents in whole-cell or perforated-patch configurations (198). We felt that this was now incontrovertible evidence that the gp91phox protein was not a proton channel. In describing in detail the absence of time-dependent outward currents that could possibly have been ascribed to HV1, we mentioned with naïve honesty, that sometimes when cells become leaky (i.e., they die), a time-dependent outward current abruptly appeared. To our embarrassment, one recalcitrant reviewer forced us to include an example of this phenomenon – the appearance of big, ugly currents at the moment the cell membrane ruptures – we were forced to publish a figure showing what cell death looks like electrophysiologically!
Despite this further evidence that gp91phox was not a proton channel, the field went in the other direction – soon all of the NOX isoforms would be proclaimed to contain proton channels. Given that these incorrect papers have already been cited over 1000 times (Google Scholar), I will not list them here. The Journal of General Physiology invited four interested parties to write “Perspectives on the Identity of the Proton Channel Involved in the Respiratory Burst” (199–202). The debate refused to die!
Clues from the enhanced gating response
One reason for the stubborn persistence of the idea that gp91phox was a proton channel resulted from some still-poorly-understood aspects of the physiological interaction between these two molecules. A very peculiar and consistent finding in stimulated phagocytes was that electron current and the slowing of tail current decay (larger τtail) always occurred together (89, 91). As we watched superimposed test currents from pulses applied every 30 s, at first there would be just a hint of slowing of the tail current. The inward tail current would return to baseline more slowly, and would not quite return all the way. This marked the onset of electron current. As mentioned above, the slow turn-on and small amplitude of electron current made it difficult to be sure of. However, when electron current appeared right on cue with the slowing of tail currents, there was no mistaking it. Another phenomenon that was (and still is) hard to explain was that enhanced gating is less pronounced in cells lacking functional NADPH oxidase, such as CGD neutrophils (90), basophils (203), K562 cells (204), or B lymphocytes which do have NADPH oxidase but at very low levels (136). There is a distinct constellation of intermediate enhanced gating effects in these cells: the increased H+ current amplitude and faster channel opening both occur normally, but τtail is scarcely slowed, and the shift of the gH-V relationship is just half that of the full-blown enhanced gating response (93). These phenomena have not been explained but strongly suggest that NADPH oxidase activity affects proton currents. One proposal is that NADPH oxidase activity produces enough protons near the membrane to alter the local pH sensed by HV1 (45, 91, 93). Calculations assuming random localization of HV1 and NADPH oxidase (45) indicate they would be too far apart in phagocyte membranes to sense much of the H+ released locally by NADPH (Fig. 1), although rapid proton translocation in the plane of the membrane (205–208) could invalidate this conclusion. On the other hand, global pHi is clearly reduced during the respiratory burst (3, 44, 45, 89, 91, 94, 181).
The final bit of evidence that at long last convinced everyone materialized only after the HVCN1 gene coding for HV1 was identified and knockout (Hvcn1KO) mice were produced. Perforated-patch recording showed that neutrophils from normal mice had proton currents (Fig. 12C), and Hvcn1KO mice did not (Fig. 12A), but the neutrophils from both Hvcn1KO and normal mice (Fig. 12B & 12D) had identical electron currents (44, 130). The electron current proved that all required components of NADPH oxidase (including gp91phox) were present and functioning, and that they did so with or without proton channels. At last we could all agree that there is one gene for HV1 and that the protein it codes for is not part of the NADPH oxidase complex.
Not another controversy!
By 2004, it was clear to almost everyone that NADPH oxidase and proton channels had a special relationship in phagocytes. NADPH oxidase produces massive quantities of ROS, causing depolarization and lowering pHi as direct consequences of its activity. Both depolarization and low pHi inhibit NOX2 activity (120, 139), and left to its own devices, the enzyme would rapidly terminate its own activity. However, both depolarization and low pHi activate proton channels, and HV1 activity counteracts both depolarization and low pHi. This yin-yang synergy could hardly be improved upon. However, the Segal laboratory at University College London threw a spanner into the works. In a paper published in Nature, the Segal group reported several unexpected and novel observations that prompted them to conclude that the predominant currents in human neutrophils and eosinophils were BK potassium currents and not proton currents. They reported that the BK channels inhibitors iberiotoxin and paxilline abolished all outward current in these cells and that the HV1 inhibitor Zn2+ had no effect. They further suggested that previous evidence based on Zn2+ effects, indicating that proton channels were essential to sustain the respiratory burst, were instead artefactual. In support of an important role for BK channels in neutrophil function, they reported that iberiotoxin inhibited the capacity of human neutrophils to kill ingested Staphylococcus aureus or Candida albicans. Collectively, these new data provided the basis for a radical shift in understanding the mechanism underpinning neutrophil antimicrobial action. Whereas decades of experimental and clinical evidence had demonstrated that ROS served as an essential factor in killing of most microbes by neutrophils, the BK channel hypothesis relegated oxidants to incidental byproducts of the NADPH oxidase, and myeloperoxidase to an enzyme that simply consumed H2O2 rather than serving as a source of the potent microbicide HOCl (209, 210). Thus it appeared that the long-accepted paradigm for neutrophil antimicrobial action had shifted.
However, the experimental observations that provided the foundation for the new model proved difficult to replicate (131, 211). The electrophysiological data were completely at odds with a dozen existing studies of human neutrophils or eosinophils (212). The reported BK amplitude was a nanoampere, which meant roughly a hundred BK channels must be open. Despite having recorded from neutrophils and eosinophils from more than 50 individuals, often under conditions favorable to detecting BK channels, we had never seen a single BK channel. A further complication was that the putative K+ conductance would prevent the depolarization that occurs during the respiratory burst (212). As shown in Fig. 10, even one open BK channel would produce hyperpolarization during the respiratory burst (42), which is contradicted by numerous studies reporting depolarization (1, 88, 124–130, 145–147, 149–151, 190–192). Seven labs worldwide independently attempted and failed to find evidence that BK channel inhibitors abolished the ability of human neutrophils to kill bacteria (213). In fact, other labs carefully examined each conclusion from the BK channel report and were unable to substantiate any, raising serious concerns about the validity of the original report (131, 211, 214). A somewhat belated investigation by University College London identified evidence of scientific misconduct and assigned it to the first author. The report concluded that he had manipulated experimental conditions to obtain false data in order to fit the hypothesis. Six years after its publication, the paper was retracted. Even so, despite the red “RETRACTED” plastered across each page, the paper has been cited at least 48 times since its retraction.
The story of the birth and death of the BK channel hypothesis differs fundamentally from the controversy over whether gp91phox was a proton channel, because the latter was a legitimate scientific question. In contrast, from the moment of its publication, the BK data presented contradictions that made the story impossible. This episode underscores both the necessity and difficulty of refuting published studies that are incorrect or irreproducible, particularly when reported in high profile journals (215).
What the discovery of the HVCN1 gene meant
The discovery of the proton channel gene in 2006 (173, 175) was a breakthrough on many levels. It provided the possibility of a knock-out mouse to test the respiratory burst paradigm and other proposed functions of HV1. Before the advent of the Hvcn1KO mouse, we were obliged to do pharmacological lesion experiments using the most potent HV1 inhibitor, which was Zn2+. Although this divalent cation does inhibit proton currents potently (172, 216), it also binds to most proteins and thus has a vast array of possible unintended effects. Convincing demonstration of the functional importance of HV1 is difficult when the most potent inhibitor is as promiscuous as Zn2+. With the HVCN1 gene (coding for HV1) identified, Hvcn1KO mice have been produced, largely confirming conclusions reached in earlier studies using Zn2+. In Hvcn1KO mice, regulation of pH both in cytoplasm (44, 130) and phagosomes is altered (132), neutrophil granule release is altered (217), an autoimmune-like phenotype is observed (137), and B cell signaling is disturbed (136). Reflecting the facilitation of ROS production by HV1 (Fig. 1) in microglia (177), the presence of HV1 (compared to Hvcn1KO) can exacerbate brain damage in an ischemic stroke model (138) and enhance model chronic demyelination (218). Intriguingly, knock-out of HV1 makes eosinophils more susceptible to cell death when NADPH oxidase is activated (129). In all cells studied to date, including mouse neutrophils (130, 134, 135), eosinophils (129), macrophages (219), B lymphocytes (136), T lymphocytes (137), and microglia (138), genetic ablation of HV1 reduces, but does not eliminate ROS production. That ROS production is not eliminated entirely might reflect the ability of other conductances to compensate charge when the preferred ion (H+) is not available (198). It has been pointed out that the incomplete suppression of ROS production makes HV1 an excellent drug target, because its inhibition could abrogate functions such as signaling in a variety of cells, while sparing the innate immune response, which could lead to susceptibility to infections (220). Patients with variant forms of CGD that retain some residual NADPH oxidase activity tend to have milder symptoms (26) and female carriers of X-linked CGD patients with only 5–10% normal neutrophils may be asymptomatic (18, 221).
Knowing the HVCN1 gene makes proteomic screening possible. Such screens have provided evidence correlating HV1 expression and B cell malignancies (136, 222–224), cystic fibrosis (225), and Crohn’s (inflammatory bowel) disease (226) in humans, hyperoxia-induced lung injury in mice (227), and vitiligo in chickens (228).
Identification of the HVCN1 gene has further transformed research by enabling site-directed mutation and other structure-function studies. These possibilities were exploited immediately by several labs experienced in mutation studies of other ion channels whose genes had been identified decades earlier. Progress on HV1 was quite rapid, and will be summarized only briefly here because of its limited relevance to our main topic. In rapid succession, it was discovered that HV1 is a dimer both in heterologous expression systems (229–232) and in situ in phagocytes (133), although each protomer has a separate conduction pathway (229, 231), and the monomer functions almost normally but with faster opening kinetics (229, 231, 233, 234); the dimer was found to open cooperatively (235–237); a knockout mouse was produced and found to appear grossly normal but with distinct deficiencies (44, 134, 135, 137, 138, 217); a “signature sequence” of amino acids was identified as RxWRxxR in the S4 helix that has enabled the discovery of several new HV1 (153); the phosphorylation site responsible for enhanced gating was identified as Thr29 on the intracellular N terminus (96, 224); and the selectivity mechanism was elucidated in which Asp-Arg interaction is crucial (153, 238–241). A crystal structure of the closed mouse HV1 has been reported (174), but the open state structure relies on homology models (239, 242–246) and EPR measurements (247, 248).
Finally, knowing the gene enables the identification of HV1 in diverse species. HV1 genes that have been confirmed by voltage-clamp studies in heterologous expression systems include: human (173), mouse and sea squirt Ciona intestinalis (175), dinoflagellates Karlodinium veneficum (153) and Lingulodinium polyedrum (154), coccolithophores Emiliania huxleyi and Coccolithus pelagicus (249), diatom Phaeodactylum tricornutum (250), insect Nicoletia phytophila (241), and sea urchin Strongylocentrotus purpuratus (251).
Future Directions
The symbiotic relationship between NADPH oxidase (NOX2) and voltage gated proton channels (HV1) in phagocytes has proven to be robust. Somewhat surprisingly, HV1 seems to play a similar role in other cells and with other NOXes (204), despite the level of ROS production typically being orders of magnitude lower (4, 252). Much work needs to be done to establish whether the HV1/NOX paradigm operates wherever a NOX isoform exists, or if charge and pH are compensated in different ways in different systems. Presumably all NOXes function by electron transfer analogous to that in NOX2, but their voltage dependence and other properties remain to be determined. The relationships among the multiple transporters that alter pH (e.g., HV1, H+-ATPase, Na+-H+ antiport, Cl−/HCO3− exchange, ClC-mediated H+/Cl− antiport; see Fig. 1) are complex and not well understood. Why do cells use one or another in any particular circumstance? Deeper understanding of the molecular mechanisms of HV1 and other transporters is important in the attempt to identify critical sites or domains and develop tools to modulate their activity. The development of drugs to modulate the activity of NADPH oxidase (4, 15, 253) and HV1 (254) is underway but far from completion. The diversity of species, phyla, and even kingdoms in which HV1 are found as well as the diverse sequences of these proteins provide a vast arena for exploring structure-function relationships at the protein level as well as biological functions at the organism level.
Acknowledgments
The author deeply appreciates invaluable contributions by Vladimir V. Cherny and Deri Morgan who played indispensible roles in most of the adventures related here; and Edgar Pick for incisive and constructive criticism and discussion. This work was supported by NSF (MCB-1242985) and NIH (GM-102336).
Footnotes
No conflict of Interest
Authors’ contributions: TD wrote the paper.
References
- 1.Henderson LM, Chappell JB, Jones OTG. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem J. 1987;246:325–329. doi: 10.1042/bj2460325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson LM, Chappell JB, Jones OTG. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem J. 1988;255:285–290. [PMC free article] [PubMed] [Google Scholar]
- 3.Henderson LM, Chappell JB, Jones OTG. Internal pH changes associated with the activity of NADPH oxidase of human neutrophils. Further evidence for the presence of an H+ conducting channel. Biochem J. 1988;251:563–567. doi: 10.1042/bj2510563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Yu L, Quinn MT, Cross AR, Dinauer MC. Gp91phox is the heme binding subunit of the superoxide-generating NADPH oxidase. Proc Natl Acad Sci U S A. 1998;95:7993–7998. doi: 10.1073/pnas.95.14.7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biberstine-Kinkade KJ, DeLeo FR, Epstein RI, LeRoy BA, Nauseef WM, Dinauer MC. Heme-ligating histidines in flavocytochrome b 558: identification of specific histidines in gp91phox . J Biol Chem. 2001;276:31105–31112. doi: 10.1074/jbc.M103327200. [DOI] [PubMed] [Google Scholar]
- 7.Nauseef WM. How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev. 2007;219:88–102. doi: 10.1111/j.1600-065X.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 8.Lapouge K, Smith SJ, Groemping Y, Rittinger K. Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox . J Biol Chem. 2002;277:10121–10128. doi: 10.1074/jbc.M112065200. [DOI] [PubMed] [Google Scholar]
- 9.Park JW, Ma M, Ruedi JM, Smith RM, Babior BM. The cytosolic components of the respiratory burst oxidase exist as a Mr approximately 240,000 complex that acquires a membrane-binding site during activation of the oxidase in a cell-free system. J Biol Chem. 1992;267:17327–17332. [PubMed] [Google Scholar]
- 10.Iyer SS, Pearson DW, Nauseef WM, Clark RA. Evidence for a readily dissociable complex of p47phox and p67phox in cytosol of unstimulated human neutrophils. J Biol Chem. 1994;269:22405–22411. [PubMed] [Google Scholar]
- 11.DeLeo FR, Quinn MT. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol. 1996;60:677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 12.Roos D, et al. Mutations in the X-linked and autosomal recessive forms of chronic granulomatous disease. Blood. 1996;87:1663–1681. [PubMed] [Google Scholar]
- 13.Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/s0092-8674(03)00314-3. [DOI] [PubMed] [Google Scholar]
- 14.Taylor RM, Foubert TR, Burritt JB, Baniulis D, McPhail LC, Jesaitis AJ. Anionic amphiphile and phospholipid-induced conformational changes in human neutrophil flavocytochrome b observed by fluorescence resonance energy transfer. Biochim Biophys Acta. 2004;1663:201–213. doi: 10.1016/j.bbamem.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Pick E. Cell-free NADPH oxidase activation assays: “in vitro veritas”. Methods Mol Biol. 2014;1124:339–403. doi: 10.1007/978-1-62703-845-4_22. [DOI] [PubMed] [Google Scholar]
- 16.Swain SD, Helgerson SL, Davis AR, Nelson LK, Quinn MT. Analysis of activation-induced conformational changes in p47phox using tryptophan fluorescence spectroscopy. J Biol Chem. 1997;272:29502–29510. doi: 10.1074/jbc.272.47.29502. [DOI] [PubMed] [Google Scholar]
- 17.Berendes H, Bridges RA, Good RA. A fatal granulomatosus of childhood: the clinical study of a new syndrome. Minn Med. 1957;40:309–312. [PubMed] [Google Scholar]
- 18.Dinauer MC, Nauseef WM, Newburger PEI. Inherited Disorders of Oxidative Phagocyte Killing, Chap. Vol. 189. New York: McGraw-Hill Inc; 2009. [Google Scholar]
- 19.Matute JD, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmes B, Page AR, Good RA. Studies of the metabolic activity of leukocytes from patients with a genetic abnormality of phagocytic function. J Clin Invest. 1967;46:1422–1432. doi: 10.1172/JCI105634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quie PG, White JG, Holmes B, Good RA. In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest. 1967;46:668–679. doi: 10.1172/JCI105568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes B, Quie PG, Windhorst DB, Good RA. Fatal granulomatous disease of childhood. An inborn abnormality of phagocytic function. Lancet. 1966;1:1225–1228. doi: 10.1016/s0140-6736(66)90238-8. [DOI] [PubMed] [Google Scholar]
- 23.Gabig TG, Schervish EW, Santinga JT. Functional relationship of the cytochrome b to the superoxide-generating oxidase of human neutrophils. J Biol Chem. 1982;257:4114–4119. [PubMed] [Google Scholar]
- 24.Borregaard N, Schwartz JH, Tauber AI. Proton secretion by stimulated neutrophils. Significance of hexose monophosphate shunt activity as source of electrons and protons for the respiratory burst. J Clin Invest. 1984;74:455–459. doi: 10.1172/JCI111442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gabig TG, Lefker BA, Ossanna PJ, Weiss SJ. Proton stoichiometry associated with human neutrophil respiratory-burst reactions. J Biol Chem. 1984;259:13166–13171. [PubMed] [Google Scholar]
- 26.Curnutte JT. Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol. 1993;67:S2–S15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- 27.Baldridge CW, Gerard RW. The extra respiration of phagocytosis. Am J Physiol. 1932;103:235–236. [Google Scholar]
- 28.Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. 1959;234:1355–1362. [PubMed] [Google Scholar]
- 29.Iyer GYN, Islam MF, Quastel JH. Biochemical aspects of phagocytosis. Nature. 1961;192:535–541. [Google Scholar]
- 30.Zatti M, Rossi F. Early changes of hexose monophosphate pathway activity and of NADPH oxidation in phagocytizing leucocytes. Biochim Biophys Acta. 1965;99:557–561. doi: 10.1016/s0926-6593(65)80213-2. [DOI] [PubMed] [Google Scholar]
- 31.Repine JE, White JG, Clawson CC, Holmes BM. Effects of phorbol myristate acetate on the metabolism and ultrastructure of neutrophils in chronic granulomatous disease. J Clin Invest. 1974;54:83–90. doi: 10.1172/JCI107752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeCoursey TE, Ligeti E. Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci. 2005;62:2173–2193. doi: 10.1007/s00018-005-5177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baehner RL, Johnston RB, Jr, Nathan DG. Comparative study of the metabolic and bactericidal characteristics of severely glucose-6-phosphate dehydrogenase-deficient polymorphonuclear leukocytes and leukocytes from children with chronic granulomatous disease. J Reticuloendothel Soc. 1972;12:150–169. [PubMed] [Google Scholar]
- 34.Gray GR, et al. Neutrophil dysfunction, chronic granulomatous disease, and non-spherocytic haemolytic anaemia caused by complete deficiency of glucose-6-phosphate dehydrogenase. Lancet. 1973;2:530–534. doi: 10.1016/s0140-6736(73)92350-7. [DOI] [PubMed] [Google Scholar]
- 35.Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 36.Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal. 2013;18:642–660. doi: 10.1089/ars.2012.4827. [DOI] [PubMed] [Google Scholar]
- 37.Harrison JE, Schultz J. Studies on the chlorinating activity of myeloperoxidase. J Biol Chem. 1976;251:1371–1374. [PubMed] [Google Scholar]
- 38.Chapman AL, Hampton MB, Senthilmohan R, Winterbourn CC, Kettle AJ. Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus . J Biol Chem. 2002;277:9757–9762. doi: 10.1074/jbc.M106134200. [DOI] [PubMed] [Google Scholar]
- 39.Foote CS, Goyne TE, Lehrer RI. Assessment of chlorination by human neutrophils. Nature. 1983;301:715–716. doi: 10.1038/301715a0. [DOI] [PubMed] [Google Scholar]
- 40.Thomas EL, Grisham MB, Jefferson MM. Myeloperoxidase-dependent effect of amines on functions of isolated neutrophils. J Clin Invest. 1983;72:441–454. doi: 10.1172/JCI110992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weiss SJ, Klein R, Slivka A, Wei M. Chlorination of taurine by human neutrophils. Evidence for hypochlorous acid generation. J Clin Invest. 1982;70:598–607. doi: 10.1172/JCI110652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy R, DeCoursey TE. Charge compensation during the phagocyte respiratory burst. Biochim Biophys Acta. 2006;1757:996–1011. doi: 10.1016/j.bbabio.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Klebanoff SJ. Oxygen metabolites from phagocytes. 3rd. Philadelphia, PA: Lippincott Williams & Wilkins; 1999. pp. 721–768. [Google Scholar]
- 44.Morgan D, et al. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc Natl Acad Sci USA. 2009;106:18022–18027. doi: 10.1073/pnas.0905565106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeCoursey TE. Voltage-gated proton channels and other proton transfer pathways. Physiol Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 46.Nauseef WM, Volpp BD, McCormick S, Leidal KG, Clark RA. Assembly of the neutrophil respiratory burst oxidase. Protein kinase C promotes cytoskeletal and membrane association of cytosolic oxidase components. J Biol Chem. 1991;266:5911–5917. [PubMed] [Google Scholar]
- 47.Ambruso DR, Bolscher BG, Stokman PM, Verhoeven AJ, Roos D. Assembly and activation of the NADPH:O2 oxidoreductase in human neutrophils after stimulation with phorbol myristate acetate. J Biol Chem. 1990;265:924–930. [PubMed] [Google Scholar]
- 48.Rotrosen D, Leto TL. Phosphorylation of neutrophil 47-kDa cytosolic oxidase factor. Translocation to membrane is associated with distinct phosphorylation events. J Biol Chem. 1990;265:19910–19915. [PubMed] [Google Scholar]
- 49.Park JW, Hoyal CR, Benna JE, Babior BM. Kinase-dependent activation of the leukocyte NADPH oxidase in a cell-free system. Phosphorylation of membranes and p47PHOX during oxidase activation. J Biol Chem. 1997;272:11035–11043. doi: 10.1074/jbc.272.17.11035. [DOI] [PubMed] [Google Scholar]
- 50.Bromberg Y, Pick E. Unsaturated fatty acids stimulate NADPH-dependent superoxide production by cell-free system derived from macrophages. Cell Immunol. 1984;88:213–221. doi: 10.1016/0008-8749(84)90066-2. [DOI] [PubMed] [Google Scholar]
- 51.Heyneman RA, Vercauteren RE. Activation of a NADPH oxidase from horse polymorphonuclear leukocytes in a cell-free system. J Leukoc Biol. 1984;36:751–759. doi: 10.1002/jlb.36.6.751. [DOI] [PubMed] [Google Scholar]
- 52.McPhail LC, Shirley PS, Clayton CC, Snyderman R. Activation of the respiratory burst enzyme from human neutrophils in a cell-free system. Evidence for a soluble cofactor. J Clin Invest. 1985;75:1735–1739. doi: 10.1172/JCI111884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dagher MC, Pick E. Opening the black box: lessons from cell-free systems on the phagocyte NADPH-oxidase. Biochimie. 2007;89:1123–1132. doi: 10.1016/j.biochi.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Curnutte JT. Activation of human neutrophil nicotinamide adenine dinucleotide phosphate, reduced (triphosphopyridine nucleotide, reduced) oxidase by arachidonic acid in a cell-free system. J Clin Invest. 1985;75:1740–1743. doi: 10.1172/JCI111885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Palicz A, Foubert TR, Jesaitis AJ, Marodi L, McPhail LC. Phosphatidic acid and diacylglycerol directly activate NADPH oxidase by interacting with enzyme components. J Biol Chem. 2001;276:3090–3097. doi: 10.1074/jbc.M007759200. [DOI] [PubMed] [Google Scholar]
- 56.Erickson RW, Langel-Peveri P, Traynor-Kaplan AE, Heyworth PG, Curnutte JT. Activation of human neutrophil NADPH oxidase by phosphatidic acid or diacylglycerol in a cell-free system. Activity of diacylglycerol is dependent on its conversion to phosphatidic acid. J Biol Chem. 1999;274:22243–22250. doi: 10.1074/jbc.274.32.22243. [DOI] [PubMed] [Google Scholar]
- 57.McPhail LC, Qualliotine-Mann D, Waite KA. Cell-free activation of neutrophil NADPH oxidase by a phosphatidic acid-regulated protein kinase. Proc Natl Acad Sci U S A. 1995;92:7931–7935. doi: 10.1073/pnas.92.17.7931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bromberg Y, Pick E. Activation of NADPH-dependent superoxide production in a cell-free system by sodium dodecyl sulfate. J Biol Chem. 1985;260:13539–13545. [PubMed] [Google Scholar]
- 59.Pick E, Bromberg Y, Shpungin S, Gadba R. Activation of the superoxide forming NADPH oxidase in a cell-free system by sodium dodecyl sulfate. Characterization of the membrane-associated component. J Biol Chem. 1987;262:16476–16483. [PubMed] [Google Scholar]
- 60.Curnutte JT, Badwey JA, Robinson JM, Karnovsky MJ, Karnovsky ML. Studies on the mechanism of superoxide release from human neutrophils stimulated with arachidonate. J Biol Chem. 1984;259:11851–11857. [PubMed] [Google Scholar]
- 61.Kakinuma K. Effects of fatty acids on the oxidative metabolism of leukocytes. Biochim Biophys Acta. 1974;348:76–85. [PubMed] [Google Scholar]
- 62.Bromberg Y, Pick E. Unsaturated fatty acids as second messengers of superoxide generation by macrophages. Cell Immunol. 1983;79:240–252. doi: 10.1016/0008-8749(83)90067-9. [DOI] [PubMed] [Google Scholar]
- 63.Badwey JA, Curnutte JT, Karnovsky ML. cis-Polyunsaturated fatty acids induce high levels of superoxide production by human neutrophils. J Biol Chem. 1981;256:12640–12643. [PubMed] [Google Scholar]
- 64.Yamaguchi T, Kaneda M, Kakinuma K. Effect of saturated and unsaturated fatty acids on the oxidative metabolism of human neutrophils. The role of calcium ion in the extracellular medium. Biochim Biophys Acta. 1986;861:440–446. doi: 10.1016/0005-2736(86)90452-9. [DOI] [PubMed] [Google Scholar]
- 65.Badwey JA, Curnutte JT, Robinson JM, Berde CB, Karnovsky MJ, Karnovsky ML. Effects of free fatty acids on release of superoxide and on change of shape by human neutrophils. Reversibility by albumin. J Biol Chem. 1984;259:7870–7877. [PubMed] [Google Scholar]
- 66.Walsh CE, Waite BM, Thomas MJ, DeChatelet LR. Release and metabolism of arachidonic acid in human neutrophils. J Biol Chem. 1981;256:7228–7234. [PubMed] [Google Scholar]
- 67.McPhail LC, Clayton CC, Snyderman R. A potential second messenger role for unsaturated fatty acids: activation of Ca2+-dependent protein kinase. Science. 1984;224:622–625. doi: 10.1126/science.6231726. [DOI] [PubMed] [Google Scholar]
- 68.Henderson LM, Moule SK, Chappell JB. The immediate activator of the NADPH oxidase is arachidonate not phosphorylation. Eur J Biochem. 1993;211:157–162. doi: 10.1111/j.1432-1033.1993.tb19882.x. [DOI] [PubMed] [Google Scholar]
- 69.Morgan D, Cherny VV, Finnegan A, Bollinger J, Gelb MH, DeCoursey TE. Sustained activation of proton channels and NADPH oxidase in human eosinophils and murine granulocytes requires PKC but not cPLA2α activity. J Physiol. 2007;579:327–344. doi: 10.1113/jphysiol.2006.124248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rubin BB, et al. Cytosolic phospholipase A2-α is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-α does not regulate neutrophil NADPH oxidase activity. J Biol Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hii CST, et al. Stimulation of p38 phosphorylation and activity by arachidonic acid in HeLa cells, HL60 promyelocytic leukemic cells, and human neutrophils. Evidence for cell type-specific activation of mitogen-activated protein kinases. J Biol Chem. 1998;273:19277–19282. doi: 10.1074/jbc.273.30.19277. [DOI] [PubMed] [Google Scholar]
- 72.Park JW, Babior BM. Activation of the leukocyte NADPH oxidase subunit p47phox by protein kinase C. A phosphorylation-dependent change in the conformation of the C-terminal end of p47phox . Biochemistry. 1997;36:7474–7480. doi: 10.1021/bi9700936. [DOI] [PubMed] [Google Scholar]
- 73.Shiose A, Sumimoto H. Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J Biol Chem. 2000;275:13793–13801. doi: 10.1074/jbc.275.18.13793. [DOI] [PubMed] [Google Scholar]
- 74.Foubert TR, Burritt JB, Taylor RM, Jesaitis AJ. Structural changes are induced in human neutrophil cytochrome b by NADPH oxidase activators, LDS, SDS, and arachidonate: intermolecular resonance energy transfer between trisulfopyrenyl-wheat germ agglutinin and cytochrome b 558 . Biochim Biophys Acta. 2002;1567:221–231. doi: 10.1016/s0005-2736(02)00619-3. [DOI] [PubMed] [Google Scholar]
- 75.Taylor RM, Riesselman MH, Lord CI, Gripentrog JM, Jesaitis AJ. Anionic lipid-induced conformational changes in human phagocyte flavocytochrome b precede assembly and activation of the NADPH oxidase complex. Arch Biochem Biophys. 2012;521:24–31. doi: 10.1016/j.abb.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 76.Vignais PV. The superoxide-generating NADPH oxidase: structural aspects and activation mechanism. Cell Mol Life Sci. 2002;59:1428–1459. doi: 10.1007/s00018-002-8520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Doussiere J, Gaillard J, Vignais PV. Electron transfer across the O2 generating flavocytochrome b of neutrophils. Evidence for a transition from a low-spin state to a high-spin state of the heme iron component. Biochemistry. 1996;35:13400–13410. doi: 10.1021/bi960916b. [DOI] [PubMed] [Google Scholar]
- 78.Berdichevsky Y, Mizrahi A, Ugolev Y, Molshanski-Mor S, Pick E. Tripartite chimeras comprising functional domains derived from the cytosolic NADPH oxidase components p47phox, p67phox, and Rac1 elicit activator-independent superoxide production by phagocyte membranes: an essential role for anionic membrane phospholipids. J Biol Chem. 2007;282:22122–22139. doi: 10.1074/jbc.M701497200. [DOI] [PubMed] [Google Scholar]
- 79.Gorzalczany Y, Sigal N, Itan M, Lotan O, Pick E. Targeting of Rac1 to the phagocyte membrane is sufficient for the induction of NADPH oxidase assembly. J Biol Chem. 2000;275:40073–40081. doi: 10.1074/jbc.M006013200. [DOI] [PubMed] [Google Scholar]
- 80.Rossi F. The O2 -forming NADPH oxidase of the phagocytes: nature, mechanisms of activation and function. Biochim Biophys Acta. 1986;853:65–89. doi: 10.1016/0304-4173(86)90005-4. [DOI] [PubMed] [Google Scholar]
- 81.Henderson LM, Chappell JB. The NADPH-oxidase-associated H+ channel is opened by arachidonate. Biochem J. 1992;283:171–175. doi: 10.1042/bj2830171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeCoursey TE, Cherny VV. Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys J. 1993;65:1590–1598. doi: 10.1016/S0006-3495(93)81198-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gordienko DV, Tare M, Parveen S, Fenech CJ, Robinson C, Bolton TB. Voltage-activated proton current in eosinophils from human blood. J Physiol. 1996;496:299–316. doi: 10.1113/jphysiol.1996.sp021686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schrenzel J, Lew DP, Krause KH. Proton currents in human eosinophils. Am J Physiol. 1996;271:C1861–C1871. doi: 10.1152/ajpcell.1996.271.6.C1861. [DOI] [PubMed] [Google Scholar]
- 85.Kapus A, Romanek R, Grinstein S. Arachidonic acid stimulates the plasma membrane H+ conductance of macrophages. J Biol Chem. 1994;269:4736–4745. [PubMed] [Google Scholar]
- 86.Suszták K, Mócsai A, Ligeti E, Kapus A. Electrogenic H+ pathway contributes to stimulus-induced changes of internal pH and membrane potential in intact neutrophils: role of cytoplasmic phospholipase A2 . Biochem J. 1997;325:501–510. doi: 10.1042/bj3250501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abramson SB, Leszczynska-Piziak J, Weissmann G. Arachidonic acid as a second messenger. Interactions with a GTP-binding protein of human neutrophils. J Immunol. 1991;147:231–236. [PubMed] [Google Scholar]
- 88.Bánfi B, et al. A novel H+ conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J Exp Med. 1999;190:183–194. doi: 10.1084/jem.190.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.DeCoursey TE, Cherny VV, DeCoursey AG, Xu W, Thomas LL. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J Physiol. 2001;535:767–781. doi: 10.1111/j.1469-7793.2001.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeCoursey TE, Cherny VV, Morgan D, Katz BZ, Dinauer MC. The gp91phox component of NADPH oxidase is not the voltage-gated proton channel in phagocytes, but it helps. J Biol Chem. 2001;276:36063–36066. doi: 10.1074/jbc.C100352200. [DOI] [PubMed] [Google Scholar]
- 91.DeCoursey TE, Cherny VV, Zhou W, Thomas LL. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc Natl Acad Sci USA. 2000;97:6885–6889. doi: 10.1073/pnas.100047297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Musset B, Cherny VV, DeCoursey TE. Strong glucose dependence of electron current in human monocytes. Am J Physiol Cell Physiol. 2012;302:C286–C295. doi: 10.1152/ajpcell.00335.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Musset B, Cherny VV, Morgan D, DeCoursey TE. The intimate and mysterious relationship between proton channels and NADPH oxidase. FEBS Lett. 2009;583:7–12. doi: 10.1016/j.febslet.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cherny VV, Henderson LM, Xu W, Thomas LL, DeCoursey TE. Activation of NADPH oxidase-related proton and electron currents in human eosinophils by arachidonic acid. J Physiol. 2001;535:783–794. doi: 10.1111/j.1469-7793.2001.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kawanabe A, Okamura Y. Effects of unsaturated fatty acids on the kinetics of voltage-gated proton channels heterologously expressed in cultured cells. J Physiol. 2016;594:595–610. doi: 10.1113/JP271274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Musset B, et al. Identification of Thr29 as a critical phosphorylation site that activates the human proton channel Hvcn1 in leukocytes. J Biol Chem. 2010;285:5117–5121. doi: 10.1074/jbc.C109.082727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bankers-Fulbright JL, Kita H, Gleich GJ, O’Grady SM. Regulation of human eosinophil NADPH oxidase activity: a central role for PKCδ. J Cell Physiol. 2001;189:306–315. doi: 10.1002/jcp.10022. [DOI] [PubMed] [Google Scholar]
- 98.Szteyn K, Yang W, Schmid E, Lang F, Shumilina E. Lipopolysaccharide-sensitive H+ current in dendritic cells. Am J Physiol Cell Physiol. 2012;303:C204–C212. doi: 10.1152/ajpcell.00059.2012. [DOI] [PubMed] [Google Scholar]
- 99.Schrenzel J, et al. Electron currents generated by the human phagocyte NADPH oxidase. Nature. 1998;392:734–737. doi: 10.1038/33725. [DOI] [PubMed] [Google Scholar]
- 100.DeChatelet LR, Shirley PS, McPhail LC, Huntley CC, Muss HB, Bass DA. Oxidative metabolism of the human eosinophil. Blood. 1977;50:525–535. [PubMed] [Google Scholar]
- 101.Shult PA, Graziano FM, Wallow IH, Busse WW. Comparison of superoxide generation and luminol-dependent chemiluminescence with eosinophils and neutrophils from normal individuals. J Lab Clin Med. 1985;106:638–645. [PubMed] [Google Scholar]
- 102.Petreccia DC, Nauseef WM, Clark RA. Respiratory burst of normal human eosinophils. J Leukoc Biol. 1987;41:283–288. doi: 10.1002/jlb.41.4.283. [DOI] [PubMed] [Google Scholar]
- 103.Klebanoff SJ, Durack DT, Rosen H, Clark RA. Functional studies on human peritoneal eosinophils. Infect Immun. 1977;17:167–173. doi: 10.1128/iai.17.1.167-173.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yazdanbakhsh M, Eckmann CM, De Boer M, Roos D. Purification of eosinophils from normal human blood, preparation of eosinoplasts and characterization of their functional response to various stimuli. Immunology. 1987;60:123–129. [PMC free article] [PubMed] [Google Scholar]
- 105.Lacy P, Abdel-Latif D, Steward M, Musat-Marcu S, Man SF, Moqbel R. Divergence of mechanisms regulating respiratory burst in blood and sputum eosinophils and neutrophils from atopic subjects. J Immunol. 2003;170:2670–2679. doi: 10.4049/jimmunol.170.5.2670. [DOI] [PubMed] [Google Scholar]
- 106.Sedgwick JB, Vrtis RF, Gourley MF, Busse WW. Stimulus-dependent differences in superoxide anion generation by normal human eosinophils and neutrophils. J Allergy Clin Immunol. 1988;81:876–883. doi: 10.1016/0091-6749(88)90945-1. [DOI] [PubMed] [Google Scholar]
- 107.Yagisawa M, et al. Superoxide release and NADPH oxidase components in mature human phagocytes: correlation between functional capacity and amount of functional proteins. Biochem Biophys Res Commun. 1996;228:510–516. doi: 10.1006/bbrc.1996.1691. [DOI] [PubMed] [Google Scholar]
- 108.Kovács I, et al. Comparison of proton channel, phagocyte oxidase, and respiratory burst levels between human eosinophil and neutrophil granulocytes. Free Radic Res. 2014;48:1190–1199. doi: 10.3109/10715762.2014.938234. [DOI] [PubMed] [Google Scholar]
- 109.Ehrlich P. Ueber die specifischen Granulationen des Blutes. Archiv für Anatomie und Physiologie Physiologische Abtheilung. 1879:571–579. [Google Scholar]
- 110.Rothenberg ME. Eosinophilia. N Engl J Med. 1998;338:1592–1600. doi: 10.1056/NEJM199805283382206. [DOI] [PubMed] [Google Scholar]
- 111.Gleich GJ. Mechanisms of eosinophil-associated inflammation. J Allergy Clin Immunol. 2000;105:651–663. doi: 10.1067/mai.2000.105712. [DOI] [PubMed] [Google Scholar]
- 112.Giembycz MA, Lindsay MA. Pharmacology of the eosinophil. Pharmacol Rev. 1999;51:213–340. [PubMed] [Google Scholar]
- 113.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 114.Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindau M, Fernandez JM. IgE-mediated degranulation of mast cells does not require opening of ion channels. Nature. 1986;319:150–153. doi: 10.1038/319150a0. [DOI] [PubMed] [Google Scholar]
- 116.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 117.Robertson AK, Cross AR, Jones OTG, Andrew PW. The use of diphenylene iodonium, an inhibitor of NADPH oxidase, to investigate the antimicrobial action of human monocyte derived macrophages. J Immunol Methods. 1990;133:175–179. doi: 10.1016/0022-1759(90)90357-2. [DOI] [PubMed] [Google Scholar]
- 118.Slayman CL, Gradmann D. Electrogenic proton transport in the plasma membrane of Neurospora . Biophys J. 1975;15:968–971. doi: 10.1016/S0006-3495(75)85877-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gradmann D, Hansen UP, Long WS, Slayman CL, Warncke J. Current-voltage relationships for the plasma membrane and its principal electrogenic pump in Neurospora crassa: I. Steady-state conditions. J Membr Biol. 1978;39:333–367. doi: 10.1007/BF01869898. [DOI] [PubMed] [Google Scholar]
- 120.DeCoursey TE, Morgan D, Cherny VV. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 2003;422:531–534. doi: 10.1038/nature01523. [DOI] [PubMed] [Google Scholar]
- 121.Nisimoto Y, Motalebi S, Han CH, Lambeth JD. The p67phox activation domain regulates electron flow from NADPH to flavin in flavocytochrome b 558 . J Biol Chem. 1999;274:22999–23005. doi: 10.1074/jbc.274.33.22999. [DOI] [PubMed] [Google Scholar]
- 122.Isogai Y, Iizuka T, Shiro Y. The mechanism of electron donation to molecular oxygen by phagocytic cytochrome b 558 . J Biol Chem. 1995;270:7853–7857. doi: 10.1074/jbc.270.14.7853. [DOI] [PubMed] [Google Scholar]
- 123.Petheő GL, Demaurex N. Voltage- and NADPH-dependence of electron currents generated by the phagocytic NADPH oxidase. Biochem J. 2005;388:485–491. doi: 10.1042/BJ20041889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Geiszt M, Kapus A, Nemet K, Farkas L, Ligeti E. Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J Biol Chem. 1997;272:26471–26478. doi: 10.1074/jbc.272.42.26471. [DOI] [PubMed] [Google Scholar]
- 125.Jankowski A, Grinstein S. A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. J Biol Chem. 1999;274:26098–26104. doi: 10.1074/jbc.274.37.26098. [DOI] [PubMed] [Google Scholar]
- 126.Rada BK, Geiszt M, Káldi K, Tímár C, Ligeti E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood. 2004;104:2947–2953. doi: 10.1182/blood-2004-03-1005. [DOI] [PubMed] [Google Scholar]
- 127.Bankers-Fulbright JL, Gleich GJ, Kephart GM, Kita H, O’Grady SM. Regulation of eosinophil membrane depolarization during NADPH oxidase activation. J Cell Sci. 2003;116:3221–3226. doi: 10.1242/jcs.00627. [DOI] [PubMed] [Google Scholar]
- 128.Demaurex N, Petheõ GL. Electron and proton transport by NADPH oxidases. Philos Trans R Soc Lond B Biol Sci. 2005;360:2315–2325. doi: 10.1098/rstb.2005.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhu X, Mose E, Zimmermann N. Proton channel HVCN1 is required for effector functions of mouse eosinophils. BMC Immunol. 2013;14:24. doi: 10.1186/1471-2172-14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.El Chemaly A, Okochi Y, Sasaki M, Arnaudeau S, Okamura Y, Demaurex N. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J Exp Med. 2010;207:129–139. doi: 10.1084/jem.20091837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Femling JK, et al. The antibacterial activity of human neutrophils and eosinophils requires proton channels but not BK channels. J Gen Physiol. 2006;127:659–672. doi: 10.1085/jgp.200609504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.El Chemaly A, Nunes P, Jimaja W, Castelbou C, Demaurex N. Hv1 proton channels differentially regulate the pH of neutrophil and macrophage phagosomes by sustaining the production of phagosomal ROS that inhibit the delivery of vacuolar ATPases. J Leukoc Biol. 2014 doi: 10.1189/jlb.0513251. [DOI] [PubMed] [Google Scholar]
- 133.Petheő GL, et al. Molecular and functional characterization of HV1 proton channel in human granulocytes. PLoS One. 2010;5:e14081. doi: 10.1371/journal.pone.0014081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Okochi Y, Sasaki M, Iwasaki H, Okamura Y. Voltage-gated proton channel is expressed on phagosomes. Biochem Biophys Res Commun. 2009;382:274–279. doi: 10.1016/j.bbrc.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 135.Ramsey IS, Ruchti E, Kaczmarek JS, Clapham DE. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc Natl Acad Sci USA. 2009;106:7642–7647. doi: 10.1073/pnas.0902761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Capasso M, et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat Immunol. 2010;11:265–272. doi: 10.1038/ni.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sasaki M, et al. Autoimmune disorder phenotypes in Hvcn1-deficient mice. Biochem J. 2013;450:295–301. doi: 10.1042/BJ20121188. [DOI] [PubMed] [Google Scholar]
- 138.Wu LJ, et al. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci. 2012;15:565–573. doi: 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morgan D, Cherny VV, Murphy R, Katz BZ, DeCoursey TE. The pH dependence of NADPH oxidase in human eosinophils. J Physiol. 2005;569:419–431. doi: 10.1113/jphysiol.2005.094748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morgan D, Cherny VV, Murphy R, Xu W, Thomas LL, DeCoursey TE. Temperature dependence of NADPH oxidase in human eosinophils. J Physiol. 2003;550:447–458. doi: 10.1113/jphysiol.2003.041525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Picco C, et al. How are cytochrome b561 electron currents controlled by membrane voltage and substrate availability? Antioxid Redox Signal. 2014;21:384–391. doi: 10.1089/ars.2013.5809. [DOI] [PubMed] [Google Scholar]
- 142.Koshkin V, Lotan O, Pick E. Electron transfer in the superoxide-generating NADPH oxidase complex reconstituted in vitro. Biochim Biophys Acta. 1997;1319:139–146. doi: 10.1016/s0005-2728(96)00154-5. [DOI] [PubMed] [Google Scholar]
- 143.Cross AR, Parkinson JF, Jones OTG. Mechanism of the superoxide-producing oxidase of neutrophils. O2 is necessary for the fast reduction of cytochrome b-245 by NADPH. Biochem J. 1985;226:881–884. doi: 10.1042/bj2260881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cherny VV, Murphy R, Sokolov V, Levis RA, DeCoursey TE. Properties of single voltage-gated proton channels in human eosinophils estimated by noise analysis and by direct measurement. J Gen Physiol. 2003;121:615–628. doi: 10.1085/jgp.200308813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cameron AR, Nelson J, Forman HJ. Depolarization and increased conductance precede superoxide release by concanavalin A-stimulated rat alveolar macrophages. Proc Natl Acad Sci USA. 1983;80:3726–3728. doi: 10.1073/pnas.80.12.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cohen HJ, Newburger PE, Chovaniec ME, Whitin JC, Simons ER. Opsonized zymosan-stimulated granulocytes-activation and activity of the superoxide-generating system and membrane potential changes. Blood. 1981;58:975–982. [PubMed] [Google Scholar]
- 147.Jones GS, VanDyke K, Castranova V. Transmembrane potential changes associated with superoxide release from human granulocytes. J Cell Physiol. 1981;106:75–83. doi: 10.1002/jcp.1041060109. [DOI] [PubMed] [Google Scholar]
- 148.Korchak HM, Weissmann G. Changes in membrane potential of human granulocytes antecede the metabolic responses to surface stimulation. Proc Natl Acad Sci USA. 1978;75:3818–3822. doi: 10.1073/pnas.75.8.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sklar LA, Jesaitis AJ, Painter RG, Cochrane CG. The kinetics of neutrophil activation. The response to chemotactic peptides depends upon whether ligand-receptor interaction is rate-limiting. J Biol Chem. 1981;256:9909–9914. [PubMed] [Google Scholar]
- 150.Whitin JC, Chapman CE, Simons ER, Chovaniec ME, Cohen HJ. Correlation between membrane potential changes and superoxide production in human granulocytes stimulated by phorbol myristate acetate. Evidence for defective activation in chronic granulomatous disease. J Biol Chem. 1980;255:1874–1878. [PubMed] [Google Scholar]
- 151.Shurin SB, Cohen HJ, Whitin JC, Newburger PE. Impaired granulocyte superoxide production and prolongation of the respiratory burst due to a low-affinity NADPH-dependent oxidase. Blood. 1983;62:564–571. [PubMed] [Google Scholar]
- 152.Fogel M, Hastings JW. Bioluminescence: mechanism and mode of control of scintillon activity. Proc Natl Acad Sci USA. 1972;69:690–693. doi: 10.1073/pnas.69.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Smith SME, et al. Voltage-gated proton channel in a dinoflagellate. Proc Natl Acad Sci USA. 2011;108:18162–18167. doi: 10.1073/pnas.1115405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rodriguez JD, et al. Characterization and subcellular localization of HV1 in Lingulodinium polyedrum confirms its role in bioluminescence. Biophys J. 2015;108:425a. [Google Scholar]
- 155.Meech R. A contribution to the history of the proton channel. Wiley Interdisciplinary Reviews: Membrane Transport and Signaling. 2012;1:533–557. doi: 10.1002/wmts.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Thomas RC, Meech RW. Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature. 1982;299:826–828. doi: 10.1038/299826a0. [DOI] [PubMed] [Google Scholar]
- 157.Byerly L, Meech R, Moody W., Jr Rapidly activating hydrogen ion currents in perfused neurones of the snail, Lymnaea stagnalis . J Physiol. 1984;351:199–216. doi: 10.1113/jphysiol.1984.sp015241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Doroshenko PA, Kostyuk PG, Martynyuk AE. Transmembrane outward hydrogen current in intracellularly perfused neurones of the snail Helix pomatia . Gen Physiol Biophys. 1986;5:337–350. [PubMed] [Google Scholar]
- 159.Barish ME, Baud C. A voltage-gated hydrogen ion current in the oocyte membrane of the axolotl, Ambystoma . J Physiol. 1984;352:243–263. doi: 10.1113/jphysiol.1984.sp015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Baud C, Barish ME. Changes in membrane hydrogen and sodium conductances during progesterone-induced maturation of Ambystoma oocytes. Dev Biol. 1984;105:423–434. doi: 10.1016/0012-1606(84)90299-9. [DOI] [PubMed] [Google Scholar]
- 161.Hutter OF, Warner AE. The pH sensitivity of the chloride conductance of frog skeletal muscle. J Physiol. 1967;189:403–425. doi: 10.1113/jphysiol.1967.sp008176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hutter OF, Warner AE. The voltage dependence of the chloride conductance of frog muscle. J Physiol. 1972;227:275–290. doi: 10.1113/jphysiol.1972.sp010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.DeCoursey TE. Hydrogen ion currents in rat alveolar epithelial cells. Biophys J. 1991;60:1243–1253. doi: 10.1016/S0006-3495(91)82158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cherny VV, Markin VS, DeCoursey TE. The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J Gen Physiol. 1995;105:861–896. doi: 10.1085/jgp.105.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.DeCoursey TE, Cherny VV. Deuterium isotope effects on permeation and gating of proton channels in rat alveolar epithelium. J Gen Physiol. 1997;109:415–434. doi: 10.1085/jgp.109.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.DeCoursey TE, Cherny VV. Effects of buffer concentration on voltage-gated H+ currents: does diffusion limit the conductance? Biophys J. 1996;71:182–193. doi: 10.1016/S0006-3495(96)79215-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.DeCoursey TE, Cherny VV. Voltage-activated proton currents in membrane patches of rat alveolar epithelial cells. J Physiol. 1995;489:299–307. doi: 10.1113/jphysiol.1995.sp021051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.DeCoursey TE, Cherny VV. Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J Gen Physiol. 1998;112:503–522. doi: 10.1085/jgp.112.4.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cherny VV, Morgan D, Musset B, Chaves G, Smith SM, DeCoursey TE. Tryptophan 207 is crucial to the unique properties of the human voltage-gated proton channel, hHV1. J Gen Physiol. 2015;146:343–356. doi: 10.1085/jgp.201511456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.DeCoursey TE, Cherny VV. Na+-H+ antiport detected through hydrogen ion currents in rat alveolar epithelial cells and human neutrophils. J Gen Physiol. 1994;103:755–785. doi: 10.1085/jgp.103.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.DeCoursey TE, Cherny VV. Voltage-activated hydrogen ion currents. J Membr Biol. 1994;141:203–223. doi: 10.1007/BF00235130. [DOI] [PubMed] [Google Scholar]
- 172.Cherny VV, DeCoursey TE. pH-dependent inhibition of voltage-gated H+ currents in rat alveolar epithelial cells by Zn2+ and other divalent cations. J Gen Physiol. 1999;114:819–838. doi: 10.1085/jgp.114.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Takeshita K, et al. X-ray crystal structure of voltage-gated proton channel. Nat Struct Mol Biol. 2014;21:352–357. doi: 10.1038/nsmb.2783. [DOI] [PubMed] [Google Scholar]
- 175.Sasaki M, Takagi M, Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 176.DeCoursey TE. Voltage-gated proton channels: molecular biology, physiology, and pathophysiology of the HV family. Physiol Rev. 2013;93:599–652. doi: 10.1152/physrev.00011.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Eder C, DeCoursey TE. Voltage-gated proton channels in microglia. Prog Neurobiol. 2001;64:277–305. doi: 10.1016/s0301-0082(00)00062-9. [DOI] [PubMed] [Google Scholar]
- 178.DeCoursey TE. Hypothesis: do voltage-gated H+ channels in alveolar epithelial cells contribute to CO2 elimination by the lung? Am J Physiol Cell Physiol. 2000;278:C1–C10. doi: 10.1152/ajpcell.2000.278.1.C1. [DOI] [PubMed] [Google Scholar]
- 179.Demaurex N, Grinstein S, Jaconi M, Schlegel W, Lew DP, Krause KH. Proton currents in human granulocytes: regulation by membrane potential and intracellular pH. J Physiol. 1993;466:329–344. [PMC free article] [PubMed] [Google Scholar]
- 180.Kapus A, Romanek R, Qu AY, Rotstein OD, Grinstein S. A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J Gen Physiol. 1993;102:729–760. doi: 10.1085/jgp.102.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nanda A, Grinstein S. Protein kinase C activates an H+ (equivalent) conductance in the plasma membrane of human neutrophils. Proc Natl Acad Sci USA. 1991;88:10816–10820. doi: 10.1073/pnas.88.23.10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nanda A, Grinstein S, Curnutte JT. Abnormal activation of H+ conductance in NADPH oxidase-defective neutrophils. Proc Natl Acad Sci USA. 1993;90:760–764. doi: 10.1073/pnas.90.2.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nanda A, Romanek R, Curnutte JT, Grinstein S. Assessment of the contribution of the cytochrome b moiety of the NADPH oxidase to the transmembrane H+ conductance of leukocytes. J Biol Chem. 1994;269:27280–27285. [PubMed] [Google Scholar]
- 184.Kuno M, Kawawaki J, Nakamura F. A highly temperature-sensitive proton current in mouse bone marrow-derived mast cells. J Gen Physiol. 1997;109:731–740. doi: 10.1085/jgp.109.6.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nordström T, et al. Regulation of cytoplasmic pH in osteoclasts. Contribution of proton pumps and a proton-selective conductance. J Biol Chem. 1995;270:2203–2212. doi: 10.1074/jbc.270.5.2203. [DOI] [PubMed] [Google Scholar]
- 186.Demaurex N, Downey GP, Waddell TK, Grinstein S. Intracellular pH regulation during spreading of human neutrophils. J Cell Biol. 1996;133:1391–1402. doi: 10.1083/jcb.133.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Morihata H, Nakamura F, Tsutada T, Kuno M. Potentiation of a voltage-gated proton current in acidosis-induced swelling of rat microglia. J Neurosci. 2000;20:7220–7227. doi: 10.1523/JNEUROSCI.20-19-07220.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nanda A, Gukovskaya A, Tseng J, Grinstein S. Activation of vacuolar-type proton pumps by protein kinase C. Role in neutrophil pH regulation. J Biol Chem. 1992;267:22740–22746. [PubMed] [Google Scholar]
- 189.Murphy R, Cherny VV, Morgan D, DeCoursey TE. Voltage-gated proton channels help regulate pHi in rat alveolar epithelium. Am J Physiol Lung Cell Mol Physiol. 2005;288:L398–L408. doi: 10.1152/ajplung.00299.2004. [DOI] [PubMed] [Google Scholar]
- 190.Rada BK, Geiszt M, Van Bruggen R, Német K, Roos D, Ligeti E. Calcium signalling is altered in myeloid cells with a deficiency in NADPH oxidase activity. Clin Exp Immunol. 2003;132:53–60. doi: 10.1046/j.1365-2249.2003.02138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kuroki M, Kamo N, Kobatake Y, Okimasu E, Utsumi K. Measurement of membrane potential in polymorphonuclear leukocytes and its changes during surface stimulation. Biochim Biophys Acta. 1982;693:326–334. doi: 10.1016/0005-2736(82)90439-4. [DOI] [PubMed] [Google Scholar]
- 192.Seeds MC, Parce JW, Szejda P, Bass DA. Independent stimulation of membrane potential changes and the oxidative metabolic burst in polymorphonuclear leukocytes. Blood. 1985;65:233–240. [PubMed] [Google Scholar]
- 193.Henderson LM, Banting G, Chappell JB. The arachidonate-activable, NADPH oxidase-associated H+ channel. Evidence that gp91-phox functions as an essential part of the channel. J Biol Chem. 1995;270:5909–5916. [PubMed] [Google Scholar]
- 194.Cherny VV, Henderson LM, DeCoursey TE. Proton and chloride currents in Chinese hamster ovary cells. Membr Cell Biol. 1997;11:337–347. [PubMed] [Google Scholar]
- 195.Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axons. J Physiol. 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhen L, King AA, Xiao Y, Chanock SJ, Orkin SH, Dinauer MC. Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox . Proc Natl Acad Sci U S A. 1993;90:9832–9836. doi: 10.1073/pnas.90.21.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Price MO, McPhail LC, Lambeth JD, Han CH, Knaus UG, Dinauer MC. Creation of a genetic system for analysis of the phagocyte respiratory burst: high-level reconstitution of the NADPH oxidase in a nonhematopoietic system. Blood. 2002;99:2653–2661. doi: 10.1182/blood.v99.8.2653. [DOI] [PubMed] [Google Scholar]
- 198.Morgan D, Cherny VV, Price MO, Dinauer MC, DeCoursey TE. Absence of proton channels in COS-7 cells expressing functional NADPH oxidase components. J Gen Physiol. 2002;119:571–580. doi: 10.1085/jgp.20018544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.DeCoursey TE, Morgan D, Cherny VV. The gp91phox component of NADPH oxidase is not a voltage-gated proton channel. J Gen Physiol. 2002;120:773–779. doi: 10.1085/jgp.20028704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Henderson LM, Meech RW. Proton conduction through gp91phox . J Gen Physiol. 2002;120:759–765. doi: 10.1085/jgp.20028708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Maturana A, Krause KH, Demaurex N. NOX family NADPH oxidases: do they have built-in proton channels? J Gen Physiol. 2002;120:781–786. doi: 10.1085/jgp.20028713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Touret N, Grinstein S. Voltage-gated proton “channels”: a spectator’s viewpoint. J Gen Physiol. 2002;120:767–771. doi: 10.1085/jgp.20028706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Musset B, et al. A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc Natl Acad Sci USA. 2008;105:11020–11025. doi: 10.1073/pnas.0800886105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Musset B, et al. NOX5 in human spermatozoa: expression, function and regulation. J Biol Chem. 2012;287:9376–9388. doi: 10.1074/jbc.M111.314955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ädelroth P, Brzezinski P. Surface-mediated proton-transfer reactions in membrane-bound proteins. Biochim Biophys Acta. 2004;1655:102–115. doi: 10.1016/j.bbabio.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 206.Teissié J, Prats M, Soucaille P, Tocanne JF. Evidence for conduction of protons along the interface between water and a polar lipid monolayer. Proc Natl Acad Sci U S A. 1985;82:3217–3221. doi: 10.1073/pnas.82.10.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Heberle J, Riesle J, Thiedemann G, Oesterhelt D, Dencher NA. Proton migration along the membrane surface and retarded surface to bulk transfer. Nature. 1994;370:379–382. doi: 10.1038/370379a0. [DOI] [PubMed] [Google Scholar]
- 208.Haines TH. Anionic lipid headgroups as a proton-conducting pathway along the surface of membranes: a hypothesis. Proc Natl Acad Sci U S A. 1983;80:160–164. doi: 10.1073/pnas.80.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Klebanoff SJ. Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science. 1970;169:1095–1097. doi: 10.1126/science.169.3950.1095. [DOI] [PubMed] [Google Scholar]
- 210.Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol. 2013;93:185–198. doi: 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Essin K, et al. Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity. Am J Physiol Cell Physiol. 2007;293:C45–C54. doi: 10.1152/ajpcell.00450.2006. [DOI] [PubMed] [Google Scholar]
- 212.DeCoursey TE. During the respiratory burst, do phagocytes need proton channels or potassium channels, or both? Sci STKE. 2004;2004:pe21. doi: 10.1126/stke.2332004pe21. [DOI] [PubMed] [Google Scholar]
- 213.DeCoursey TE. Electrophysiology of the phagocyte respiratory burst. Focus on “Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity”. Am J Physiol Cell Physiol. 2007;293:C30–C32. doi: 10.1152/ajpcell.00093.2007. [DOI] [PubMed] [Google Scholar]
- 214.Essin K, et al. BK channels in innate immune functions of neutrophils and macrophages. Blood. 2009;113:1326–1331. doi: 10.1182/blood-2008-07-166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.DeCoursey TE. It’s difficult to publish contradictory findings. Nature. 2006;439:784. doi: 10.1038/439784b. [DOI] [PubMed] [Google Scholar]
- 216.Mahaut-Smith MP. The effect of zinc on calcium and hydrogen ion currents in intact snail neurones. J Exp Biol. 1989;145:455–464. doi: 10.1242/jeb.145.1.455. [DOI] [PubMed] [Google Scholar]
- 217.Okochi Y, et al. The voltage-gated proton channel Hv1/VSOP inhibits neutrophil granule release. J Leukoc Biol. 2016;99:7–19. doi: 10.1189/jlb.3HI0814-393R. [DOI] [PubMed] [Google Scholar]
- 218.Liu J, et al. Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J Neurochem. 2015;135:347–356. doi: 10.1111/jnc.13242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS., Jr Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39:697–710. doi: 10.1016/j.immuni.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Capasso M, DeCoursey TE, Dyer MJS. pH regulation and beyond: unanticipated functions for the voltage-gated proton channel, HVCN1. Trends Cell Biol. 2011;21:20–28. doi: 10.1016/j.tcb.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Roos D, et al. Chronic granulomatous disease with partial deficiency of cytochrome b 558 and incomplete respiratory burst: variants of the X-linked, cytochrome b 558-negative form of the disease. J Leukoc Biol. 1992;51:164–171. doi: 10.1002/jlb.51.2.164. [DOI] [PubMed] [Google Scholar]
- 222.Boyd RS, Dyer MJS, Cain K. Proteomic analysis of B-cell malignancies. J Proteomics. 2010;73:1804–1822. doi: 10.1016/j.jprot.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 223.Boyd RS, Jukes-Jones R, Walewska R, Brown D, Dyer MJS, Cain K. Protein profiling of plasma membranes defines aberrant signaling pathways in mantle cell lymphoma. Mol Cell Proteomics. 2009;8:1501–1515. doi: 10.1074/mcp.M800515-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Hondares E, et al. Enhanced activation of an amino-terminally truncated isoform of the voltage-gated proton channel HVCN1 enriched in malignant B cells. Proc Natl Acad Sci USA. 2014;111:18078–18083. doi: 10.1073/pnas.1411390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Conese M, et al. Evaluation of genome-wide expression profiles of blood and sputum neutrophils in cystic fibrosis patients before and after antibiotic therapy. PLoS One. 2014;9:e104080. doi: 10.1371/journal.pone.0104080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Haglund S, Almer S, Peterson C, Soderman J. Gene expression and thiopurine metabolite profiling in inflammatory bowel disease - novel clues to drug targets and disease mechanisms? PLoS One. 2013;8:e56989. doi: 10.1371/journal.pone.0056989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Howden R, et al. Cardiac physiologic and genetic predictors of hyperoxia-induced acute lung injury in mice. Am J Respir Cell Mol Biol. 2012;46:470–478. doi: 10.1165/rcmb.2011-0204OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Shi F, Kong BW, Song JJ, Lee JY, Dienglewicz RL, Erf GF. Understanding mechanisms of vitiligo development in Smyth line of chickens by transcriptomic microarray analysis of evolving autoimmune lesions. BMC Immunol. 2012;13:18. doi: 10.1186/1471-2172-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Koch HP, Kurokawa T, Okochi Y, Sasaki M, Okamura Y, Larsson HP. Multimeric nature of voltage-gated proton channels. Proc Natl Acad Sci USA. 2008;105:9111–9116. doi: 10.1073/pnas.0801553105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lee SY, Letts JA, Mackinnon R. Dimeric subunit stoichiometry of the human voltage-dependent proton channel Hv1. Proc Natl Acad Sci USA. 2008;105:7692–7695. doi: 10.1073/pnas.0803277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tombola F, Ulbrich MH, Isacoff EY. The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron. 2008;58:546–556. doi: 10.1016/j.neuron.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Smith SME, DeCoursey TE. Consequences of dimerization of the voltage-gated proton channel. Prog Mol Biol Transl Sci. 2013;117:335–360. doi: 10.1016/B978-0-12-386931-9.00012-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fujiwara Y, et al. The cytoplasmic coiled-coil mediates cooperative gating temperature sensitivity in the voltage-gated H+ channel Hv1. Nat Commun. 2012;3:816. doi: 10.1038/ncomms1823. [DOI] [PubMed] [Google Scholar]
- 234.Musset B, Smith SM, Rajan S, Cherny VV, Morgan D, DeCoursey TE. Oligomerization of the voltage gated proton channel. Channels (Austin) 2010;4:260–265. doi: 10.4161/chan.4.4.12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gonzalez C, Koch HP, Drum BM, Larsson HP. Strong cooperativity between subunits in voltage-gated proton channels. Nat Struct Mol Biol. 2010;17:51–56. doi: 10.1038/nsmb.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tombola F, Ulbrich MH, Kohout SC, Isacoff EY. The opening of the two pores of the Hv1 voltage-gated proton channel is tuned by cooperativity. Nat Struct Mol Biol. 2010;17:44–50. doi: 10.1038/nsmb.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Musset B, et al. Zinc inhibition of monomeric and dimeric proton channels suggests cooperative gating. J Physiol. 2010;588:1435–1449. doi: 10.1113/jphysiol.2010.188318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Dudev T, et al. Selectivity mechanism of the voltage-gated proton channel, HV1. Sci Rep. 2015;5:10320. doi: 10.1038/srep10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Morgan D, et al. Peregrination of the selectivity filter delineates the pore of the human voltage-gated proton channel hHV1. J Gen Physiol. 2013;142:625–640. doi: 10.1085/jgp.201311045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Musset B, Smith SME, Rajan S, Morgan D, Cherny VV, DeCoursey TE. Aspartate 112 is the selectivity filter of the human voltage-gated proton channel. Nature. 2011;480:273–277. doi: 10.1038/nature10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Chaves G, Derst C, Franzen A, Mashimo Y, Machida R, Musset B. Identification of an HV1 voltage-gated proton channel in insects. FEBS J. 2016 doi: 10.1111/febs.13680. [DOI] [PubMed] [Google Scholar]
- 242.Chamberlin A, Qiu F, Wang Y, Noskov SY, Peter Larsson H. Mapping the gating and permeation pathways in the voltage-gated proton channel Hv1. Journal of Molecular Biology. 2015;427:131–145. doi: 10.1016/j.jmb.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kulleperuma K, et al. Construction and validation of a homology model of the human voltage-gated proton channel hHV1. J Gen Physiol. 2013;141:445–465. doi: 10.1085/jgp.201210856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wood ML, Schow EV, Freites JA, White SH, Tombola F, Tobias DJ. Water wires in atomistic models of the Hv1 proton channel. Biochim Biophys Acta. 2012;1818:286–293. doi: 10.1016/j.bbamem.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Pupo A, Baez-Nieto D, Martinez A, Latorre R, González C. Proton channel models: Filling the gap between experimental data and the structural rationale. Channels (Austin) 2014;8:180–192. doi: 10.4161/chan.28665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ramsey IS, Mokrab Y, Carvacho I, Sands ZA, Sansom MSP, Clapham DE. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat Struct Mol Biol. 2010;17:869–875. doi: 10.1038/nsmb.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.DeCoursey TE. Structural revelations of the human proton channel. Proc Natl Acad Sci U S A. 2015;112:13430–13431. doi: 10.1073/pnas.1518486112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Li Q, et al. Resting state of the human proton channel dimer in a lipid bilayer. Proc Natl Acad Sci USA. 2015;112:E5926–E5935. doi: 10.1073/pnas.1515043112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Taylor AR, Chrachri A, Wheeler G, Goddard H, Brownlee C. A voltage-gated H+ channel underlying pH homeostasis in calcifying coccolithophores. PLoS Biol. 2011;9:e1001085. doi: 10.1371/journal.pbio.1001085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Taylor AR, Brownlee C, Wheeler GL. Proton channels in algae: reasons to be excited. Trends Plant Sci. 2012;17:675–684. doi: 10.1016/j.tplants.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 251.Okamura Y, Fujiwara Y, Sakata S. Gating mechanisms of voltage-gated proton channels. Annu Rev Biochem. 2015;84:685–709. doi: 10.1146/annurev-biochem-060614-034307. [DOI] [PubMed] [Google Scholar]
- 252.Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009;11:1349–1356. doi: 10.1089/ars.2008.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Smith SME, et al. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem Biol. 2012;19:752–763. doi: 10.1016/j.chembiol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Seredenina T, Demaurex N, Krause KH. Voltage-gated proton channels as novel drug targets: From NADPH oxidase regulation to sperm biology. Antioxid Redox Signal. 2015;23:490–513. doi: 10.1089/ars.2013.5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Cross AR, Rae J, Curnutte JT. Cytochrome b -245 of the neutrophil superoxide-generating system contains two nonidentical hemes. Potentiometric studies of a mutant form of gp91phox . J Biol Chem. 1995;270:17075–17077. doi: 10.1074/jbc.270.29.17075. [DOI] [PubMed] [Google Scholar]
- 256.DeCoursey TE. Interactions between NADPH oxidase and voltage-gated proton channels: why electron transport depends on proton transport. FEBS Lett. 2003;555:57–61. doi: 10.1016/s0014-5793(03)01103-7. [DOI] [PubMed] [Google Scholar]
- 257.Canton J, Khezri R, Glogauer M, Grinstein S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell. 2014;25:3330–3341. doi: 10.1091/mbc.E14-05-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.DeCoursey TE. Voltage-gated proton channels find their dream job managing the respiratory burst in phagocytes. Physiology (Bethesda) 2010;25:27–40. doi: 10.1152/physiol.00039.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Grinstein S, Romanek R, Rotstein OD. Method for manipulation of cytosolic pH in cells clamped in the whole cell or perforated-patch configurations. Am J Physiol. 1994;267:C1152–C1159. doi: 10.1152/ajpcell.1994.267.4.C1152. [DOI] [PubMed] [Google Scholar]
- 260.Babior BM, Curnutte JT, McMurrich BJ. The particulate superoxide-forming system from human neutrophils. Properties of the system and further evidence supporting its participation in the respiratory burst. J Clin Invest. 1976;58:989–996. doi: 10.1172/JCI108553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Clark RA, Leidal KG, Pearson DW, Nauseef WM. NADPH oxidase of human neutrophils. Subcellular localization and characterization of an arachidonate-activatable superoxide-generating system. J Biol Chem. 1987;262:4065–4074. [PubMed] [Google Scholar]
- 262.Cox JA, Jeng AY, Blumberg PM, Tauber AI. Comparison of subcellular activation of the human neutrophil NADPH-oxidase by arachidonic acid, sodium dodecyl sulfate (SDS), and phorbol myristate acetate (PMA) J Immunol. 1987;138:1884–1888. [PubMed] [Google Scholar]
- 263.Suzuki Y, Lehrer RI. NAD(P)H oxidase activity in human neutrophils stimulated by phorbol myristate acetate. J Clin Invest. 1980;66:1409–1418. doi: 10.1172/JCI109994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Light DR, Walsh C, O’Callaghan AM, Goetzl EJ, Tauber AI. Characteristics of the cofactor requirements for the superoxide-generating NADPH oxidase of human polymorphonuclear leukocytes. Biochemistry. 1981;20:1468–1476. doi: 10.1021/bi00509a010. [DOI] [PubMed] [Google Scholar]
- 265.Gabig TG, Babior BM. The O2 -forming oxidase responsible for the respiratory burst in human neutrophils. Properties of the solubilized enzyme. J Biol Chem. 1979;254:9070–9074. [PubMed] [Google Scholar]
- 266.Mori H, et al. Regulatory mechanisms and physiological relevance of a voltage-gated H+ channel in murine osteoclasts: phorbol myristate acetate induces cell acidosis and the channel activation. J Bone Miner Res. 2003;18:2069–2076. doi: 10.1359/jbmr.2003.18.11.2069. [DOI] [PubMed] [Google Scholar]
- 267.Cherny VV, Thomas LL, DeCoursey TE. Voltage-gated proton currents in human basophils. Biologicheskie Membrany. 2001;18:458–465. [Google Scholar]
- 268.Masia R, Krause DS, Yellen G. The inward rectifier potassium channel Kir2.1 is expressed in mouse neutrophils from bone marrow and liver. Am J Physiol Cell Physiol. 2015;308:C264–C276. doi: 10.1152/ajpcell.00176.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Jin C, et al. HV1 acts as a sodium sensor and promotes superoxide production in medullary thick ascending limb of Dahl salt-sensitive rats. Hypertension. 2014;64:541–550. doi: 10.1161/HYPERTENSIONAHA.114.03549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Eder C, Fischer HG, Hadding U, Heinemann U. Properties of voltage-gated currents of microglia developed using macrophage colony-stimulating factor. Pflügers Arch. 1995;430:526–533. doi: 10.1007/BF00373889. [DOI] [PubMed] [Google Scholar]
- 271.Schilling T, Eder C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 2007;1186:21–28. doi: 10.1016/j.brainres.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 272.Klee R, Heinemann U, Eder C. Changes in proton currents in murine microglia induced by cytoskeletal disruptive agents. Neurosci Lett. 1998;247:191–194. doi: 10.1016/s0304-3940(98)00322-x. [DOI] [PubMed] [Google Scholar]
- 273.Klee R, Heinemann U, Eder C. Voltage-gated proton currents in microglia of distinct morphology and functional state. Neuroscience. 1999;91:1415–1424. doi: 10.1016/s0306-4522(98)00710-6. [DOI] [PubMed] [Google Scholar]
- 274.Morihata H, Kawawaki J, Sakai H, Sawada M, Tsutada T, Kuno M. Temporal fluctuations of voltage-gated proton currents in rat spinal microglia via pH-dependent and -independent mechanisms. Neurosci Res. 2000;38:265–271. doi: 10.1016/s0168-0102(00)00170-x. [DOI] [PubMed] [Google Scholar]
- 275.Schilling T, Gratopp A, DeCoursey TE, Eder C. Voltage-activated proton currents in human lymphocytes. J Physiol. 2002;545:93–105. doi: 10.1113/jphysiol.2002.028878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.El Chemaly A, et al. A voltage-activated proton current in human cardiac fibroblasts. Biochem Biophys Res Commun. 2006;340:512–516. doi: 10.1016/j.bbrc.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 277.Lishko PV, Botchkina IL, Fedorenko A, Kirichok Y. Acid extrusion from human spermatozoa is mediated by flagellar voltage-gated proton channel. Cell. 2010;140:327–337. doi: 10.1016/j.cell.2009.12.053. [DOI] [PubMed] [Google Scholar]
- 278.Sánchez JC, Powell T, Staines HM, Wilkins RJ. Electrophysiological demonstration of voltage- activated H+ channels in bovine articular chondrocytes. Cell Physiol Biochem. 2006;18:85–90. doi: 10.1159/000095171. [DOI] [PubMed] [Google Scholar]
- 279.Bernheim L, Krause RM, Baroffio A, Hamann M, Kaelin A, Bader CR. A voltage-dependent proton current in cultured human skeletal muscle myotubes. J Physiol. 1993;470:313–333. doi: 10.1113/jphysiol.1993.sp019860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kuno M, et al. Temperature dependence of proton permeation through a voltage-gated proton channel. J Gen Physiol. 2009;134:191–205. doi: 10.1085/jgp.200910213. [DOI] [PMC free article] [PubMed] [Google Scholar]